 Xiaoya Zhao1†Yan Huang1†Siai Zhou1Jiaming Ao1Hui Cai1*Katsunori Tanaka2,3,4Yukishige Ito2,5
Xiaoya Zhao1†Yan Huang1†Siai Zhou1Jiaming Ao1Hui Cai1*Katsunori Tanaka2,3,4Yukishige Ito2,5 Akihiro Ishiwata2*
Akihiro Ishiwata2* Feiqing Ding1*
Feiqing Ding1*- 1School of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Shenzhen, China
- 2RIKEN Cluster for Pioneering Research, Saitama, Japan
- 3School of Materials and Chemical Technology, Tokyo Institute of Technology, Tokyo, Japan
- 4Alexander Butlerov Institute of Chemistry, Kazan Federal University, Kazan, Russian Federation
- 5Graduate School of Science, Osaka University, Osaka, Japan
Glycosylation is one of the major forms of protein post-translational modification. N-glycans attached to proteins by covalent bonds play an indispensable role in intercellular interaction and immune function. In human bodies, most of the cell surface glycoproteins and secreted glycopeptides are modified with complex-type N-glycans. Thus, for analytical or medicinal purposes, efficient and universal methods to provide homogeneous complex-type N-glycans have been an urgent need. Despite the extremely complicated structures, tremendous progress in the synthesis of N-glycans has been achieved. On one hand, chemical strategies are shown to be effective to prepare core oligosaccharides of N-glycans by focusing on stereoselective glycosylations such as β-mannosylation and α-sialylation, as well as the methodology of the N-glycan assembly. On the other hand, chemoenzymatic strategies have also become increasingly powerful in recent years. This review attempts to highlight the very recent advancements in chemical and chemoenzymatic strategies for eukaryotic complex-type N-glycans.
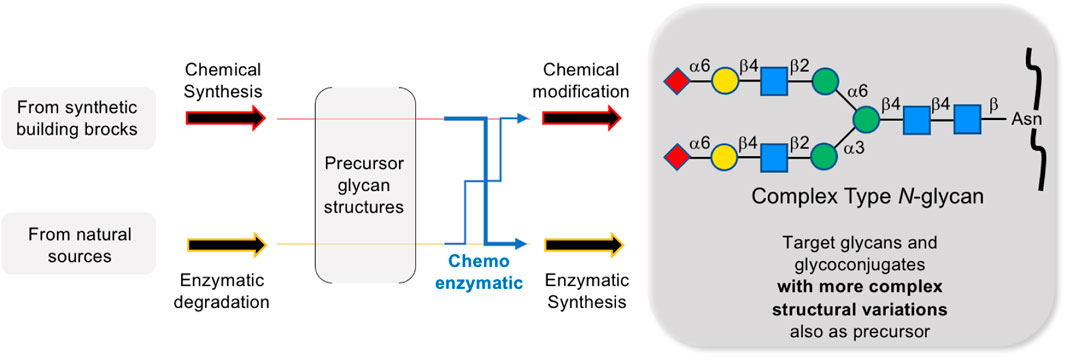
GRAPHICAL ABSTRACT
Introduction
Glycosylation is one of the major post-translational modifications of protein, playing an important role in protein folding, transport, and localization (Varki et al., 2015). It affects protein secretion and stability and participates in cell adhesion and signal transduction. N-glycosylation is the most common protein modification, and the attached glycans called N-glycans are linked to the asparagine residues in the consensus amino acid sequence (Asn-X-Thr/Ser, X ≠ Pro) at the reducing terminal N-acetyl-glucosamine (GlcNAc). According to the differences in non-reducing terminal residues, three subtypes can further be derived, high-mannose type, hybrid type, and complex type. Typical structures are shown in Figure 1A. For medical use, the sialylated complex-type glycans are seen quite often as found in secretory and cell surface glycoproteins. The principal modification can directly improve the in vivo stability and physiological activity of glycoproteins, including extending half-life, participating in intermolecular recognition, and enhancing drug targeting compared to the unmodified ones (Reily et al., 2019; Shirakawa et al., 2021b).
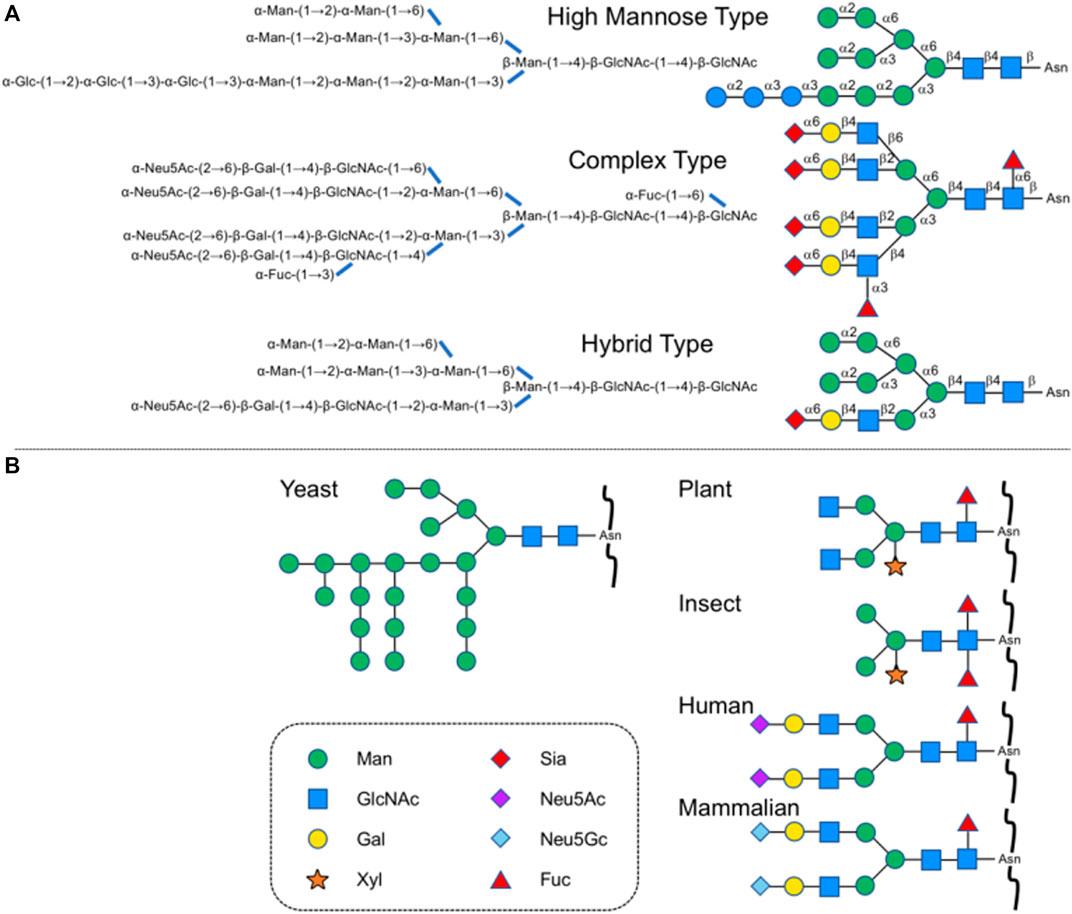
FIGURE 1. (A) Typical N-glycan subtypes in yeast and a mammalian cell. (B) Typical N-glycan structures in other species.
In recent years, a variety of glycoproteins have been approved for the treatment and diagnosis of human diseases, such as erythropoietin (EPO), granulocyte colony-stimulating factor (GCSF), β-interferon, certain cancer biomarkers, and monoclonal antibodies for public health purposes as similar biological products (SBPs) with acceptable levels of quality, safety and efficacy (Buettner et al., 2018). Large-scale production has been realized through glycoprotein expression by yeast, plant, insect, and Chinese hamster ovary (CHO) cells, but structures are quite different from those of glycans on human proteins (Figure 1B).
At present, many N-linked glycoproteins are produced through biosynthesis of recombinant protein in cell expression systems of insects, plants, yeast, and mammals, in which the required sets of glycosyl transfer systems including enzymes for the preassemble of dolichol pyrophosphate-linked tetradecasaccharide, oligosaccharyltransferase, ER and Golgi glycosidases, UDP glucose glucosyltransferase, and Golgi glycosyltransferases as well as many lectins, have already existed. However, the available proteins obtained by this method are mostly asialo form with lacking other modifications such as fucosylation to N-glycan and afford heterogeneous products due to the uncompleted biosynthesis to be a desired N-glycan structures (Figure 2) (Vinikangas et al., 2021). Since the heterogeneity of medicinal N-glycans affects clinical application, the preparation of humanized and homogeneous glycoforms is important for both research and therapeutic purposes. Meanwhile, isolation from natural resources requires rather complicated protocols with rather difficult purifications and is inadequate for researchers to study diverse structures, especially unnatural forms of N-glycans.
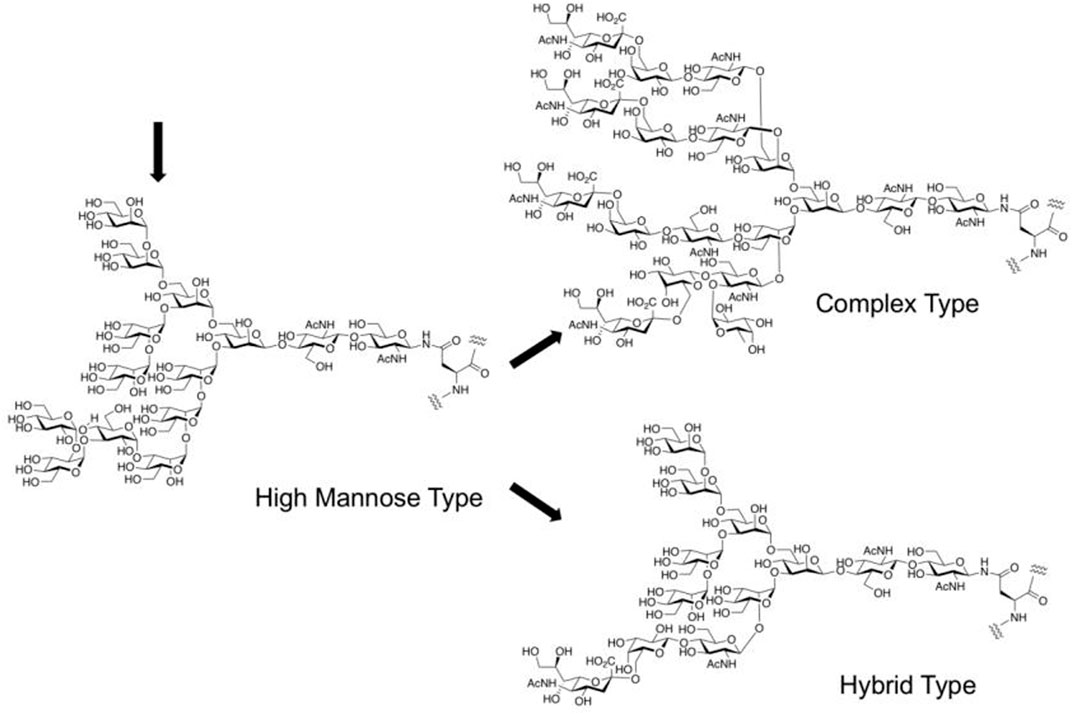
FIGURE 2. Biosynthesis of N-glycan in a cell expression system.
