
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 08 April 2022
Sec. Chemical Physics and Physical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.859750
This article is part of the Research Topic Electronic and Nuclear Quantum Dynamics of Molecules in Intense Laser Fields View all 13 articles
 Tomoyuki Endo1,2
Tomoyuki Endo1,2 Karl Michael Ziems3,4
Karl Michael Ziems3,4 Martin Richter3Friedrich G. Fröbel3
Martin Richter3Friedrich G. Fröbel3 Akiyoshi Hishikawa5,6Stefanie Gräfe3,4François Légaré1*
Akiyoshi Hishikawa5,6Stefanie Gräfe3,4François Légaré1* Heide Ibrahim1*
Heide Ibrahim1*We have investigated the dissociation mechanisms of the prototypical heavy polar molecule OCS into the two break-up channels of the dication, OCS2+ → O+ + CS+ and OC+ + S+, in phase-locked two-color intense laser fields. The branching ratio of the breaking of the C–O and C–S bonds followed a pronounced 2π-oscillation with a modulation depth of 11%, depending on the relative phase of the two-color laser fields. The fragment ejection direction of both break-up channels reflects the anisotropy of the tunneling ionization rate, following a 2π-periodicity, as well. The two dissociation pathways in the C–S bond breaking channel show different phase dependencies of the fragment ejection direction, which are assigned to post-ionization dynamics. These observations, resulting from the excitation with asymmetric two-color intense laser fields, supported by state-of-the-art theoretical simulations, reveal the importance of post-ionization population dynamics in addition to tunneling ionization in the molecular fragmentation processes, even for heavy polar molecules.
Coherent control of molecular dynamics in real-time is one of the ultimate goals of chemistry. One can envision the production of novel chemical substances and chemical reactions without undesired side-products. Coherent reaction control with photons such as the pump-dump technique (Tannor and Rice, 1985; Shen et al., 1999) adapting the field to the instantaneous dynamics (Malinovsky et al., 1997; Gräfe et al., 2005) or interference between reaction pathways (Brumer and Shapiro, 1986; Wang et al., 1996) have been demonstrated in the past decades. Though these methods are powerful to achieve desired chemical reactions, possible reaction outcomes are limited by the structures of the potential energy surface (PES) along the reaction coordinates.
The development of ultrashort laser techniques opened-up new paths for direct reaction control by tailored intense laser fields (Rabitz, 2006). In intense laser fields (typically
Recently, simple pulse shaping techniques based on breaking the inversion symmetry of an electric field have been employed instead of complex tailored laser fields. Carrier-envelope-phase (CEP) stabilized few-cycle laser fields and phase-locked two-color laser fields are widely used to investigate the underlying mechanisms of such reaction control (Alnaser and Litvinyuk, 2017). Control of the fragment ejection direction has been demonstrated for a wide range of non-polar (symmetric) molecules, from the simplest diatomic molecule H2 (D2) (Kling et al., 2006; Roudnev and Esry, 2007; Ray et al., 2009; Znakovskaya et al., 2012; Wanie et al., 2016), summarized in (Ibrahim et al., 2018), to poly-atomic hydrocarbons (Xie et al., 2012, 2014; Alnaser et al., 2014; Miura et al., 2014; Song et al., 2015; Doblhoff-Dier et al., 2016), the tri-atomic CO2 molecule (Endo et al., 2016, 2017), as well as CF4 as part of this special issue (Hasegawa et al., 2022). Several mechanisms have been proposed such as the interference of dissociation pathways for
In the case of polar (asymmetric) molecules on the contrary, the fragment ejection direction in asymmetric laser fields has been exclusively explained by the anisotropy of the tunneling ionization rate, which is determined by the shape of the ionizing molecular orbital (usually the highest occupied molecular orbital, HOMO) and the molecule’s permanent dipole moment (Ohmura and Tachiya, 2008; Holmegaard et al., 2010; Dimitrovski et al., 2011; Li et al., 2011; Wu et al., 2012; Ohmura et al., 2014; Li et al., 2016; Wustelt et al., 2018; Yue et al., 2018; Endo et al., 2019; Ohmura et al., 2019), rather than by post-ionization interactions. Although post-ionization interactions should be important even for the dissociation of polar molecules, their contributions are generally buried under the strong anisotropy of the tunneling ionization rate. A deeper understanding of both the ionization step and post-ionization interactions is required to achieve flexible and mighty reaction control.
In the present study, we investigated the break-up processes of carbonyl sulfide, OCS, in phase-locked two-color intense laser fields by using the Coulomb explosion imaging technique (Wales et al., 2014; Endo et al., 2020; Zhao et al., 2021). OCS is the prototype of a heavy polar molecule (not containing hydrogen atoms) and has been widely investigated, including a molecular movie of its alignment (Karamatskos et al., 2019). In addition, we have demonstrated the applicability of reaction control based on post-ionization interactions to the dynamics of heavy atoms by using a simple pulse shaping technique. The effects of post-ionization interactions between polar molecules and phase-locked two-color intense laser fields, and the resulting molecular dynamics are discussed on the basis of the two dissociation channels OCS2+, OCS2+ → O+ + CS+/OC+ + S+, and compared to state-of-the-art theoretical simulations using real-time real-space time-dependent density functional theory (rtTDDFT) and semi-classical surface-hopping dynamics.
