 Nur Intan Saidaah Mohamed Yusof
Nur Intan Saidaah Mohamed Yusof Zafirah Liyana Abdullah1
Zafirah Liyana Abdullah1 Fazlin Mohd Fauzi
Fazlin Mohd Fauzi- 1Faculty of Pharmacy, Universiti Teknologi MARA Selangor, Selangor, Malaysia
- 2Haematology Unit, Cancer Research Centre, Institute for Medical Research, National Institutes of Health Complex, Selangor, Malaysia
- 3Collaborative Drug Discovery Research, Faculty of Pharmacy, Universiti Teknologi MARA Selangor, Selangor, Malaysia
Drug development in Alzheimer’s disease (AD) suffers from a high attrition rate. In 2021, 117 agents tested in phases I and II and 36 agents tested in phase III were discontinued. Natural product compounds may be good lead compounds for AD as they contain functional groups that are important for binding against key AD targets such as β-secretase enzyme (BACE1). Hence, in this study, 64 flavonoids collected from rigorous literature search and screening that have been tested from 2010 to 2022 against BACE1, which interferes in the formation of amyloid plaque, were analyzed. The 64 unique flavonoids can be further classified into five core fragments. The flavonoids were subjected to clustering analysis based on its structure, and each representative of the clusters was subjected to molecular docking. There were 12 clusters formed, where only 1 cluster contained compounds from two different core fragments. Several observations can be made where 1) flavanones with sugar moieties showed higher inhibitory activity compared to flavanones without sugar moieties. The number of sugar moieties and position of glycosidic linkage may also affect the inhibitory activity. 2) Non-piperazine-substituted chalcones when substituted with functional groups with decreasing electronegativity at the para position of both rings result in a decrease in inhibitory activity. Molecular docking indicates that ring A is involved in hydrogen bond, whereas ring B is involved in van der Waals interaction with BACE1. 3) Hydrogen bond is an important interaction with the catalytic sites of BACE1, which are Asp32 and Asp228. As flavonoids contain favorable structures and properties, this makes them an interesting lead compound for BACE1. However, to date, no flavonoids have made it through clinical trials. Hence, these findings may aid in the design of highly potent and specific BACE1 inhibitors, which could delay the progression of AD.
Introduction
Alzheimer’s disease (AD) is a disorder characterized by the progressive degeneration of the structure and function of the central nervous system (Plaingam et al., 2017). Globally, individuals with this disorder have steadily increased in numbers, where in 2016, there were 43.8 million cases reported, which is expected to increase to 131.5 million by 2050. In Malaysia, it is estimated that in 2016, there were about 50,000 individuals diagnosed with AD, and this number is projected to increase to 123,000 by 2030 (Feigin et al., 2019). AD is an irreversible disease where it will slowly affect memory and thinking skills. If the disease is not properly managed, the person diagnosed with AD may not be able to carry out daily activities (Nazarko, 2019). In 2021, drug development for all types of AD suffered from a high attrition rate (around 95%), where 117 agents tested in phases I and II and 36 agents tested in phase III were discontinued (Cummings et al., 2021). In 2020, clinical trials for early-to-mild and mild-to-moderate phases of AD involving semagacestat, bapineuzumab, and solanezumab were halted due to lack of improvement in cognitive function. While these drugs showed good results in phases I and II such as reducing the CSF biomarker level, little or no significant improvement in cognitive and functional endpoints were observed during phase III. An increase in dose did not display a change in the cognitive and functional endpoints but resulted in several severe toxic effects such as vascular edema in bapineuzumab (Huang et al., 2020). Currently, only six drugs are approved by the Food and Drug Administration (FDA), which are donepezil, rivastigmine, galantamine, memantine, combination capsule of memantine with donepezil, and aducanumab (Cummings et al., 2021). Aducanumab is the first disease-modifying drug that has been approved by the FDA in June 2021. Although aducanumab clears the amyloid plaque, its efficacy in improving cognitive function is still lacking in evidence (Walsh et al., 2021). This highlights the insufficient evidence of the clinical benefit as well as the narrow therapeutic index of potential AD drugs as some of the challenges faced in the development of AD drugs (Mehta et al., 2017).
The neuronal cell death that occurs in AD is due to the molecular and cellular changes in the brain. It is characterized by the presence of extracellular amyloid-β (Aβ) proteins in senile plaques and intracellular deposits of tau (τ) proteins in neurofibrillary tangles (NFTs) that affect memory and cognition (Hampel et al., 2020). Amyloid precursor protein (APP) is an integral membrane protein found in various tissues, such as synapses of neurons. APP acts as a regulator of synaptic formation and repair, anterograde neuronal transport, and iron export. In healthy neurons, APP is cleaved by an enzyme known as α-secretase through the non-amyloidogenic pathway and produces amyloid precursor protein alpha-secretase (APPsα) ectodomain and membrane-bound carboxyl-terminal fragment (CTF) known as C83. Then, C83 is cleaved by γ-secretase to form p3 and APP intracellular domain (AICD) (Chen et al., 2017). The activity of α-secretase is associated with proteases such as tumor necrosis factor-alpha converting enzyme (TACE), disintegrin, and metalloproteinase domain-containing protein 9 (ADAM9) and ADAM10 (Chen et al., 2017).
However, in AD, β-secretase enzyme (BACE1) and γ-secretase cleave APP to form amyloid beta (Aβ), an insoluble peptide through the amyloidogenic pathway. APP is first cleaved by BACE1 to produce two protein fragments, which are APPsβ (secreted ectodomain) and C99 (membrane-bound fragment) (Ashrafian et al., 2020). C99 will then be cleaved by γ-secretase, which is composed of proteins such as anterior pharynx defective 1 (APH1), presenilin enhancer 2 (PEN2), nicastrin and presenilin (PS1 or PS2), and forms AICD and C-terminus of Aβ peptides. The insoluble Aβ peptide, which is made up of 42 amino acids, can aggregate and form amyloid plaques in the brain, a pathological hallmark of AD (Ashrafian et al., 2020). Hence, inhibiting BACE1 could interfere in the formation of amyloid plaque since it is the initial and rate-limiting step of Aβ production. Several inhibitors against BACE1 have been developed over the past few years (Abeysinghe et al., 2020; Hampel et al., 2021; Zhumanova et al., 2021); however, most of them have failed during preclinical trials. BACE1 has a large catalytic pocket, and hence the inhibitors need to be large enough to interact with key amino acid residues of the BACE1 active site (Coimbra et al., 2018). However, large-sized inhibitors may not be able to cross the blood–brain barrier (BBB), as only low molecular weight and lipophilic compounds are able to cross the BBB, unless aided by transporters (Pardridge et al., 2005; Banks, 2009).
BACE1 is structurally homologous to other aspartic proteases of the pepsin family such as BACE2 and cathepsin D (CTSD) where the inhibition of these enzymes can increase the production of Aβ and protein deposits in several organs. Hernández et al. (2016) performed alignment studies, molecular dynamics simulations, and docking studies on these three aspartic proteases to study their differences. Based on the result, selective BACE1 inhibition can be achieved through strong electrostatic interactions with Asp32 and Asp228 of the catalytic site, in addition to a large number of hydrogen bonds, π–π interaction, and van der Waals interaction.
BACE1 is also limited by its highly flexible catalytic site. It has been observed that the presence or absence of an inhibitor within the active site influences the flap’s conformation in BACE1. A better understanding of the binding of substrates and inhibitors to BACE1 is required before developing an inhibitor (Xu et al., 2012). Chakraborty et al. (2011) studied the dynamic transition of BACE1 employing normal mode analysis (NMA) using a simplified elastic network model (ENM). The dynamic transition of BACE1 from an open to a closed conformation can be observed from this study using the combined approach of cavity and volume calculation obtained from the dynamics of BACE1 encoded by normal mode. In the open conformation, a large catalytic cavity allowed the binding of a wide range of substrates with the help of the 10s and F loops. Both loops are part of the cavity, and the F loop forms the C-terminal lining of the cavity. Meanwhile, in the closed conformation, the F loop detaches from the cavity, and the 10s loop moves upward and lines the cavity, which causes the cavity to squeeze and tightly hold the substrate. The effect of myricetin on the BACE1 enzyme was also studied to explain the pharmacophoric feature of BACE1 inhibitors using molecular docking. The docking results showed that myricetin forms a strong interaction with BACE1 in the closed conformation compared to that in the open conformation. In the closed conformation of BACE1, myricetin formed interactions with the enzyme through hydrogen bonding by binding to the catalytic aspartic acid residues (Asp228) and forming van der Waals interactions with Asp32 and Ser35. Myricetin also binds with other residues such as Pro70, Val69, Tyr71, and Thr72 through hydrogen bonds.
In addition, BACE1 inhibitors also need to overcome potential drug–drug interactions from activity against CYP450 enzymes and potential hERG channel inhibition. Inhibition of the hERG channel could cause QT interval prolongation that may lead to ventricular arrhythmias known as Torsades de Pointes (TdP) (Coimbra et al., 2018). Malone and Hancox (2020) evaluated evidence from the scientific literature to find potential association between AD drugs (donepezil, galantamine, and rivastigmine) and the risk of QT interval prolongation and Torsades de Pointes (TdP). From the study, one of the AD drugs, which is donepezil, showed a risk of producing QT interval prolongation and Torsades de Pointes (TdP).