Isolation of N-glycans from natural resources remains one of the most common ways to acquire a large amount of N-glycan production (Figure 3). Asn-linked oligosaccharides containing biantennary complex-type N-glycans from egg yolk immunoglobulin after hydrazinolysis had been reported (Matsuura et al., 1993). The ammonia-catalyzed release method of ovalbumin and soy protein was reported recently without side reactions and degradation of core N-glycan structures (Yang et al., 2020). Sialylglycopeptide (SGP) was also found in hen’s egg yolk and digested by PNGase, one of the most highly specific glycosidases extensively used in enzymatic isolation (Seko et al., 1997). Chemical modification of SGP to various other glycoproteins has been developed by Kajihara’s group, which achieved semi-synthesis of poly-LacNAc containing complex-type biantennary oligosaccharide (Maki et al., 2017) and was recently applied to triantennary erythropoietin (EPO) N-glycan (Maki et al., 2020).
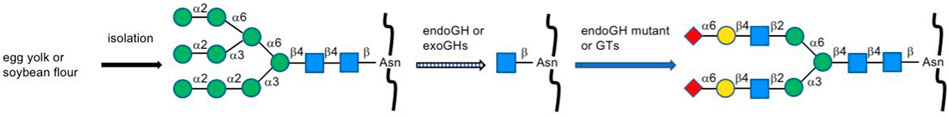
FIGURE 3. Isolation of N-glycans from natural resources.
Chemical and Chemoenzymatic Strategies Toward Eukaryotic Complex-Type N-Glycans
Chemical Assembly of Complex-Type N-Glycans
As shown in the introduction briefly, the extreme complexity of N-glycan structures, especially in terms of the regioselectivity and stereoselectivity control, strongly induces the current situation with no universal methods and strategies for chemical synthesis of N-glycan, although there have been widely studied all over the world. One of the issues for the chemical synthesis of N-glycan is stereoselective glycosylations including β-D-mannosylation in the core structure of all types of N-glycans, and α-sialylation at the non-reducing terminal end of the complex and the hybrid-type N-glycans.
The earliest glycosylation reaction dates back to the late 19th century. An example of simple phenolic glycosides synthesis, glycosylation between glycosyl chloride/bromide and nucleophilic potassium phenoxide, was first reported by Michael in 1879 (Michael and Norton, 1879). A great deal of work has been done since then in the development of glycosylation methods and dozens of novel glycosyl donors have been reported in succession, such as halide donors, semi-aldehyde glycosyl donors, trichloroacetimidate donors (TCAI, Schmidt donors), thioglycoside donors, glycal donors, glycosyl phosphate donors, and o-acetoxy benzoate glycosyl donors (Koenigs and Knorr, 1901; Danishefsky and Bilodeau, 1996; Garcia and Gin, 2000; Schmidt et al., 1986; Fügedi et al., 1987; Plante et al., 2001). With the emergence of these glycosyl donors, a variety of activation systems have also been developed to optimize the yield and selectivity, which are still playing important roles in the synthesis of various glycans. Among the commonly use donors such as halide donors, Schmidt’s donors, and thioglycosides, it is worth mentioning that, sulfides groups of thioglycosides can act as anomeric protecting groups, and sometimes orthogonal activated leaving groups as well, which give a unique value to thioglycosides, especially in the application to a liquid-phase one-pot synthesis of glycans.
The stereoselective construction of β-D-mannosidic bonds has always been a hot topic as one of the most difficult issues in glycochemistry (Tsutsui et al., 2020; Ding et al., 2018; Zhong et al., 2021). Stereo electronic effects such as the anomeric effect and C (2)-OH axial substitution of mannose is both beneficial to the formation of α-configuration products. For the construction of the core structure of N-glycan, a variety of effective methods has been reported. To sum up, there are three main chemical strategies so far for the construction of β-D-mannosidic bonds to synthesize complex-type N-glycans, including the β-glycosylation-inversion strategy (Matsuo et al., 1999), intramolecular aglycon delivery (IAD) (Barresi and Hindsgaul, 1991), and 4,6-O-benzylidene protecting strategy (Crich’s mannosylation) (Crich and Sun, 1996) (Figure 4). Because of the efficiency of the simple intermolecular reaction, and the predominant β-selectivity of Crich’s mannosylation (for a recent example of β-glycosylation-double inversion strategy, see Ishii et al., 2018) (for an example of an improved IAD, see Ishiwata et al., 2008), 4,6-O-benzylidene-protected glycosyl donors were trends to be selected, although the other methods also afford β-D-mannose in the acceptable stereoselectivity. Furthermore, some efforts have also been made to optimize this strategy (For example, the gold(I)-catalyzed glycosylation reaction with glycosyl ortho-alkynyl benzoates as donors, see: Zhu and Yu, 2015).
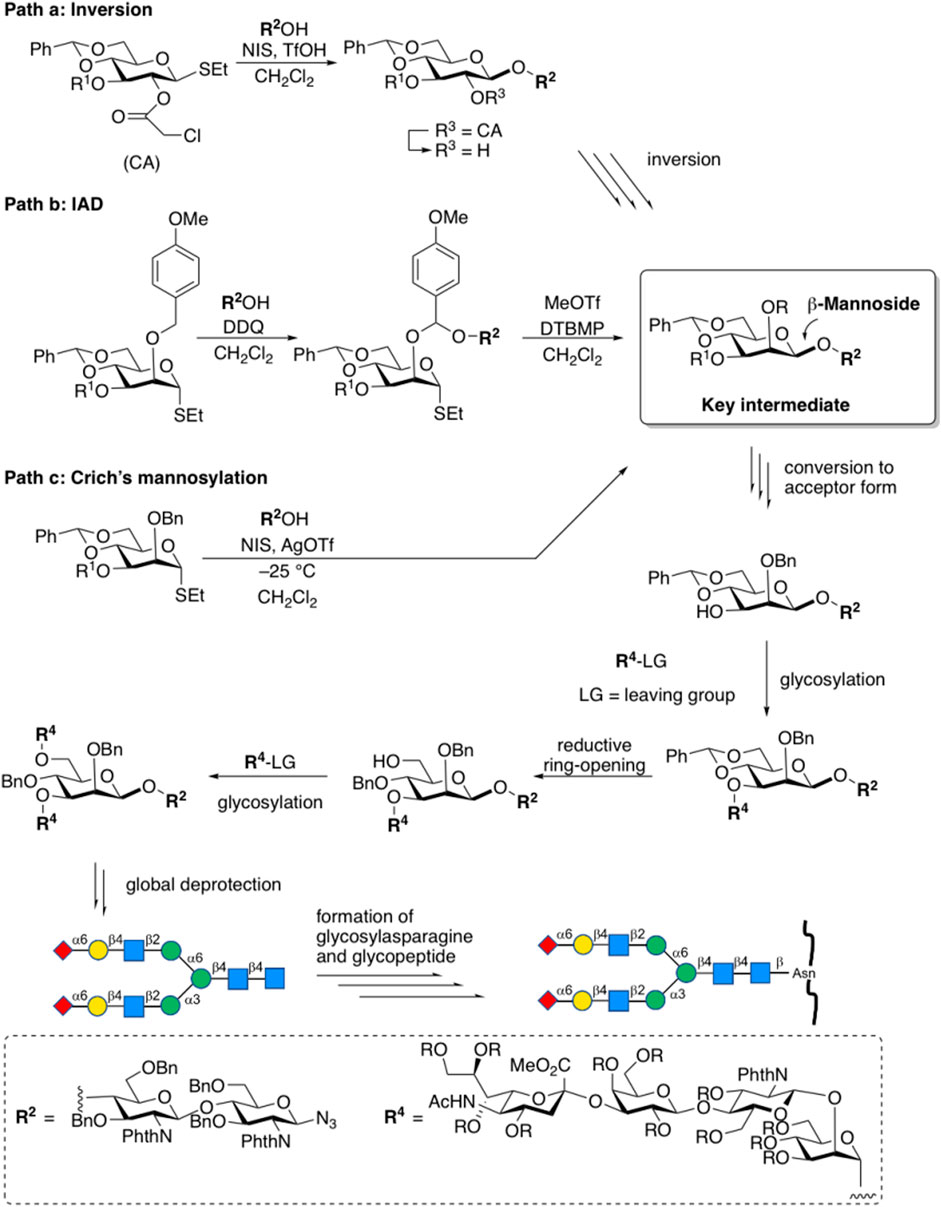
FIGURE 4. Chemical strategies for the construction of β-d-mannosidic bonds in complex-type N-glycans.
The non-reducing terminal modification of N-glycans with sialic acids has a great impact on the physiological and pathological function of glycoproteins. Sialic acids on proteins of mammalian cell surface can be divided into N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc), which link to 3 or 6 positions of D-galactopyranose residue of N-glycan through α-glycosidic linkages (Shirakawa et al., 2021a). The stereoselective formation of sialo side possessing a 3-deoxy-keto-pyranoside structure is extremely difficult since natural sialo side in N-glycan is in equatorial glycoside form, which can’t be controlled by both anomeric effect and neighboring group participation when missing stereo-directing hydroxy group at 3-position of sialic acid. Scientists have applied chemical sialylation by effective stereoselective methodology including the effect of solvent (Kanie et al., 1988), the introduction of auxiliary at C3-position, functionality at C5-N and C1-O positions (for a recent review, see Vibhute et al., 2021), C4-O-,C5-N-oxazoline (Tanaka et al., 2020) and C1-O-,5-N-macrocyclic constrain (Komura et al., 2019) to the synthesis of the non-reducing terminal structure of N-glycan as well as cell surface glycolipid and polysialic acid structures. Ando’s group designed bicyclic sialyl donors tethered by macrocycle formation between C1 and C5 to realize selectively α-sialylation in 2019, through constrained oxocarbenium ions, which could be applied to a wide range of substrates from simple aglycone to glycosyl acceptors, giving high yields and complete stereo control (Komura et al., 2019). In the recent example for N-glycan structure, 1-O-picolinyl-5-azido thiosialosides have been used as α-selective glycosyl donors followed by using a “latent-active” protocol such as MPEP (o-(p-methoxyphenyl ethynyl)phenyl) glycosylation strategies for further elongation of reducing side residues (Chen et al., 2019).
