 Sin Hang Crystal Chan1
Sin Hang Crystal Chan1 Jarred M. Griffin2
Jarred M. Griffin2 Connor A. Clemett2
Connor A. Clemett2 Margaret A. Brimble1,3,4
Margaret A. Brimble1,3,4 Simon J. O’Carroll2*
Simon J. O’Carroll2* Paul W. R. Harris1,3,4*
Paul W. R. Harris1,3,4*- 1School of Chemical Sciences, The University of Auckland, Auckland, New Zealand
- 2Centre for Brain Research, Department of Anatomy and Medical Imaging, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand
- 3School of Biological Sciences, The University of Auckland, Auckland, New Zealand
- 4Maurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Auckland, New Zealand
Peptide5 is a 12–amino acid mimetic peptide that corresponds to a region of the extracellular loop 2 (EL2) of connexin43. Peptide5 regulates both cellular communication with the cytoplasm (hemichannels) and cell-to-cell communication (gap junctions), and both processes are implicated in neurological pathologies. To address the poor in vivo stability of native peptide5 and to improve its activity, twenty-five novel peptide5 mimetics were designed and synthesized. All the analogues underwent biological evaluation as a hemichannel blocker and as a gap junction disruptor, and several were assessed for stability in human serum. From this study, it was established that several acylations on the N-terminus were tolerated in the hemichannel assay. However, the replacement of the L-Lys with an N-methylated L-Lys to give H-VDCFLSRPTE-N-MeKT-OH showed good hemichannel and gap junction activity and was more stable in human serum. The cyclic peptide variants generally were not tolerated in either the hemichannel and gap junction assay although several possessed outstanding stability in human serum.
1 Introduction
Connexin43 (Cx43) is a member of the gap junction protein family and is found widely throughout the body (Oyamada et al., 2005). Cx43 plays a crucial role in tissue function, allowing cell-to-cell communication via the formation of gap junctions (Márquez-Rosado et al., 2012). Connexin proteins are membrane-bound proteins that consist of cytoplasmic N- and C-termini, four transmembrane domains, a cytoplasmic loop, and two highly conserved extracellular loops (EL1 and EL2) (Ribeiro-Rodrigues et al., 2017). Within the cell membrane, six connexin proteins oligomerize to form a connexon, and connexons on adjacent cell membranes align and dock via EL1 and EL2 to form an intercellular gap junction channel (Li et al., 1996). The gap junctions allow for the movement of ions and other small molecules between cells, which is crucial for cell function and survival (Krysko et al., 2005). Within the central nervous system, Cx43 gap junctions form a syncytium and provide a high degree of cell-to-cell communication, especially in endothelial and glial cells (Sarrouilhe et al., 2017). The injury or inflammation of the central nervous system leads to the upregulation and opening of unopposed connexons (hemichannels) (Xing et al., 2019). This creates a conduit between the intra- and extra-cellular environment allowing the movement of water and small molecules. The opening of connexin hemichannels negatively impacts a cell’s ability to osmoregulate (Quist et al., 2000; Rodríguez-Sinovas et al., 2007; Danesh-Meyer et al., 2012), leading to cell edema (Quist et al., 2000), excitotoxic neuronal cell death (Froger et al., 2010), glial cell activation (Li et al., 1996), vascular hemorrhage (De Bock et al., 2011), and ultimately the spread of secondary damage (Davidson et al., 2013.; Decrock et al., 2015). The opening of Cx43 hemichannels has been shown to be a major contributor to a number of CNS pathologies including retinal ischemia–reperfusion injury (Danesh-Meyer et al., 2012), spinal cord injury (O’Carroll et al., 2008; O’Carroll et al., 2013; Tonkin et al., 2015), preterm ischemia and asphyxiation (Davidson et al., 2012; Davidson et al., 2014), and infection (Eugenin et al., 2011). As such, targeting Cx43 hemichannels has potential as a treatment for neurological conditions.
One approach to target Cx43 hemichannels has been the use of peptidomimetics that target the EL1 and EL2 extracellular loops of Cx43 (gap26, gap27, and peptide5) (Evans, 2015) or intracellular regions of the protein (gap19, ACT-1) (D’hondt et al., 2013). The extracellular targeting of peptide5, H2N-VDCFLSRPTEKT-CO2H (O’Carroll et al., 2008), has been shown to selectively target either hemichannels or gap junctions in a concentration-dependent manner, with concentrations as low as 5 µM selectively inhibiting hemichannels and higher concentrations (e.g., 500 µM), inhibiting both gap junctions and hemichannels in vitro. This ability to selectively target pathological hemichannels whilst not interfering with the normal function of gap junctions presents the opportunity for therapeutic applications and Peptide5 has shown benefit in reducing inflammation, vascular breakdown, neuronal survival, and improved functional outcomes in a number of models of CNS injury such as brain ischemia–reperfusion injuries (Davidson et al., 2012, 2014), retinal injury and disease (Danesh-Meyer et al., 2012; Guo et al., 2016; Mugisho et al., 2019; Kuo et al., 2020), and spinal cord injury (O’Carroll et al., 2013; Mao et al., 2017). Despite these advances, further work is required to maximize both the efficacy and serum stability of peptide5-based therapeutic candidates to demonstrate their potential as agents to treat human diseases. Generally speaking, short native, unmodified peptides (< 20 amino acids) are rapidly cleared or degraded in vivo due to the action of proteases and/or filtration by the kidneys. The peptides are also limited in that they are unable to cross cell membranes due to polar functional groups and low lipophilicity leading to low bioavailability. A variety of chemical modifications have been employed to render peptides more suitable for human therapeutics (Muttenthaler et al., 2021) including lipidation (Zhang and Bulaj, 2012), glycosylation (van Witteloostuijn et al., 2016), cyclization (Bechtler and Lamers, 2021), and modification of individual amino acids (Gentilucci et al., 2010); these improvements are exemplified by the ever-increasing clinical application of peptides for use in cancer, infectious diseases, and diabetes (D’Aloisio et al., 2021; de la Torre and Albericio, 2020).
We report herein our efforts in designing, synthesizing, and evaluating 25 novel linear and cyclic peptide5 analogues (Figure 1) as inhibitors of connexin hemichannels and gap junctions as well as assessing their stability in human serum. All peptides were readily generated via solid-phase peptide synthesis. It was found that most of the modifications to peptide5 resulted in the loss of activity in both a hemichannel inhibition assay and a gap junction dye spread assay. Several linear peptide5 analogs exhibiting N-terminal acylation did show activity comparable to peptide5 in their ability to block hemichannels, while all the cyclic peptide5 congeners failed to demonstrate significant hemichannel activity compared to the peptide5 control. Several D- or N-methylated variants of linear peptide5 were both shown to have gap junction activity similar to that of peptide5 and were significantly more stable in human serum.
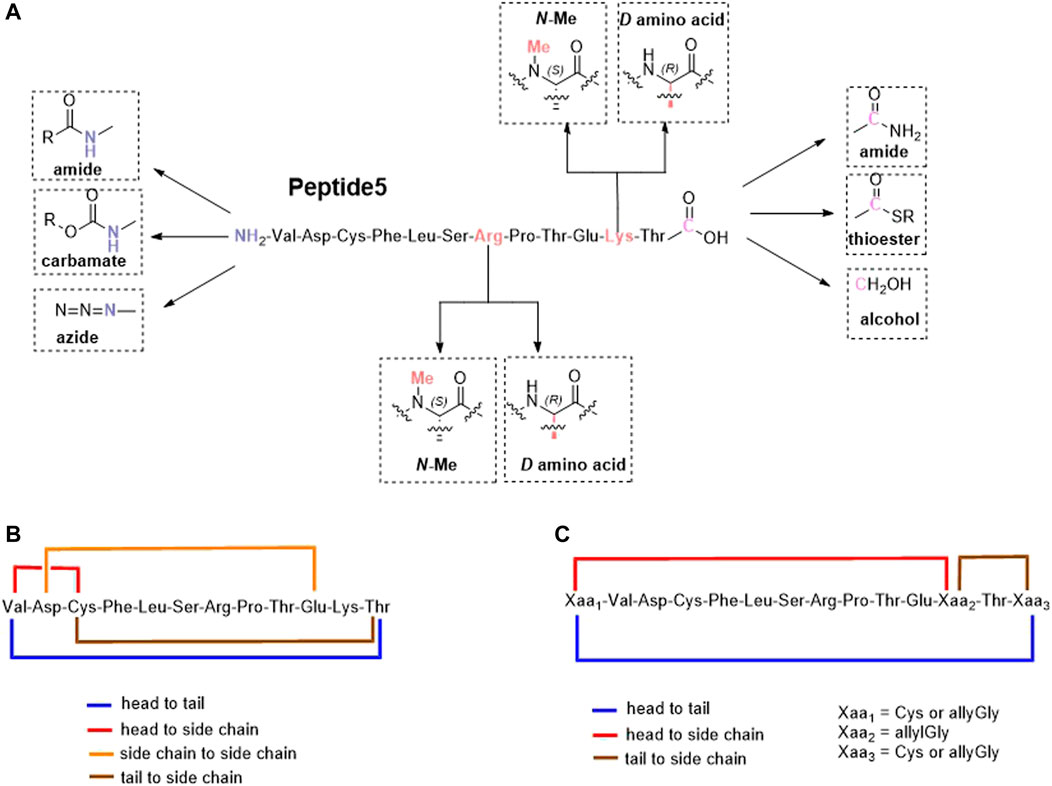
FIGURE 1. Proposed linear (A) and cyclic variants using either existing amino acids (B) or introduced handles (C) of the connexin inhibitory peptide, peptide5.
2 Results
In our previous studies on peptide5 (1), we constructed a 42-peptide library of S-lipidated linear analogues modified at six different amino acid positions (Yang et al., 2020) that were informed by the results of our preliminary alanine scan study (Kim et al., 2017a). While it was shown that lipidation at certain residues was tolerated and, in some cases improved connexon hemichannel blocking, (rat) serum stability of the lipidated peptides was not improved relative to peptide5 which had a short half-life of just 5 min. This suggested that the individual amino acids are responsible for the degradation of peptide5 and that modification of specific amino acids was required to improve the stability, whilst maintaining connexin blocking activity.
Lipidation is only one of several methods employed to improve the stability, permeability, and half-life of peptides that include the incorporation of non-canonical amino acids, D- and N-methylated amino acids, and modification of the C- and N-termini including cyclization (Erak et al., 2018). Cyclization, in particular, is also known to rigidify the peptide backbone, reducing the number of possible conformers and promoting binding to its intended target (Philippe et al., 2021). An exquisite example of this is the FDA-approved macrocyclic peptide cilengitide which displayed a 10-fold increase in binding compared to its linear form. (Kumagai et al., 1991). Based on the aforementioned considerations, we proposed to synthesize peptide5 analogues as depicted in Figure 1. These analogues included modification of both the C- and N-termini and replacement of enzymatically susceptible Arg and Lys by their D- and N-methylated variants. The cyclic peptide5 analogues were prepared using the side chains and the C- and N-termini already present in the primary sequence or employing introduced chemical handles to affect cyclization.