The 2.5 kHz Titanium-Sapphire laser beam line of the Advanced Laser Light Source (ALLS) user facility in Varennes, Canada, was used to perform ion coincidence three-dimensional (3D) momentum imaging measurements. Two-color laser fields were obtained in a Mach-Zehnder interferometer after second harmonic generation, as schematically shown in Figure 1.
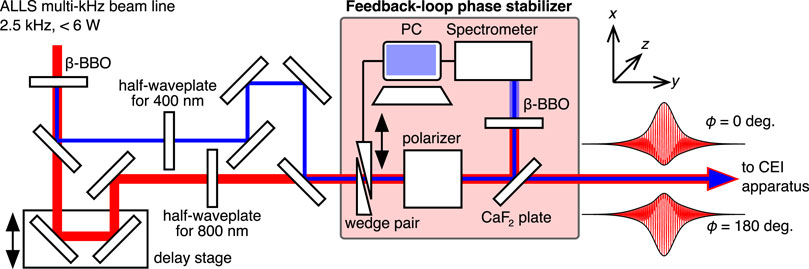
FIGURE 1. Schematic of the experimental setup to generate phase-locked two-color laser fields. The output of a Ti:Sapphire laser amplifier system (ω, 800 nm, 2.5 kHz) was introduced into a β-barium borate (BBO, Type I) crystal to generate second-harmonic pulses (2ω, 400 nm). ω and 2ω pulses were separated by a dielectric mirror. The polarization direction of both pulses was rotated by a half-waveplate and extracted by a polarizer to parallelize the polarization directions along the x-axis in the laboratory frame and tune the field intensity individually. The time-delay between both pulses was controlled by a linear delay stage before ω and 2ω pulses were collinearly re-combined. A pair of fused silica wedges was used to fine-tune the relative phase ϕ, which was locked by a feedback loop utilizing the interference spectrum of the 2ω pulses. Schematic shapes of the phase-locked two-color laser electric fields (ϕ =0° and 180°) are shown.
The electric field of the resulting two-color laser field, composed of the fundamental (carrier frequency of ω) and parallel polarized second harmonic (2ω) pulses, can be expressed as
where Fω and F2ω are the electric field amplitudes of the fundamental and second harmonic fields, respectively, and ϕ is the relative phase between both fields. The relative phase was determined keeping the fragment ejection direction consistent with previous OCS studies (Ohmura and Tachiya, 2008; Ohmura et al., 2014, 2019); ϕ is zero when the maximum amplitude side of F(t) points towards the positive x direction in the laboratory frame (see Figure 1).
To stabilize ϕ during long data acquisition times, the obtained two-color pulses were partly reflected by a CaF2 wedge. The reflected pulses were subsequently introduced into a second β-BBO crystal (Type I). The interference spectrum of both second harmonics generated by the first and the second BBO crystals was used to lock ϕ by a feedback loop with a pair of fused silica wedges mounted on a motorized translational stage (Endo et al., 2016, 2017). The root mean square of the measured phase deviation was less than 14°, corresponding to 0.05 fs over 24 h. The intensity of each laser field,
The obtained two-color laser fields were focused on an effusive molecular beam of a gas mixture (He 95% + OCS 5%) by a concave mirror (f = 100 mm) placed in an ultrahigh vacuum chamber. The generated ions were accelerated by electrodes in velocity map configuration to a position sensitive detector with delay-line anodes (RoentDek Handels GmbH). The 3D momentum of the ith ion
with mi being the mass of the ith fragment ion.
Numerical simulations based on rtTDDFT and semi-classical surface-hopping dynamics were performed separately for the ionization and dissociation steps. First, we simulated the ionization of OCS in two-color laser fields under the fixed nuclei approximation, since the effects of bond stretching during ionization is not significant in the present experimental conditions. Second, to consider nuclear dynamics of OCS2+ in two-color laser fields, we simulated the dissociation processes starting from the selected electronic states.