The assembly strategy of glycans, in other words, by using existing glycosylation methods to construct complex-type glycan structures is another important part of glycochemistry. In 2009, Danishefsky et al. adopted a linear strategy to synthesize the I-type antenna sialylated N-glycans and coupled core heptasaccharide with mannose and fucose units (Wang et al., 2009), and Sun et al. recently reported the linear strategy in fewer steps with higher selectivity (Chen et al., 2019) (Figure 5). On the other hand, Wang et al. developed a three-component one-pot synthetic strategy to assembly the core hyperbranched hexasaccharide of N-glycans linked to chloroviruses ATCV-1 using thioglycosides as building blocks, specifically attaching two xylose donors and a rhamnose donor to a trisaccharide, with regio- and stereo-selectivity (Wang Y. S. et al., 2019). Most recently, a convergent strategy was applied to synthesize the representative glycoform in homogeneous human E-cadherin N-linked glycopeptides containing a core tri- or tetra-saccharide and biantennary moiety, which attached the antennas to N-glycan by coupling glycosyl fluoride or thioglycoside donors with core oligosaccharide, and showed stereoselectivity controls (Zeng et al., 2020).
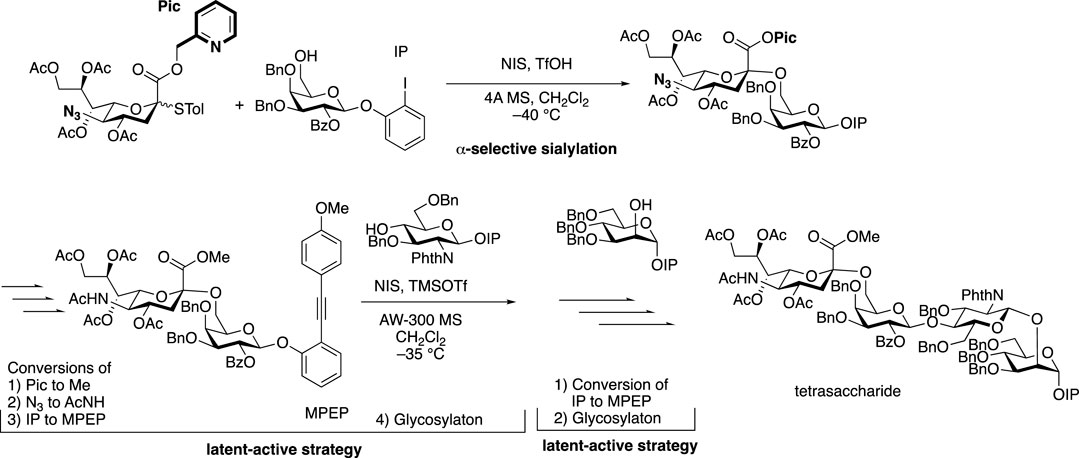
FIGURE 5. α-Sialylation for α-2,6-linkage to galactose residue followed by latent-active strategies toward the synthesis of tetrasaccharide branch (type-I) in complex-type N-glycan.
In 2019, the microfluidic systems combined with convergent synthetic routes had been applied to produce complex-type N-glycans (Manabe, 2019). In this strategy, glycosylated fragments are reproducibly obtained under strict control of reaction conditions by using microflow systems, followed by the assembly of fragments into desired oligosaccharide backbone structures through convergent synthetic routes. Overall, the synthesis of N-glycans could be accomplished in relatively short steps. The key to this strategy is how to achieve a satisfactory level of efficiency in glycosylation among less reactive large fragments. It shows that amide groups (NHAc) form intermolecular hydrogen bonds to reduce the reactivity. The glycosylation reactivity could be markedly improved by protecting them as imides (NAc2) (Demchenko and Boons, 1998). A relatively high yield of the desired product can be achieved by using ether solvent for coordination stabilization of the oxocarbenium ion intermediate even in poorly reactive glycosylation. In addition, this strategy has successfully improved the stereoselectivity by carefully altering protection strategies. The strategy was applied to the synthesis of H antigen trisaccharide (Manabe, 2020) as well as 3- and 6-α-sialylated tetraantennary N-glycans for H1N1 neuraminidase recognition (Manabe, 2021) very recently (Figure 6).

FIGURE 6. Microfluidic assembly of complex-type N-glycans.
The automated solid-phase strategy first reported in 2001 (Sabbavarapu Seeberger et al., 2001) has been applied to the N-glycan core structures (Le Mai Hoanget al., 2019) (Figure 7). As with the automated synthesis of peptides or nucleic acids, glycosyl acceptors are linked to resins via a connecting arm, which is called a linker. Each desired linkage is completed by adding a glycosyl donor, and then the temporary protecting group has to be removed. The procedure is repeated and the solid support serves to keep the growing chain in a form that can be removed from the reaction mixture by filtration. The linker is disconnected upon completion of the synthesis. Finally, the fully assembled product is cut from the resin after purifying by simple filtration. Since donors in each step have overdosed, acceptors supported on resins could be completely glycosylated as far as possible, which leads to high overall efficiency in theory. Using this automated glycan assembly (AGA) strategy, the automatic synthesis has been applied to the chemical synthesis of core pentasaccharide of N-glycans, arabinomannan polysaccharides (Pardo-Vargas et al., 2019), chitooligosaccharides (Tyrikos-Ergas et al., 2021), galactofuranoses (Sabbavarapu and Seeberger, 2021) and starch and glycogen polysaccharides (Zhu et al., 2021).
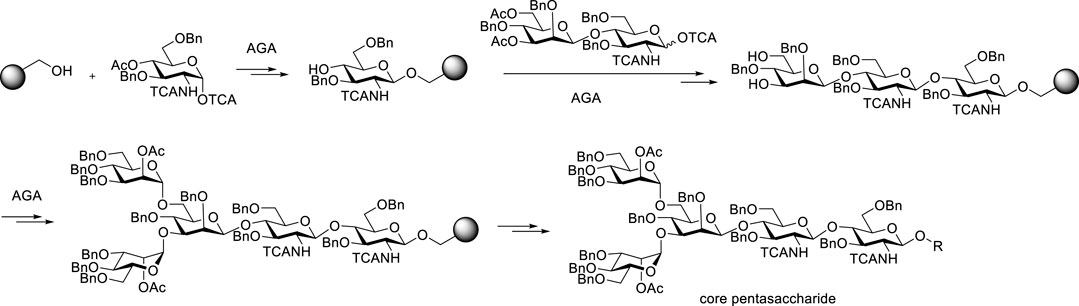
FIGURE 7. Automated glycan assembly of core pentasaccharide present in complex-type N-glycans.
Chemoenzymatic Strategies
Chemists have begun exploring the combination of biocatalytic and modern chemical methods to provide complex carbohydrates in a more efficient way since the 1990s. For example, Unverzagt reported the synthesis of a sialylated biantennary N-glycan in 2002 (Unverzagtet al., 2002). Such a chemoenzymatic approach not only shows the flexibility of chemical methods but also the high stereoselectivity and regioselectivity of enzymatic reactions. In general, there are two types of chemoenzymatic methods based on the sequence of reaction events. One starts from the chemical synthesis to generate substrate analogs for enzymatic extension, and the other is to apply enzymatic synthesis first before chemical diversification takes place. In the early stage, many studies focused on the chemoenzymatic synthesis of simple symmetrical N-glycans (recent review, see Chao et al., 2020; Li and Wang, 2018).
Top-Down Strategy From Synthetic Large Glycan (One Large Precursor) Using Glycosidases
In 2013, Ito’s group first reported a “top-down” chemoenzymatic strategy to construct a high-mannose type N-glycan library from a designed precursor, a tetradecasaccharide modified with Glc, Gal, and GlcNAc terminal sugar residues, followed by trimming this precursor into a variety of high-mannose type glycans by glycosidases. For instance, D-glucose moiety was removed by glucosidase Ⅱ and D-galactose by β-galactosidase, while N-acetyl-D-glucosamine was removed by β-HexNAc’ase (Koizumi et al., 2013) (Figure 8). They then developed the second generation precursor in 2016, substituted acetal protecting group directly on Mannose residue instead of Gal protection in that branch, due to the difficulty of galactosidase digestion (Fujikawa et al., 2016). So far, this strategy has been employed in the synthesis of high-mannose type N-glycans. Wang et al. also applied a top-down strategy starting from Soybean flour Man9GlcNAc2Asn and Hen egg yolk sialylglycopeptide (SGP) to high-mannose and complex-type N-glycans (Wang Y. S. et al., 2019).
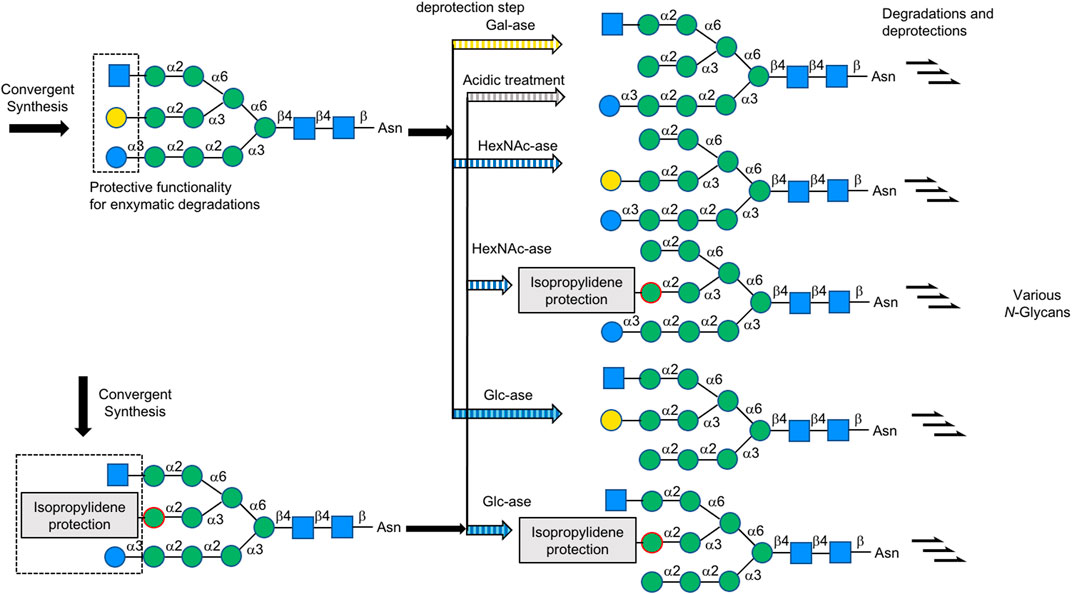
FIGURE 8. Top to down strategy for complex-type N-glycans.
