
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 31 March 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.860398
This article is part of the Research Topic Frontiers in Chemistry: Editors Showcase 2021 View all 50 articles
 Shengzhou Jin1†
Shengzhou Jin1† Jia-Ying Wang1†
Jia-Ying Wang1† Yao Tang2†
Yao Tang2† Hossein Rouh2
Hossein Rouh2 Sai Zhang2Ting Xu1Yu Wang1Qingkai Yuan2Daixiang Chen3Daniel Unruh2
Sai Zhang2Ting Xu1Yu Wang1Qingkai Yuan2Daixiang Chen3Daniel Unruh2 Guigen Li1,2*
Guigen Li1,2*New multilayer 3D chiral molecules have been designed and synthesized asymmetrically through the strategy of center-to-multilayer folding chirality control and double Suzuki couplings. Individual diastereoisomers were readily obtained and separated via flash column chromatography. The key diastereoisomer was further converted into corresponding enantiomers. These enantiomers possess electron-deficient aromatic bridges layered with top and bottom aromatic scaffolds. X-ray structural analysis has unambiguously confirmed the configuration, and intermolecular packing results in regular planar patterns in solid crystals. The synthesis was achieved in a total of ten steps starting from commercially available starting materials.
Chiral domains widely exist in natural and biological products and have been utilized in chiral catalysts and ligands for asymmetric transformations (Corey et al., 2012; Ito and Nozaki, 2010; Taniguchi et al., 2011; Smith and Jones, 2008). Asymmetric synthesis of both natural and unnatural enantiopure compounds through chiral controllers has been actively pursued for more than half a century (Ito and Nozaki, 2010). This topic will continue to be prevalent in academic laboratories and pharmaceutical and materials industries (Smith and Jones, 2008). Among asymmetric methodologies, those protocols by taking advantage of planar and helical chirality including corresponding chiral π-stacked scaffolds are particularly attractive to chemical and materials communities and have been well documented in the literature (Chen and Shen, 2017; Stará and stary, 2020; Anger et al., 2012). For example, chiral ferrocene ligands, which feature a unique sandwich-like structure, have been implemented in asymmetric catalysis (Dai et al., 2003; Arrayás et al., 2006). Double-layered enantiopure [2.2]paracyclophanes have also been employed as chiral ligands for transition metal–catalyzed asymmetric reactions (Mukhopadhyay et al., 2012; Spuling et al., 2018). Similarly, chiral π-layered [n]helicene derivatives have been employed for materials research regarding polarized organic electronics, optoelectronic chiroptical properties, etc. (Chen and Shen, 2017). In biological systems, DNA is one of the most instrumental biomolecules notably featuring multilayer paired chirality (Moser and Dervan, 1987; Gong et al., 2018; Knouse et al., 2018). The design and synthesis of chiral targets with novel multilayer structures for chemical and biological research have still been a significant and ongoing challenge.
Recently, our group has reported multilayer 3D folding chirality (Wu et al., 2019; Liu et al., 2020; Wu et al., 2020; Zhang and kurti, 2021; Wu et al., 2020) allowing pushing–releasing flexibility of structures, which is different from the rigid layered chirality documented in the literature. This multilayer folding chirality was inspired by our GAP chemistry and technology (Wu et al., 2020; Zhang and kurti, 2021). Figure 1A depicts the C–N bond–based multilayer targets prepared via double Buchwald–Hartwig cross-couplings (Wu et al., 2019; Liu et al., 2020) and featuring pseudo-C2-symmetry. Figure 1B illustrates the C–C bond–based multilayer 3D chiral counterparts, prepared via asymmetric double Suzuki–Miyaura cross-couplings (Wu et al., 2020; Wu et al., 2020; Zhang and kurti, 2021). Interestingly, when the aforementioned chiral compounds were irradiated with 365 nm UV light, various colors in solutions and macro-chirality patterns in solid states were witnessed with the naked eyes without the aid of a scanning device (Wu et al., 2020). Chiral separation via chiral HPLC and chiral auxiliary–based method was conducted for multilayer targets presented in Figure 1A and Figure 1B, respectively.