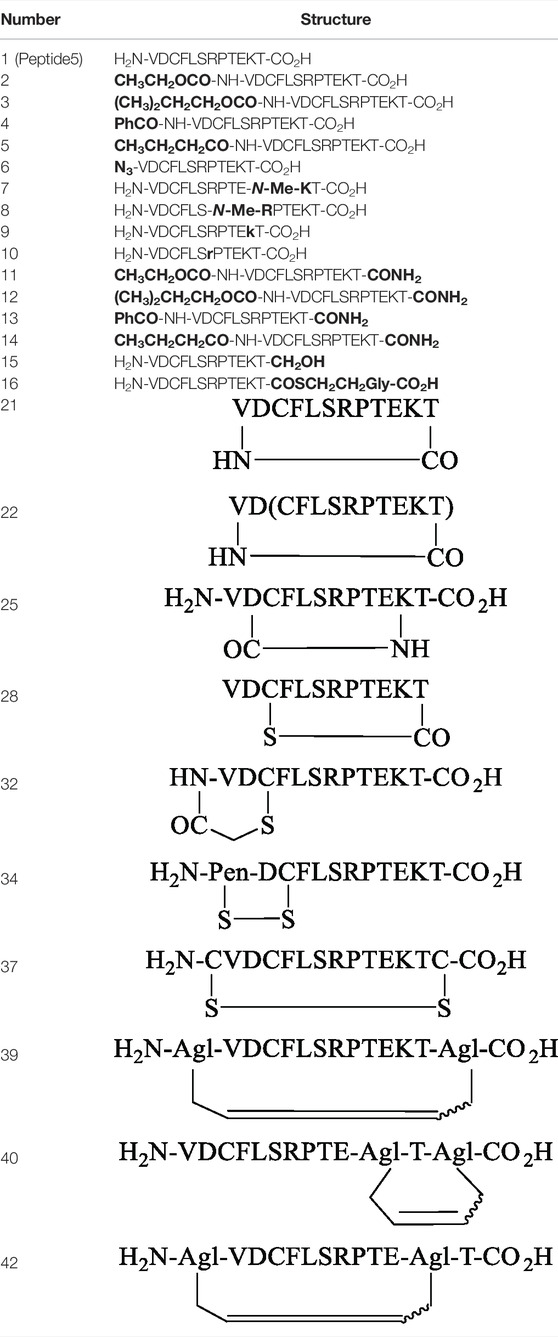
TABLE 1. A summary of all linear and cyclic variants of the connexon43 inhibitor peptide. Peptide5 prepared. The chemical changes are highlighted in bold.
2.1 Synthesis of linear peptides modified at the C- and N-termini, and incorporation of non-proteogenic amino acids
We commenced by designing and synthesizing a set of linear peptides that 1) were modified at the N-terminus by acylation or by converting the amine group to an azide; 2) replaced the C terminal carboxy group with a carboxamide and capping of the N-terminus by acylation; 3) replaced the trypsin-susceptible amino acids, Arg-7 and Lys-11 with their D- or N-methylated congeners; and 4) replaced the C terminal carboxy group with a carboxamide, a thioester, or an alcohol. The synthesis of these sixteen analogues is depicted in Figure 2. These modifications were in line with our alanine scan data which demonstrated that replacement of each amino acid by alanine had little effect on gap junction uncoupling (Kim et al., 2017b).
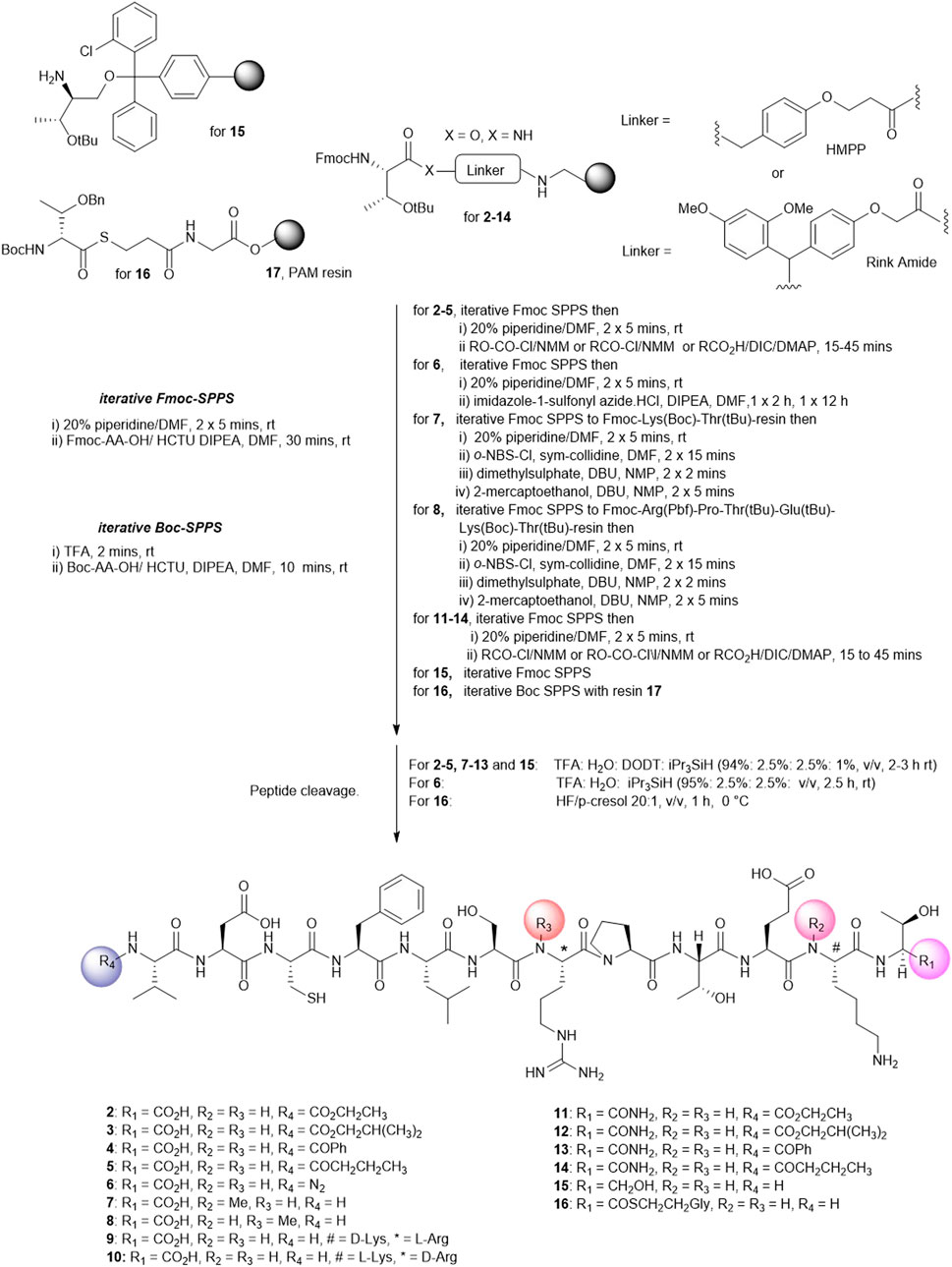
FIGURE 2. Solid phase synthesis strategy to prepare linear Peptide5 analogues modified at the C- and Ntermin and D and N-Me variants.
The analogues of Peptide5 capped at the N-terminus (2–5) were readily prepared on Fmoc-Thr (tBu) esterified hydroxymethylphenoxypropionic acid (HMPP) linker functionalized on aminomethyl polystyrene resin. Following the peptide chain assembly, the final Fmoc group was removed and the N-terminal amino group reacted with either ethyl chloroformate/N-methylmorpholine (NMM), isobutyl chloroformate/NMM, benzoyl chloride/NMM, or butyric acid in the presence of the acylating reagents diisoproylcarbodiimide (DIC)/catalytic dimethylaminopyridine (DMAP) to effect amidation. In all cases, a single treatment was required, and all compounds were recovered in >95% purity following HPLC purification (see Supplementary Material).
For azide analogue 6, the azide transfer reagent, imidazole-1-sulfonyl azide.HCl (Goddard-Borger and Stick, 2007), reacted directly with the N-terminal amino group of the resin-bound peptide. In this case, two treatments were required for complete conversion, and importantly, no thiol scavenger (DODT) was used for the peptide cleavage cocktail to mitigate the potential reduction of the azide to the amine.
N-methylated Arg-7 and Lys-11 peptides (7 and 8) could be accessed by adopting the on-resin N-methylation protocol developed by Kessler (Chatterjee et al., 2012). Following Fmoc removal at the amino acid site of putative N-methylation, the amino group was nosylated affording a sulfonamide. Methylation of the sulfonamide (dimethylsulfate/DBU) and removal of the nosyl group (2-mecaptoethanol/DBU) yielded the N-methylated peptides. The elongation of the peptide sequence, resin cleavage, and HPLC purification gave 7 and 8 in high purity. No difficulties were encountered when coupling the subsequent Fmoc amino acid to the sterically hindered, resin-bound N-methylated amino acid. For the synthesis of peptide5 containing d-amino acids at Lys-11 (9) and Arg-7 (10), Fmoc-D-Lys (Boc)-OH or Fmoc-D-Arg (Pbf)-OH was used at the appropriate positions during the Fmoc SPPS assembly.
Finally, the peptide analogues modified at the C-terminus (11–16) were prepared on the appropriate linker resin. Carboxamides (11–14) were assembled on a Rink amide linker using Fmoc SPPS and were N-terminally modified according to the protocols outlined for peptides 2–5. C-terminal alcohol 15 was directly accessible from the 2-chlorotrityl resin functionalized with tert-butyl–protected Fmoc-threoninol using Fmoc SPPS. Thioester 16 was best prepared using the thioester generating resin 17 (Camarero et al., 2000) and employing Boc SPPS to avoid aminolysis of the thioester linkage, a known concern when employing piperidine as an Fmoc deblocking reagent during Fmoc synthesis (Li et al., 1998).
2.1.1 Synthesis of Cyclic Peptides Using Pre-Existing Amino Acids
We next attempted to cyclize the C- and N-termini of the maximally side-chain–protected peptide 18 to obtain cyclic analogue 21. In turn, protected peptide 18 was prepared on the hyper acid-labile 2-chloro trityl resin (Figure 3A). However, using HBTU or BOP-Cl/DMAP as condensation reagents was unsuccessful and none of the expected head-to-tail cyclic products was detected after side-chain deprotection and LC-MS analysis. This is most likely due to the difficult cyclization site that contains the bulky, secondary amino acids, valine and threonine. We therefore repositioned the cyclization site to the Asp-Cys junction as we anticipated that using native chemical ligation (NCL) (Dawson et al., 1994), the chemoselective amide bond-forming reaction between a cysteinyl peptide and peptide thioester would furnish the desired cyclic peptide 21 (Figure 3B).