Elongation and Branching Strategy From Synthetic Small Glycan Using Glycosyltransferases
A lot of explorations have been done by Boons on the chemoenzymatic synthesis of complex-type N-glycans. In his previous explorations, Boons developed a common glycan precursor first by using modern chemical methods, followed by the enzymatic extension of elongation and branching of N-glycans. For example, in 2013, a general strategy for chemoenzymatic synthesis of asymmetrically branched N-glycans was reported, and 14 tri-antennary N-glycans were obtained. In this strategy, the precursor decasaccharide was prepared from the core pentasaccharide that at potential branching positions was modified by orthogonal protecting groups and then modified branch-specifically by glycosyltransferases (Wang et al., 2013). Recently, Boons’ group synthesized N-glycan heptasaccharide precursors of the Parasite Schistosoma mansoni by chemical glycosylation method, achieved xylose modification by β-1,2-xylosyltransferase XYLT, and transferred to multi-antennary glycans enzymatically via β-Man-ase, B4GalT1 and FUT5 (Srivastava et al., 2021) (Figure 9). Weiss et al. also applied the synthesis of the rare biantennary N-glycans with Gal-β1,4-linked bisecting GlcNAc motif found in IgG by using a chemical modular approach to assemble the core bisected N-glycans followed by simple enzymatic antenna modifications with β1,4-galactosyltransferase (Weiss et al., 2020). A previous application of this strategy was also shown in order to obtain various N-glycan structures including Neu5Ac/Gc and core-fucoses. It features the initial convergent chemical assembly of core oligosaccharide precursors and following enzymatic extension catalyzed by an alternative combination of four robust glycosyltransferases, PmST1m, Hpα1,3FT, B4GALT1, and Pd2,6ST, to yield a library of complex-type N-glycans. (Li et al., 2015).
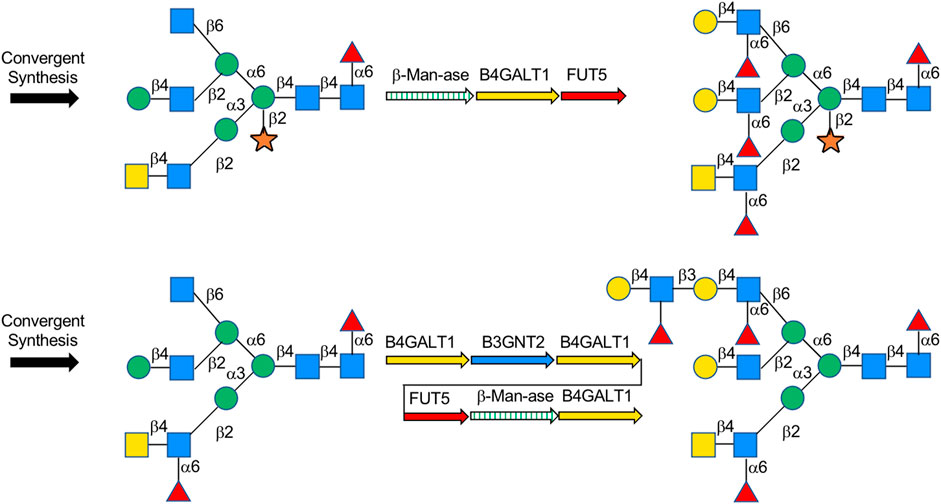
FIGURE 9. Chemoenzymatic synthesis of complex-type N-glycans.
Shivatare et al. prepared N-glycans of hybrid, high-mannose, and complex types for microarray and binding analysis of HIV antibodies first by chemical glycosylation to obtain building blocks, followed by a modular chemoenzymatic strategy to add antennas using various glycosyltransferases including β-1,4-galactosyltransferases, α-2,3/2,6-sialyltransferases and α-1,3/1,2-fucosyltransferases (Shivatare et al., 2016) (Figure 10).
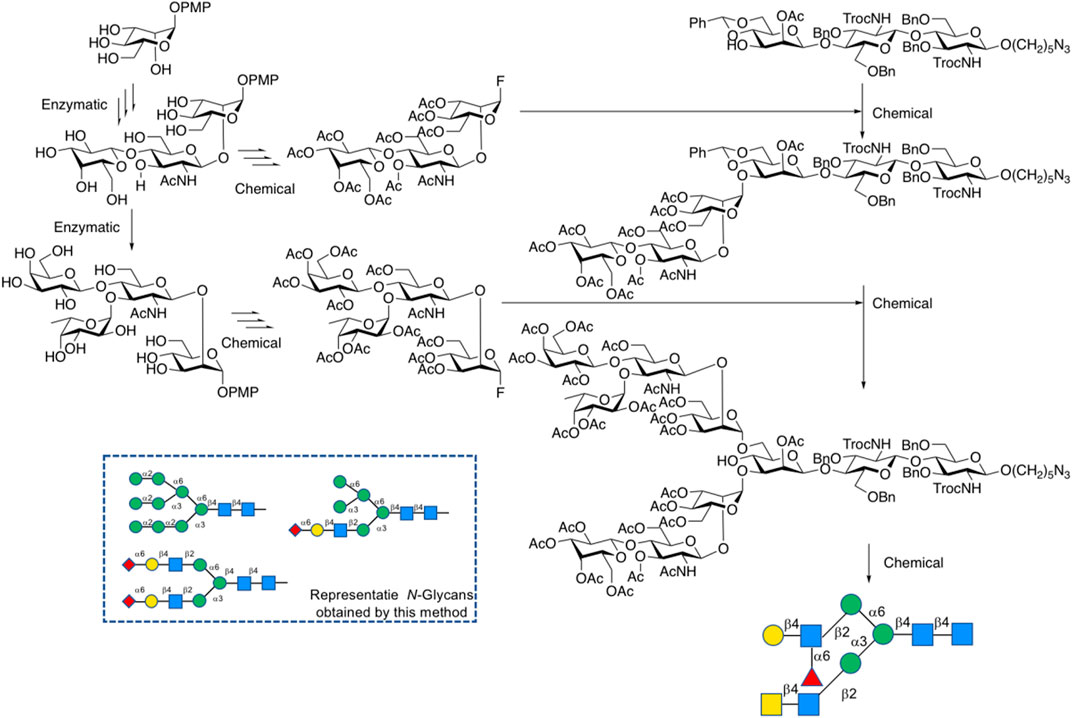
FIGURE 10. Chemo-enzymatic synthesis of modules.
Pawar et al. constructed N-glycan libraries by synthesizing oligosaccharide building blocks chemically in Asymmetric Glycan as Common-substrate Strategy (AGCS), followed by introducing fucose and sialic motifs to core structures via glycosyltransferases FUT, α-2,3-STGal, or α-2,6-STGal (Pawar et al., 2020) (Figure 11).
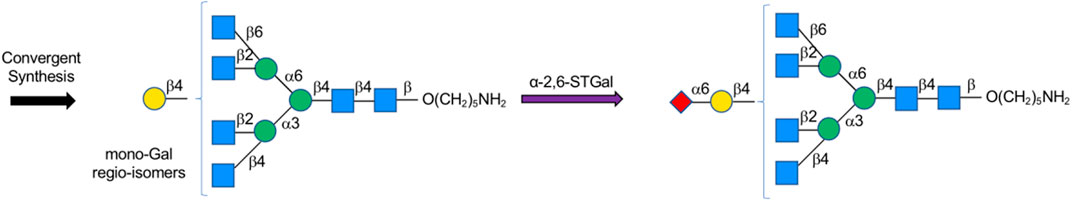
FIGURE 11. Asymmetric glycan common-substrate (AGCS).
Cummings et al. enzymatically generated a multi-antennary oligosaccharide library containing 32 complex-type N-glycans from a natural source, in which recombinant glycosyltransferases (B4GALT1, ST3GAL4, ST6GAL1, MGAT, and FUT8) were used to expand synthesis from a precursor. Desired sialylated, fucosylated, and mannosylated terminal modifications and branching were realized on a biantennary GlcNAc-terminated structure (Gao et al., 2019) (Figure 12).
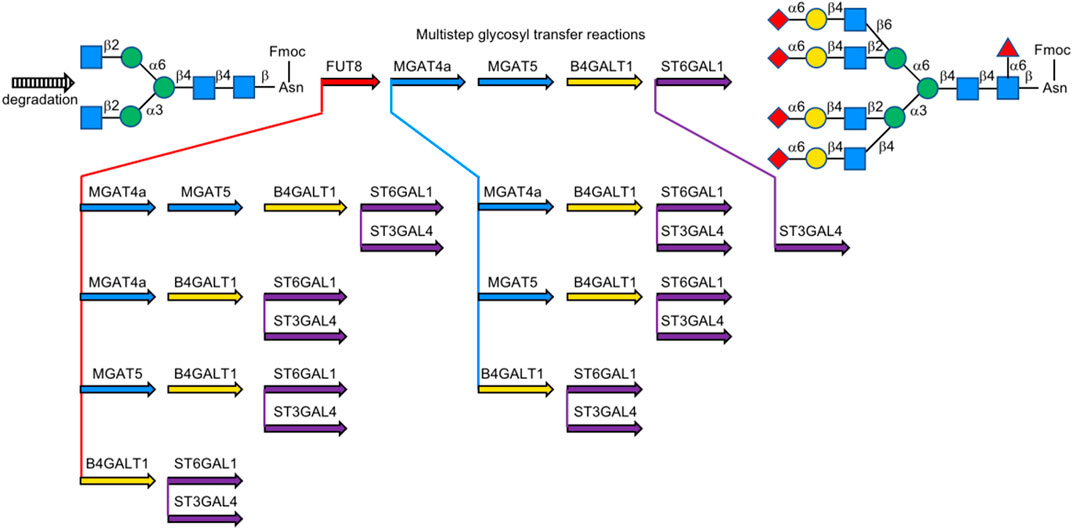
FIGURE 12. Generation of complex-type naturally occurring N-glycans library.