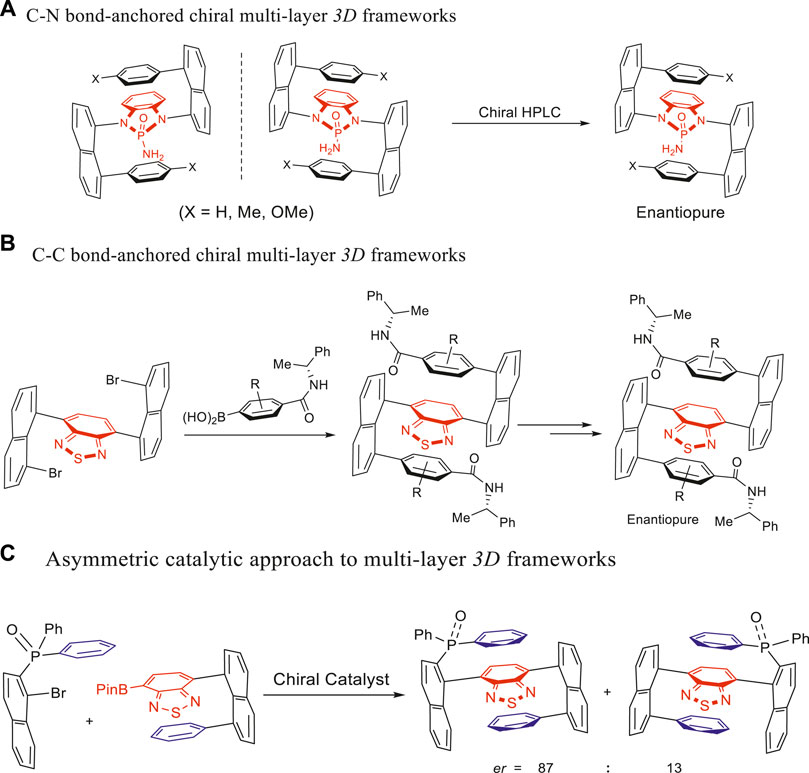
FIGURE 1. Strategy and progress of assembling multilayer 3D chirality. (A) C–N bond–anchored chiral multilayer 3D frameworks. (B) C–C bond–anchored chiral multilayer 3D frameworks. (C) Asymmetric catalytic approach to multilayer 3D frameworks.
Asymmetric catalysis using chiral amide–phosphine ligands resulted in another category of C–C bond–based multilayer 3D chiral targets as shown in Figure 1C (Wu et al., 2020). These compounds would demonstrate new potentials as chiral phosphine ligands for transition metal–mediated reactions upon removing oxygen from the P=O group. Interestingly, these targets presented a new chiral framework containing a pseudo- or pro-chiral center and orientational or rotational axis. The pseudo-chiral center on the phosphorus atom was attached by one naphthalenyl ring and two identical aryl groups but differentiated by planar packing. This pseudo- or pro-chiral center can be extended to other centers of the tetrahedron (e.g., C and Si) or higher polyhedrons in the future. Obviously, atropisomers’ operation along the C–P bond axis made the orientation chirality possible. Three potential rotamers are generated by three moieties of diarylphosphine oxide scaffolds (two differentiated aromatic rings and one P=O group).
We also designed and assembled triple-columned and multiple-layered chiral folding polymers via asymmetric catalytic polymerization based on the use of various new monomers (Tang et al., 2022; Wang et al., 2022). The resulting chiral folding polymers exhibited remarkable optical properties in aggregated states (photoluminescence in solids and aggregation-induced emission in solutions), as well as reversible redox properties in electrochemical performance.
In this work, we would like to report the design and synthesis of enantiomers possessing multilayer 3D folding chirality by utilizing a bridge of electron-deficient scaffolds. The extended central bridge layer can completely avoid possible racemization via rotational operations (Scheme 1) due to the extended aromatic system. The synthetic strategy of central-to-multilayer folding chirality and double Suzuki couplings was proven effective in stereocontrol and chemical yields.
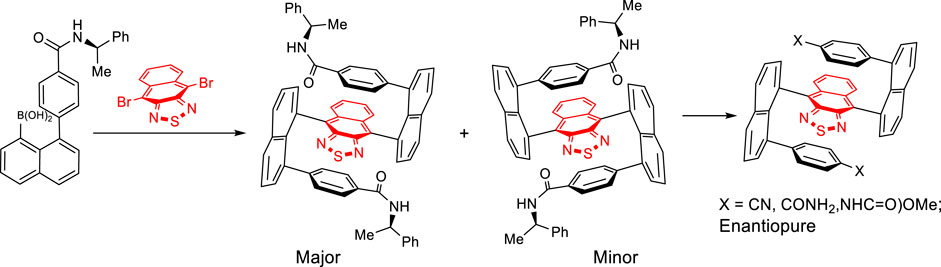
SCHEME 1. Asymmetric synthesis of multilayer chiral folding molecules.