Natural product compounds may be potential candidates for BACE1 inhibitors. Several natural product compounds have been shown to bind simultaneously to the catalytic amino acid residues of BACE1, which are Asp228 and Asp32. Wagle et al. (2019) performed molecular docking between BACE1 (pdb id: 2wjo) and isolated compounds from Cirsium japonicum var. maackii, namely luteolin, luteolin 5-O-β-D-glucopyranoside, and luteolin 7-O-β-D-glucopyranoside. The result showed that luteolin 5-O-β-D-glucopyranoside and luteolin 7-O-β-D-glucopyranoside interacted with both Asp228 and Asp32 residues via hydrogen bonds. Additionally, natural product compounds have low activity against the hERG channel due to the presence of aromatic rings (Kratz et al., 2017; Babiaka et al., 2020). Moreover, natural product compounds have the ability to cross the blood–brain barrier (BBB) as they have physicochemical properties that are favorable for BBB permeation (Carecho et al., 2020). Low-molecular-weight polyphenol molecules can cross the BBB through passive permeation and carrier-mediated transport. Flavonoids are able to cross the BBB through transporters such as ATP-binding cassette (ABC) transporters, organic anion transporters (OATs), and organic anion transporting polypeptides (OATPs) (Lu et al., 2014; Stieger et al., 2017; Williamson et al., 2018).
One of the essential classes of natural product compounds is flavonoids. It can be found in several parts of a plant and is a secondary metabolite. The main structure of flavonoids contains two aromatic rings (ring A and ring B) connected by three carbon atoms, which form an oxygenated heterocycle (ring C) (Figure 1). There are about eight subclasses of flavonoids, which are flavones, flavonols, flavanols, isoflavonoids, chalcones, anthocyanins, flavanones, and neoflavanoids. The subclasses are categorized based on the basic flavan ring system, alkylation, glycosylation, and hydroxylation (Vauzour et al., 2008; Panche et al., 2016). The presence of chromone structure in the backbone of many flavonoids is an important feature in several anti-HIV, anti-inflammatory, antibacterial, and anticancer drugs as well as those used in neurodegenerative diseases, inflammatory diseases, and diabetes (Reis et al., 2017).
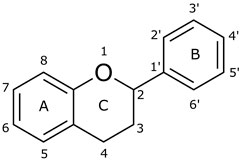
FIGURE 1. Main structure of flavonoid, which contains two aromatic rings (A and B) and linked via a heterocyclic ring (ring C).
Several flavonoids from various plants such as mycertin, kaempferol, morin, apigenin, luteolin, and polymethoxyflavones have been studied for their inhibitory activity against BACE1. A study from Youn et al. (2016a) showed that 5,7-dimethoxyflavone (DMF), 5,7,4′-trimethoxyflavone (TMF), and 3,5,7,3′,4′-pentamethoxyflavone (PMF) exhibited strong BACE1 inhibitory activities with no suppression of other enzyme activities such as α-secretase and other serine proteases. This indicates that these three flavonoid compounds are relatively specific and selective toward the BACE1 enzyme. Moreover, the study by Zhumanova et al. (2021) also reported that O-methylated quercetins, a flavonoid isolated from the aerial part of the endemic Caragana balchaschensis (Kom.) Pojark, were significantly effective in inhibiting BACE1 with IC50 values ranging from 1.2 to 6.5 μM. Additionally, flavonoids such as kaempferol and quercetin have diverse bioactivities and are known to exert neuroprotective effects and inhibit BACE1 activity. A number of flavonoids have also been investigated for anti-acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) activities. In a study, macluraxanthone was found to be the most potent and specific inhibitor of both AChE and BChE, with IC50 values of 8.47 and 29.8 μM, respectively (Khan et al., 2009).
Despite their favorable structure and properties, to date, no flavonoids have advanced further in clinical trials as BACE1 inhibitors. Hence, the aim of this study was to analyze the data published between 2010 and 2022 regarding flavonoids and their activity against the BACE1 enzyme. Specifically, the two-dimensional (2D) structural patterns as well as binding patterns of the flavonoids are analyzed, with the aim of discovering the functional relationship between structure and activity. Understanding the structure–activity relationship between flavonoids and BACE1 is important in designing highly potent and specific BACE1 inhibitors.
Materials and Methods
In the data collection that is similar to that of a systematic review process, two authors (Nur Intan Saidaah; NS, and (Zafirah Liyana Abdullah; ZA) independently reviewed and evaluated all articles found in the literature search, including titles, abstracts, and keywords based on the inclusion and exclusion criteria. If there was any disagreement between the authors about the articles reviewed, a third author (Fazlin Mohd Fauzi; FF) reviewed the article and made the final decision.
Keyword Identification
A comprehensive literature search was performed through several databases such as Science Direct, Scopus, and PubMed using search strings (Table 1). The search was limited to articles published from 2010 to 2022, and only research and journal articles were selected as the document type. In this study, 389 articles were retrieved in the first round of electronic literature searches. After removing review papers, conference proceedings, letters, meta-analyses, abstracts, emails, and unrelated topics (n = 299), 90 articles were identified for screening.

TABLE 1. Search string used for each database to search for relevant literatures.
Screening
In the screening stage, 90 articles were screened based on their titles and abstracts. The articles were screened based on the following criteria: 1) studies that did not include the BACE1 enzyme inhibition assay and 2) studies that did not involve flavonoids as compounds against BACE1. Duplicate articles were also removed. A total of 74 articles were excluded based on these criteria.
Eligibility
A total of 16 articles were evaluated for their eligibility. All articles were examined based on inclusion and exclusion criteria (Table 2). Four studies were excluded because the studies either involved plant extracts, only in silico experiments were conducted without in vitro validation, no full text was available, and/or IC50 values that were not available in M (Table 2). Finally, 12 studies with 64 flavonoids were selected after 2 duplicates were removed.

TABLE 2. Inclusion and exclusion criteria used in the screening phase of the literature search.
Data Extraction
Information of all 64 flavonoids from 12 studies were extracted, which include authors, year of publication, the structure of flavonoids, and BACE1 inhibitory activities. A chemical annotation of the flavonoids such as SMILES was identified and tabulated in an Excel spreadsheet, which was then converted into a CSV format for further analysis.
Molecular Properties and Clustering
The molecular properties of the flavonoids were calculated based on their structure in DataWarrior (www.openmolecules.org). Next, the flavonoids were clustered based on their chemical structure. All of the extracted information in the CSV file was uploaded into the DataWarrior software. Under the Chemistry tab, the Calculate Properties was selected. Properties for drug-likeness such as cLogP, H-acceptors, H-donors, and total surface area were chosen. Automatic structure–activity relationship (SAR) analysis was performed under the Chemistry tab to determine core fragments and functional groups of flavonoid compounds. In this analysis, SMILES was used to generate the scaffold, with Murcko scaffold selected as the scaffold type. After core fragments and functional groups were determined, a cluster analysis was performed. In the same Chemistry tab, the Cluster Compounds or Reactions was selected. Clustering starts with the construction of a complete similarity matrix of all flavonoids that was calculated based on the default descriptor, FragFp, which is similar to MDL keys and consists of 512 predefined structural fragments. The flavonoids with the highest similarity were included in the first cluster. Then, the similarity matrix is updated by removing all similarity values related to the first two flavonoids. This is replaced by a new set of similarities between the cluster center and other compounds. The new values were calculated as mean of the two original similarity values. The process continues by merging the most similar flavonoid and terminated when the highest similarity falls below 0.8.
Molecular Docking
Representative compounds from each cluster were subjected to molecular docking against BACE1. The crystal structure of BACE1 (pdb id: 2wjo, 2.50 Å) was obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB). Using Discovery Studio Visualizer Software, heteroatoms and water were removed, and polar hydrogen was added to the BACE1 structure. BACE1 was loaded onto the PyRx software and set as a macromolecule. The three-dimensional (3D) structures of each representative compound and the reference ligand (2,2,4-trihydroxychalcone; CID: 5811533) were obtained from the PubChem Compound Database (NCBI). These files were uploaded to the Open Babel tab in PyRx software, and energy minimization was selected for all the ligands. The force field used in the Open Babel software package is by default the universal force field. The docking of the BACE1 and flavonoid compounds was simulated using PyRx Software (AutoDock Vina) to determine the binding energy for each flavonoid with BACE1. The Lamarckian genetic algorithm (LGA) method was used for all calculations between the protein and ligand. The docking site on the protein target was defined by establishing a grid box with the dimensions of X: 61.0616, Y: 51.4261, and Z: 59.9516 Å, with a center of X: 16.0076, Y: 39.7409, and Z: 40.5572 Å. The visualization of BACE1 with flavonoid compounds was visualized using Discovery Studio Visualizer (Trott and Olson, 2010; Dallakyan and Olson, 2015).
Results
General Findings
From the 12 articles that were filtered, 66 flavonoids were collected and included information such as chemical structure, molecular properties, and IC50 values. Two duplicates were found and removed, which were naringenin and baicalein. In total, 64 compounds were filtered (Figure 2). For each of the 64 flavonoids, their core fragments were identified, which include flavone, flavanone, chalcone, rotenoid, and isoflavone. For one compound, its core fragment could not be identified, which was neocyclomorusin.