We employed a state-of-the-art numerical description of the strong-field response of OCS (and OCS+) based on the rtTDDFT as realized in the Octopus program package (Andrade et al., 2012; Tancogne-Dejean et al., 2020). Shortly, the 16 outermost electrons, within the time-dependent Kohn–Sham orbitals, are propagated numerically in real-time and real-space using finite element methods. From these orbitals, the time-dependent electron density is constructed. For the calculations, a spherical box with a radius of R = 20 a. u. was used, including a complex absorbing potential at its boundary with rabs = 5 a.u. We confirmed that increasing the box radius to R = 30 a. u. did not change the results substantially (a maximum deviation in the total number of emitted electrons Δnemit ∼ 0.002,
For the exchange-correlation potential, we chose the local-density approximation (LDA) functional with average-density self-interaction correction (ADSIC). The highest occupied DFT Kohn–Sham orbitals included in our calculations can be specified as follows: the HOMO-5, HOMO-4, HOMO-3, and HOMO-2 are of σ-type, while the (doubly degenerate) HOMO-1 and HOMO are of π-type. We performed five different calculations for relative phases of ϕ = 0°, 45°, 90°, 135°, 180°. The laser pulses were implemented as in Eq. 1, with Fω = 0.0755 a.u. (corresponding to an intensity Iω = 2 × 1014 W/cm2), a pulse width of 20 fs, and a wavelength of 800 nm. For the 2ω field, a field strength of 0.0407 a.u. (corresponding to an intensity I2ω = 0.29Iω = 0.58 × 1014 W/cm2), a pulse width of 50 fs, and a wavelength of 400 nm are chosen. The pulse duration used in the simulation is shorter than that in the experiment due to the limitation of computational costs. However, as mentioned below in Section 3, since ionization mainly occurs around the peak of the laser electric field and dissociation from the excited state of OCS2+ proceeds in a few tens of femtoseconds, the pulse durations of 20 and 50 fs are long enough to compare with the experiment quantitatively. As the orbitals strongly mix in the presence of the laser field, we focus our evaluation on the projection of time-dependent Kohn–Sham wavefunctions on ground-state wave-functions at the end of the simulation time, when the pulse has vanished. By doing so, we can monitor the fraction of ionized electrons that originated from σ and π orbitals, respectively.
This procedure allows us to assign, which orbitals are depopulated in the field, and, consequently, which cationic (and dicationic) electronic states are formed upon ionization.
The ground state equilibrium structure of neutral OCS was optimized by means of CASSCF(12, 10)/aug-cc-pVTZ level of theory (Aquilante et al., 2016) state-averaged over the first six singlet roots using the OpenMolcas suite program (Roos et al., 1980; Godbout et al., 1992). A subsequent vibrational analysis was carried out to verify that a minimum on the PES was obtained. The active orbitals were chosen by using the full set of valence orbitals (16 electrons in 12 orbitals) and omitting the two lowest σ orbitals with main contributions from 1s atomic orbitals of the oxygen and sulfur atom.
For the dynamical calculations of OCS2+, the semi-classical program package SHARC 2.1 (Richter et al., 2011; Mai et al., 2018) interfaced with OpenMolcas (Aquilante et al., 2016) was employed. The electronic structure properties were calculated using state-average CASSCF(10, 10)/cc-pVDZ for the first 20 roots in the triplet manifold, while the nuclei are treated classically. Non-adiabatic effects are realized in the SHARC code via jumps between electronic surfaces, and the two-color field is incorporated explicitly as off-diagonal element in the electronic Hamiltonian used for the propagation of the electronic wave function. The initial trajectories were Wigner sampled around the equilibrium structure (obtained as described above) of the neutral OCS and started in the triplet ground state of OCS2+, X3Σ−, and the excited 13Π state assuming an instantaneous excitation. For each of the three investigated relative phases of ϕ = 0°, 180°, 270°, an ensemble of 20 trajectories was started. The classic propagation was carried out for 75 fs with a time step of 0.1 fs centred around the light pulse. The KER was calculated by tracking the relevant mass-weighted molecular fragment in position space.
For all calculations, a reproducible and transferable computational environment was set up with the Nix package manager using NixOS-QChem (Kowalewski and Seeber, 2022) (commit 206dcba) and Nixpkgs (NixOS, 2021) (commit 9775b39).
The electronic configuration of neutral OCS in the ground state is (core)14(6σ)2(7σ)2(8σ)2 (9σ)2(2π)4(3π)4(4π)0. The potential energy curves of the lower lying electronic states, relevant to the two dissociation channels, OC+ + S+ and O+ + CS+, are depicted as a function of the bond lengths in Figure 2. The double ionization potential of OCS at the equilibrium structure (Franck-Condon region), reached by removing two valence electrons from the 3π orbital, is 30.3 eV (Langford et al., 1991). Since the electronic ground state of OCS2+ is the metastable X3Σ− state (Kaneyasu et al., 2015) and its lifetime is theoretically predicted to be on the order of 10160 s (Ridard et al., 1988), the dissociation does not occur directly from the ground state of OCS2+. In our nuclear dynamics simulation in intense laser fields, dissociation from the (3π)−2 states does not occur within our simulation time. Note that the molecular states are described in terms of single orbital occupations, which correspond to the dominant contributions.