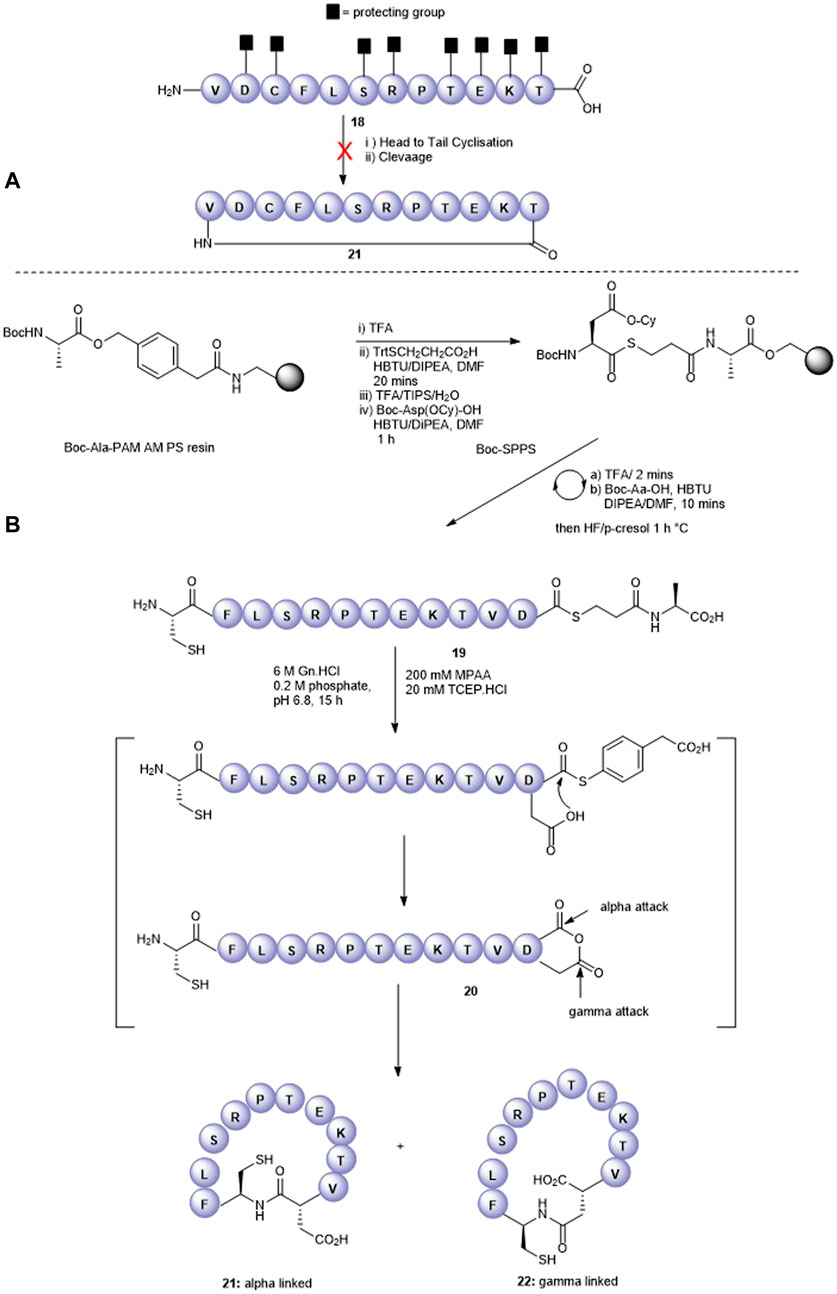
FIGURE 3. Unsuccessful cyclization attempt to prepare cyclic peptide5 21 (A) and the successful strategy employing native chemical ligation (B).
The linear thioester precursor, CFLSRPTEKTVD-COS-CH2CH2Ala-OH 19, was prepared satisfactorily using Boc SPPS and then directly subjected to NCL conditions (0.2 M phosphate, 6 M Gn.HCl, 20 mM TCEP.HCl, and 200 mM 4-mercaptophenyl acetic acid [MPAA]) for 15 h which afforded a separable mixture (2:1) of the desired cyclic product 21 (calc (M + H)+ 1378.6, found 1378.4) and undesired product 22 (calc (M + H)+ 1378.6, found 1378.5) (Figure 3B). This was not unexpected as ligation between Asp and Cys has been reported (Villain et al., 2003) to result in the formation of the unnatural β amide bond as a by-product (20%–30%) (Dang et al., 2013) by the formation of an intermediate succinic anhydride 20 which can undergo aminolysis at either carbonyl group. Fortunately, in the present work, the undesired isomeric by-product 22 was easily separated from the desired product 21 using RP-HPLC.
Having successfully prepared the head-to-tail cyclic variant of peptide5 (21), we next focused on constraining the peptide via its amino acid side chains. An alanine scan (Kim et al., 2017a) revealed that all amino acids could be modified with no significant loss in gap junction uncoupling, although some replacements abolished hemichannel blocking activity. We first pursued the preparation of an amide linkage between Asp and Lys (Figure 4). This is most easily accomplished on the solid support whereby the pseudo-dilution effect minimizes unwanted dimerization or oligomerization that prevails under solution-phase conditions. Furthermore, the coupling reaction can be conveniently monitored by the Kaiser test that determines any free primary amines.
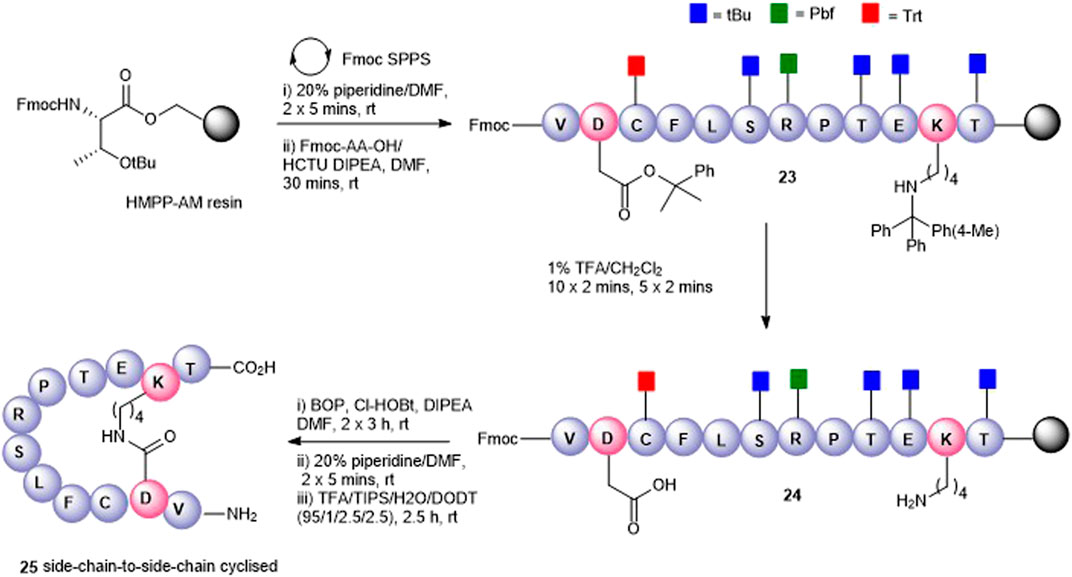
FIGURE 4. Synthesis of the Asp2-Lys11 side-chain–to–side-chain cyclized peptide5 (25).
To maximize synthetic efficiency, the 4-methyltriphenylmethyl (Mtt)– and 2-phenylisopropyl (2-PhiPr)–protecting groups were used on the Lys and Glu side chains, respectively, as these can be removed simultaneously with diluted TFA, selectively unmasking the amine and carboxylic acid functionalities for subsequent lactamisation leaving other side-chain–protecting groups unmodified. Thus, resin-bound peptide 23 was assembled by Fmoc SPPS on Fmoc-Thr (OtBu)-HMPP resin using 20% piperidine in DMF for Fmoc removal and HCTU/DIPEA for coupling. Orthogonally protected Fmoc-Lys (Mtt)-OH and Fmoc-Asp(O-2-PhiPr) were incorporated uneventfully and the Mtt and 2-Ph-iPr side-chain–protecting groups both cleaved upon repetitive treatments with 1% TFA in CH2Cl2 to give 24. The key macrolactamization step was then explored on resin using PyBOP/Cl-HOBt, DIC/HOAt, DIC/Oxyma, or BOP/Cl-HOBt as condensation reagents. Employing PyBOP- or DIC-based couplings led to aspartimide formation (PyBOP), no reaction or incomplete reaction (DIC/HOAt), or a complex mixture (DIC/Oxyma). PyBOP/Cl-HOBt (2 × 3 h) proved to be the optimal coupling reagents and affording the desired cyclic peptide 25 in >90% conversion as judged by LC-MS (calc (M + H)+ 1378.6, found 1378.6) that was easily purified by RP-HPLC.
The attempts to form the head-to-side-chain macrolactamisation 26 by on-resin coupling of the N-terminal amino group and the side-chain carboxylic acid of Glu using the orthogonal protecting strategy as described above met with failure (Figure 5). Extensive oligomerization was observed using several different coupling conditions and the use of a lower loading resin, or PEG-based resins had little effect on the success of the reaction.
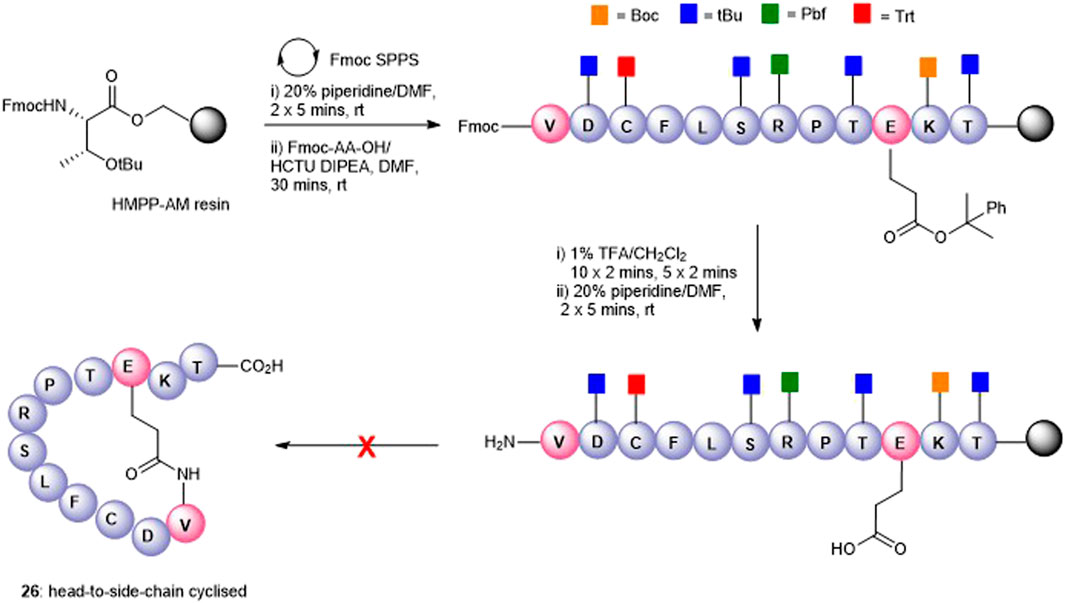
FIGURE 5. Unsuccessful synthesis of Glu10-Val1 side-chain-to-head cyclized peptide5 26.