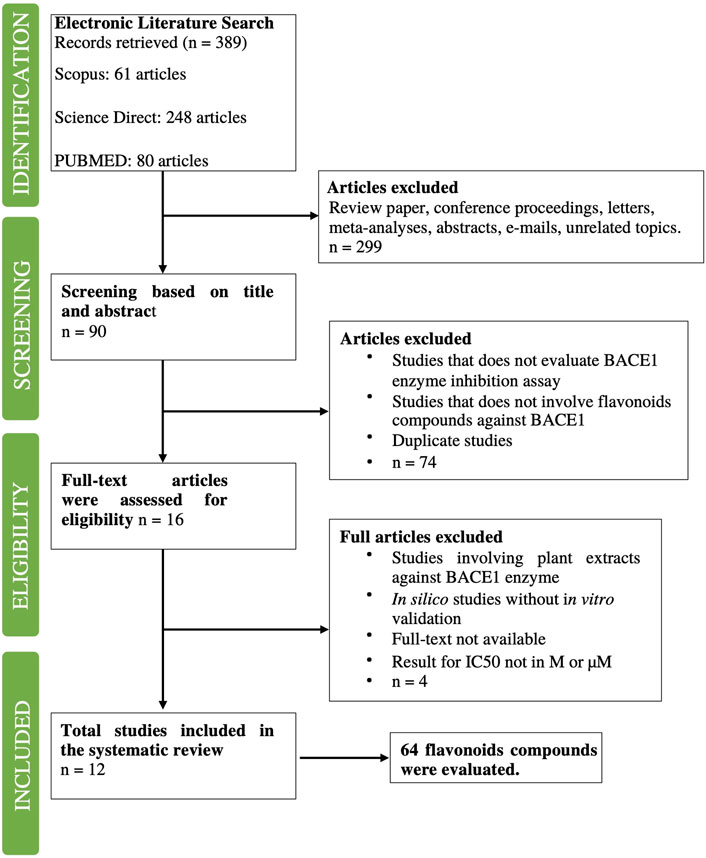
FIGURE 2. Flowchart of the literature search conducted and number of articles filtered at each stage. The process is similar to that of a systematic review.
Clustering Analysis Based on Core Fragment and Functional Groups of Flavonoids
Using DataWarrior, the 64 flavonoids were clustered based on the similarity of their structures, which resulted in 12 clusters. A full detail of each compound and their cluster is listed in Table 3, and a graph showing the distribution of the IC50 for each compound in the cluster is shown in Figure 3. Each cluster contains only compounds with one distinct core fragment, with the exception of cluster 4 where there are compounds with either a flavanone or flavone core fragment, as well as the presence of at least one sugar moiety. Flavonoids from these 12 studies exhibited IC50 values ranging from 1.2 to 147 μM against the BACE1 enzyme.
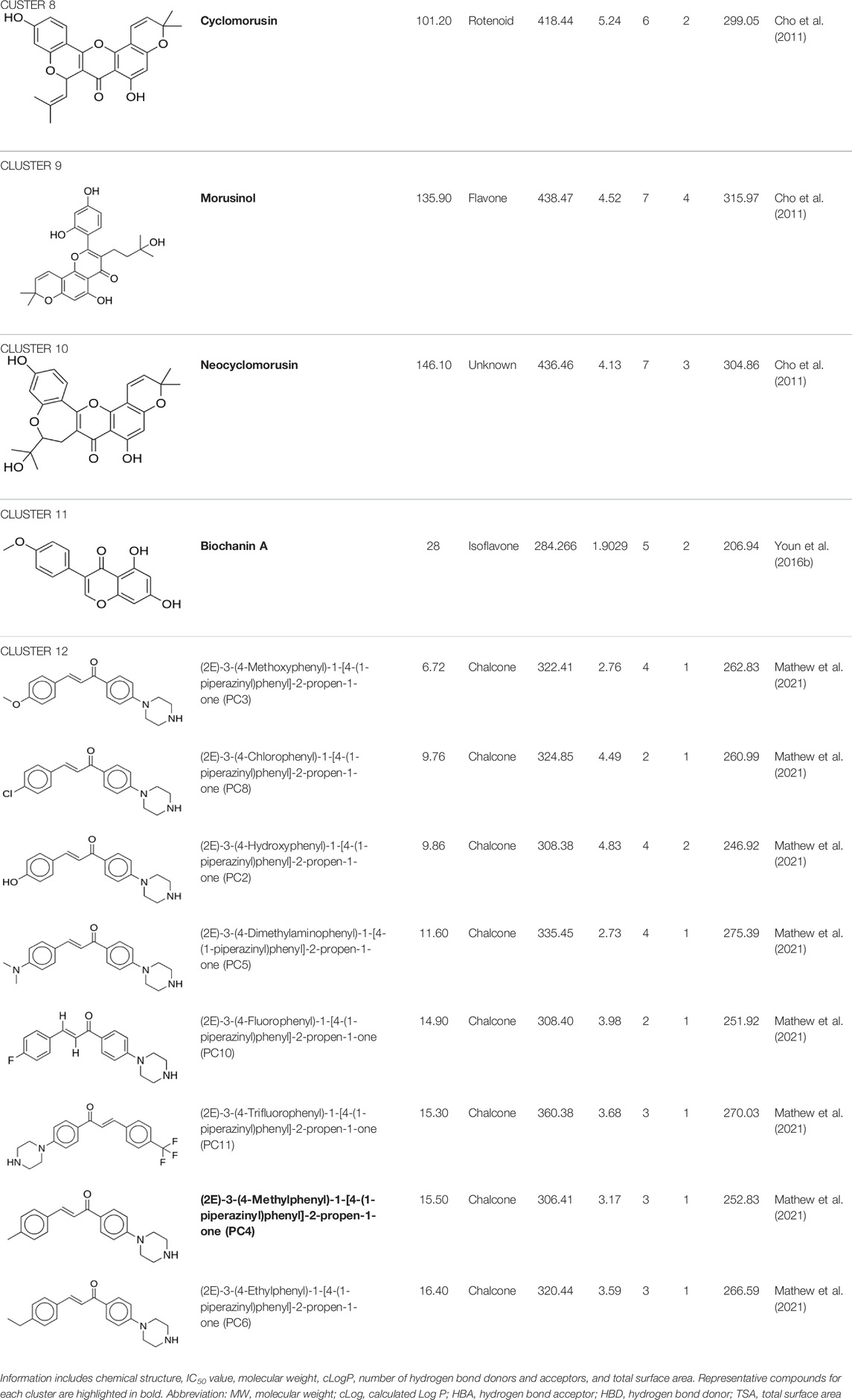
TABLE 3. Information of the 64 flavonoid compounds that were filtered and classified according to their cluster.
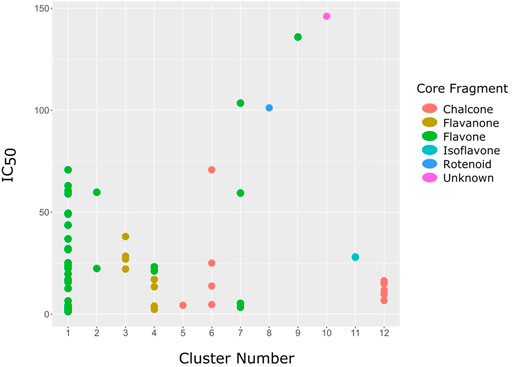
FIGURE 3. The graph showing the distribution of IC50 values for compounds in each cluster. The graph shows that each cluster contain one distinct core fragment, with the exception of Cluster 4, which contains both Flavone and Flavanone. Flavanone and Chalcone showed the obvious correlation between structure and bioactivity. Flavanones in Cluster 4 contain sugar moieties but is absent in Flavanones in Cluster 3, and the IC50 values of both clusters do not overlap.
From Figure 3, it can be seen that clusters 1, 2, 7, and 9 contain compounds with the flavone core exclusively with the highest number of compounds in cluster 1 (31 compounds). Additionally, cluster 4 also contains two compounds with the flavone core. The standard structure of flavone consists of a double bond between C2 and C3, with a ketone at C4. The flavones showed cLogP values of 0.21–7.5, a total surface area of 191–422 m2/g, and a molecular weight of 270–541 g/mol. Flavones in clusters 1, 2, 7, and 9 contain around 0–5 hydrogen bond donors and 3–8 hydrogen acceptors, but flavones in cluster 4 contain 6 hydrogen bond donors and 12 hydrogen bond acceptors. The higher hydrogen bond acceptors and donors of compounds in cluster 4 may be attributed to the presence of a sugar moiety. The IC50 values of compounds with the flavone core fragment were below 100 μM except for mormin in cluster 7 (103.5 μM) and morusinol in cluster 9 (135.9 μM). Compound 3,7,3′-tri-O-methylquercetin from cluster 1 showed the lowest IC50 value, which was 1.20 μM. 3,7,3′-tri-O-methylquercetin consists of one hydroxyl group at C5 and one methoxy group at C7 (ring A). It also has a hydroxyl group at C4’ and a methoxy group at C5′ (ring B). In addition, the presence of a methoxy group at C3 (ring C) may contribute to the inhibition of BACE1. It can be seen from Figure 3 that compounds with the flavone core fragment do not have an obvious correlation between the 2D structure and bioactivity, where the clusters show distinct structures but the ranges of IC50 values overlap between the clusters.