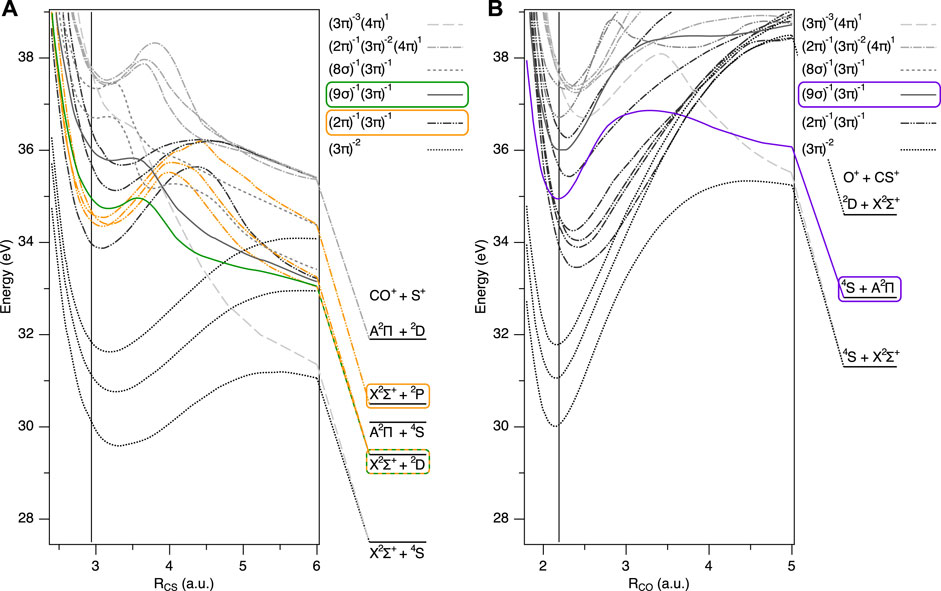
FIGURE 2. Potential energy curves of lower lying electronic states of OCS2+ and corresponding dissociation limits, dissociating to (A) OC+ + S+ (RCO =2.185 a. u.) and (B) O+ + CS+ (RCS =2.95 a. u.) (Brites et al., 2008). The bending angle is fixed at 180°. Vertical lines correspond to the equilibrium geometry of neutral OCS in the ground state. Dotted lines correspond to (3π)−2 states (X3Σ−, a1Δ, b1Σ+), dash-double dotted lines correspond to (2π)−1(3π)−1 states (11Σ−, 13Δ, 11Σ+, 23Σ−, 21Δ, 21Σ+), solid lines correspond to (9σ)−1(3π)−1 states (13Π, 11Π), broken lines correspond to (8σ)−1(3π)−1 states (23Π, 21Π), dash-dotted lines correspond to (2π)−1(3π)−2(4π)1 states (21Σ−, 23Δ, 23Σ+), and dashed lines correspond to (3π)−3(4π)1 states (15Σ−). The latter two states include electron excitation to a vacant 4π orbital. The potential energy curves and corresponding dissociation limits mainly responsible for the KER peaks in Figure 3 are highlighted in green, orange, and purple.
As observed in the previous experiments of OCS in one-color intense laser fields (Bryan et al., 2006; Ma et al., 2019), OCS2+ preferentially dissociates into OC+ + S+ rather than O+ + CS+. The total yield of the OC+ + S+ channel is more than one order of magnitude larger than that of the O+ + CS+ channel in our experimental conditions. This can be explained by the fact that the products of the C–S bond breaking are more stable than the ones of the C–O bond breaking (Masuoka et al., 1992). The potential energies of the lowest state at the dissociation limits are 27.5 eV for OC+ (X2Σ+) + S+ (4S) vs 31.3 eV for O+ (4S) + CS+ (X2Σ+) (Brites et al., 2008). In addition, the potential barrier for the C–S bond breaking is much smaller than the one for the C–O bond breaking, as shown in Figure 2. According to photoionization experiments by Masuoka and Koyano (Masuoka and Koyano, 1991), the onset of the major OC+ + S+ dissociation channel is at a threshold energy of 33.5 eV, while more than 40 eV are necessary for the O+ + CS+ dissociation channel.
We start the discussion with the major dissociation channel, OCS2+ → OC+ + S+, compare it to previous studies of OCS in intense laser fields (Holmegaard et al., 2010; Dimitrovski et al., 2011; Ohmura et al., 2014, 2019; Ma et al., 2019; Zhao et al., 2021), and discuss its dissociation mechanisms. Next, we focus on the minor dissociation channel, OCS2+ → O+ + CS+. We then investigate the newly observed shoulder peak in the major dissociation channel and discuss the effects of post-ionization interactions. Lastly, we discuss the control of the branching ratio between the C–O and C–S bond breaking channels.
The phase-averaged total KER (Ekin) spectrum of the OC+ + S+ channel in phase-locked two-color intense laser fields (Iω+2ω = 2 × 1014 W/cm2, α = 0.19) is shown in Figure 3A. The Ekin spectrum shows a main peak at 5.2 eV (highlighted in green) and a shoulder structure at 4 eV (highlighted in orange).