Satisfactorily, a side-chain-to-tail mode of macrocyclization was effected by the selective condensation of Cys with the C-terminal threonyl aryl thioester at neutral pH (Figure 6). The precursor and less reactive alkyl thioester, 14, was quantitatively transformed into the more reactive aryl thioester 27 (calc (M+2H)2 + 773.8, found 773.4) using the water-soluble aryl thiol, MPAA (Johnson and Kent, 2006) at pH 6.5 after 6 h and isolated by SPE. We anticipated that at neutral pH in the absence of added thiols, the cysteine thiol would undergo transthioesterfication with the activated C-terminus in an irreversible manner. Thus, 27 (2 mM) was dissolved in 0.2 M phosphate/6 M Gn.HCl, at pH = 6.7, and the cyclization was monitored by RP-HPLC. After 2 h, the linear polypeptide was quantitatively converted into the side-chain-to-tail macrocyclic thiolactone 28 (calc (M+2H)2 + 689.8, found 689.4) as the sole product that was easily recovered by RP-HPLC.

FIGURE 6. Synthesis of the Cys3-Thr12 thioester-linked side-chain-to-tail cyclized peptide5 (28).
2.1.2 Synthesis of Cyclic Peptides Using Introduced Handles
Disulfide bonds, the covalent linkage formed between two cysteines, are naturally occurring structure-stabilizing motifs that are important for maintaining the structure and function of proteins and peptides. Not surprisingly, they have been also been employed for peptide cyclization to confer structural rigidity on synthetic peptides, leading to higher affinity to target receptors (Góngora-Benítez et al., 2014). Peptide5 contains a single cysteine residue (Cys3) that could be used as a cyclization handle, though our alanine scanning data suggested that this residue could not be altered without some loss of hemichannel activity. However, the N-terminal valine (Val1) residue could be modified as the substitution of Val1 to Ala caused little effect in activity (Kim et al., 2017b) in both hemichannel and gap junction assays. Furthermore, a truncated peptide5, SRPTEKT, was equally effective as a gap junction inhibitor, suggesting that the N-terminal residues, VDCFL, could be modified. Therefore, we used Cys3 to 1) create a thioether bridge by intramolecular chemo-selective alkylation of Cys-SH with the N-terminus capped with a 2-bromoacetyl group or 2) to form a disulfide bond between Cys-SH and β,β-dimethylcysteine (penicillamine, Pen) introduced as a substitute for Val1. A disulfide-linked head-to-tail cyclic peptide was also proposed by introducing a cysteine adjacent to both the C- and N-termini as cyclization handles enabling subsequent oxidation to the disulfide.
The thioether macrocycle (32, Figure 7) was prepared by acylation of the resin-bound N-terminus (29) with bromoacetic acid anhydride (Robey and Fields, 1989), followed by cleavage from the resin to afford the N-bromoacetylated peptide 31 (calc (M+2H)2 + 759.2, found 758.3). Spontaneous cyclization then took place by exposing the crude peptide to an aqueous buffer (pH = 6.9) to afford the thioether 32 (calc (M+2H)2 + 717.8, found 718.4) in 20% yield.
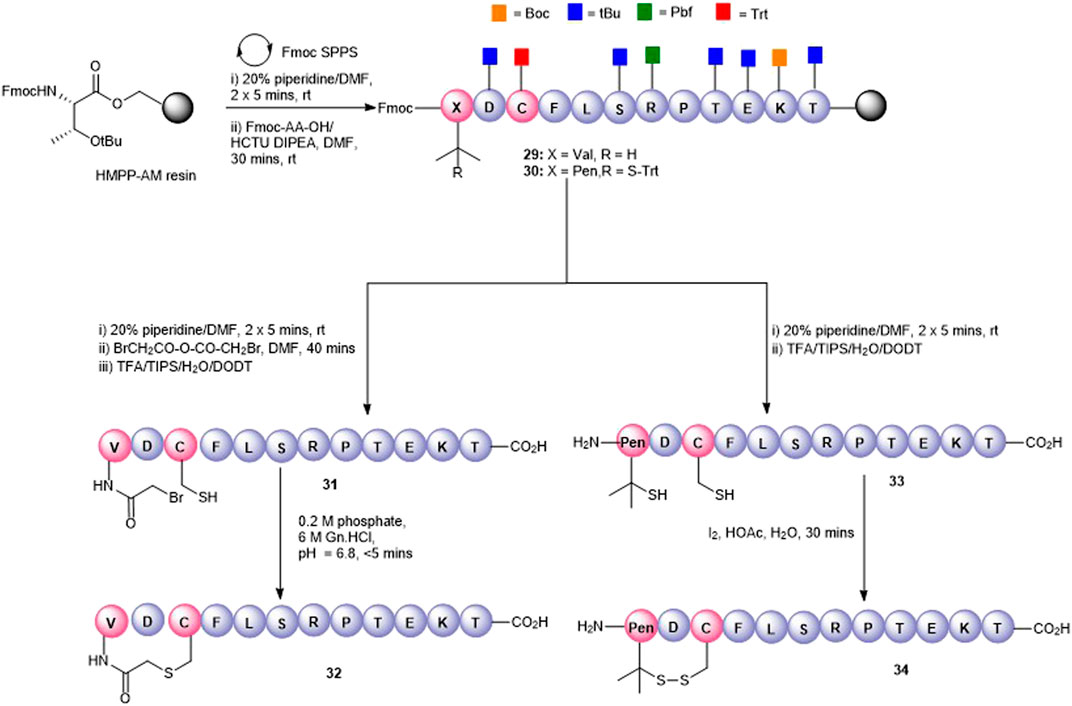
FIGURE 7. Synthesis of Val1-Cys3 thioester-linked cyclized peptide5 (32) and a Pen1-Cys3 disulfide-bonded cyclic peptide5 (34).
To effect the disulfide head-to-side-chain–linked macrocycle (34, Figure 7), commercially available Fmoc-Pen (Trt)-OH was coupled with the resin-bound peptide in lieu of Fmoc-Val to afford 30. Following final Fmoc removal and recovery of the peptide by cleavage from the resin, the crude peptide (33) was subjected to several oxidative folding conditions. Stirring in aqueous buffer (0.1 M tris, pH = 7.9) at room temperature or heating at 37°C gave no reaction or undesired by-products. Using 4,4′-dithiodipyridine (DTDP) in DMSO or the more reactive 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) in 4:1 HOAc:water gave mostly unoxidized peptide after 24 h and unknown by-products. Employing I2 in 4:1 HOAc:water resulted in quantitative conversion to the expected disulfide 34 (calc. M+2H)2 + 713.8, found 713.4) which was consistent with the expected loss of 2 Da.
Finally, a disulfide head-to-tail peptide5 analogue (37, Figure 8) was accessed from peptide 35 prepared on a 2-chlorotrityl resin. In this case, Cys3 required orthogonal protection on the sulfhydryl so that the disulfide bond between the two Cys at either end of the peptide could be formed selectively. In contrast to the oxidation of 33, oxidation of 35 proceeded readily in the air (0.1 M Tris buffer, pH = 8.1) with no exogenous reagents required to afford 36 (calc (M+2H)2 + 825.9, found 825.5). A noticeable shift in HPLC retention time (see SI) was observed for the disulfide-containing peptide 36 compared to the free thiol-containing peptide 35. The subsequent unmasking of Cys3 with TFA/TFMSA afforded 37 (calc. M+2H)2 + 800.9, found 800.4) in 22% yield.
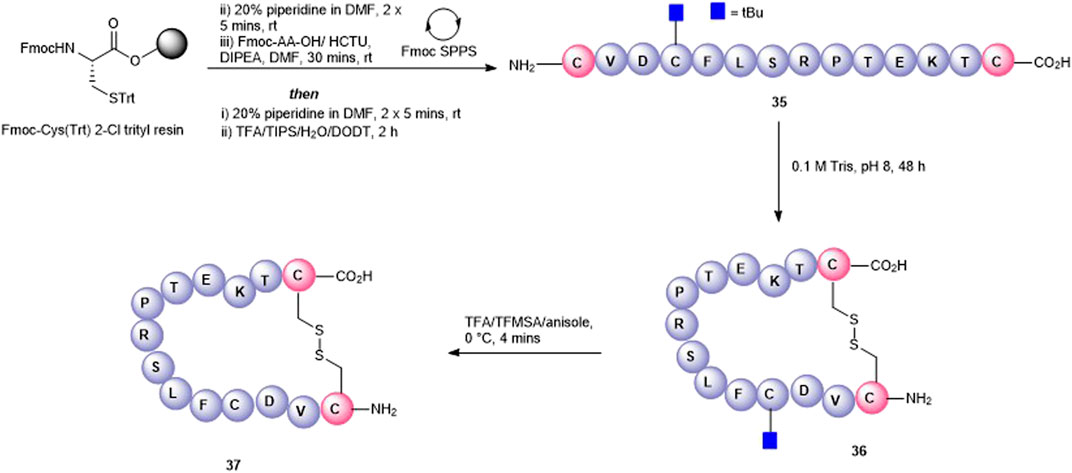
FIGURE 8. Synthesis of a disulfide-linked head-to-tail macrocycle 37 via introduction of Cys at the C- and N-termini.
Olefin metathesis has been demonstrated as an exquisite way to prepare cyclic peptides containing all carbon bridges (coined “stapling”) (Miller et al., 1996) and is most often used to stabilize alpha-helical peptides (Schafmeister et al., 2000). In our case, we employed olefin metathesis to prepare three further cyclic peptide5 variants; a head-to-tail analogue 39, a head-to-side chain analogue 42, and a tail-to-side-chain analogue 40. Fmoc-(S)-allylGly (AgI), a commercially available chiral alkenyl amino acid, was employed to introduce the required olefin at positions adjacent to the N- and C-termini and at Lys-11, a position known to tolerate substitution in gap junction unblocking.