We started this project by synthesizing 4,9-dibromo-2,1,3-naphthothiadiazole (3) at first (Scheme 2). As shown in Scheme 1, 3 was obtained by treating 2,3-diaminonaphthalene (1) with bromine in acetic acid at room temperature followed by intramolecular cyclization with thionyl chloride in mixed solvents of pyridine and chloroform. The next step was furnished by stirring the reaction mixture at room temperature for 2 h and then refluxing for 8 h to give compound 3 in a two-step yield of 39% (Wu et al., 2020; Wu et al., 2021).
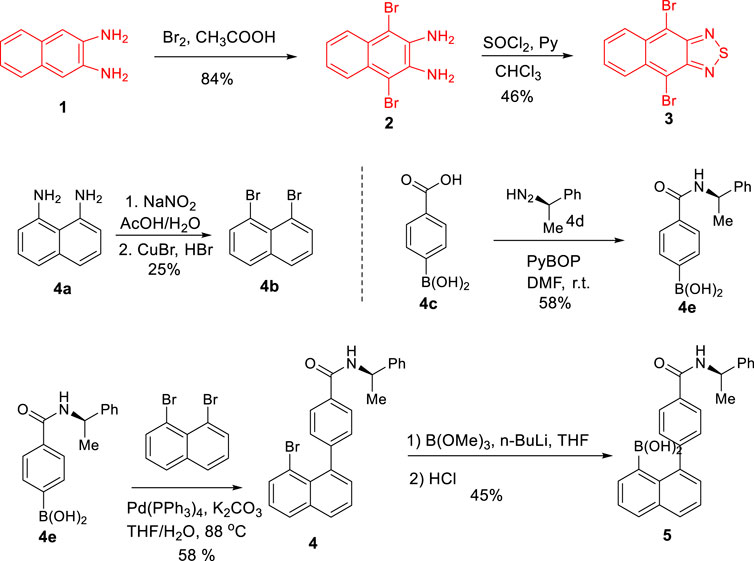
SCHEME 2. Synthesis of building blocks of 3 and 5.
1,8-Dibromonaphthalene (4b) was generated through oxidative cyclization by treating naphthalene-1,8-diamine (4a) with sodium nitrite in an aqueous solution in the presence of acetic acid to afford 1H-naphtho[1,8-de][1,2,3]triazine (Beletskaya et al., 2007). The resulting hetero five-membered ring of triazine was opened by continuously treating with sodium nitrite and then with a mixture of CuBr in 47% HBr solution to give 4b in a yield of 25% (Noland et al., 2011).
Precursor 4e, (R)-(4-((1-phenylethyl)carbamoyl)phenyl)boronic acid, was obtained according to a classical procedure (Coste et al., 1990). In this preparation, 4-carboxybenzeneboronic acid (4c) (1.0 equiv) was treated with PyBOP (2.0 equiv) in DMF stirring for a few minutes, followed by adding (R)-(1-phenylethyl)diazene (2.0 equiv) into the reaction mixture. The carbonyl coupling was completed within 14 h at room temperature before quenching. Work-up and purification via column chromatography gave 4e in a chemical yield of 58%.
Precursor 4, (R)-4-(8-bromonaphthalen-1-yl)-N-(1-phenylethyl)benzamide, was prepared by performing Suzuki coupling of (R)-(4-((1-phenylethyl)carbamoyl)phenyl)boronic acid (4e) with 1,8-dibromonaphthalene (4b) under the standard catalytic condition with a moderate yield of 58%. (R)-(8-(4-((1-Phenylethyl)carbamoyl)phenyl)naphthalen-1-yl)boronic acid (5) was obtained by treating (R)-4-(8-bromonaphthalen-1-yl)-N-(1-phenylethyl)benzamide 4 with n-BuLi followed by the reaction with B(OMe)3 under Ar protection (Ishihara et al., 2002). After aqueous HCl hydrolyzed the resulting boronic ester, the precursor 5 was obtained via flash silica gel chromatography in 45% yield as a yellow powder solid.
For the synthesis of diastereoisomers, 6a and 6b, double Suzuki couplings turned out to be the crucial step for asymmetrically constructing the two C–C bonds (Wu et al., 2020). Investigations on the optimization of the coupling reaction of 3 with 5 became necessary. At first, the reaction was conducted using Pd(PPh3)4 as the catalyst and K2CO3 as the base in THF/H2O at 90°C for 36 h under argon protection (Table 1, entry 1). Surprisingly, almost no product was detected under this condition. To further optimize the reaction, various conditions, such as toluene/H2O, Et2O/H2O, and 1,4-dioxane/H2O as the solvent and K2CO3 as the base (Table 1, entries 2–4) and toluene as the solvent and K3PO4 as the base (Table 1, entry 5), were examined. These changes did not show satisfactory improvements either. To our delight, when Pd(OAc)2/PPh3 was employed as the catalyst, and xylene/H2O as the solvent at 130°C for 36 h, the yield was improved to 32% (Table 1, entry 6). Subsequently, as the reaction time was extended to 60 h, the yield increased to 45% (Table 1, entry 7).