Cluster 4 is the only cluster that includes compounds with different core fragments where it contains four flavanones and two flavones. Unlike flavone, flavanone compounds have no double bond between C2 and C3, but all compounds in cluster 4 contain at least one sugar moiety. The IC50 of compounds in cluster 4 ranges from 2.34 to 23.2 μM where didymin in Cluster 4 showed the most potent BACE1 inhibition with an IC50 of 2.34 μM, which may be due to the presence of disaccharides at ring A. Although poncirin also has disaccharides at ring A, the different position of the glycosidic linkage may explain the IC50 value of poncirin being slightly higher than that of didymin. Prunin, which only has one sugar moiety, showed a lower IC50 value than hesperidin, which has two sugar moieties, similar to didymin. The substitution of the hydroxide at ring B at different positions may explain the difference in the IC50 value observed. Additionally, the two flavones in the cluster, 3,7-di-O-methylquercetin-4′ -O-glucoside and 3,7-di-O-methylquercetin-3′-O-glucoside, had the highest IC50 values of 21.2 and 23.2 μM, respectively. Compounds with the flavanone core also can be found in cluster 3 but without the presence of a sugar moiety. The physicochemical properties of flavanone core fragments between clusters 3 and 4 are comparatively different. In cluster 3, the range of cLogP was between 2 and 2.8 (vs. −0.18–0.17) with a total surface area of 186–215 m2/g (vs. 294–411 m2/g) and a molecular weight ranging from 256 to 303 g/mol (vs. 434–611 g/mol). In cluster 3, IC50 values ranged from 22 to 39 μM. When comparing flavanones between clusters 3 and 4, it showed the most obvious relationship between 2D structure and bioactivity where flavanones with sugar moieties produce lower IC50 values.
Thirteen flavonoids with the chalcone core fragment were retrieved from the literature screening. Chalcone structures contain an α, β-unsaturated ketone with two aromatic rings (rings A and B). These chalcones can be further categorized as piperazine- and non-piperazine-substituted chalcones. The piperazine-substituted chalcones (8 compounds) were all grouped in cluster 12, while the non-piperazine-substituted chalcones were grouped in clusters 5 (1 compound) and 6 (4 compounds). The SAR of the chalcones was not obvious; however, this relationship is more obvious among the non-piperazine-substituted chalcones. Cardamonin (cluster 5) showed the highest inhibitory activity among all chalcones, with an IC50 of 4.35 μM. In contrast, compounds in cluster 6 showed IC50 above 4.7 μM with compounds 2j ((E)-1-(4-bromo-2-hydroxy-5-iodophenyl)-3-(4-fluorophenyl)propanone), 2h ((E)-1-(4-chloro-2-hydroxy-5-iodophenyl)-3-(4-methoxyphenyl)propanone), 2n ((E)-3-(4-fluorophenyl)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)prop-2-en-1-one), and 2p ((E)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one) showing IC50 values of 4.703, 13.82, 25.07, and 70.79 μM, respectively. Cardamonin contains a hydroxyl group at the ortho and para positions of ring A and no substitution in ring B. For compounds in cluster 6, all compounds are substituted at the ortho, para, and meta positions of ring A and the para position of ring B. All compounds in cluster 6 contain iodine at the meta position of ring A. Compound 2j ((E)-1-(4-bromo-2-hydroxy-5-iodophenyl)-3-(4-fluorophenyl)propanone), which has the lowest IC50, is substituted with bromine and fluorine at the para position of rings A and B, respectively. Substitution with functional groups with decreasing electronegativity at the para position of rings A and B seems to result in an increase in IC50 values. When the bromine in 2j ((E)-1-(4-bromo-2-hydroxy-5-iodophenyl)-3-(4-fluorophenyl)propanone) is replaced with chlorine in ring A and fluorine is replaced with methoxy in ring B, this produces compound 2h ((E)-1-(4-chloro-2-hydroxy-5-iodophenyl)-3-(4-methoxyphenyl)propanone) with a higher IC50 value. When methoxy is substituted at the para position of ring A, this produces compounds with higher IC50 values, as seen in 2n ((E)-3-(4-fluorophenyl)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)prop-2-en-1-one) and 2p ((E)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one). Both compounds differ in the substitution at the para position of ring B, where 2n ((E)-3-(4-fluorophenyl)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)prop-2-en-1-one) is substituted with a fluorine and recorded a lower IC50 value than 2p ((E)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one), which is substituted with a methoxy group. This suggests that the potency of non-piperazine-substituted chalcone decreases with the substitution of functional groups with decreasing electronegativity. Additionally, no substitution at ring B seems to correlate to higher activity, and similar to ring A, substitution at the para position with functional groups with decreasing electronegativity results in an increase in the IC50 value. In terms of the pharmacokinetic profile, the cLogP values for the non-piperazine-substituted chalcone was in the range of 2.5–4.22, with a total surface area of 212–255 m2/g and a molecular weight of 270–448 g/mol. The number of hydrogen bond acceptors in these clusters was around 2–4, while the number of hydrogen bond donors was between 1 and 2. In regards to the piperazine-substituted chalcones, although the piperazine-substituted chalcones only differ by the substitution at the para position of ring B, no correlation can be established between the properties of the functional groups, i.e., electronegativity or lipophilicity and IC50 values. The lowest IC50 value in the piperazine-substituted chalcones was observed in compound PC3 ((2E)-3-(4-methoxyphenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 6.72 μM), where a methoxy is substituted at the para position of ring B. This is followed by PC8 ((2E)-3-(4-chlorophenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 9.76), PC2 ((2E)-3-(4-hydroxyphenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 9.86 μM), PC5 ((2E)-3-(4-dimethylaminophenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 11.6 μM), PC10 ((2E)-3-(4-fluorophenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 14.9 μM), PC11 ((2E)-3-(4-trifluorophenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 15.3 μM), PC4 ((2E)-3-(4-methylphenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 15.5 μM), and PC6 ((2E)-3-(4-ethylphenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one; 16.4 μM). In terms of the pharmacokinetic profile, the cLogP values for the piperazine-substituted chalcone was in the range of 2.7–4.9, with a total surface area of 246–276 m2/g and a molecular weight of 306–360 g/mol, which are comparable to the non-piperazine-substituted chalcones.
The remaining three clusters contained only one compound each, where cyclomorusin, neocyclomorusin, and biochanin A were clustered in clusters 8, 10, and 11, respectively, each representing a distinct core fragment. Neocyclomorusin from cluster 10 showed the highest IC50 value, which was 146.1 μM.
Molecular Docking of Compound Representative From Each Cluster
The binding interactions of the representative compounds from each cluster with BACE1 were analyzed through molecular docking using AutoDock Vina and visualized using Discovery Studio Visualizer. The 12 compounds were D3 (3′,7-diethoxy-5-hydroxy-4′-methoxyflavone), 3,5,7,3′,4′-pentamethoxyflavone, naringenin, didymin, cardomin, 2n ((E)-3-(4-fluorophenyl)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)prop-2-en-1-one), morusin, cyclomorusin, morusinol, neocyclomorusin, biochanin A, and PC4((2E)-3-(4-methylphenyl)-1-[4-(1-piperazinyl)phenyl]-2-propen-1-one). All these flavonoids were docked with the crystal structure of human BACE1 (pdb id: 2wjo) that was obtained from the PDB database, and 2,2,4-trihydroxychalcone was used as the reference ligand for this study. The results are shown in Figures 4A–Z and tabulated in Table 4.