Strategy Using Endo-Type Enzymes and Mutants
Endo-β-N-acetylglucosaminidases, known as endo-type enzymes of wild types and mutants, are often used in the synthesis of N-linked glycopeptides, which perform activities of deglycosylation by breaking between GlcNAcs in N-linked chitobiosemoiety, and transglycosylation to GlcNAc-linked peptides. These enzymes are found in a wide range of species, such as Endo-A from Arthrobacter, Endo-M from Mucor hiemalis, and Endo-S from Streptococcus pyogenes. Since endo-α-N-acetylglucosaminidases are only specific to O-glycans, endo-β-N-acetylglucosaminidases (ENGases) are considered the main endo-type to prepare homogeneous N-linked glycoprotein. For the transfer of oligosaccharides back to the cleaved structure, the mutants mostly use glycan oxazolines as donor substrates and have stereoselective control on substrates. Recently, Wang et al. applied this procedure to synthesize HIV-1 glycopeptides through the preparation of GlcNAc-peptide precursor by using solid-phase peptide synthesis (SPPS) and followed by the connection of N-glycan to the precursor using the N-glycan oxazoline donor via endo-glycosidase (Endo-A or Endo-M) mutant catalysis (Zong et al., 2020) (Figure 13). They also reported recombinant Endo-S and mutants that proceeded deglycosylation to convert structurally heterogeneous sugar chains to GlcNAc-IgG acceptor and remodeling to homogeneously N-glycosylated Fc fragments on rituximab (Wang L.-X. et al., 2019) (Figure 14). Dong et al. introduced N-linked oligosaccharides including GlcNAc or GlcNAc2 by chemical glycosylation with asparagine residue on peptide precursor as well as complex-type sialyl N-glycan by transfer using Endo-M mutant onto IL-17A peptide (Li H. et al., 2021) (Figure 15). It is notable as another previous example of this strategy that the chemo-enzymatic synthesis of N-glycopeptide structural motif of haptoglobin glycopeptides had been achieved by a total chemical synthesis of the highly branched oligosaccharide oxazoline donors through fragment couplings and an alternative wild-type Endo A-catalyzed transglycosylation of the oxazolines in the presence of bisecting GlcNAc to GlcNAc-introduced haptoglobin glycopeptide fragment (Yang et al., 2018). Interestingly, wild-type Endo A could be also applicable to the preparation of an N-linked glycoprotein that contains M6P-terminated glycans catalysis for the treatment of lysosomal storage disorders (LSDs) (Priyanka, et al., 2016).
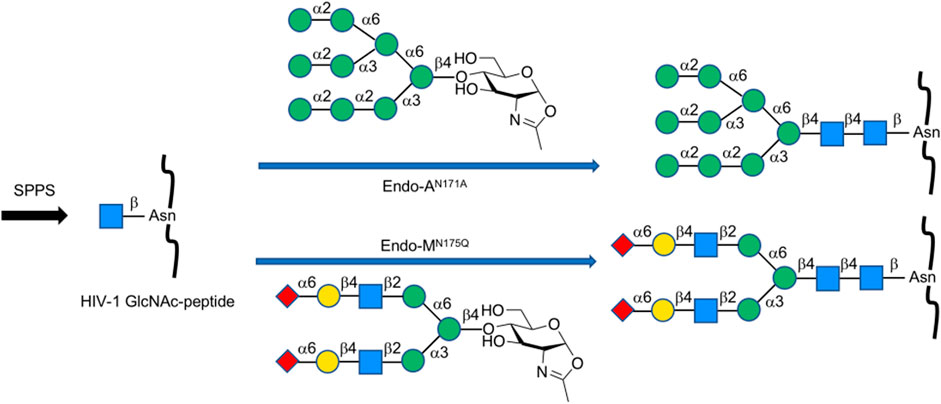
FIGURE 13. Chemoenzymatic synthesis of homogeneous N-glycan-linked HIV-1 glycopeptides.
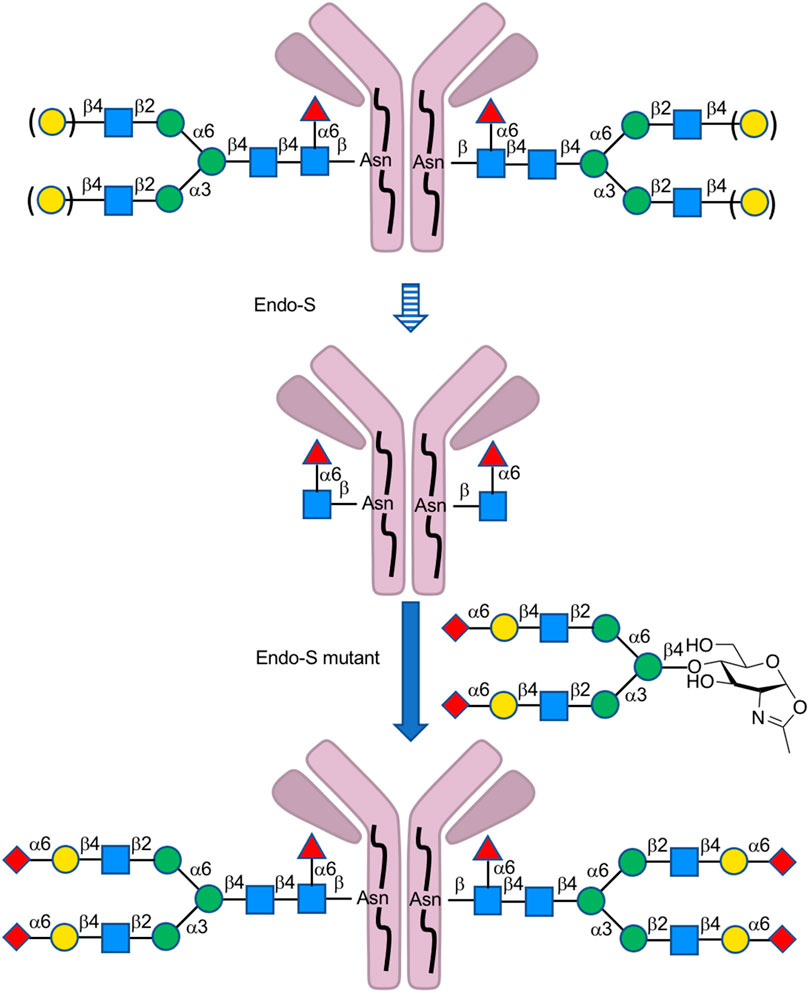
FIGURE 14. Preparation of homogeneous N-glycan-linked antibody-mediated by Endo-type enzymes.
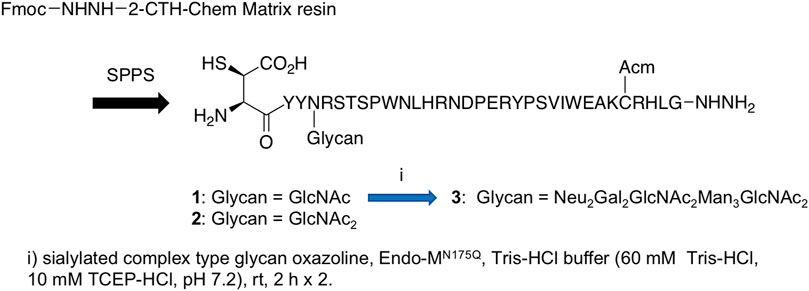
FIGURE 15. Preparation of IL-17A glycopeptides.
OST Strategy
Oligosaccharyltransferases (OST) usually exist in prokaryotes (bacteria and archaea) and eukaryotes in the form of complex, playing a key role in N-glycosylation, with the catalysis of transferring oligosaccharide chains onto protein substrates. Single subunit OST was found in the bacterial pathway of glycoprotein biosynthesis, such as PglB from Campylobacter jejuni (Wacker et al., 2002; Deshpande et al., 2008; Lizak et al., 2011; Jinnelov et al., 2017; Poljak et al., 2017; Mohanty et al., 2020; Schjoldager et al., 2020) and TbSTT3A from Trypanosoma brucei for N-glycan (Wilson et al., 2021). The archaeal pathway also includes the single subunit OST such as AglB (Meyer and Albers, 2014; recent review, see Eichler, 2020), whose X-ray structure from Archaeoglobusfulgidus has been reported very recently (Taguchi et al., 2021). The most studied PglB can transfer the highly conserved heptasaccharide composed of GalNAc5Glc1Bac1 (Bac: bacillosamine, 2,4-acetamide-2.4.6-trideoxy-D-glucose) to proteins in the periplasm of C. jejuni. Chemical and enzymatic synthesis of N-glycans and glycopeptide synthesis using OST have been already reported (Glover et al., 2005; Amin et al., 2007; Lee et al., 2009; Lukose et al., 2015; Ishiwata et al., 2015) (Figure 16).
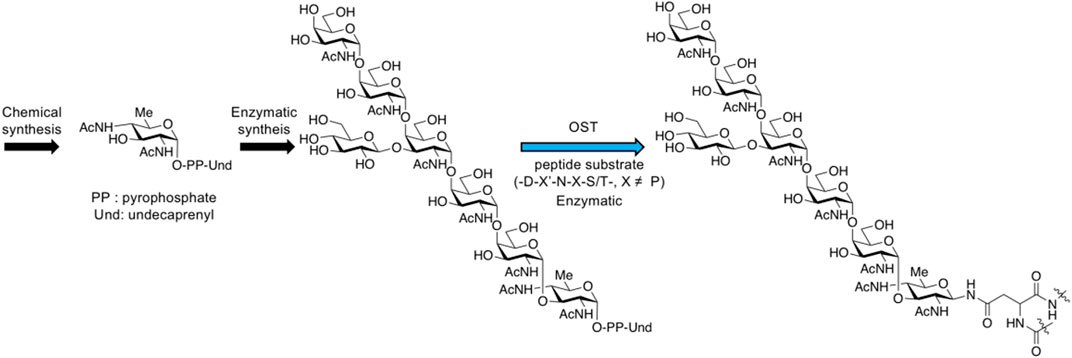
FIGURE 16. N-glycan synthesis through bacterial biosynthesis including oligosaccharyltransferase reaction.
In humans, mutations of gene encoding OST complex may cause some diseases, like congenital disorders of glycosylation (CDGs) (Ganetzky et al., 2017), making OST an important enzyme for human glycosyl transfer to maintain normal physiological functions, but there are no examples for using mammalian OST complex to produce the good quantity of N-glycoprotein effectively (Kay et al., 2019).