TABLE 1. Optimization of the reaction conditions for the palladium-catalyzed Suzuki coupling of 3 with 5.
As shown in Scheme 3, the treatment of 6a with thionyl chloride at 80°C for 8 h afforded 7a in 85% yield (Lv et al., 2016). At room temperature, oxidative hydrolysis of 7a by H2O2 and K2CO3 gave 8a in 25% yield (Mothana et al., 2010). The yield was improved to 55% after the unreacted starting material 6a was recovered. Hofmann rearrangement of 7a by PhI(OAc)2 and KOH in CH3OH afforded 9a in 80% yield (Zhang et al., 2016).
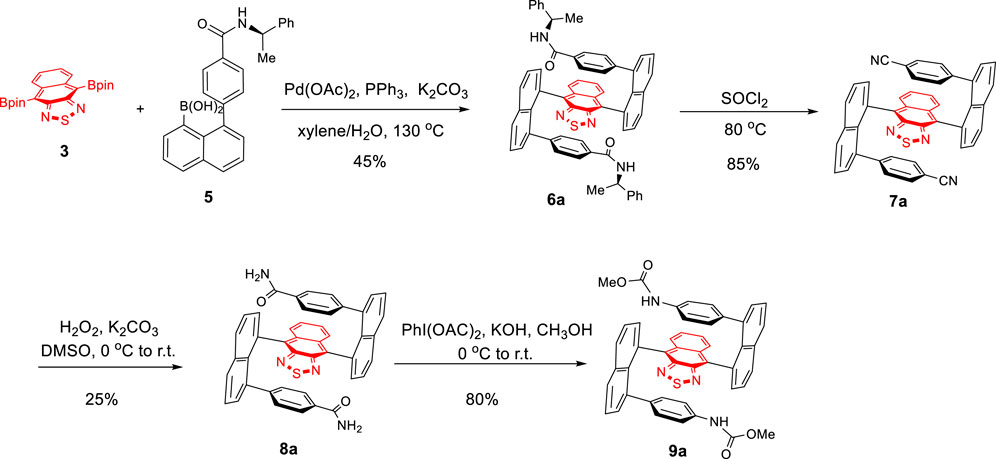
SCHEME 3. Asymmetric synthesis of multilayer 3D target 9a.
Product 6 was obtained as a red solid showing the diastereoselectivity of two diastereoisomers, 6a–6b, as 1.6:1 dr. Two isomers were readily separated from the silica gel column. Single crystals of both 6a and 6b were grown by slowly diffusing hexane into the dichloromethane solution. X-ray crystal structure analysis confirmed the configurations of 6a and 6b (Supplementary Material and Figure 2A). This structure shows the two naphthalene columns are nearly perpendicular to the central naphthothiadiazole ring. This structural arrangement is slightly different from that of a previously bicyclic bridge-based counterpart in which two naphthalene piers are positioned at a dihedral angle of about 60° (Wu et al., 2020). The up and down planes of the present molecules are almost parallel to the central layer in a similar arrangement to the aforementioned bicyclic bridge-based structural system (Figure 2A). The enantiomers’ packing in a single X-ray unit cell represents an interesting arrangement in which the intermolecular distances between proximate aromatic rings are very similar to intramolecular distances (Figure 2B). Additionally, the phenyl ring of one of the amide groups is oriented nearly parallel to the naphthalene piers on two sides. Interestingly, the packing between two crystal unit cells displays a similar distance to that inside a single unit cell. This pattern involves 12 layers in one packing column (Figure 2C).