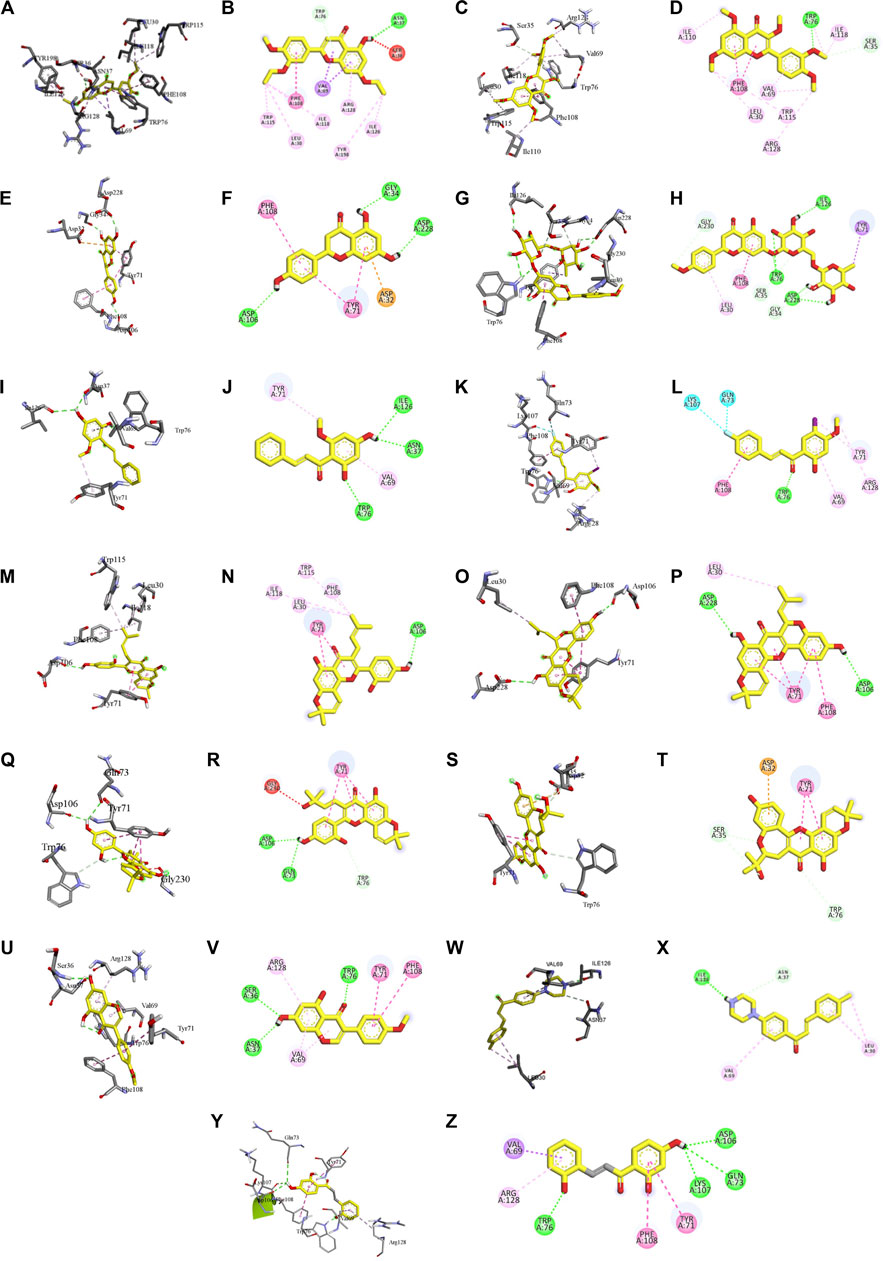
FIGURE 4. Visualization of the protein–ligand interaction between 11 flavonoids, which represents the 11 clusters against BACE1 from molecular docking. The flavonoids were D3 (3′,7-diethoxy-5-hydroxy-4′-methoxyflavone (A,B), 3,5,7,3′,4′-pentamethoxyflavone (C,D), naringenin (E,F), didymin (G,H), cardomin (I,J), 2n (K,L), morusin (M,N), cyclomorusin (O,P), morusinol (Q,R), neocyclomorusin (S,T), biochanin A (U,V), and PC4 (4′-piperazinoacetophenone-CH3; (W,X) reference ligand: 2,2,4-trihydroxychalcone (Y,Z).
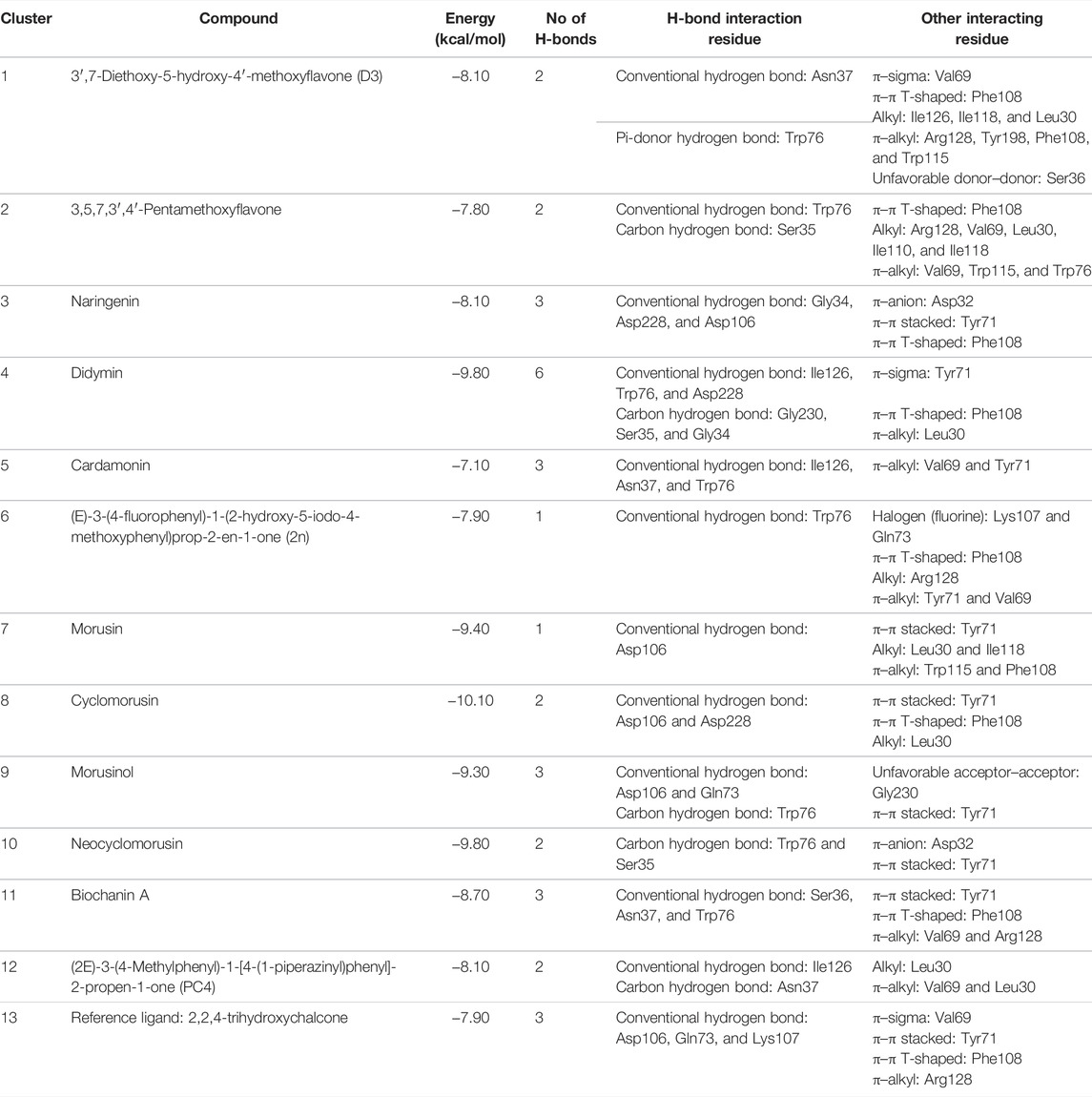
TABLE 4. Result of the molecular docking of 12 compounds, which are representative of each cluster. Information includes the binding scores, number of hydrogen bond formed, residues involved in hydrogen bond, and others.
Based on Table 4, cyclomorusin (cluster 8) was predicted to bind to BACE1 with the lowest binding score, which is −10.10 kcal/mol. Cyclomorusin also has a lower binding score compared to the reference ligand (2,2,4-trihydroxychalcone), which was −7.90 kcal/mol. The hydroxyl group of cyclomorusin formed two hydrogen bonds with Asp106 (ring B) and also catalytic aspartic residues, Asp228 (ring A) of BACE1. All three rings of cyclomorusin also formed a π–π stacking interaction with Tyr71 of BACE1. Leu30 residue of BACE1 also interacts with the methyl group of the pentacyclic structure. However, the IC50 value of cyclomorusin was in contrast with the docking result, where cyclomorusin showed lower potency in inhibiting BACE1 compared to other flavonoids (101.2 μM).
Another compound, neocyclomorusin, which belongs to cluster 10, also showed a low binding score, which is −9.80 kcal/mol. Similar to cylcomorusin, neocyclomorusin showed lower potency in inhibiting BACE1 (146.1 μM) than the rest. Uniquely, these two compounds have a similar pentacyclic structure formed by the ring closure of the prenyl group. As can be seen in Figures 4S–T, the embedded oxygen in the pentacyclic structure of neocyclomorusin binds to Ser35 of BACE1 through a hydrogen bond. Moreover, the π–anion interaction was observed between ring B of neocyclomorusin and Asp32 of BACE1. This interaction of the π–anion is stronger than the hydrogen bond (Bartlett et al., 2013). The presence of these structures in neocyclomorusin may contribute to the lower binding energy against BACE1.
From all the representative clusters, didymin (cluster 4) had the highest number of hydrogen bonds, which was 6 (Ile126, Trp76, Asp228, Gly230, Ser35, and Gly34) compared to the reference ligand, where there were only three hydrogen bond interactions with Asp106, Gln73, and Lys107. Didymin showed a binding energy of −9.40 kcal/mol where hydroxyl groups from the two sugar molecules form the most hydrogen interactions with the amino acid residues of BACE1. One of the hydroxyl groups interacts with the catalytic aspartic residue, Asp228. Other interactions such as π–sigma (Tyr71), π–π T-shaped (Phe108), and π–alkyl (Leu30) were present between didymin and BACE1. Only the π–sigma interaction was observed between the residue of Tyr71 with the methyl group of the sugar moiety. Other π–π interactions involved the aromatic rings of didymin.
Compound 2n ((E)-3-(4-fluorophenyl)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)prop-2-en-1-one) is the only representative compound that has a halogen functional group. The fluorine group forms a van der Waals interaction with Lys107 and Gln73. Molecular docking results also showed that D3 (3′,7-diethoxy-5-hydroxy-4′-methoxyflavone), Didymin, cardamonin, 2n ((E)-3-(4-fluorophenyl)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)prop-2-en-1-one), morusinol, neocyclomorusin, and biochanin A share a common hydrogen bond interaction with Trp76. Moreover, all of the representative flavonoids including the reference ligand shared a common π–π interaction with Tyr71 except for 3,5,7,3′,4′-pentamethoxyflavone.