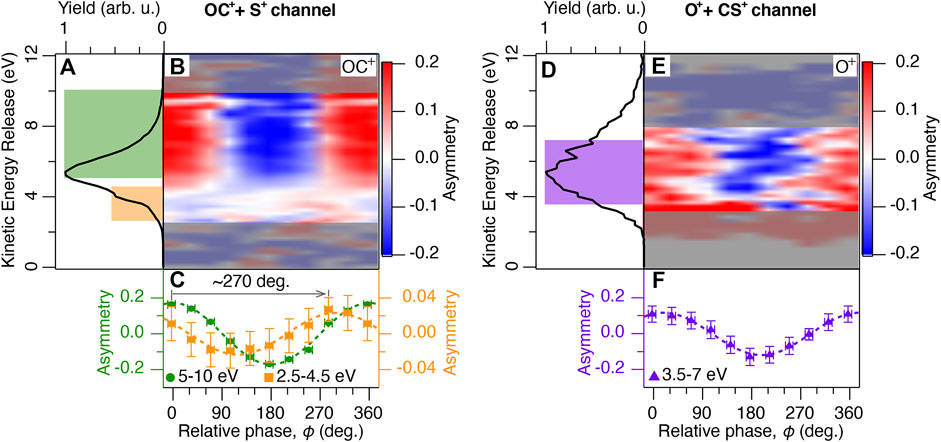
FIGURE 3. (A) The total kinetic energy release Ekin spectrum of the OC+ + S+ channel averaged over the relative phase ϕ between the ω and 2ω laser fields. The KER spectrum is calculated from the fragments ejected within 45 deg. with respect to the laser polarization direction. The colored areas are linked to the different dissociation pathways indicated in Figure 2. (B) Two-dimensional plot of the asymmetry parameter as a function of Ekin and ϕ. The asymmetry parameter is calculated from the OC+ fragment yields. Noisy data outside of the region of interest is masked grey shaded. (C) Asymmetry parameter of the OC+ fragments integrated over 5–10 eV (green circles) and 2.5–4.5 eV (orange squares) are plotted as a function of ϕ. The dotted lines are sine fits. (D) Same as (A), but for the O+ + CS+ channel. (E) Same as (B), but calculated from the O+ fragment yields. (F) Same as (C), but the O+ fragments are integrated over 3.5–7 eV (purple triangles).
The peak around 5 eV was also observed in a synchrotron experiment (Masuoka et al., 1992) and assigned to the fragments generated via the electronically excited states of OCS2+ such as (9σ)−1(3π)−1 or (2π)−1(3π)−1. Note that in a previous study using intense laser fields (Ma et al., 2019), the largest peak was however observed at a smaller KER of 4 eV instead of the 5.2 eV in our work. This difference can be attributed to the fact that different double ionization channels open up depending on the laser field intensity. In high intensity fields (
Three possible double ionization mechanisms in intense laser fields have been widely accepted, as indicated in Figure 4. One of them is SDI as mentioned above, others are identified as non-sequential double ionization (NSDI). SDI plays an important role in a higher intensity regime, because the first ionization step can occur on the rising edge of laser pulses and the residual laser fields are intense enough to remove the second electron by field ionization. However, in a lower intensity regime, the first ionization step occurs around the peak of the laser pulse. The residual field is not strong enough to lead to a second field ionization, therefore subsequent ionization is assisted by an electron recollision process. The simplest type of NSDI is recollision impact ionization (RII), in which the recolliding electron has gained sufficient energy to ionize the second electron. In the RII mechanism, since electron impact ionization occurs within half an optical cycle (1.3 fs at 800 nm) after the first ionization step, there is no time for bond stretching to occur. Another NSDI mechanism is known as recollision excitation with subsequent tunneling ionization (RESI), in which the recolliding electron can promote molecular ions to the excited states. The second electron will be subsequently ionized by the residual laser fields because of a smaller effective ionization potential. Due to the time delay between the first and second ionization step, the bond can slightly stretch in the RESI mechanism. Xu et al. (2020) clarified that NSDI is the dominant mechanism to generate OCS2+ at a laser field intensity below 1 × 1014 W/cm2 and SDI becomes dominant as the laser intensity increases.