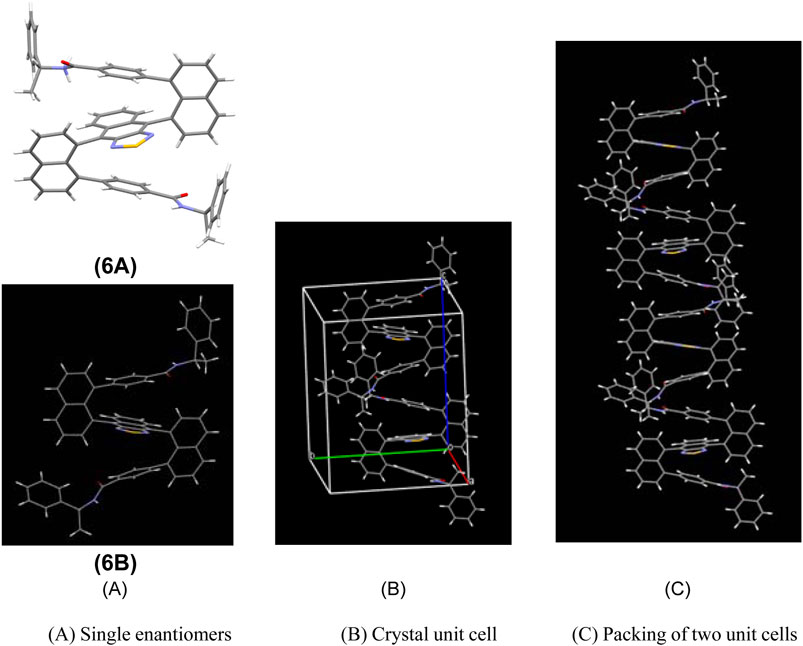
FIGURE 2. Single-crystal structure of 6a and 6b and intermolecular packing. (A) Single enantiomers. (B) Crystal unit cell. (C) Packing of two unit cells.
As shown in Figure 3A, 6a–9a exhibited a similar spectral behavior, featuring a sharp absorption band between 285 and 350 nm and one broader band between 420 nm and 560 nm. In 6a, 8a, and 9a, the π–π conjugation between the aromatic rings and the adjacent carbonyl groups leads to an extended conjugated system. A significant red shift was observed in 9a, which moved to a longer wavelength by about 15 nm than 6a. It is worth noting that the peak intensity of 8a was sharply lower than the intensity of other derivatives.
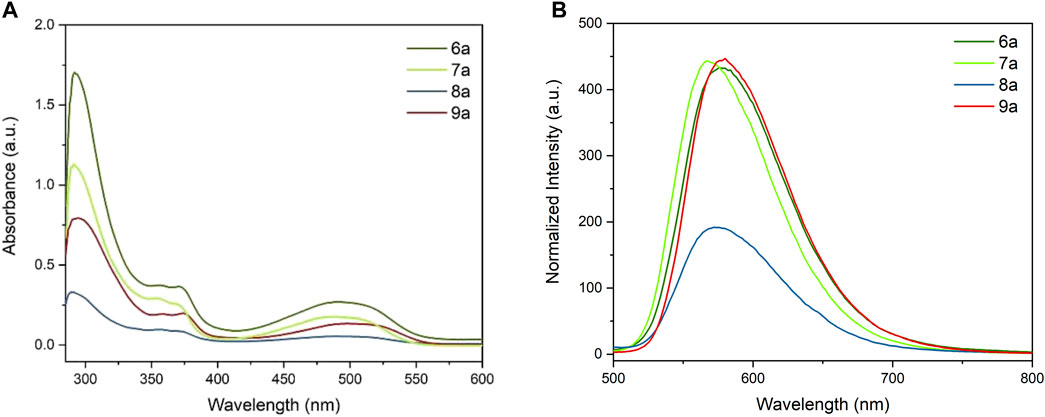
FIGURE 3. (A) UV–Vis absorbance of 6a–9a (0.05 mg/ml) in THF. (B) Normalized fluorescent spectra of 6a–9a (0.05 mg/ml) in THF. Excitation wavelength for 6a–9a: 323 nm.
The fluorescence emission studies of 6a–9a were performed in THF by performing excitation at the same wavelength, and all derivatives displayed a broad emission band around 520–740 nm (Figure 3B). Both 6a and 9a showed a strong fluorescence emission at about 576 nm. The nitrile group with a stronger electron-withdrawing effect made the maximum fluorescent emission peaks shift to a shorter wavelength (568 nm, compared to 6a and 9a). Like the absorption spectrum, the emission intensity of 8a was diminished mainly, which was half of that of the other derivatives. Based on the spectral observation on absorption and emission, it can be concluded that different electron-withdrawing groups influence the conjugating system in various manners, resulting in an increase/decrease in π–π* energy gaps. The different effects on conjugations are probably one of the reasons for the shift of absorption to higher wavelengths and lowered emission; however, the coordination of those groups with the solvent could also contribute to the observed change in both spectrums.
A mixture of 3.45 ml of bromine (20.7 g, 85.5 mmol) and 90 ml of glacial acetic acid was added dropwise into a solution of 2,3-diaminonaphthalene (5.0 g, 31.6 mmol) in 140 ml of glacial acetic acid, with vigorous stirring at room temperature. After 8 h, water was added to the solution. The precipitate was filtered off and washed subsequently with glacial acetic acid and water. After drying, a brown powder (8.4 g, 84%) was obtained.