Enzymatic and Chemoenzymatic Strategy From Natural Resources (such as SGP) Using Glycosidases With/Without Glycosyltransferases
Chemical synthesis of precursors for enzymatic modification is still not efficient enough because it often requires many steps including cumbersome protection/deprotection, resulting in product loss, and hindering the preparation of various types of asymmetric N-glycans. Researchers have discovered starting materials from natural sources, such as sialylglycopeptide (SGP), and have greatly optimized the isolation method for gram quantities of homogeneous SGP from commercially available egg yolk powder. Hamilton et al. generated a library containing complex-type asymmetric and multi-antennary N-glycans for microarray analysis, by deglycosylation of biosynthetic precursor glycoproteins and lipid-linked oligosaccharides from natural sources (yeast- and bacteria-derived precursors) and structurally remodeling by GnTⅠ, GnTⅡ, and GnTⅣ as well as the following early mammalian glycosylation pathway (Hamilton et al., 2017). Wang et al. reported chemoenzymatic strategy on natural N-glycans from soybean flour and SGP from egg yolks and trimmed enzymatically by α-mannosidases to generate high-mannose glycan library also for microarray analysis (Toonstra et al., 2018).
In 2019, Boons et al. described a biomimetic strategy called Stop and Go strategy, in which SGP can be converted through degradations and glycosyl transfer reactions including the introduction of nonreactive chemically modified residue and its chemical modifications to activate form from unreactive one and further enzymatic steps into multi-antennary N-glycans that at each arm can be uniquely extended by glycosyltransferases to obtain highly complex asymmetrically branched N-glycans. By using this strategy, Boons has successfully accomplished the chemoenzymatic synthesis of several complex-type N-glycans found in human pathological tissues, like bi-antennary N-glycans observed on ovarian cancer cell lines as well as tetra-antennary N-glycans observed on human cytolytic T lymphocytes (Liu et al., 2019) (Figure 17).
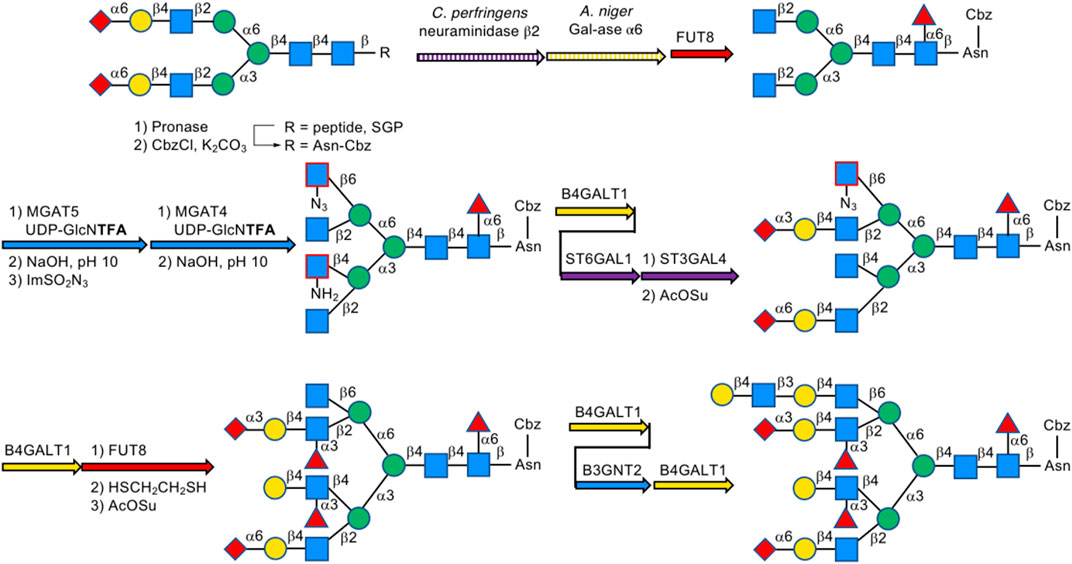
FIGURE 17. Stop and Go strategy for multi-antennary N-glycans; For MGAT4/5&B3GNT2, B4GALT1, FUT8, and ST3/6GAL1/4, UDP-GlcNAc, UDP-Gal, GDP-Fuc, and CMP-Neu5Ac were used as the donor, respectively, unless otherwise noted.
In 2021, Wang et al. prepared the complex-type asialo-nonasaccharide of SARS-Cov-2 Spike Receptor-Binding Domain (RBD) using Boon’s reported enzymatically trimming approach on SGP in 2017, after enzymolysis of neuraminidase and pronase to remove Neu5Ac and peptide, followed by transferring into glycosyl amine under Kochetkov amination condition and coupling with an acid group of RBD peptide fragment (Ye et al., 2021).
Concluding Remarks
Most of the human cell surface and secreted proteins are modified by complex-type N-glycans, which not only affect the structure and function of proteins but also participate in the signal transduction of tumor cells. In the field of biomedical applications, complex-type N-glycans and their glycoconjugates show broad prospects, such as anti-tumor kits and vaccines. However, unlike other important biopolymers such as DNA/RNA and proteins, the biosynthesis of glycans is neither driven by templates nor genetically encoded. This biosynthetic nature of N-glycan as a secondary metabolite leads directly to the micro-heterogeneity of the naturally obtained glycans, which means that it is difficult to obtain enough homogeneous complex-type N-glycans by means of separation and isolation. In order to learn more about the properties of N-linked glycopeptides and use them in the production of peptide vaccines, it is necessary to develop a universal and efficient method to prepare homogeneous N-glycans in addition to further optimization of purification methods from natural sources. Over the past few decades, researchers have made lots of efforts for the chemical and chemoenzymatic strategies for well-defined N-glycans. Though great progress has been made, there are still problems and difficulties to be solved urgently, which requires researchers to explore continuously in this field as the recent examples are shown here. Further improving efficiency and simplifying procedures are quite significant.
Author Contributions
XZ, YH, AI, and FD wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported partly by the Fundamental Research Funds for the Central Universities (No. 31610011 to FD), the province natural science fund of Guangdong (No. 42030015 to FD; No. 2022A1515011109 to HC), the Sun Yat-sen University Startup fund (No.18841224 to FD) for financial support, and the grant-in-Aid for Specially Promoted Research (No. 16H06290 to YI and AI) and Scientific Research (no. 18K05345 and 19H00929 to AI) from the Japan Society for the Promotion of Science.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amin, M. N., Ishiwata, A., and Ito, Y. (2007). Synthesis of N-Linked Glycan Derived from Gram-Negative Bacterium, Campylobacter Jejuni. Tetrahedron 63, 8181–8198. doi:10.1016/j.tet.2007.05.126
Barresi, F., and Hindsgaul, O. (1991). Synthesis of .beta.-mannopyranosides by Intramolecular Aglycon Delivery. J. Am. Chem. Soc. 113, 9376–9377. doi:10.1021/ja00024a057
Buettner, M. J., Shah, S. R., Saeui, C. T., Ariss, R., and Yarema, K. J. (2018). Improving Immunotherapy through Glycodesign. Front. Immunol. 9, 2485. doi:10.3389/fimmu.2018.02485
Chao, Q., Ding, Y., Chen, Z.-H., Xiang, M.-H., Wang, N., and Gao, X.-D. (2020). Recent Progress in Chemo-Enzymatic Methods for the Synthesis of N-Glycans. Front. Chem. 8, 513. doi:10.3389/fchem.2020.00513
Chen, J., Hansen, T., Zhang, Q. J., Liu, D. Y., Sun, Y., Yan, H., et al. (2019). 1‐Picolinyl‐5‐azido Thiosialosides: Versatile Donors for the Stereoselective Construction of Sialyl Linkages. Angew. Chem. Int. Ed. 58, 17000–17008. doi:10.1002/anie.201909177
Crich, D., and Sun, S. (1996). Formation of β-Mannopyranosides of Primary Alcohols Using the Sulfoxide Method. J. Org. Chem. 61, 4506–4507. doi:10.1021/jo9606517
Danishefsky, S. J., and Bilodeau, M. T. (1996). Glycals in Organic Synthesis: The Evolution of Comprehensive Strategies for the Assembly of Oligosaccharides and Glycoconjugates of Biological Consequence. Angew. Chem. Int. Ed. Engl. 35, 1380–1419. doi:10.1002/anie.199613801
Demchenko, A. V., and Boons, G.-J. (1998). A Novel and Versatile Glycosyl Donor for the Preparation of Glycosides of N-Acetylneuraminic Acid. Tetrahedron Lett. 39, 3065–3068. doi:10.1016/s0040-4039(98)00359-1
Deshpande, N., Wilkins, M. R., Packer, N., and Nevalainen, H. (2008). Protein Glycosylation Pathways in Filamentous Fungi. Glycobiology 18, 626–637. doi:10.1271/bbb.7008010.1093/glycob/cwn044
Ding, F., Ishiwata, A., and Ito, Y. (2018). Stereodivergent Mannosylation Using 2-O-(ortho-Tosylamido)benzyl Group. Org. Lett. 20, 4833–4837. doi:10.1021/acs.orglett.8b01979
Eichler, J. (2020). N‐glycosylation in Archaea-New Roles for an Ancient Posttranslational Modification. Mol. Microbiol. 114, 735–741. doi:10.1111/mmi.14569
Fugedi, P., Garegg, P. J., Lunn, H., and Norberg, T. (1987). Thioglycosides as Glycosylating Agents in Oligosaccharide Synthesis. Glycoconjugate J. 4, 97–108. doi:10.1007/BF01049447
Fujikawa, K., Seko, A., Takeda, Y., and Ito, Y. (2016). Approaches toward High-mannose-type Glycan Libraries. Chem. Rec. 16, 35–46. doi:10.1002/tcr.201500222
Ganetzky, R., Reynoso, F. J., and He, M. (2017). Congenital Disorders of Glycosylation. Biomarkers in Inborn Errors of Metabolism. Chapter 15, 343–360. doi:10.1016/B978-0-12-802896-4.00014-6, ,
Gao, C., Hanes, M. S., Byrd-Leotis, L. A., Wei, M., Jia, N., Kardish, R. J., et al. (2019). Unique Binding Specificities of Proteins toward Isomeric Asparagine-Linked Glycans. Cel Chem. Biol. 26, 535–547. doi:10.1016/j.chembiol.2019.01.002
Garcia, B. A., and Gin, D. Y. (2000). Dehydrative Glycosylation with Activated Diphenyl Sulfonium Reagents. Scope, Mode of C(1)-Hemiacetal Activation, and Detection of Reactive Glycosyl Intermediates. J. Am. Chem. Soc. 122, 4269–4279. doi:10.1021/ja993595a