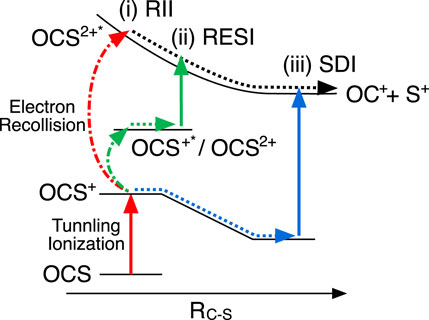
FIGURE 4. Schematic of sequential and non-sequential double ionization and dissociation processes of OCS along the C-S bond stretching coordinate. Three possible pathways for population to the excited states of OCS2+ (OCS2+*); (i) Recollision impact ionization (RII) leading to direct ionization to OCS2+* by electron recollision, (ii) recollisional excitation with subsequent ionization (RESI) causing indirect population to OCS2+* via intermediate states (OCS+* or OCS2+), and (iii) sequential double ionization (SDI) leading to ionization from OCS+, involving bond stretching. Due to the different bond stretching in the intermediate states and the shape of the PES of OCS2+*, the observed KERs appear in the order (i)
In the present study, we observed the generation of fragments via the excited states of OCS2+ (OCS2+*) instead of the yields of the parent ion OCS2+. Therefore, the threshold intensity for SDI would be shifted towards larger intensity due to the larger potential energy of OCS2+* compared to the ground state of OCS2+. Here, three double ionization mechanisms, including the population of the excited state of OCS2+, should be considered as shown in Figure 4: i) direct ionization to the excited states of OCS2+* by electron recollision, that is RII to the excited states, ii) recollisional excitation (ionization) to OCS+* (OCS2+) with subsequent ionization (excitation) to OCS2+*, in analogy to RESI, and iii) SDI to the excited states of OCS2+.
To clarify contributions of these mechanisms, the laser field intensity dependence of the main peak position is shown in Figure 5A. We observe a larger peak shift in the lower field intensity region (
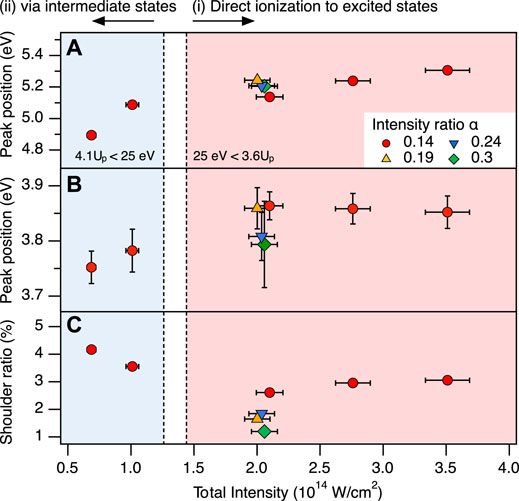
FIGURE 5. Intensity dependence of the KER spectrum. (A) Position of the main peak in the OC+ + S+ channel as a function of the total laser field intensity (Iω+2ω). (B) Position of the shoulder peak in the OC+ + S+ channel. (C) Ratio of the shoulder peak in the OC+ + S+ channel. Circles, triangles, up-side down triangles, and diamonds indicate the different intensity ratios of α = 0.14, 0.19, 0.24, and 0.3, respectively. Colored areas correspond to the intensity of (i) direct ionization to the 13Π state of OCS2+ from the ground state of OCS+ by electron recollision (Iω+2ω >1.44 × 1014 W/cm2) and (ii) indirect excitation via the electronically excited states of OCS+ or the electronic ground state of OCS2+ (Iω+2ω <1.26 × 1014 W/cm2) in two-color laser fields at α = 0.14 (see the text for details).
In addition, Wales et al. (2014) showed that the peak position of the KER spectra of the O+ + C+ + S+ channel from OCS3+ did not change with pulse duration below 200 fs; in contrast to the peak position from the OCS4+ break-up channel, which strongly depended on the pulse duration. The effects of bond stretching during ionization would be larger in higher charged states, because the bond can stretch at each charged state, causing further ionization of the molecules to an even higher charged state. Since the intensity of the fundamental pulse is much stronger than that of the second harmonic pulse in the present study, the pulse duration of the resulting two-color laser fields can be considered to be comparable to that of the fundamental pulse (100 fs). Bending is likely to occur under the present experimental condition, however, it would play a minor role in the two-body breaking channel we focused on in this study. Another concern might be the effect of photo-absorption of the 2ω field, due to its higher photon energy. We observe however, that the KER spectra in one-color and two-color laser fields show the same main and shoulder structure independent of α (see Supplementary Figure S3). Moreover, in longer wavelength (1700 nm + 850 nm) laser fields, similar main and shoulder peaks are observed at identical position compared to the shorter wavelength (800 nm + 400 nm). These results suggest that field ionization is the dominant mechanism and photo-absorption can be excluded.
From the discussion above and based on the PESs in Figure 2, we assign the main peak at 5.2 eV to dissociation via the electronically excited states 11Π and 13Π of OCS2+, populated by the RII mechanism. The electronic configuration of these states is (9σ)−1(3π)−1, leading to OC+ (X2Σ+) and S+ (2D) highlighted in green in Figure 2A. The states highlighted in Figure 2 represent the ones mainly contributing to the dynamics observed. We point out that, in general, for almost every triplet state, there is a corresponding singlet state of almost identical electronic configuration and similar fragmentation behavior (leading to almost identical KERs). Experimentally, we can not distinguish between singlet and triplet states, thus, also for the theoretical analysis, we concentrate on one spin species only (the triplet states). The shoulder peak is discussed in the separate Subsection 3.3.