A solution of 2 (8.1 g, 25.6 mmol) in 280 ml of chloroform was added dropwise into the mixture of thionyl chloride (12.7 ml, 20.8 g, 174.8 mmol) in 280 ml of chloroform and 31 ml of pyridine, with vigorous stirring in an ice-water bath. After the dropwise addition, the solution was stirred for 2 h at room temperature and refluxed overnight. Evaporation of the solvent and purification by column chromatography on silica gel with dichloromethane–hexane as the eluent were performed, and an orange-color product (4.5 g, 46%) was obtained.
4e (3.7 g, 13.9 mmol), 4b (4.0 g, 13.9 mmol), Pd(PPh3)4 (0.8 g, 0.7 mmol), and anhydrous K2CO3 (5.8 g, 42.0 mmol) were dissolved into THF/H2O (100 ml/20 ml), and the solution mixture was degassed and charged with argon. The solution was heated to 88°C and stirred overnight. After the reaction was completed, the resulting mixture was evaporated, and the residue was diluted with EtOAc. The organic phase was washed twice with water and brine, dried over Na2SO4, and concentrated under reduced pressure. Work-up via column chromatography (DCM: acetone = 10/1) gave 4 as a white solid (3.5 g, 58% yield).
In a dried and argon-flushed round bottom flask with a stir bar, bromide substrate 4 (10 mmol) was dissolved into anhydrous THF and stirred for 5 min at −78°C. 1.6 M n-butyllithium (25 mmol) solution was added dropwise with syringe and stirred at −78°C for 0.5 h. Then, B(OMe)3 (40 mmol) was added dropwise at −78°C; the reaction mixture was warmed up to RT and stirred for 8 h. Then, 1 M HCl (10 mmol) was added, and the mix was stirred for 6 h. After monitoring by TLC analysis, extraction with EA, and drying for column (Hexane/EA = 5/1 to 1/1), we obtained the crude solid, which can be directly used in the next step.
3 (60 mg, 1 equiv), chiral boronic acid 5 (200 mg, 3 equiv), Pd(OAc)2 (8 mg, 0.2 equiv), PPh3 (14 mg, 0.6 equiv), and K2CO3 (73 mg, 6 equiv) were dissolved into xylene/H2O (5 ml/1 ml), and the solution mixture was degassed with argon. The resulting solution was heated to 130°C and stirred for 60 h. Work-up via column chromatography (DCM/acetone, 20/1) afforded the pure isomer 6 as a red solid (70 mg, 45% yield).
6 (100 mg) was dissolved into CHCl3 (8 ml), SOCl2 (5 ml), and DMF (one drop). The resulting solution was heated to 80°C for 8 h. The reaction was monitored by TLC analysis and worked up for column (DCM/acetone = 20/1) to afford the pure product 7 as a red solid (62 mg, 85% yield).
To the solution of 7 (50 mg, 0.08 mmol) in DMSO (2 ml), cooled in an ice bath, were added 30% H2O2 (2 ml) and anhydrous K2CO3 (1.5 g, 11 mmol). The mixture was stirred and allowed to warm up to room temperature. After 30 min, distilled water (2 ml) and DMSO (2 ml) were added and cooled, and the mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with EtOAc and quenched with saturated NH4Cl (2 ml). The organic phase was washed twice with water and brine, dried over Na2SO4, and concentrated under reduced pressure. Work-up via column chromatography (DCM:CH3OH = 5/1) afforded 8 as a red solid (13 mg, 25% yield).
To a solution of KOH (10 mg, 0.17 mmol) in methanol (5 ml) was added 8 (25 mg, 0.04 mmol). The mixture was stirred at room temperature until a homogeneous solution was obtained and then cooled to 0°C in an ice-water bath. Diacetoxyiodobenzene (23 mg, 0.07 mmol) was added in one portion. The mixture was stirred at ice bath temperature for 6 h. TLC was checked, the solvent was evaporated, and the column was run (DCM: acetone = 20: 1) to yield 9 as a red solid (9 mg, 80% yield).
In conclusion, we have reported the design and asymmetric synthesis of new multilayer 3D chiral molecules through the strategy of central-to-multilayer folding chirality and double Suzuki couplings. Individual diastereopure isomers have been obtained and separated via flash column chromatography. The major key isomer has been converted into corresponding enantiomers. These enantiomers possess electron-deficient aromatic bridges and top and bottom layers. The absolute configuration of the products has been unambiguously determined by X-ray structural analysis. Intermolecular packing results in a regular planar pattern in crystals. The synthesis was achieved in a total of ten steps starting from commercially available starting materials.