The head-to-tail cyclic variant 39 was obtained through ring-closing metathesis (RCM) of peptidyl-resin 38 containing two allylglycine (Agl) residues adjacent to the N- and C-terminal amino acids using 30 mol% Hoveyda-Grubbs’ II catalyst in DMF/CH2Cl2 100°C for 2 h (Figure 9). These conditions had previously been employed by us in the synthesis of carba analogues of dianthin G (Amso et al., 2016). The side-chain–protecting groups were removed using the standard TFA cleavage cocktail to afford crude product. Although the expected product 39 was observed by HPLC and MS (calc [M + H]+ 1561.8 found 1560.0), the complicated HPLC profile precluded subsequent purification. The reaction conditions were modified to two 1-h treatments at 100°C using fresh 30 mol% Hoveyda-Grubbs’ II catalyst in each instance. This resulted in a better HPLC profile and the cyclic product (39) was easily purified.
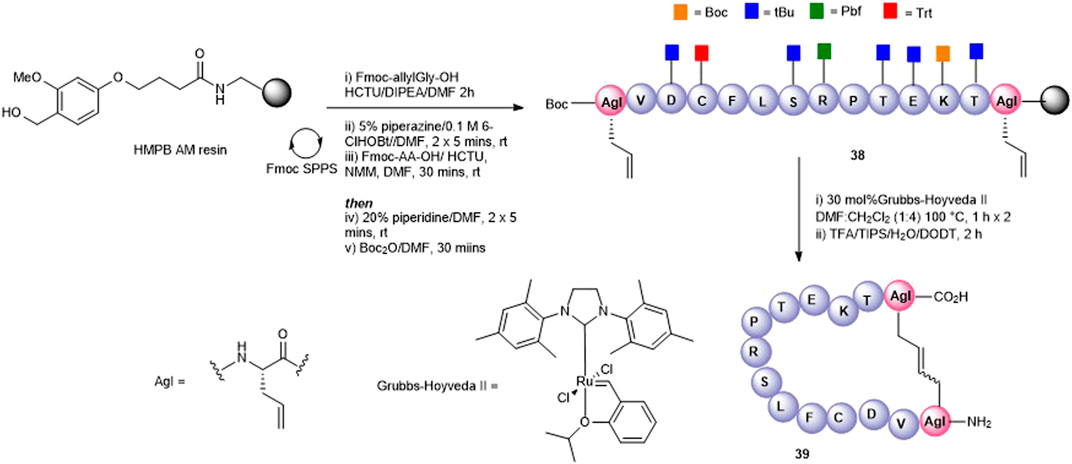
FIGURE 9. Synthesis of an all-carba head-to-tail macrocycle 37 via the introduction allylGly (AgI) at the C- and N-termini and ring-closing metathesis.
A tail-to-side-chain carba analogue (40) was prepared by RCM of peptidyl resin (41) containing an allylGly at the C-terminus and at Lys11 (Figure 10). A single treatment using 30 mol% Hoveyda–Grubbs’ II catalyst at 100°C for 2 h afforded the cyclized side-chain–protected product which upon cleavage from the resin and side-chain deprotection using the TFA cocktail yielded the cyclic RCM product 40 (calc [M + H]+ 1433.7, found 1433.5) which was recovered by semi-preparative HPLC. The clean HPLC profile in this instance may be reflective of the smaller macrocyclic ring formed in this analogue.
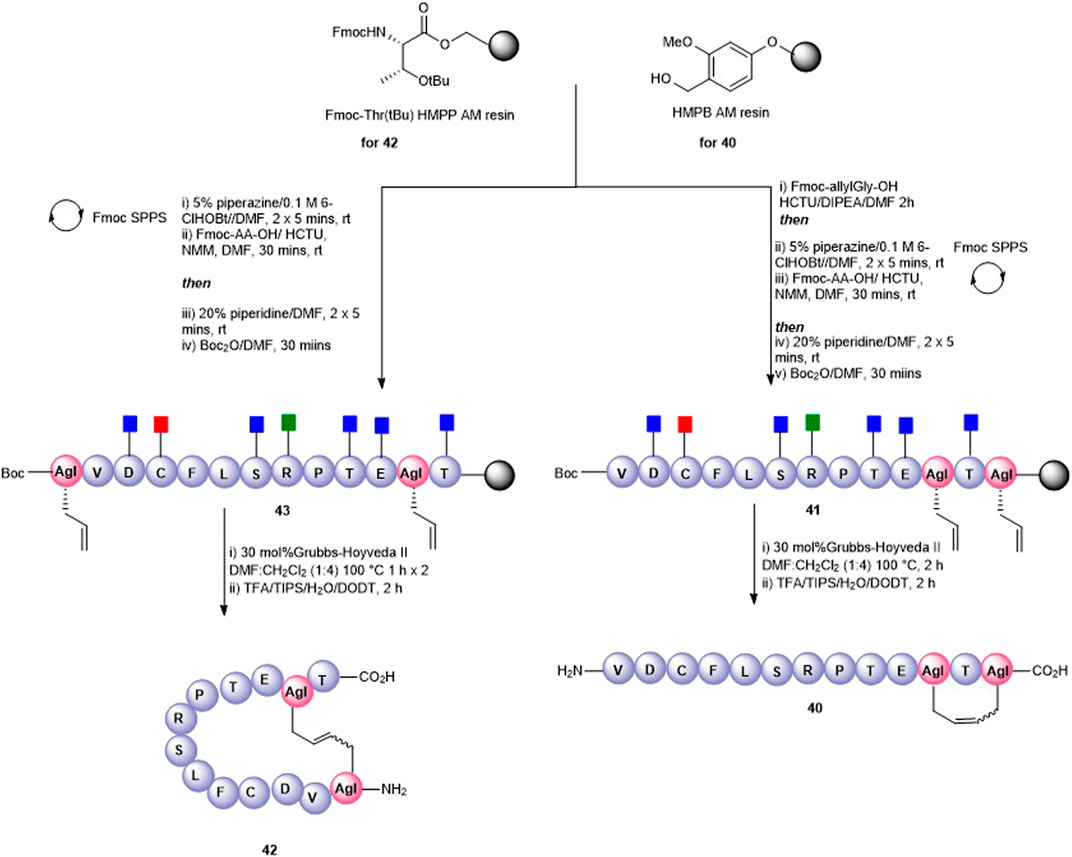
FIGURE 10. Synthesis of an all-carba head-to-side-chain macrocycle 42 and a tail-to-side-chain variant 40 by ring-closing metathesis.
Finally, a head-to-side-chain variant (42) was accessed through RCM of peptidyl resin 43 containing an allylGly at Lys11 and adjacent to the N-terminus (Figure 10). In this case, two 1-h treatments at 100°C each in the presence of 30 mol% Hoveyda–Grubbs’ II catalyst were required for complete conversion to the desired macrocycle 42. Following resin cleavage, an extremely clean HPLC chromatogram was observed (see SI) with the MS of the major peak being consistent for 42 (calc [M + H]+ 1434.7, found 1434.6).
For the three carba analogues 39, 42, and 40 obtained by RCM, it was postulated that a mixture of E and Z isomers were present. No attempt was made to determine the E/Z ratio or to separate out each individual isomer, and the analogues underwent biological testing as the presumed E/Z mixture.
2.2 Biological assay of linear and cyclic peptides to inhibit gap junctions and hemichannel opening
To test the effect of the peptide modifications on peptide5 (1) activity against hemichannels, a model of ischemic insult on a cell culture model of the brain microvasculature was used to induce Cx43 hemichannel opening and ATP release (Supplementary Material, Method 4). The cells were incubated in hypoxic-acidic ion-shifted ringers (HAIR) solution to generate disrupted pH and ion balance leading to ATP leak from the cells, which can be sampled and quantified luminometrically using a commercially available firefly luciferase assay (Molecular Probes). Consistent with our previous study, treatment with HAIR solution induced an approximately three-fold increase of ATP in the culture medium. The peptide5 (1) treatment caused a reduction of ATP release over untreated HAIR controls (49.4 ± 6.9%; p = <0.001. One-way ANOVA, Dunnet’s multiple comparisons post-hoc test) as did carbenoxolone (Cbx; 42.5 ± 3.4%; p = <0.001), an inhibitor of both Cx43 hemichannels and gap junctions (Figure 11).
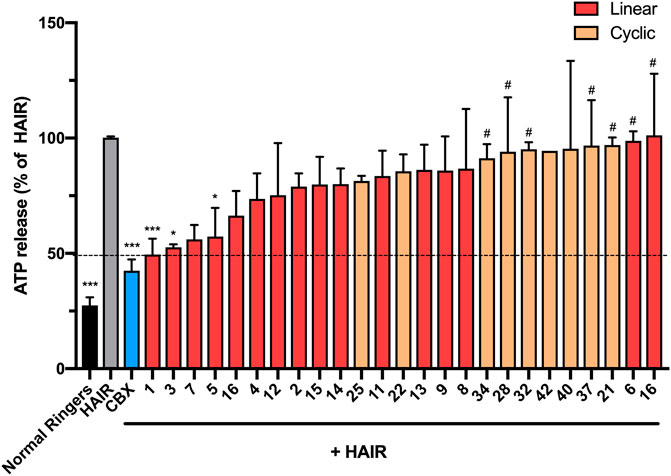
FIGURE 11. N-terminal, C-terminal, and cyclic modifications affect peptide5 (1) function against ischemic injury–induced ATP release from hCMVEC cells in vitro. N-terminal modified peptides 3 and 5 displayed efficacy equaling peptide5 (*p < 0.05, ***p < 0.001, respectively). N-terminal modified peptide 6 and the d-arginine–containing peptide 10 equaled the ATP release of the HAIR control indicating a complete lack of activity (#p < 0.05). None of the cyclic peptides led to a reduction in ATP release that was significantly different from HAIR control, and a number of peptides (34, 28, 32, and 37) demonstrated a level of ATP release significantly greater than peptide5, indicating loss of function of these peptides, #p < 0.05. All values represent the mean, SEM. One-way ANOVA, Dunnett’s comparison test against HAIR (*) or peptide5 (#).
Of the 15 linear analogues (2–16) of peptide5 (1) tested, the majority of these modifications did not reduce the release of ATP to comparable levels as peptide5 (1) (Figure 11) although generally, any modification to the native peptide5 (1) resulted in reduced or abolished activity. The addition of either an isobutyloxycarbonyl (3) or butanoyl (5) group lead to a statistically significant reduction in ATP release (52.6 ± 1.3%; p < 0.05 and 57.3 ± 12.4%; p < 0.05, respectively), equaling the efficacy of peptide5 (1) (Figure 11). Peptide 7 also appeared to reduce ATP release to a similar level as Peptide5 (1), but due to limited peptide, this could only be used for two experimental repeats and was not found to be statistically significant. Other N-terminal modifications involving the addition of an ethyloxycarbonyl (2), benzoyl (4), or masking of the amine as an azide (6) did not reduce ATP release compared to HAIR control, and ATP release from cells treated with modified peptide 6 was significantly increased compared to peptide5 (1). C-terminal modifications involving the removal of the carbonyl group (15) or using a thioester (16) also did not reduce ATP release. The addition of a C-terminal amide group (peptides 11–14) in conjunction with the N-terminal modifications used for peptides 2–5 did not reduce ATP release compared to HAIR, demonstrating the addition of a C-terminal amide group prevents the reduction in ATP release observed with peptides 3 and 5. Substitution of the lysine and arginine amino acids with an N-methyl-lysine (7), N-methyl-arginine (8), d-lysine (9), or d-arginine (10) did not reduce ATP release compared to HAIR-treated cells, and ATP release from cells treated with modified peptide 10 was significantly increased compared to peptide5 (1). None of the cyclic variants reduced the ATP levels (Figure 11). Rather, several of these showed a complete lack of activity compared to peptide5 (28, 32, 34, and 37; p < 0.05). However, a number of the peptides (28, 32, 34, and 37) showed significantly increased ATP release compared to peptide5 (1). This indicated that cyclization of peptide5 critically hinders the hemichannel blocking activity.