To evaluate the fragment ejection direction quantitatively, the asymmetry parameter A is defined as a function of Ekin and ϕ,
where Y+ and Y− are the fragment yields with positive and negative momenta along the laser polarization direction (x-axis), respectively. Here, the fragment yields are evaluated by the counts of fragments ejected within 45° with respect to the laser polarization direction to reduce artificial blurring of the asymmetry due to the molecular rotation around the boundary of Y+ and Y−. The two-dimensional plot of the asymmetry parameter calculated from the OC+ fragment yields is shown in Figure 3B. A clear asymmetry was observed above 5 eV which showed a 2π-oscillatory behavior when changing the relative phase as observed in previous studies based on the fragments’ flight time measurements without coincidence detection (Ohmura et al., 2014, 2019). When a dissociation process is fast enough, the orientation of the parent ions generated by tunneling ionization reflects the ejection direction of fragment ions. Therefore, such strong asymmetry of the fragment ejection direction can be explained by the asymmetry of the ionization rate, which is determined by the shape of the HOMO and the effective ionization potential (Holmegaard et al., 2010). When the laser field is parallel to the difference in permanent dipole moment vectors of neutral and ionized states, the effective ionization potential becomes lower than in the field-free case and the ionization rate increases. Vice versa, in the case of an anti-parallel electric field, the ionization rate decreases since the effective ionization potential becomes higher than the field-free one. As a result of this competition between the shape of the HOMO and the effective ionization potential, the ionization rate increases when the laser field points to the O atom side and the electron ejects from the S atom side in circularly polarized laser fields (Holmegaard et al., 2010). However, in linearly polarized two-color laser fields (Ohmura et al., 2014, 2019), the effects of the permanent dipole moment are not strong enough to flip the ionization rate of OCS, as can be deduced from the shape of the HOMO (see Figure 6C), with larger electron density on the S atom than on the O atom side. That is, the ionization rate increases when the laser field points towards the O atom, and the electron ejects from the S atom side. Then, the OC+ fragments are preferentially ejected to the larger amplitude side of the two-color laser fields (positive direction of x-axis at ϕ = 0°).
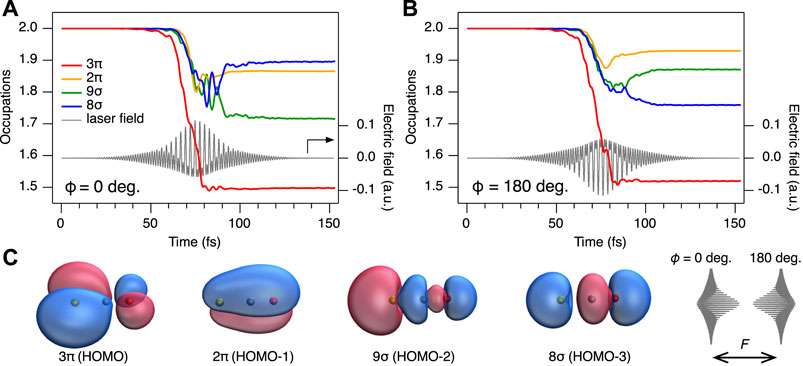
FIGURE 6. Results of the rtTDDFT calculations. (A) Time-dependent orbital occupations for ϕ = 0°. (B) same for ϕ = 180°. The temporal shapes of the electric fields are shown as solid grey lines. Note that 3π and 2π are doubly degenerate. (C) Orbitals of HOMO to HOMO-3. HOMO and HOMO-1 are π orbitals, while HOMO-2 and HOMO-3 are σ orbitals. For π orbitals, only one variant is shown, the other variant is a simple rotation around the molecular axis by 90°. Yellow atom indicate sulfur, blue atoms = carbon and red atoms = oxygen. F denotes the laser polarization direction. Schematic shapes of the phase-locked two-color laser electric fields (ϕ =0° and 180°) are shown.