The original contributions presented in the study are included in the article/Supplementary Material, and further inquiries can be directed to the corresponding author.
GL designed the project, and DC assisted GL on project designing. GL and YT wrote the paper. J-YW, YT, HR, SZ, TX, and QY performed the experiments and analysis. DC participated in polishing the manuscript.
We would like to acknowledge the financial support from the National Natural Science Foundation of China (Nos. 22071102 and 91956110) and Robert A. Welch Foundation (D-1361-20210327, United States).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.860398/full#supplementary-material
Anger, E., Srebro, M., Vanthuyne, N., Toupet, L., Rigaut, S., Roussel, C., et al. (2012). Ruthenium-Vinylhelicenes: Remote Metal-Based Enhancement and Redox Switching of the Chiroptical Properties of a Helicene Core. J. Am. Chem. Soc. 134, 15628–15631. doi:10.1021/ja304424t
Arrayás, R. G., Adrio, J., and Carretero, J. C. (2006). Recent Applications of Chiral Ferrocene Ligands in Asymmetric Catalysis. Angew. Chem. Int. Ed. 45, 7674–7715. doi:10.1002/anie.200602482
Beletskaya, I., Sigeev, A., Peregudov, A., and Petrovskii, P. (2007). Catalytic Sandmeyer Bromination. Synthesis 2007, 2534–2538. doi:10.1055/s-2007-983784
Chen, C.-F., and Shen, Y. (2017). Helicene Chemistry: From Synthesis to Applications. Springer. doi:10.1007/978-3-662-53168-6
Coste, J., Le-Nguyen, D., and Castro, B. (1990). PyBOP: A New Peptide Coupling Reagent Devoid of Toxic By-Product. Tetrahedron Lett. 31, 205–208. doi:10.1016/s0040-4039(00)94371-5
Dai, L.-X., Tu, T., You, S.-L., Deng, W.-P., and Hou, X.-L. (2003). Asymmetric Catalysis with Chiral Ferrocene Ligands. Acc. Chem. Res. 36, 659–667. doi:10.1021/ar020153m
Gong, H., Li, J., Xu, A., Tang, Y., Ji, W., Gao, R., et al. (2018). An Electron Transfer Path Connects Subunits of a Mycobacterial Respiratory Supercomplex. Science 362, 6418. doi:10.1126/science.aat8923
Ishihara, K., Ohara, S., and Yamamoto, H. (2002). 3,4,5-Trifluorophenyl)Boronic Acid-Catalyzed Amide Formation from Carboxylic Acids and Amines: N-Benzyl-4- Phenylbutyramide. Org. Synth. 79, 176–183. doi:10.1002/0471264180.os079.21
Knouse, K. W., deGruyter, J. N., Schmidt, M. A., Zheng, B., Vantourout, J. C., Kingston, C., et al. (2018). Unlocking P(V): Reagents for Chiral Phosphorothioate Synthesis. Science 361, 1234–1238. doi:10.1126/science.aau3369
Liu, Y., Wu, G., Yang, Z., Rouh, H., Katakam, N., Ahmed, S., et al. (2020). Multi-layer 3D Chirality: New Synthesis, AIE and Computational Studies. Sci. China Chem. 63, 692–698. doi:10.1007/s11426-019-9711-x
Lv, Z., Wang, B., Hu, Z., Zhou, Y., Yu, W., and Chang, J. (2016). Synthesis of Quinazolines from N,N′-Disubstituted Amidines via I2/KI-Mediated Oxidative C-C Bond Formation. J. Org. Chem. 81, 9924–9930. doi:10.1021/acs.joc.6b02100
Moser, H. E., and Dervan, P. B. (1987). Sequence-specific Cleavage of Double Helical DNA by Triple helix Formation. Science 238, 645–650. doi:10.1126/science.3118463
Mothana, S., Grassot, J.-M., and Hall, D. G. (2010). Multistep Phase-Switch Synthesis by Using Liquid-Liquid Partitioning of Boronic Acids: Productive Tags with an Expanded Repertoire of Compatible Reactions. Angew. Chem. Int. Edition 49, 2883–2887. doi:10.1002/anie.200906710
Mukhopadhyay, S., Jagtap, S. P., Coropceanu, V., Brédas, J.-L., and Collard, D. M. (2012). π-Stacked Oligo(phenylene Vinylene)s Based on Pseudo-geminal Substituted [2.2]Paracyclophanes: Impact of Interchain Geometry and Interactions on the Electronic Properties. Angew. Chem. Int. Ed. 51, 11629–11632. doi:10.1002/anie.201205738
Noland, W. E., Narina, V. S., and Britton, D. (2011). Synthesis and Crystallography of 8-Halonaphthalene-1-Carbonitriles and Naphthalene-1,8-Dicarbonitrile. J. Chem. Res. 35, 694–697. doi:10.3184/174751911x13222107572093
Smith, D. A., and Jones, R. M. (2008). The Sulfonamide Group as a Structural Alert: a Distorted story? Curr. Opin. Drug Discov. Devel. 11, 72–79.