We next investigated the ability of peptide5 variants to inhibit gap junctional communication using a scrape loading assay. In this assay, the cells in culture are loaded with a dye (Lucifer Yellow dilithium salt (LY), M.W. 457.25 Da) of molecular size (<1 kDa) that is able to transit through the gap junctions and the degree of dye spread is then measured by counting the number of cells that contain LY (Supplementary Material, Method 4) 1. Consistent with our previous study, peptide5 (1) and Cbx reduced the degree of dye spread following incubation with LY (56.16 ± 2.1%; p < 0.0001 and 16.2 ± 1.1%; p < 0.0001, respectively) compared to the untreated controls (Figure 12). Of the 15 linear analogues (2–16) of peptide5 (1) tested, two reduced LY dye spread. These were peptide 7, containing an N-methyl lysine substitution, and 9, containing a d-lysine substitution which caused a statistically significant reduction in the number of LY-positive cells (64.3 ± 7.2%; p < 0.001 and 74.7 ± 10.4%; p < 0.01, respectively), and these were not statistically different from peptide5. The corresponding peptides with N-methyl arginine (8) and d-arginine (10) did not reduce the degree of dye spread compared to control, and dye spread in cells treated with modified peptide 10 was significantly increased compared to peptide5 (1). A number of additional peptides including those with N-modifications (2, 4, and 6), C-modification (15), and a C-terminal amide group in conjunction with the N-terminal modifications (11 and 14) also showed significantly increased dye spread relative to peptide5 (1).
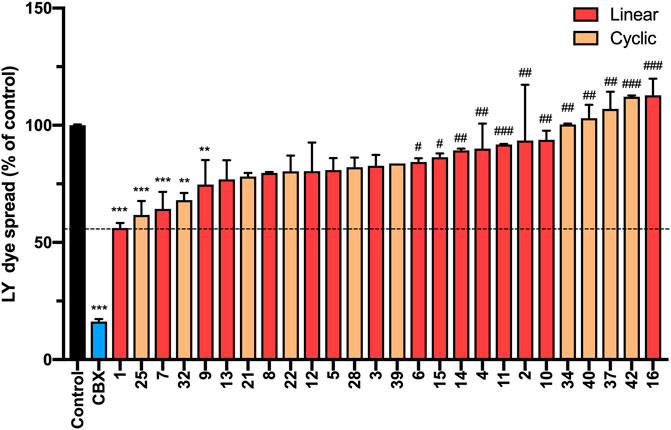
FIGURE 12. N-terminal, C-terminal, and cyclic modifications affect peptide5 (1) function against gap junctional coupling in hCMVEC cells in vitro. Positive LY dye transfer between coupled gap junction channels is represented as a percentage of the untreated control. LY dye transfer was significantly reduced compared to untreated control following treatment with Cbx or peptide5 (1) (****p < 0.0001). The peptides substituted with an N-methyl lysine (7) or d-lysine (9) prevented the spread of LY to a level comparable to peptide5 (1) (**p < 0.01, ***p < 0.001, respectively). A number of modified peptides demonstrated a level of LY dye transfer significantly greater than peptide5 and not different from control, indicating complete loss of function of these peptides, #p < 0.05. The cyclic peptides 25 and 32 significantly reduced LY transfer compared to control but not peptide5 **p < 0.01, ***p < 0.001. The cyclic peptides 40, 34, and 42 demonstrated a level of LY dye transfer significantly greater than peptide5 and not different from control, indicating loss of function of these peptides, ###p < 0.001, ####p < 0.0001. All values represent the mean, SEM. One-way ANOVA, Dunnett’s comparison test against control (*) or peptide5 (#).
Of the 8 cyclic peptides tested, peptides 25 and 32 caused a statistically significant reduction in the number of LY-positive cells (61.7 ± 5.9%; p < 0.001 and 68.0 ± 3.0%; p < 0.01, respectively) compared to control that was not statistically different from peptide5 (1) (Figure 12), while peptides 34 and 42 had levels of dye spread that were significantly greater than that observed with peptide5 (1).
2.3 Serum Stability of Selected Analogs
One of the limitations of using peptide5 (1) as a therapeutic is its poor half-life. The half-lives of peptide therapeutics can be significantly improved by N- and C-terminal functional group masking, cyclization, and conversion of susceptible amide bonds using D- or N-methylated amino acids, and these methods have been successfully used to progress peptides to the clinic (Yao et al., 2018; Khatri et al., 2019). Our previous studies of lipidated peptide5 demonstrated that the native sequence was completely degraded in rat serum in less than 5 min (Yang et al., 2020). In the present work, we instead employed the more relevant human serum and established that peptide5 (1) was relatively stable and degraded completely within 10 h. We then evaluated the N-methylated variants 7 and 8, D amino acid variants 9 and 10, and the two carbo cyclic peptides 39 and 42. Each peptide was incubated with 25% aq. human serum at 37°C, and 6 aliquots were extracted within 48 h. The stability of each peptide upon exposure to human serum was then analyzed via analytical RP HPLC, and the amount of peptide still present at each time point was recorded relative to t = 0 h. The results are presented in Figure 13.
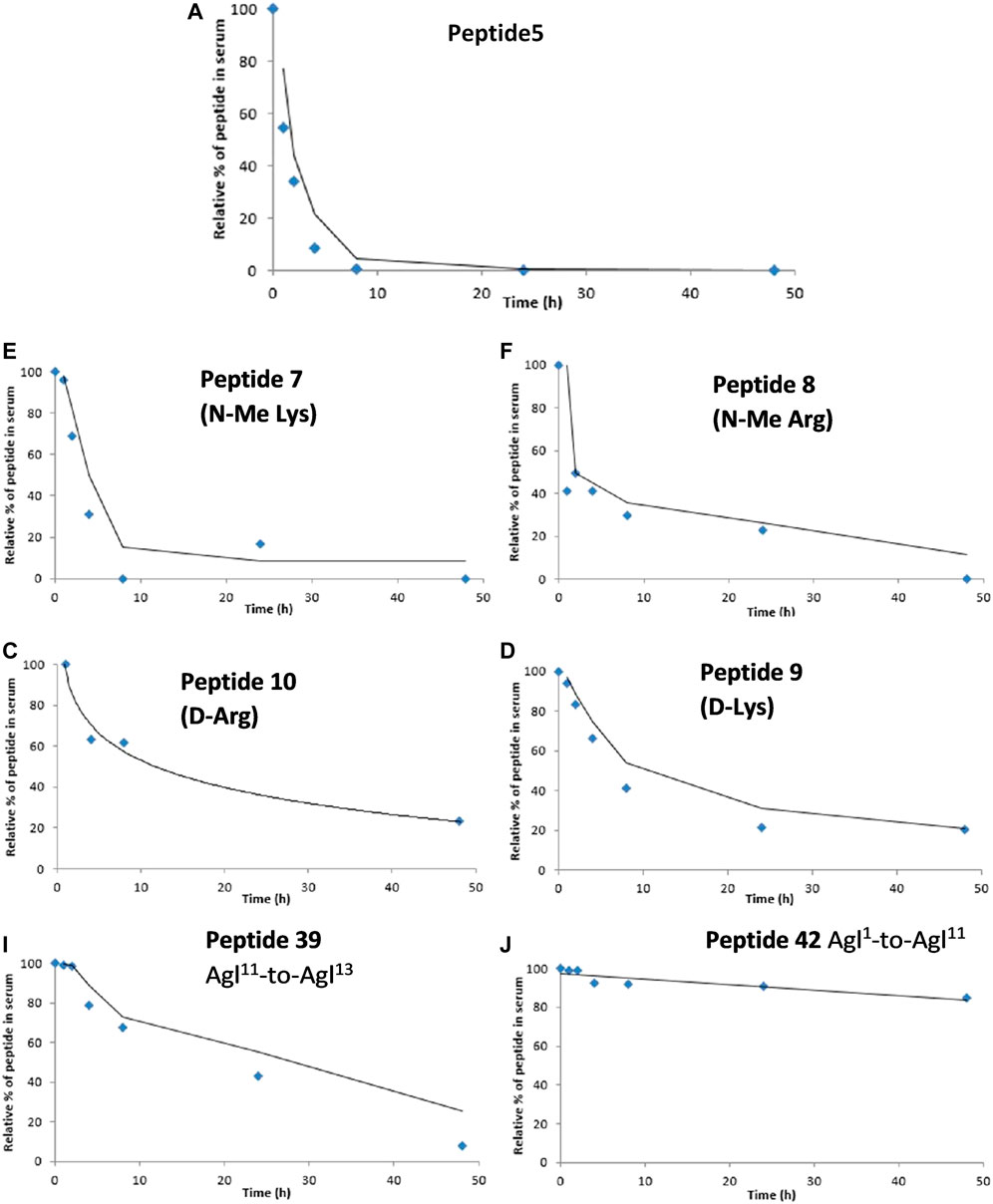
FIGURE 13. Degradation studies of peptide5 and selected analogues in 25% human serum at 37°C via monitoring by HPLC at 210 nm.
N-methylated peptides, 7 and 8, showed a similar degradation profile to peptide5 (1), although the rate of degradation was slower. Peptide5 (1) containing either a D-Arg or D-Lys showed good stability with ca. 30% remaining after 48 h suggesting that Ser-L-Arg and Glu-L-Lys sites were susceptible to enzymatic cleavage and that replacement with the D amino acids conferred improved stability. The side-chain-to-tail carba peptide5 analogue 39 exhibited good stability over 48 h, but the head-to-tail analogue 42, was extremely resistant to enzymatic degradation, and ca. 90% of the peptide remained intact at the completion of the experiment.