Discussion
Due to its favorable structures and properties, flavonoids have been considered a potential BACE1 inhibitor. However, to date, no flavonoids have advanced into clinical trials as BACE1 inhibitors. Here, an analysis of flavonoids that has been tested as BACE1 inhibitors from studies published from 2010 to 2022 was conducted to discover the pharmacophoric features of flavonoids against BACE1. In the clustering analysis, flavonoids were clustered based on their core fragments and binding interactions between flavonoids and BACE1 were analyzed using molecular docking. These two analyses were performed to determine the relationship between the chemical structure and inhibitory activity of flavonoids against BACE1. From the results of the clustering and molecular docking of 64 flavonoids compiled, several observations can be made.
First, compounds with the flavanone core fragments showed an apparent relationship between the 2D structure and bioactivity. Flavanone core fragments were divided into two clusters, where compounds in the two clusters differed by the presence of sugar moieties. The sugar moiety may increase inhibition toward BACE1, as seen in Table 3, where most of the flavonoids in cluster 4 have low IC50 values compared to those in cluster 3. This has also been discussed by Ali et al. (2019), where the number and position of sugar moieties and the different positions of the glycosidic linkages of flavanone may affect the inhibitory activity of BACE1. There are also a few studies that demonstrated that the presence of a sugar moiety in natural product compounds contributes to the inhibitory activity of BACE1 such as terpenoids isolated from Dipsacus radix, rubrofusarin, and derivatives from Cassia obtusifolia Linn (Shrestha et al., 2018; Wang et al., 2021).
The non-piperazine-substituted chalcone fragment also showed an obvious relationship between the 2D structure and bioactivity. Substitution at the para position of rings A and B with functional groups with decreasing electronegativity seems to result in an increase in the IC50 value. When ring A is substituted with a hydroxyl group at the meta position of ring A and there is no substitution on ring B, such as in the case of cardamonin, this produces the lowest IC50 value among the chalcones. Molecular docking shows that cardamonin formed three hydrogen bonds with BACE1 compared to only one by compound 2n ((E)-3-(4-fluorophenyl)-1-(2-hydroxy-5-iodo-4-methoxyphenyl)prop-2-en-1-one). In a study by Ma et al. (2011), a series of hydroxychalcones were evaluated for their inhibitory activities against BACE1. The structure–activity relationship from this study showed that the inhibitory activity of the chalcone against BACE1 was governed by the hydroxyl substituents on rings A and B of the chalcone, where the most potent chalcone was substituted with four hydroxyl groups (IC50 = 0.27 μM). The high potency may be attributed to the ability of the compounds to form hydrogen bonds with the catalytic site. Van der Waals interaction may also play an important part in the interaction between chalcone and BACE1. When compounds in cluster 6 were substituted at the para position in rings A and B with decreasing electronegativity (Br > Cl > CH3O and F > CH3O, respectively), the IC50 value increases. In addition, flavonoids with chalcone core fragments did not violate the “Lipinski’s rule,” which shows they have good absorption and bioavailability. Chalcone is also known as “privileged structures” as both natural and synthetic chalcone derivatives have shown compelling biological activities with clinical potential against different types of diseases (Zhuang et al., 2017).
Thirdly, the number and position of sugar moieties attached to the flavanone core play an important role in inhibiting BACE1. These sugar moieties can be linked to an aglycone as monosaccharides, disaccharides, or oligosaccharides. In this study, all flavanones in cluster 4 have different numbers of sugar moieties and positions attached to the core fragment. Didymin, which contains disaccharides, showed the most potent inhibitory activity against BACE1 compared to other flavanones in cluster 4. Moreover, the hydroxyl group from one of the sugar moieties in didymin interacts with catalytic aspartic residues of BACE1 (Asp228). Although poncirin also contains two disaccharides, it exhibited a different level of potency against BACE1 due to a different position of the glycosidic linkage. These results are supported by Shrestha et al. (2018), where the activity of rubrofusarin and its derivatives against AChE and BACE1 was investigated. The study found that the glucose moiety at position C6 of nor-rubrofusarin is responsible for the inhibitory activity against BACE1. Moreover, the presence of two sugar moieties inhibited AChE nine times more than a compound having no sugar moiety. Choi et al. (2016) also showed that sugar molecules of ginsenosides had the most interactions with the residues of the BACE1 active site through hydrogen bond and van der Waals interactions.
The majority of BACE1 inhibitors seem to have a large number of hydrogen bond donors and acceptors and form strong hydrogen bonds with Asp32 and Asp228 of BACE1 (Hernández et al., 2016). From this study, didymin has the most hydrogen bonds compared to other representative flavonoids and formed hydrogen bonds with Asp228. This is indicated by the low binding energy score (9.80 kcal/mol) and high inhibitory activity (IC50 = 2.34 μM) of didymin. This finding is supported by molecular docking studies that were performed to study the effects of flavonols and flavones as BACE1 inhibitors. In the BACE1 target interaction, myricetin was found to form the largest number of hydrogen bonds with BACE1. Two hydrogen bonds were formed with Asp32 through C3-OH of ring C. Several other hydrogen bonds were formed between Trp198 and C4′-OH and C5′-OH of ring B and between Gln73 and C7-OH of ring A (Shimmyo et al., 2008). A study by Shrestha et al. (2018) also showed that nor-rubrofusarin 6-O-β-D-glucoside had a binding energy of −8.34 kcal/mol in the allosteric inhibition mode with BACE1 and formed six hydrogen bonds with Gln303, Gln304, Glu339, and Gly156. In addition, the inhibition of BACE1 with nor-rubrofusarin 6-O-β-D-glucoside by catalytic inhibition showed a binding energy of -6.61 kcal/mol with six hydrogen bonds formed with Asp32, Trp76, Asn37, Ile26, and Tyr198 (Shrestha et al., 2018). Although didymin formed the most hydrogen bonds with BACE1, it violates two of “Lipinski’s rule,” as it has 14 hydrogen bond acceptors and 7 hydrogen bond donors due to the presence of the sugar moiety.
Another unique structure of flavonoid that needs to be highlighted is the pentacyclic structure that can be found in cyclomorusin and neocyclomorusin. The extra ring in both structures may contribute to the low binding energy score observed. However, previous study has reported that the pentacyclic structure of both compounds resulted in lower potency against BACE1, where a free hydroxyl group is needed for the BACE1 activity (Cho et al., 2011). Further study needs to be done on the pentacyclic structure to understand whether this structure significantly contributes to the BACE1 inhibitory activity. It should be noted that molecular dynamics stimulation would further enhance our understanding on the binding interaction between flavonoids and BACE1, which could not be performed in this study and should be performed in future studies. Molecular dynamics simulation provides more details on the movement of every atom in a protein compared to molecular docking, which only generates the binding mode of a ligand to a protein and predicts the number of possible conformations. In molecular dynamics simulation, it predicts important biomolecular processes such as conformational change, ligand binding, protein folding, and the position of all atoms at a very fine temporal resolution (Hollingsworth and Dror, 2018).
Despite various studies supporting flavonoids as potential BACE1 inhibitors, there are also significant challenges with the isolation, purification, and pharmacokinetic properties of the flavonoids. Flavonoids are very difficult to isolate, as only a small amount can be obtained at one time. Additionally, flavonoids are highly metabolized, with low solubility and poor oral absorption (Amawi et al., 2017). However, there are several approaches that could address these issues. In regard to its isolation, nano-harvesting has been applied to increase product yield. Kurepa et al. (2014) utilized nanoparticles such as anatase TiO2 to conjugate enediol and catechol group-rich flavonoids. This technique eliminates the use of organic solvents and yielded a higher percentage of flavonoid compounds (Kurepa et al., 2014). Moreover, structural modifications can be performed to increase the solubility and stability of flavonoids. The replacement of hydroxyl with an ethyl group in quercetin was shown to improve its stability against metabolic enzymes by preventing oxidative degradation. Insertion of the ethyl group also increases the lipophilicity of quercetin from 10.7% to 18.8% (Grande et al., 2016). Micro- and nano-delivery systems could also help to increase the bioavailability of flavonoid such as the use of nano-emulsions and nano-crystals (Amawi et al., 2017). Yi et al. (2017) developed a novel silybin nano-crystal using high-pressure homogenization. It was found that the release rate of silybin nano-crystal was faster in vitro and showed a higher peak concentration in vivo compared to the silybin coarse powder. This demonstrated that the nano-crystal technique increases bioavailability and is a promising oral drug delivery system for poorly soluble drugs such as flavonoids (Yi et al., 2017).