Glover, K. J., Weerapana, E., and Imperiali, B. (2005). In Vitro assembly of the Undecaprenylpyrophosphate-Linked Heptasaccharide for Prokaryotic N-Linked Glycosylation. Proc. Natl. Acad. Sci. U.S.A. 102, 14255–14259. doi:10.1073/pnas.0507311102
Hamilton, B. S., Wilson, J. D., Shumakovich, M. A., Fisher, A. C., Brooks, J. C., Pontes, A., et al. (2017). A Library of Chemically Defined Human N-Glycans Synthesized from Microbial Oligosaccharide Precursors. Sci. Rep. 7 (1), 15907. doi:10.1038/s41598-017-15891-8
Ishii, N., Ogiwara, K., Sano, K., Kumada, J., Yamamoto, K., Matsuzaki, Y., et al. (2018). Specificity of Donor Structures for Endo -β-N -Acetylglucosaminidase-Catalyzed Transglycosylation Reactions. ChemBioChem 19, 136–141. doi:10.1002/cbic.201700506
Ishiwata, A., Munemura, Y., and Ito, Y. (2008). NAP Ether Mediated Intramolecular Aglycon Delivery: A Unified Strategy for 1,2-Cis-Glycosylation. Eur. J. Org. Chem. 2008, 4250–4263. doi:10.1002/ejoc.200800249
Ishiwata, A., Taguchi, Y., Lee, Y. J., Watanabe, T., KohdaIto, D. Y., and Ito, Y. (2015). N-glycosylation with Synthetic Undecaprenyl Pyrophosphate-Linked Oligosaccharide to Oligopeptides by PglB Oligosaccharyltransferase fromCampylobacter Jejuni. ChemBioChem 16, 731–737. doi:10.1002/cbic.201402658
Jinnelov, A., Ali, L., Tinti, M., Güther, M. L. S., and Ferguson, M. A. J. (2017). Single-subunit Oligosaccharyltransferases of Trypanosoma Brucei Display Different and Predictable Peptide Acceptor Specificities. J. Biol. Chem. 292, 20328–20341. doi:10.1074/jbc.M117.810945
Kanie, O., Kiso, M., and Hasegawa, A. (1988). Glycosylation Using Methylthioglycosides ofN-Acetylneuraminic Acid and Dimethyl(Methylthio)Sulfonium Triflate. J. Carbohydr. Chem. 7, 501–506. doi:10.1080/07328308808058938
Kay, E., Cuccui, J., and Wren, B. W. (2019). Recent Advances in the Production of Recombinant Glycoconjugate Vaccines. npj Vaccin. 4, 16. doi:10.1038/s41541-019-0110-z
Koenigs, W., and Knorr, E. (1901). Ueber einige Derivate des Traubenzuckers und der Galactose. Ber. Dtsch. Chem. Ges. 34, 957–981. doi:10.1002/cber.190103401162
Koizumi, A., Matsuo, I., Takatani, M., Seko, A., Hachisu, M., Takeda, Y., et al. (2013). Top-Down Chemoenzymatic Approach to a High-mannose-type Glycan Library: Synthesis of a Common Precursor and its Enzymatic Trimming. Angew. Chem. Int. Ed. 52, 7426–7431. doi:10.1002/anie.201301613
Komura, N., Kato, K., Udagawa, T., Asano, S., Tanaka, H.-N., Imamura, A., et al. (2019). Constrained Sialic Acid Donors Enable Selective Synthesis of α-glycosides. Science 364, 677–680. doi:10.1126/science.aaw4866
Le Mai Hoang, K., Pardo-Vargas, A., Zhu, Y., Yu, Y., Loria, M., Delbianco, M., et al. (2019). Traceless Photolabile Linker Expedites the Chemical Synthesis of Complex Oligosaccharides by Automated Glycan Assembly. J. Am. Chem. Soc. 141, 9079–9086. doi:10.1021/jacs.9b03769
Lee, Y. J., Ishiwata, A., and Ito, Y. (2009). Synthesis of Undecaprenyl Pyrophosphate-Linked Glycans as Donor Substrates for Bacterial Protein N-Glycosylation. Tetrahedron 65, 6310–6319. doi:10.1016/j.tet.2009.06.032
Li, C., and Wang, L.-X. (2018). Chemoenzymatic Methods for the Synthesis of Glycoproteins. Chem. Rev. 118 (17), 8359–8413. doi:10.1021/acs.chemrev.8b00238
Li, H., Zhang, J., An, C., and Dong, S. (2021). Probing N-Glycan Functions in Human Interleukin-17A Based on Chemically Synthesized Homogeneous Glycoforms. J. Am. Chem. Soc. 143, 2846–2856. doi:10.1021/jacs.0c12448
Li, L., Liu, Y., Ma, C., Qu, J., Calderon, A. D., Wu, B., et al. (2015). Efficient Chemoenzymatic Synthesis of an N-Glycan Isomer Library. Chem. Sci. 6 (10), 5652–5661. doi:10.1039/C5SC02025E
Liu, L., Prudden, A. R., Capicciotti, C. J., Bosman, G. P., Yang, J.-Y., Chapla, D. G., et al. (2019). Streamlining the Chemoenzymatic Synthesis of Complex N-Glycans by a Stop and Go Strategy. Nat. Chem 11, 161–169. doi:10.1038/s41557-018-0188-3
Lizak, C., Gerber, S., Numao, S., Aebi, M., and Locher, K. P. (2011). X-ray Structure of a Bacterial Oligosaccharyltransferase. Nature 474, 350–355. doi:10.1038/nature10151
Lukose, V., Whitworth, G., Guan, Z., and Imperiali, B. (2015). Chemoenzymatic Assembly of Bacterial Glycoconjugates for Site-specific Orthogonal Labeling. J. Am. Chem. Soc. 137, 12446–12449. doi:10.1021/jacs.5b07146
Maki, Y., Mima, T., Okamoto, R., Izumi, M., and Kajihara, Y. (2017). Semisynthesis of Complex-type Biantennary Oligosaccharides Containing Lactosamine Repeating Units from a Biantennary Oligosaccharide Isolated from a Natural Source. J. Org. Chem. 83, 443–451. doi:10.1021/acs.joc.7b02485
Maki, Y., Okamoto, R., Izumi, M., and Kajihara, Y. (2020). Chemical Synthesis of an Erythropoietin Glycoform Having a Triantennary N-Glycan: Significant Change of Biological Activity of Glycoprotein by Addition of a Small Molecular Weight Trisaccharide. J. Am. Chem. Soc. 142, 20671–20679. doi:10.1021/jacs.0c08719
Manabe, Y. (2019). Efficient Syntheses of Complex-type N-Glycans Using Microflow Systems and Convergent Synthetic Routes. Tigg 31, E99–E108. doi:10.4052/tigg.1768.1E
Matsuo, I., Isomura, M., and Ajisaka, K. (1999). Synthesis of an Asparagine-Linked Core Pentasaccharide by Means of Simultaneous Inversion Reactions. J. Carbohydr. Chem. 18, 841–850. doi:10.1080/07328309908544039
Matsuura, F., Ohta, M., Murakami, K., and Matsuki, Y. (1993). Structures of Asparagine Linked Oligosaccharides of Immunoglobulins (IgY) Isolated from Egg-Yolk of Japanese Quail. Glycoconjugate J. 10, 202–213. doi:10.1007/BF00702201
Meyer, B. H., and Albers, S. V. (2014). AglB, Catalyzing the Oligosaccharyl Transferase Step of the archaealN‐glycosylation Process, Is Essential in the Thermoacidophilic crenarchaeonSulfolobus Acidocaldarius. Microbiologyopen 3, 531–543. doi:10.1002/mbo3.185
Michael, A., and Norton, L. (1879). On the Action of Iodine Monochloride Upon Aromatic Amines. J. Am. Chem. Soc. 1 (11), 484–485. doi:10.1021/ja02151a603
Mohanty, S., P Chaudhary, B., and Zoetewey, D. (2020). Structural Insight into the Mechanism of N-Linked Glycosylation by Oligosaccharyltransferase. Biomolecules 10, 624. doi:10.3390/biom10040624
Nashida, J., Nishi, N., Takahashi, Y., Hayashi, C., Igarashi, M., Takahashi, D., et al. (2018). Systematic and Stereoselective Total Synthesis of Mannosylerythritol Lipids and Evaluation of Their Antibacterial Activity. J. Org. Chem. 83, 7281–7289. doi:10.1021/acs.joc.8b00032
Pardo-Vargas, A., Bharate, P., Delbianco, M., and Seeberger, P. H. (2019). Automated Glycan Assembly of Arabinomannan Oligosaccharides from Mycobacterium tuberculosis. Beilstein J. Org. Chem. 15, 2936–2940. doi:10.3762/bjoc.15.288
Pawar, S., Hsu, L., Narendar Reddy, T., Ravinder, M., Ren, C.-T., Lin, Y.-W., et al. (2020). Synthesis of Asymmetric N-Glycans as Common Core Substrates for Structural Diversification through Selective Enzymatic Glycosylation. ACS Chem. Biol. 15, 2382–2394. doi:10.1021/acschembio.0c00359
Plante, O. J., Palmacci, E. R., Andrade, R. B., and Seeberger, P. H. (2001). Oligosaccharide Synthesis with Glycosyl Phosphate and Dithiophosphate Triesters as Glycosylating Agents. J. Am. Chem. Soc. 123, 9545–9554. doi:10.1021/ja016227r
Plante, O. J., Palmacci, E. R., and Seeberger, P. H. (2001). Automated Solid-phase Synthesis of Oligosaccharides. Science 291, 1523–1527. doi:10.1126/science.1057324
Poljak, K., Breitling, J., Gauss, R., Rugarabamu, G., Pellanda, M., and Aebi, M. (2017). Analysis of Substrate Specificity of Trypanosoma Brucei Oligosaccharyltransferases (OSTs) by Functional Expression of Domain-Swapped Chimeras in Yeast. J. Biol. Chem. 292, 20342–20352. doi:10.1074/jbc.M117.811133
Priyanka, P., Parsons, T. B., Miller, A., Platt, F. M., and Fairbanks, A. J. (2016). Chemoenzymatic Synthesis of a Phosphorylated Glycoprotein. Angew. Chem. Int. Ed. 55, 5058–5061. doi:10.1002/anie.201600817
Reily, C., Stewart, T. J., RenfrowNovak, M. B. J., and Novak, J. (2019). Glycosylation in Health and Disease. Nat. Rev. Nephrol. 15, 346–366. doi:10.1038/s41581-019-0129-4
Sabbavarapu, N. M., and Seeberger, P. H. (2021). Automated Glycan Assembly of Oligogalactofuranosides Reveals the Influence of Protecting Groups on Oligosaccharide Stability. J. Org. Chem. 86, 7280–7287. doi:10.1021/acs.joc.1c00505