2.4 Discussion
Our goal was to apply a peptidomimetic strategy to improve the half-life of connexin channel inhibitor peptide5 (1), whist maintaining its ability to bind to connexin hemichannels and to moderate gap junctions. In the hemichannel assay, N-lipidated peptide5 analogues with short alkyl lipids (3 and 5) showed the comparable ability of peptide5 to block ATP release whilst an N-terminal azido (6) or D-Arg (10) variants were both poor at blocking hemichannels. This suggests that only certain N-terminal modifications are tolerated. Thus, whereas a short N-terminal lipid may interact with the lipid membrane to anchor peptide5 (1), the conversion of the amino group to an azide was detrimental, suggesting that the N-terminal amino group is important. At physiological pH, the amine group is protonated; when acylated or present as an azide, the positive charge is lost which may be compensated by the lipid at the N-terminus that can bind to the lipid membrane. The D-Arg substitution (10) also resulted in poor activity in the hemichannel assay. Given that alanine scan data showed that L-Arg could be substituted by L-Ala, this would suggest a stereochemical requirement as well as an interaction of the guanidyl group present in Arg for effective peptide5 (1) binding. Of the cyclic peptides prepared, none were able to reduce ATP lost to a level seen with the Cbx control or with peptide5 (1). While this may be logical given that we have shown that alanine substitutions at Asp2, Cys3, Leu5, Arg7, Pro8, and Lys11 were not tolerated in this assay, the head-to-tail cyclic amide analogues 21 and 22 and the disulfide cyclic variant 37 retain all side-chain functionality. This suggests that a less rigid linearized sequence is optimal for binding to Cx43.
In the gap junction assay, both an N-MeLys (7) and a D-Lys (9) substitution resulted in gap junction uncoupling comparable to that of peptide5 (1), and both exhibited improved human serum stability relative to peptide5. It should be noted that the N-Me-Lys congener (7) also appeared effective in the hemichannel assay and was the next best analogue after the N-lipidated analogues 3 and 5. The incorporation of a D-Arg resulted in poor activity in line with the hemichannel data, thus confirming that an L-Arg is critical for both hemichannel blocking and gap junction uncoupling.
Examination of the cyclic peptides to inhibit the gap junctions revealed that side-chain Asp4-Lys11, side-chain–cyclized peptide 25, and Cys3-Val1 cyclic peptide 32 were not as effective as peptide5 (1) but were better than most other cyclic peptides. As detailed in our alanine scan data, all positions in peptide5 (1) were able to be modified without adversely affecting gap junction blocking. However, given that the other cyclic compounds were poor, this suggests that positioning and composition of the cycle are important. The disulfide-containing cyclic compound 37 which retains all the side-chain amino acid functionality as well as the amino and carboxylate groups at the N- and C-termini, respectively, was a poor gap junction inhibitor as was the all-carba analogue 42. Both 42 and 37 contain significantly larger macrocyclic rings compared to 25 and 32, indicative of a preference for a smaller ring size to be optimal for cyclic variants of peptide5 (1). Nonetheless, the results demonstrated herein establish that cyclic variants of peptide5 (1) are able to retain some gap junction activity which was not the case for hemichannel blocking.
The goal of increasing the half-life of peptide5 whilst retaining activity has been met. The simple substitution of L- and D-Arg with D- or N-Me amino acids overall improved their stability. The half-life of peptide5 is estimated from Figure 13 to be ca. 2–3 h; its D-Arg congener was 5-fold higher with 50% remaining after 14–15 h, and the D-Lys peptide, 4-fold higher than peptide5 with 50% remaining after 12–13 h. Whilst not as noticeable, the N-Me-Lys variant also showed a modest increase in stability relative to peptide5. The cyclic all-carba analogues 39 and 42 were the most stable in human serum. These peptides are the most chemically modified with a 4-atom carbon bridge linking the peptide backbone with an additional rigidifying double bond. While this improved their stability, this was at the expense of both hemichannel and gap junction activity. There may therefore be scope to examine other stapling methodologies that include less rigid structures.
3 Conclusion
The abovementioned data have highlighted that a linearized peptide5 is the key design principle to ensure dual hemichannel and connexin activity. In the linear peptide5 analogues prepared, only those that had a small modification at the N-terminus, namely, an isobutyloxycarbonyl or butanoyl functional group, exhibited similar activity to peptide5 in the hemichannel assay. The only other exception was an N-MeLys replacement for the canonical L-Lys; remarkably, a D-Arg substitution for L-Arg abolished hemichannel activity completely, corroborating the sensitive relationship between individual amino acid substitutions and loss of activity. Converting the linear sequence to cyclic analogues containing several different cyclization modes was not tolerated in the hemichannel assay although an Asp-Lys side-chain–cyclized peptide performed well in the gap junction assay. Further work focusing on the development of peptide-based mimetics of peptide5 (1) should thus concentrate on the modification of the linear peptide5 sequences containing an N-acylated terminus or an N-alkylated Lys rather than cyclic variants.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
SCC designed and performed the peptide synthesis and peptide stability. JG and CC designed and performed the biological assays. MB oversaw the peptide synthesis. SJO designed and oversaw the biological assays and co-wrote the manuscript. PWH designed and performed the peptide synthesis and co-wrote the manuscript.
Funding
The authors wish to thank the Maurice Wilkins Centre for Molecular Biodiscovery for financial support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.877618/full#supplementary-material
References
Amso, Z., Kowalczyk, R., Park, Y.-E., Watson, M., Lin, J.-m., Musson, D. S., et al. (2016). Synthesis and In Vitro Bone Cell Activity of Analogues of the Cyclohexapeptide Dianthin G. Org. Biomol. Chem. 14, 6231–6243. doi:10.1039/C6OB00983B
Bechtler, C., and Lamers, C. (2021). Macrocyclization Strategies for Cyclic Peptides and Peptidomimetics. RSC Med. Chem. 12, 1325–1351. doi:10.1039/D1MD00083G
Camarero, J. A., Adeva, A., and Muir, T. W. (2000). 3-thiopropionic Acid as a Highly Versatile Multidetachable Thioester Resin Linker. Lett. Pept. Sci. 7, 17–21. doi:10.1007/bf02443557
Chatterjee, J., Laufer, B., and Kessler, H. (2012). Synthesis of N-Methylated Cyclic Peptides. Nat. Protoc. 7, 432–444. doi:10.1038/nprot.2011.450
D’Aloisio, V., Dognini, P., Hutcheon, G. A., and Coxon, C. R. (2021). PepTherDia: Database and Structural Composition Analysis of Approved Peptide Therapeutics and Diagnostics. Drug Discov. Today 26, 1409–1419. doi:10.1016/j.drudis.2021.02.019
Danesh-Meyer, H. V., Kerr, N. M., Zhang, J., Eady, E. K., O'Carroll, S. J., Nicholson, L. F. B., et al. (2012). Connexin43 Mimetic Peptide Reduces Vascular Leak and Retinal Ganglion Cell Death Following Retinal Ischaemia. Brain 135, 506–520. doi:10.1093/brain/awr338
Dang, B., Kubota, T., Mandal, K., Bezanilla, F., and Kent, S. B. H. (2013). Native Chemical Ligation at Asx-Cys, Glx-Cys: Chemical Synthesis and High-Resolution X-Ray Structure of ShK Toxin by Racemic Protein Crystallography. J. Am. Chem. Soc. 135, 11911–11919. doi:10.1021/ja4046795
Davidson, J. O., Drury, P. P., Green, C. R., Nicholson, L. F., Bennet, L., and Gunn, A. J. (2014). Connexin Hemichannel Blockade Is Neuroprotective after Asphyxia in Preterm Fetal Sheep. PLoS One 9, e96558. doi:10.1371/journal.pone.0096558
Davidson, J. O., Green, C. R., B. Nicholson, L. F., O'Carroll, S. J., Fraser, M., Bennet, L., et al. (2012). Connexin Hemichannel Blockade Improves Outcomes in a Model of Fetal Ischemia. Ann. Neurol. 71, 121–132. doi:10.1002/ana.22654
Dawson, P. E., Muir, T. W., Clark-Lewis, I., and Kent, S. B. H. (1994). Synthesis of Proteins by Native Chemical Ligation. Science 266, 776–779. doi:10.1126/science.7973629
De Bock, M., Culot, M., Wang, N., Bol, M., Decrock, E., De Vuyst, E., et al. (2011). Connexin Channels Provide a Target to Manipulate Brain Endothelial Calcium Dynamics and Blood-Brain Barrier Permeability. J. Cereb. Blood Flow. Metab. 31, 1942–1957. doi:10.1038/jcbfm.2011.86
de la Torre, B. G., and Albericio, F. (2020). Peptide Therapeutics 2.0. Molecules 25, 2293. doi:10.3390/molecules25102293
Decrock, E., De Bock, M., Wang, N., Bultynck, G., Giaume, C., Naus, C. C., et al. (2015). Connexin and Pannexin Signaling Pathways, an Architectural Blueprint for CNS Physiology and Pathology? Cell. Mol. Life Sci. 72, 2823–2851. doi:10.1007/s00018-015-1962-7
D’hondt, C., Iyyathurai, J., Wang, N., Gourdie, R. G., Himpens, B., Leybaert, L., et al. (2013). Negatively Charged Residues (Asp378 and Asp379) in the Last Ten Amino Acids of the C-Terminal Tail of Cx43 Hemichannels Are Essential for Loop/tail Interactions. Biochem. Biophysical Res. Commun. 432, 707–712. doi:10.1016/j.bbrc.2013.01.066
Erak, M., Bellmann-Sickert, K., Els-Heindl, S., and Beck-Sickinger, A. G. (2018). Peptide Chemistry Toolbox - Transforming Natural Peptides into Peptide Therapeutics. Bioorg. Med. Chem. 26, 2759–2765. doi:10.1016/j.bmc.2018.01.012