Spuling, E., Sharma, N., Samuel, I. D. W., Zysman-Colman, E., and Bräse, S. (2018). (Deep) Blue Through-Space Conjugated TADF Emitters Based on [2.2]paracyclophanes. Chem. Commun. 54, 9278–9281. doi:10.1039/c8cc04594a
Stará, I. G., and Starý, I. (2020). Helically Chiral Aromatics: The Synthesis of Helicenes by [2 + 2 + 2] Cycloisomerization of π-Electron Systems. Acc. Chem. Res. 53, 144–158. doi:10.1021/acs.accounts.9b00364
Tang, Y., Jin, S., Zhang, S., Wu, G., Wang, J., Xu, T., et al. (2022). Research. AAAS/Science partner journal, 9847949. doi:10.34133/2022/9847949
Taniguchi, K., Maeda, R., Ando, T., Okumura, T., Nakazawa, N., Hatori, R., et al. (2011). Chirality in Planar Cell Shape Contributes to Left-Right Asymmetric Epithelial Morphogenesis. Science 333, 339–341. doi:10.1126/science.1200940
Wang, J. Y., Tang, Y., Wu, G. Z., Zhang, S., Rouh, H., Jin, S., et al. (2022). Asymmetric Catalytic Assembly of Triple‐Columned and Multilayered Chiral Folding Polymers Showing Aggregation‐Induced Emission (AIE). Chem. A Eur. J 28, e202200183. doi:10.1002/chem.202200183
Wu, G., Liu, Y., Rouh, H., Ma, L., Tang, Y., Zhang, S., et al. (2021). Asymmetric Catalytic Approach to Multilayer 3D Chirality. Chem. Eur. J. 27, 7977. doi:10.1002/chem.202101318
Wu, G., Liu, Y., Yang, Z., Jiang, T., Katakam, N., Rouh, H., et al. (2020). Enantioselective Assembly of Multi-Layer 3D Chirality. Natl. Sci. Rev. 7, 588–599. doi:10.1093/nsr/nwz203
Wu, G., Liu, Y., Yang, Z., Katakam, N., Rouh, H., Ahmed, S., et al. (2019). Multilayer 3D Chirality and its Synthetic Assembly. Research 2019, 1–11. doi:10.34133/2019/6717104
Wu, G., Liu, Y., Yang, Z., Ma, L., Tang, Y., Rouh, H., et al. (2021). Triple-Columned and Multiple-Layered 3D Polymers: Design, Synthesis, Aggregation-Induced Emission (AIE), and Computational Study. Research 2021, 3565791. doi:10.34133/2021/3565791
Zhang, C.-L., Gao, Z.-H., Liang, Z.-Q., and Ye, S. (2016). N-heterocyclic Carbene-Catalyzed Synthesis of Multi-Substituted Benzenes from Enals and α-Cyano-β-methylenones. Adv. Synth. Catal. 358, 2862–2866. doi:10.1002/adsc.201600531
Keywords: multilayer folding chirality, asymmetric synthesis, asymmetric Suzuki–Miyaura coupling, central-to-folding chirality, planar packing
Citation: Jin S, Wang J-, Tang Y, Rouh H, Zhang S, Xu T, Wang Y, Yuan Q, Chen D, Unruh D and Li G (2022) Central-to-Folding Chirality Control: Asymmetric Synthesis of Multilayer 3D Targets With Electron-Deficient Bridges. Front. Chem. 10:860398. doi: 10.3389/fchem.2022.860398
Received: 22 January 2022; Accepted: 07 March 2022;
Published: 31 March 2022.
Edited by:
Iwao Ojima, Stony Brook University, United StatesReviewed by:
Ramon Rios, University of Southampton, United KingdomCopyright © 2022 Jin, Wang, Tang, Rouh, Zhang, Xu, Wang, Yuan, Chen, Unruh and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guigen Li, Z3VpZ2VuLmxpQHR0dS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.