Conclusion
In this study, an analysis was conducted on flavonoids that have been tested against BACE1 enzymes from 2010 to 2022. Specifically, the structure–activity relationship analysis was conducted, involving clustering and molecular docking. Several key findings from this study include: 1) flavanones with sugar moieties showed higher inhibitory activity than those without sugar moieties. Additionally, the number of sugar moieties and the position of glycosidic linkage affect the inhibitory activity. 2) Non-piperazine-substituted chalcones when substituted with functional groups with decreasing electronegativity at the para position of both rings result in a decreased inhibitory activity. Molecular docking indicates that ring A is involved in hydrogen bonding, whereas ring B is involved in van der Waals interaction with BACE1. 3) Hydrogen bond is an important interaction with the catalytic site of BACE1. It should be noted that the SAR analysis performed in this study is limited to 2D similarities only. Further studies involving other similarity metrics are warranted. Additionally, molecular dynamic studies are also warranted to study the behavior of BACE1 with flavonoids in full atomic detail and at very fine temporal resolution. Hence, these findings may aid in the design of highly potent and specific BACE1 inhibitors, which could aid in delaying the progression of AD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author Contributions
NY—conceptualization, data analysis, and writing—review and editing. ZA—data analysis and writing—review and editing. NO—writing—review and editing. FF—conceptualization, data analysis, and writing—review and editing.
Funding
This research was supported by the Ministry of Education, Malaysia, through the Fundamental Research Grant Scheme [FRGS/1/2021/SKK0/UITM/02/19 and 600-RMC/FRGS 5/3 (044/2021)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abeysinghe, A. A. D. T., Deshapriya, R. D. U. S., and Udawatte, C. (2020). Alzheimer's Disease; a Review of the Pathophysiological Basis and Therapeutic Interventions. Life Sci. 256, 117996. doi:10.1016/j.lfs.2020.117996
Ali, M. Y., Jannat, S., Edraki, N., Das, S., Chang, W. K., Kim, H. C., et al. (2019). Flavanone Glycosides Inhibit β-site Amyloid Precursor Protein Cleaving Enzyme 1 and Cholinesterase and Reduce Aβ Aggregation in the Amyloidogenic Pathway. Chemico-Biological Interact. 309, 108707. doi:10.1016/j.cbi.2019.06.020
Amawi, H., Ashby, C. R., and Tiwari, A. K. (2017). Cancer Chemoprevention through Dietary Flavonoids: What's Limiting? Chin. J. Cancer 36, 1–13. doi:10.1186/s40880-017-0217-4
Ashrafian, H., Zadeh, E. H., and Khan, R. H. (2021). Review on Alzheimer's Disease: Inhibition of Amyloid Beta and Tau Tangle Formation. Int. J. Biol. Macromol. 167, 382–394. doi:10.1016/j.ijbiomac.2020.11.192
Babiaka, S. B., Nia, R., Abuga, K. O., Mbah, J. A., Nziko, V. d. P. N., Paper, D. H., et al. (2020). Antioxidant Potential of Flavonoid Glycosides from Manniophyton Fulvum Müll. (Euphorbiaceae): Identification and Molecular Modeling. Sci. Afr. 8, e00423. doi:10.1016/j.sciaf.2020.e00423
Banks, W. A. (2009). Characteristics of Compounds that Cross the Blood-Brain Barrier. BMC Neurol. 9 (1), S3–S5. doi:10.1186/1471-2377-9-S1-S3
Bartlett, G. J., Newberry, R. W., Vanveller, B., Raines, R. T., and Woolfson, D. N. (2013). Interplay of Hydrogen Bonds and N→π* Interactions in Proteins. J. Am. Chem. Soc. 135 (49), 18682–18688. doi:10.1021/ja4106122
Carecho, R., Carregosa, D., and Dos Santos, C. N. (2021). Low Molecular Weight (poly)Phenol Metabolites across the Blood-Brain Barrier: The Underexplored Journey. Brain. plast. 6 (2), 193–214. doi:10.3233/BPL-200099
Chakraborty, S., Kumar, S., and Basu, S. (2011). Conformational Transition in the Substrate Binding Domain of β-secretase Exploited by NMA and its Implication in Inhibitor Recognition: BACE1-Myricetin a Case Study. Neurochem. Int. 58 (8), 914–923. doi:10.1016/j.neuint.2011.02.021
Chen, G.-f., Xu, T.-h., Yan, Y., Zhou, Y.-r., Jiang, Y., Melcher, K., et al. (2017). Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 38 (9), 1205–1235. doi:10.1038/aps.2017.28
Cho, J. K., Ryu, Y. B., Curtis-Long, M. J., Kim, J. Y., Kim, D., Lee, S., et al. (2011). Inhibition and Structural Reliability of Prenylated Flavones from the Stem Bark of Morus Lhou on β-secretase (BACE-1). Bioorg. Med. Chem. Lett. 21 (10), 2945–2948. doi:10.1016/j.bmcl.2011.03.060
Choi, R. J., Roy, A., Jung, H. J., Ali, M. Y., Min, B.-S., Park, C. H., et al. (2016). BACE1 Molecular Docking and Anti-alzheimer's Disease Activities of Ginsenosides. J. Ethnopharmacol. 190, 219–230. doi:10.1016/j.jep.2016.06.013
Coimbra, J. R. M., Marques, D. F. F., Baptista, S. J., Pereira, C. M. F., Moreira, P. I., Dinis, T. C. P., et al. (2018). Highlights in BACE1 Inhibitors for Alzheimer's Disease Treatment. Front. Chem. 6, 178. doi:10.3389/fchem.2018.00178
Cummings, J. L., Goldman, D. P., Simmons‐Stern, N. R., and Ponton, E. (2021). The Costs of Developing Treatments for Alzheimer's Disease: A Retrospective Exploration. Alzheimer's Dementia 18 (3), 469–477. doi:10.1002/alz.12450
Dallakyan, S., and Olson, A. J. (2015). Small-Molecule Library Screening by Docking with Pyrx. Biol. Methods Protoc. 1263, 243–250. doi:10.1007/978-1-4939-2269-7_19
Feigin, V. L., Nichols, E., Alam, T., Bannick, M. S., Beghi, E., Blake, N., et al. (2019). Global, Regional, and National Burden of Neurological Disorders, 1990-2016: a Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18 (5), 459–480. doi:10.1016/S1474-4422(18)30499-X
Grande, F., Parisi, O. I., Mordocco, R. A., Rocca, C., Puoci, F., Scrivano, L., et al. (2016). Quercetin Derivatives as Novel Antihypertensive Agents: Synthesis and Physiological Characterization. Eur. J. Pharm. Sci. 82, 161–170. doi:10.1016/j.ejps.2015.11.021
Hampel, H., Caraci, F., Cuello, A. C., Caruso, G., Nisticò, R., Corbo, M., et al. (2020). A Path toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer's Disease. Front. Immunol. 11, 456. doi:10.3389/fimmu.2020.00456
Hampel, H., Vassar, R., De Strooper, B., Hardy, J., Willem, M., Singh, N., et al. (2021). The β-Secretase BACE1 in Alzheimer's Disease. Biol. Psychiatry 89 (8), 745–756. doi:10.1016/j.biopsych.2020.02.001
Han, J., Ji, Y., Youn, K., Lim, G., Lee, J., Kim, D. H., et al. (2019). Baicalein as a Potential Inhibitor against BACE1 and AChE: Mechanistic Comprehension through In Vitro and Computational Approaches. Nutrients 11 (11), 2694. doi:10.3390/nu11112694
Hernández-Rodríguez, M., Correa-Basurto, J., Gutiérrez, A., Vitorica, J., and Rosales-Hernández, M. C. (2016). Asp32 and Asp228 Determine the Selective Inhibition of BACE1 as Shown by Docking and Molecular Dynamics Simulations. Eur. J. Med. Chem. 124, 1142–1154. doi:10.1016/j.ejmech.2016.08.028
Hollingsworth, S. A., and Dror, R. O. (2018). Molecular Dynamics Simulation for All. Neuron 99 (6), 1129–1143. doi:10.1016/j.neuron.2018.08.011
Huang, L.-K., Chao, S.-P., and Hu, C.-J. (2020). Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 27 (1), 1–13. doi:10.1186/s12929-019-0609-7
Khan, M. T. H., Orhan, I., Şenol, F. S., Kartal, M., Şener, B., Dvorská, M., et al. (2009). Cholinesterase Inhibitory Activities of Some Flavonoid Derivatives and Chosen Xanthone and Their Molecular Docking Studies. Chemico-Biological Interact. 181 (3), 383–389. doi:10.1016/j.cbi.2009.06.024
Kratz, J. M., Grienke, U., Scheel, O., Mann, S. A., and Rollinger, J. M. (2017). Natural Products Modulating the HERG Channel: Heartaches and Hope. Nat. Prod. Rep. 34 (8), 957–980. doi:10.1039/C7NP00014F
Kurepa, J., Nakabayashi, R., Paunesku, T., Suzuki, M., Saito, K., Woloschak, G. E., et al. (2014). Direct Isolation of Flavonoids from Plants Using Ultra‐small Anatase TiO 2 Nanoparticles. Plant J. 77 (3), 443–453. doi:10.1111/tpj.12361