Schjoldager, K. T., Narimatsu, Y., Joshi, H. J., and Clausen, H.(2020). Global View of Human Protein Glycosylation Pathways and functionsNew Methods for the Synthesis of Glycosides and Oligosaccharides—Are There Alternatives to the Koenigs-Knorr Method? Nat. Rev. Mol. Cel. Biol.Angew. Chem. Int. Ed. Engl. 2125, 729212–749235. doi:10.1038/s41580-020-00294-x
Schmidt, R. R. (1986). New Methods for the Synthesis of Glycosides and Oligosaccharides—Are There Alternatives to the Koenigs-Knorr Method? Chem. Int. Ed. Engl. 25, 212–235. doi:10.1002/anie.198602121
Seko, A., Koketsu, M., Nishizono, M., Enoki, Y., Ibrahim, H. R., Juneja, L. R., et al. (1997). Occurrence of a Sialylglycopeptide and Free Sialylglycans in Hen's Egg Yolk. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1335, 23–32. doi:10.1016/s0304-4165(96)00118-3
Shirakawa, A., Manabe, Y., and Fukase, K. (2021b). Recent Advances in the Chemical Biology of N-Glycans. Molecules 26, 1040. doi:10.3390/molecules26041040
Shirakawa, A., Manabe, Y., Marchetti, R., Yano, K., Masui, S., Silipo, A., et al. (2021a). Chemical Synthesis of Sialyl N ‐Glycans and Analysis of Their Recognition by Neuraminidase. Angew. Chem. Int. Ed. 60, 24686–24693. doi:10.1002/anie.202111035
Shivatare, S. S., Chang, S.-H., Tsai, T.-I., Tseng, S. Y., Shivatare, V. S., Lin, Y.-S., et al. (2016). Modular Synthesis of N-Glycans and Arrays for the Hetero-Ligand Binding Analysis of HIV Antibodies. Nat. Chem 8, 338–346. doi:10.1038/nchem.2463
Srivastava, A. D., Unione, L., Bunyatov, M., Gagarinov, I. A., Delgado, S., Abrescia, N. G. A., et al. (2021). Chemoenzymatic Synthesis of Complex N ‐Glycans of the Parasite S. Mansoni to Examine the Importance of Epitope Presentation on DC‐SIGN Recognition. Angew. Chem. Intl Edit 60, 19287–19296. doi:10.1002/anie.202105647
Taguchi, Y., Yamasaki, T., Ishikawa, M., Kawasaki, Y., Yukimura, R., Mitani, M., et al. (2021). The Structure of an Archaeal Oligosaccharyltransferase Provides Insight into the Strict Exclusion of Proline from the N-Glycosylation Sequon. Commun. Biol. 4, 941. doi:10.1038/s42003-021-02473-8
Tanaka, M., Sato, K., Yoshida, R., Nishi, N., Oyamada, R., Inaba, K., et al. (2020). Diastereoselective Desymmetric 1,2-Cis-Glycosylation of Meso-Diols via Chirality Transfer from a Glycosyl Donor. Nat. Commun. 11, 2431. doi:10.1038/s41467-020-16365-8
Toonstra, C., Wu, L., Li, C., Wang, D., and Wang, L.-X. (2018). Top-Down Chemoenzymatic Approach to Synthesizing Diverse High-Mannose N-Glycans and Related Neoglycoproteins for Carbohydrate Microarray Analysis. Bioconjug. Chem. 29, 1911–1921. doi:10.1021/acs.bioconjchem.8b00145
Tsutsui, M., Sianturi, J., Masui, S., Tokunaga, K., Manabe, Y., and Fukase, K. (2020). Efficient Synthesis of Antigenic Trisaccharides ContainingN-Acetylglucosamine: Protection of NHAc as NAc2. Eur. J. Org. Chem. 2020, 1802–1810. doi:10.1002/ejoc.201901809
Tyrikos‐Ergas, T., Bordoni, V., Fittolani, G., Chaube, M. A., Grafmüller, A., Seeberger, P. H., et al. (2021). Systematic Structural Characterization of Chitooligosaccharides Enabled by Automated Glycan Assembly. Chem. Eur. J. 27, 2321–2325. doi:10.1002/chem.202005228
Unverzagt, C., André, S., Seifert, J., Kojima, S., Fink, C., Srikrishna, G., et al. (2002). Structure−Activity Profiles of Complex Biantennary Glycans with Core Fucosylation and With/without Additional α2,3/α2,6 Sialylation: Synthesis of Neoglycoproteins and Their Properties in Lectin Assays, Cell Binding, and Organ Uptake. J. Med. Chem. 45, 478–491. doi:10.1021/jm0110237
Varki, A., Cummings, R. D., Esko, J. D., Freeze, H. H., Stanley, P., Bertozzi, C. R., et al. (2015). Essentials of Glycobiology. NY: Cold spring harbor.
Vibhute, A. M., Komura, N., Tanaka, H. N., Imamura, A., and Ando, H. (2021). Advanced Chemical Methods for Stereoselective Sialylation and Their Applications in Sialoglycan Syntheses. Chem. Rec. 21, 3194–3223. doi:10.1002/tcr.202100080
Viinikangas, T., Khosrowabadi, E., and Kellokumpu, S. (2021). N-glycan Biosynthesis: Basic Principles and Factors Affecting its Outcome. Exp. Suppl. 112, 237–257. doi:10.1007/978-3-030-76912-3_7
Wacker, M., Linton, D., Hitchen, P. G., Nita-Lazar, M., Haslam, S. M., North, S. J., et al. (2002). N-linked Glycosylation in Campylobacter Jejuni and its Functional Transfer into E. coli. Science 298, 1790–1793. doi:10.1126/science.298.5599.1790
Wang, L.-X., Tong, X., and Li, T. (2019). Transglycosylation of Endoglycosidase Endo-S and Endo-S Mutants for a One-Pot Antibody Glycosylation Remodeling. US20190002945A1
Wang, P., Zhu, J., Yuan, Y., and Danishefsky, S. J. (2009). Total Synthesis of the 2,6-Sialylated Immunoglobulin G Glycopeptide Fragment in Homogeneous Form. J. Am. Chem. Soc. 131, 16669–16671. doi:10.1021/ja907136d
Wang, Y. S., Wu, Y., Xiong, D. C., and Ye, X. S. (2019). Total Synthesis of a Hyperbranched N ‐Linked Hexasaccharide Attached to ATCV‐1 Major Capsid Protein without Precedent. Chin. J. Chem. 37, 42–48. doi:10.1002/cjoc.201800533
Wang, Z., Chinoy, Z. S., Ambre, S. G., Peng, W., McBride, R., de Vries, R. P., et al. (2013). A General Strategy for the Chemoenzymatic Synthesis of Asymmetrically Branched N -Glycans. Science 341, 379–383. doi:10.1126/science.1236231
Weiss, M., Ott, D., Karagiannis, T., Weishaupt, M., Niemietz, M., Eller, S., et al. (2020). Efficient Chemoenzymatic Synthesis of N‐Glycans with a β1,4‐Galactosylated Bisecting GlcNAc Motif. Chembiochem 21, 3212–3215. doi:10.1002/cbic.202000268
Wilson, M. P., Garanto, A., Pinto e Vairo, F., Ng, B. G., Ranatunga, W. K., Ventouratou, M., et al. (2021). Active Site Variants in STT3A Cause a Dominant Type I Congenital Disorder of Glycosylation with Neuromusculoskeletal Findings. Am. J. Hum. Genet. 108, 2130–2144. doi:10.1016/j.ajhg.2021.09.012
Yang, M., Wei, M., Wang, C., Lu, Y., Jin, W., Gao, X., et al. (2020). Separation and Preparation of N-Glycans Based on Ammonia-Catalyzed Release Method. Glycoconj. J. 37, 165–174. doi:10.1007/s10719-020-09909-z
Yang, W., Ramadan, S., Orwenyo, J., Kakeshpour, T., Diaz, T., Eken, Y., et al. (2018). Chemoenzymatic Synthesis of Glycopeptides Bearing Rare N-Glycan Sequences with or without Bisecting GlcNAc. Chem. Sci. 9, 8194–8206. doi:10.1039/c8sc02457j
Ye, F., Zhao, J., Xu, P., Liu, X., Yu, J., Shangguan, W., et al. (2021). Synthetic Homogeneous Glycoforms of the SARS‐CoV‐2 Spike Receptor‐Binding Domain Reveals Different Binding Profiles of Monoclonal Antibodies. Angew. Chem. Int. Ed. 60, 12904–12910. doi:10.1002/anie.202100543
Zeng, C., Sun, B., Cao, X., Zhu, H., Oluwadahunsi, O. M., Liu, D., et al. (2020). Chemical Synthesis of Homogeneous Human E-Cadherin N-Linked Glycopeptides: Stereoselective Convergent Glycosylation and Chemoselective Solid-phase Aspartylation. Org. Lett. 22, 8349–8353. doi:10.1021/acs.orglett.0c02971
Zhong, X., Zhou, S., Ao, J., Guo, A., Xiao, Q., Huang, Y., et al. (2021). Zinc(II) Iodide-Directed β-Mannosylation: Reaction Selectivity, Mode, and Application. J. Org. Chem. 86, 16901–16915. doi:10.1021/acs.joc.1c02091
Zhu, Y., Delbianco, M., and Seeberger, P. H. (2021). Automated Assembly of Starch and Glycogen Polysaccharides. J. Am. Chem. Soc. 143, 9758–9768. doi:10.1021/jacs.1c02188
Zhu, Y., and Yu, B. (2015). Highly Stereoselective β-Mannopyranosylation via the 1-α-Glycosyloxy-isochromenylium-4-Gold(I) Intermediates. Chem. Eur. J. 21, 8771–8780. doi:10.1002/chem.201500648
Keywords: N-glycans, β-mannosylation, α-sialylation, chemical assembly, chemo-enzymatic strategies
Citation: Zhao X, Huang Y, Zhou S, Ao J, Cai H, Tanaka K, Ito Y, Ishiwata A and Ding F (2022) Recent Chemical and Chemoenzymatic Strategies to Complex-Type N-Glycans. Front. Chem. 10:880128. doi: 10.3389/fchem.2022.880128
Received: 21 February 2022; Accepted: 12 April 2022;
Published: 26 May 2022.
Edited by:
Matthew A. Coleman, University of California, Davis, United StatesReviewed by:
Madhusudhan Reddy Gadi, Georgia State University, United StatesDecai Xiong, Peking University, China
Copyright © 2022 Zhao, Huang, Zhou, Ao, Cai, Tanaka, Ito, Ishiwata and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Cai, Y2FpaHVpNUBtYWlsLnN5c3UuZWR1LmNu; Akihiro Ishiwata, YWlzaGl3YUByaWtlbi5qcA==; Feiqing Ding, ZGluZ2ZxM0BtYWlsLnN5c3UuZWR1LmNu
†These authors have contributed equally to this work