Eugenin, E. A., Clements, J. E., Zink, M. C., and Berman, J. W. (2011). Human Immunodeficiency Virus Infection of Human Astrocytes Disrupts Blood-Brain Barrier Integrity by a Gap Junction-dependent Mechanism. J. Neurosci. 31, 9456–9465. doi:10.1523/JNEUROSCI.1460-11.2011
Evans, W. H. (2015). Cell Communication across Gap Junctions: a Historical Perspective and Current Developments. Biochem. Soc. Trans. 43, 450–459. doi:10.1042/BST20150056
Froger, N., Orellana, J. A., Calvo, C.-F., Amigou, E., Kozoriz, M. G., Naus, C. C., et al. (2010). Inhibition of Cytokine-Induced Connexin43 Hemichannel Activity in Astrocytes Is Neuroprotective. Mol. Cell. Neurosci. 45, 37–46. doi:10.1016/j.mcn.2010.05.007
Gentilucci, L., De Marco, R., and Cerisoli, L. (2010). Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-natural Amino Acids, Pseudo-peptide Bonds, and Cyclization. Curr. Pharm. Des. 16, 3185–3203. doi:10.2174/138161210793292555
Goddard-Borger, E. D., and Stick, R. V. (2007). An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-Sulfonyl Azide Hydrochloride. Org. Lett. 9, 3797–3800. doi:10.1021/ol701581g
Góngora-Benítez, M., Tulla-Puche, J., and Albericio, F. (2014). Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 114, 901–926. doi:10.1021/cr400031z
Guo, C. X., Mat Nor, M. N., Danesh-Meyer, H. V., Vessey, K. A., Fletcher, E. L., O'Carroll, S. J., et al. (2016). Connexin43 Mimetic Peptide Improves Retinal Function and Reduces Inflammation in a Light-Damaged Albino Rat Model. Invest. Ophthalmol. Vis. Sci. 57, 3961–3973. doi:10.1167/iovs.15-16643
Johnson, E. C. B., and Kent, S. B. H. (2006). Insights into the Mechanism and Catalysis of the Native Chemical Ligation Reaction. J. Am. Chem. Soc. 128, 6640–6646. doi:10.1021/Ja058344i
Khatri, B., Nuthakki, V. R., and Chatterjee, J. (2019). “Strategies to Enhance Metabolic Stabilities,” in Cyclic Peptide Design. Editor G. Goetz (New York, NY: Springer New York), 17–40. doi:10.1007/978-1-4939-9504-2_2
Kim, Y., Griffin, J. M., Harris, P. W. R., Chan, S. H. C., Nicholson, L. F. B., Brimble, M. A., et al. (2017a). Characterizing the Mode of Action of Extracellular Connexin43 Channel Blocking Mimetic Peptides in an In Vitro Ischemia Injury Model. Biochimica Biophysica Acta (BBA) - General Subj. 1861, 68–78. doi:10.1016/j.bbagen.2016.11.001
Kim, Y., Griffin, J. M., Harris, P. W. R., Chan, S. H. C., Nicholson, L. F. B., Brimble, M. A., et al. (2017b). Characterizing the Mode of Action of Extracellular Connexin43 Channel Blocking Mimetic Peptides in an In Vitro Ischemia Injury Model. Biochimica Biophysica Acta (BBA) - General Subj. 1861 (2), 68–78. doi:10.1016/j.bbagen.2016.11.001
Krysko, D. V., Leybaert, L., Vandenabeele, P., and D’Herde, K. (2005). Gap Junctions and the Propagation of Cell Survival and Cell Death Signals. Apoptosis 10, 459–469. doi:10.1007/s10495-005-1875-2
Kumagai, H., Tajima, M., Ueno, Y., Giga-Hama, Y., and Ohba, M. (1991). Effect of Cyclic RGD Peptide on Cell Adhesion and Tumor Metastasis. Biochem. Biophysical Res. Commun. 177, 74–82. doi:10.1016/0006-291X(91)91950-H
Kuo, C., Green, C. R., Rupenthal, I. D., and Mugisho, O. O. (2020). Connexin43 Hemichannel Block Protects against Retinal Pigment Epithelial Cell Barrier Breakdown. Acta Diabetol. 57, 13–22. doi:10.1007/s00592-019-01352-3
Li, H., Liu, T. F., Lazrak, A., Peracchia, C., Goldberg, G. S., Lampe, P. D., et al. (1996). Properties and Regulation of Gap Junctional Hemichannels in the Plasma Membranes of Cultured Cells. J. Cell Biol. 134, 1019–1030. doi:10.1083/jcb.134.4.1019
Li, X., Kawakami, T., and Aimoto, S. (1998). Direct Preparation of Peptide Thioesters Using an Fmoc Solid-phase Method. Tetrahedron Lett. 39, 8669–8672. doi:10.1016/s0040-4039(98)01868-1
Mao, Y., Tonkin, R. S., Nguyen, T., O'Carroll, S. J., Nicholson, L. F. B., Green, C. R., et al. (2017). Systemic Administration of Connexin43 Mimetic Peptide Improves Functional Recovery after Traumatic Spinal Cord Injury in Adult Rats. J. Neurotrauma 34, 707–719. doi:10.1089/neu.2016.4625
Márquez-Rosado, L., Solan, J. L., Dunn, C. A., Norris, R. P., and Lampe, P. D. (2012). Connexin43 Phosphorylation in Brain, Cardiac, Endothelial and Epithelial Tissues. Biochimica Biophysica Acta (BBA) - Biomembr. 1818, 1985–1992. doi:10.1016/j.bbamem.2011.07.028
Miller, S. J., Blackwell, H. E., and Grubbs, R. H. (1996). Application of Ring-Closing Metathesis to the Synthesis of Rigidified Amino Acids and Peptides. J. Am. Chem. Soc. 118, 9606–9614. doi:10.1021/ja961626l
Mugisho, O. O., Green, C. R., Squirrell, D. M., Bould, S., Danesh-Meyer, H. V., Zhang, J., et al. (2019). Connexin43 Hemichannel Block Protects against the Development of Diabetic Retinopathy Signs in a Mouse Model of the Disease. J. Mol. Med. 97, 215–229. doi:10.1007/s00109-018-1727-5
Muttenthaler, M., King, G. F., Adams, D. J., and Alewood, P. F. (2021). Trends in Peptide Drug Discovery. Nat. Rev. Drug Discov. 20, 309–325. doi:10.1038/s41573-020-00135-8
O'Carroll, S. J., Alkadhi, M., Nicholson, L. F. B., and Green, C. R. (2008). Connexin43 Mimetic Peptides Reduce Swelling, Astrogliosis, and Neuronal Cell Death after Spinal Cord Injury. Cell Commun. Adhesion 15, 27–42. doi:10.1080/15419060802014164
O’Carroll, S. J., Gorrie, C. A., Velamoor, S., Green, C. R., and Nicholson, L. F. B. (2013). Connexin43 Mimetic Peptide Is Neuroprotective and Improves Function Following Spinal Cord Injury. Neurosci. Res. 75, 256–267. doi:10.1016/j.neures.2013.01.004
O. Davidson, J., R. Green, C., Bennet, L., F.B. Nicholson, L., Danesh-Meyer, H., J. O'Carroll, S., et al. (2013). A Key Role for Connexin Hemichannels in Spreading Ischemic Brain Injury. Curr. Drug Targets 14, 36–46. doi:10.2174/138945013804806479
Oyamada, M., Oyamada, Y., and Takamatsu, T. (2005). Regulation of Connexin Expression. Biochimica Biophysica Acta (BBA) - Biomembr. 1719, 6–23. doi:10.1016/j.bbamem.2005.11.002
Philippe, G. J. B., Craik, D. J., and Henriques, S. T. (2021). Converting Peptides into Drugs Targeting Intracellular Protein-Protein Interactions. Drug Discov. Today 26, 1521–1531. doi:10.1016/j.drudis.2021.01.022
Quist, A. P., Rhee, S. K., Lin, H., and Lal, R. (2000). Physiological Role of Gap-Junctional Hemichannels. J. Cell Biol. 148, 1063–1074. doi:10.1083/jcb.148.5.1063
Ribeiro-Rodrigues, T. M., Martins-Marques, T., Morel, S., Kwak, B. R., and Girão, H. (2017). Role of Connexin 43 in Different Forms of Intercellular Communication - Gap Junctions, Extracellular Vesicles and Tunnelling Nanotubes. J. Cell Sci. 130, 3619–3630. doi:10.1242/jcs.200667
Robey, F. A., and Fields, R. L. (1989). Automated Synthesis of N-Bromoacetyl-Modified Peptides for the Preparation of Synthetic Peptide Polymers, Peptide-Protein Conjugates, and Cyclic Peptides. Anal. Biochem. 177, 373–377. doi:10.1016/0003-2697(89)90068-7
Rodríguez-Sinovas, A., Cabestrero, A., López, D., Torre, I., Morente, M., Abellán, A., et al. (2007). The Modulatory Effects of Connexin 43 on Cell Death/survival beyond Cell Coupling. Prog. Biophysics Mol. Biol. 94, 219–232. doi:10.1016/j.pbiomolbio.2007.03.003
Sarrouilhe, D., Dejean, C., and Mesnil, M. (2017). Connexin43- and Pannexin-Based Channels in Neuroinflammation and Cerebral Neuropathies. Front. Mol. Neurosci. 10, 320. doi:10.3389/fnmol.2017.00320
Schafmeister, C. E., Po, J., and Verdine, G. L. (2000). An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 122, 5891–5892. doi:10.1021/Ja000563a
Tonkin, R. S., Mao, Y., O’Carroll, S. J., Nicholson, L. F. B., Green, C. R., Gorrie, C. A., et al. (2015). Gap Junction Proteins and Their Role in Spinal Cord Injury. Front. Mol. Neurosci. 7, 102. doi:10.3389/fnmol.2014.00102
van Witteloostuijn, S. B., Pedersen, S. L., and Jensen, K. J. (2016). Half-Life Extension of Biopharmaceuticals Using Chemical Methods: Alternatives to PEGylation. ChemMedChem 11, 2474–2495. doi:10.1002/cmdc.201600374
Villain, M., Gaertner, H., and Botti, P. (2003). Native Chemical Ligation with Aspartic and Glutamic Acids asC-Terminal Residues: Scope and Limitations. Eur. J. Org. Chem., 2003, 3267–3272. doi:10.1002/ejoc.200300032
Xing, L., Yang, T., Cui, S., and Chen, G. (2019). Connexin Hemichannels in Astrocytes: Role in CNS Disorders. Front. Mol. Neurosci. 12, Available at: https://www.frontiersin.org/article/10.3389/fnmol.2019.00023 (Accessed May 11, 2022). doi:10.3389/fnmol.2019.00023
Yang, S.-H., Clemett, C. A., Brimble, M. A., O'Carroll, S. J., and Harris, P. W. R. (2020). Synthesis and Biological Evaluation of S-Lipidated Lipopeptides of a Connexin 43 Channel Inhibitory Peptide. RSC Med. Chem. 11, 1041–1047. doi:10.1039/D0MD00172D
Yao, J.-F., Yang, H., Zhao, Y.-Z., and Xue, M. (2018). Metabolism of Peptide Drugs and Strategies to Improve Their Metabolic Stability. Curr. Drug. Metab. 19, 892–901. doi:10.2174/1389200219666180628171531
Keywords: connexin43, gap junctions, hemichannels, peptide5, peptide mimetics
Citation: Crystal Chan SH, Griffin JM, Clemett CA, Brimble MA, O’Carroll SJ and Harris PWR (2022) Synthesis and Biological Evaluation of Termini-Modified and Cyclic Variants of the Connexin43 Inhibitor Peptide5. Front. Chem. 10:877618. doi: 10.3389/fchem.2022.877618
Received: 16 February 2022; Accepted: 20 May 2022;
Published: 13 September 2022.
Edited by:
John D. Wade, University of Melbourne, AustraliaReviewed by:
Richard J Clark, The University of Queensland, AustraliaThomas Handley, University of Melbourne, Australia
Copyright © 2022 Crystal Chan, Griffin, Clemett, Brimble, O’Carroll and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon J. O’Carroll, cy5vY2Fycm9sbEBhdWNrbGFuZC5hYy5ueg==; Paul W. R. Harris, cGF1bC5oYXJyaXNAYXVja2xhbmQuYWMubno=