Lee, S., Youn, K., Lim, G., Lee, J., and Jun, M. (2018). In Silico docking and In Vitro Approaches towards BACE1 and Cholinesterases Inhibitory Effect of Citrus Flavanones. Molecules 23 (7), 1509. doi:10.3390/molecules23071509
Ma, L., Yang, Z., Li, C., Zhu, Z., Shen, X., and Hu, L. (2011). Design, Synthesis and SAR Study of Hydroxychalcone Inhibitors of Human β-secretase (BACE1). J. Enzyme Inhibition Med. Chem. 26 (5), 643–648. doi:10.3109/14756366.2010.543420
Malone, K., and Hancox, J. C. (2020). QT interval prolongation and Torsades de Pointes with donepezil, rivastigmine and galantamine. Ther. Adv. Drug Saf. 11, 204209862094241. doi:10.1177/2042098620942416
Mathew, B., Oh, J. M., Baty, R. S., Batiha, G. E.-S., Parambi, D. G. T., Gambacorta, N., et al. (2021). Piperazine-substituted Chalcones: A New Class of MAO-B, AChE, and BACE-1 Inhibitors for the Treatment of Neurological Disorders. Environ. Sci. Pollut. Res. 28 (29), 38855–38866. doi:10.1007/s11356-021-13320-y
Mehta, D., Jackson, R., Paul, G., Shi, J., and Sabbagh, M. (2017). Why Do Trials for Alzheimer's Disease Drugs Keep Failing? A Discontinued Drug Perspective for 2010-2015. Expert Opin. Investigational Drugs 26 (66), 735–739. doi:10.1080/13543784.2017.1323868
Mphahlele, M., Agbo, E., and Gildenhuys, S. (2018). Synthesis and Evaluation of the 4-Substituted 2-Hydroxy-5-Iodochalcones and Their 7-Substituted 6-Iodoflavonol Derivatives for Inhibitory Effect on Cholinesterases and β-Secretase. Int. J. Mol. Sci. 19 (12), 4112. doi:10.3390/ijms19124112
Nazarko, L. (2019). Dementia 2. Alzheimer's Disease: Diagnosis, Treatment and Managment. Br. J. Healthc. Assistants 13 (7), 329–335. doi:10.12968/bjha.2019.13.7.329
Panche, A. N., Diwan, A. D., and Chandra, S. R. (2016). Flavonoids: an Overview. J. Nutr. Sci. 5, 1–15. doi:10.1017/jns.2016.41
Pardridge, W. M. (2005). The Blood-Brain Barrier: Bottleneck in Brain Drug Development. Neurotherapeutics 2 (1), 3–14. doi:10.1602/neurorx.2.1.3
Plaingam, W., Sangsuthum, S., Angkhasirisap, W., and Tencomnao, T. (2017). Kaempferia Parviflora Rhizome Extract and Myristica Fragrans Volatile Oil Increase the Levels of Monoamine Neurotransmitters and Impact the Proteomic Profiles in the Rat hippocampus: Mechanistic Insights into Their Neuroprotective Effects. J. Traditional Complementary Med. 7 (4), 538–552. doi:10.1016/j.jtcme.2017.01.002
Reis, J., Gaspar, A., Milhazes, N., and Borges, F. (2017). Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 60 (19), 7941–7957. doi:10.1021/acs.jmedchem.6b01720
Shimmyo, Y., Kihara, T., Akaike, A., Niidome, T., and Sugimoto, H. (2008). Flavonols and Flavones as BACE-1 Inhibitors: Structure-Activity Relationship in Cell-free, Cell-Based and In Silico Studies Reveal Novel Pharmacophore Features. Biochimica Biophysica Acta (BBA) - General Subj. 1780 (5), 819–825. doi:10.1016/j.bbagen.2008.01.017
Shrestha, S., Seong, S., Paudel, P., Jung, H., and Choi, J. (2018). Structure Related Inhibition of Enzyme Systems in Cholinesterases and BACE1 In Vitro by Naturally Occurring Naphthopyrone and its Glycosides Isolated from Cassia Obtusifolia. Cassia obtusifoliaMolecules 23 (1), 69. doi:10.3390/molecules23010069
Stieger, B., Mahdi, Z. M., and Jäger, W. (2017). Intestinal and Hepatocellular Transporters: Therapeutic Effects and Drug Interactions of Herbal Supplements. Annu. Rev. Pharmacol. Toxicol. 57, 399–416. doi:10.1146/annurev-pharmtox-010716-105010
Ting Chan, X. L., Xu, C., Ng, W., Zhu, L., and Zhou, F. (2014). The Interactions of Herbal Compounds with Human Organic Anion/Cation Transporters. J. Pharmacogenomics Pharmacoproteomics 05 (5), 142. doi:10.4172/2153-0645.1000142
Tran, T.-S., Tran, T.-D., Tran, T.-H., Mai, T.-T., Nguyen, N.-L., Thai, K.-M., et al. (2020). Synthesis, In Silico and In Vitro Evaluation of Some Flavone Derivatives for Acetylcholinesterase and BACE-1 Inhibitory Activity. Molecules 25 (18), 4064. doi:10.3390/molecules25184064
Trott, O., and Olson, A. J. (2009). Autodock Vina: Improving the Speed and Accuracy of Docking with A New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31 (2), 21334. doi:10.1002/jcc.21334
Vauzour, D., Vafeiadou, K., Rodriguez-Mateos, A., Rendeiro, C., and Spencer, J. P. E. (2008). The Neuroprotective Potential of Flavonoids: a Multiplicity of Effects. Genes Nutr. 3 (3-4), 115–126. doi:10.1007/s12263-008-0091-4
Wagle, A., Seong, S. H., Shrestha, S., Jung, H. A., and Choi, J. S. (2019). Korean Thistle (Cirsium Japonicum Var. Maackii (Maxim.) Matsum.): A Potential Dietary Supplement against Diabetes and Alzheimer's Disease. Molecules 24 (3), 649. doi:10.3390/molecules24030649
Walsh, S., Merrick, R., Milne, R., and Brayne, C. (2021). Aducanumab for Alzheimer's Disease? BMJ 374, n1682. doi:10.1136/bmj.n1682
Wang, Z. Y., Zhang, X. D., and Whang, W. K. (2021). The Effect of Terpenoids of Dipsacus Asperoides against Alzheimer's Disease and Development of Simultaneous Analysis by High Performance Liquid Chromatography. Nat. Product. Commun. 16 (9), 1934578X2110446–11. doi:10.1177/1934578X211044603
Williamson, G., Kay, C. D., and Crozier, A. (2018). The Bioavailability, Transport, and Bioactivity of Dietary Flavonoids: A Review from a Historical Perspective. Compr. Rev. Food Sci. Food Saf. 17 (5), 1054–1112. doi:10.1111/1541-4337.12351
Xu, Y., Li, M.-j., Greenblatt, H., Chen, W., Paz, A., Dym, O., et al. (2012). Flexibility of the Flap in the Active Site of BACE1 as Revealed by Crystal Structures and Molecular Dynamics Simulations. Acta Crystallogr. D. Biol. Cryst. 68 (1), 13–25. doi:10.1107/S0907444911047251
Yi, T., Liu, C., Zhang, J., Wang, F., Wang, J., and Zhang, J. (2017). A New Drug Nanocrystal Self-Stabilized Pickering Emulsion for Oral Delivery of Silybin. Eur. J. Pharm. Sci. 96, 420–427. doi:10.1016/j.ejps.2016.08.047
Youn, K., and Jun, M. (2019). Biological Evaluation and Docking Analysis of Potent BACE1 Inhibitors from Boesenbergia Rotunda. Boesenb. rotundaNutrients 11 (3), 662. doi:10.3390/nu11030662
Youn, K., Lee, J., Ho, C.-T., and Jun, M. (2016a). Discovery of Polymethoxyflavones from Black Ginger ( Kaempferia Parviflora ) as Potential β-secretase (BACE1) Inhibitors. J. Funct. Foods 20, 567–574. doi:10.1016/j.jff.2015.10.036
Youn, K., Park, J.-H., Lee, J., Jeong, W.-S., Ho, C.-T., and Jun, M. (2016b). The Identification of Biochanin A as a Potent and Selective β-Site App-Cleaving Enzyme 1 (Bace1) Inhibitor. Nutrients 8 (10), 637. doi:10.3390/nu8100637
Youn, K., Yu, Y., Lee, J., Jeong, W.-S., Ho, C.-T., and Jun, M. (2017). Polymethoxyflavones: Novel β-Secretase (BACE1) Inhibitors from Citrus Peels. Nutrients 9 (9), 973. doi:10.3390/nu9090973
Zhuang, C., Zhang, W., Sheng, C., Zhang, W., Xing, C., and Miao, Z. (2017). Chalcone: a Privileged Structure in Medicinal Chemistry. Chem. Rev. 117 (12), 7762–7810. doi:10.1021/acs.chemrev.7b00020
Keywords: flavonoids, Alzheimer’s disease, BACE1, molecular docking, structure–activity relationship (SAR)
Citation: Mohamed Yusof NIS, Abdullah ZL, Othman N and Mohd Fauzi F (2022) Structure–Activity Relationship Analysis of Flavonoids and Its Inhibitory Activity Against BACE1 Enzyme Toward a Better Therapy for Alzheimer’s Disease. Front. Chem. 10:874615. doi: 10.3389/fchem.2022.874615
Received: 12 February 2022; Accepted: 16 May 2022;
Published: 27 June 2022.
Edited by:
Hongyun Lu, Zhejiang University, ChinaReviewed by:
Lhassane Ismaili, Université Bourgogne Franche-Comté, FranceDaryono Hadi Tjahjono, Bandung Institute of Technology, Indonesia
Copyright © 2022 Mohamed Yusof, Abdullah, Othman and Mohd Fauzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fazlin Mohd Fauzi, ZmF6bGluNTQ2NUB1aXRtLmVkdS5teQ==