The total KER spectrum of the O+ + CS+ channel is shown in Figure 3D. In contrast to the OC+ + S+ channel, no low-energy shoulder peak is observed. The two-dimensional plot of the asymmetry parameter calculated from the O+ fragment yields is shown in Figure 3E. No clear dependence on the KER is observed. The integrated asymmetry between 3.0 and 7.5 eV follows a similar phase dependence as the main peak of the OC+ + S+ channel, as shown in Figure 3F. Such similarity indicates that this broad peak can be assigned to the fragments originating from the excited 11Π or 13Π states. These states are the same ones contributing to the main peak of the OC+ + S+ channel, and dissociate to e.g. O+ (4S) and CS+ (A2Π) highlighted in purple in Figure 2B. The anisotropy of fragments from the excited 11Π or 13Π states reflects the angular dependence of the ionization rate because of the prompt dissociation through small potential barriers. Although the asymmetry amplitude slightly decreases compared to the main peak in the OC+ + S+ channel, which we attribute to the effects of molecular rotation due to a higher potential barrier along the O–C bond breaking. This result indicates that the orientation of the parent OCS2+ ions is mainly determined by the ionization step, even for different dissociation channels as discussed in the previous studies of polar molecules (Ohmura and Tachiya, 2008; Holmegaard et al., 2010; Dimitrovski et al., 2011; Li et al., 2011; Wu et al., 2012; Ohmura et al., 2014; Li et al., 2016; Wustelt et al., 2018; Yue et al., 2018; Endo et al., 2019; Ohmura et al., 2019).
In the main OC+ + S+ dissociation channel, we have observed a shoulder peak at around 4 eV, next to the main peak, as indicated in orange in Figure 3A. This shoulder structure was not observed in synchrotron radiation experiments (Masuoka and Koyano, 1991; Masuoka et al., 1992). From the following discussion and nuclear dynamics simulation, we assign this peak to dissociation via the (2π)−1(3π)−1 states highlighted in orange in Figure 2A.
The peak position of the shoulder peak shifts in lower intensity region rather than in higher intensity region as well as the main peak. This shift indicates that the shoulder peak can be assigned to dissociation via the excited states of OCS2+, in a way similar to the main peak. To identify the origin of this shoulder peak from another point of view, we compare its ratio to the main peak depending on the laser field intensity in Figure 5C. From Xu et al. (2020) we know, that the SDI mechanism becomes dominant at higher intensities. If the shoulder peak can be attributed to fragments generated by the SDI process, its yield would significantly increase with increasing intensity. However, we observe that the yield in the OC+ + S+ channel remains almost constant as the total laser field increases. We therefore conclude that the occurrence of both the shoulder peak and the main peak are mainly caused by the RII mechanism that does not involve bond stretching.
Another possible pathway for the occurrence of the shoulder peak would be due to interactions in neutral states such as enhanced ionization induced by structural deformation. In general, enhanced ionization causes a decrease of the observed KER (Ibrahim et al., 2018); the main peak, however, appears at the same energy here and also in the case of a synchrotron experiment (Masuoka et al., 1992) and bond stretching in neutral states was not observed in previous studies (Wales et al., 2014; Ma et al., 2019). From these observations, we conclude that interactions in neutral states are not significant here and that the ionization process, vertical ionization in the Franck-Condon region, is essentially the same as in previous studies. However, interactions after ionization/during dissociation, such as potential deformation or population transfer play important roles in the dissociation of polar molecules, as we discuss in the next sections.
The shoulder peak is not only present, it also shows a different phase dependence in its asymmetry parameter A, compared to the main peak. Its asymmetry amplitude is about 5 times weaker, and the relative phase leading to maximum asymmetry is shifted by 270° (-90°). This shift is directly apparent in the integrated asymmetry parameters between 5 and 10 eV for the main peak and between 2.5 and 4.5 eV for the shoulder peak in Figure 3C. Such shift indicates that contributions from two (or more) components are present whose phase difference is not an integer multiple of 180°. We draw this conclusion, because two components, phase-shifted by 180°, would only cause a dependence of the overall asymmetry amplitude on their ratio but no phase shift would occur.
The weak amplitude can be attributed to competition between the contributions from the main and shoulder peaks with different phase dependence, leading to a cancellation in modulation contrast. The observed phase shift, however, cannot be explained by the simple tunneling ionization picture discussed above for the main peak. The ionization rate becomes largest at a relative phase of 0° or 180°, where the peak of the electric field amplitude is maximum. In the extreme case of diametrical effects of the orbital shapes and dipole moments, e.g. ionization from lower lying orbitals, the asymmetry would be shifted by 180°. The observed phase shift of 270° therefore means that other mechanisms, occurring after the actual ionization, such as electron recollisional excitation, potential deformation, or population transfer between electronic states determine the fragment’s asymmetry. One of the possible scenarios is based on electron recolliding excitation and potential deformation as suggested in the asymmetric dissociation of CO2 (Endo et al., 2017): The excitation probability due to electron impact depends on the kinetic energy and the incident direction of the recollisional electron, which are determined by both the field intensity and the relative phase (Endo et al., 2019). This means that the population distribution among electronic states in the ionic states manifold depends on the relative phase. In addition, potential deformations would be more prominent in higher electronic states, which have diffuse wavefunctions. The sum of these contributions from different electronic states can lead to the observed phase shift of 270° in the fragment ejection direction. However, in the case of CO2, the asymmetry amplitudes are on the order of a few percent, while in this study, they reach values of up to 0.16 (Ekin > 5 eV) and decrease to 0.03 (Ekin < 4.5 eV). This suggests that both shoulder- and main peak have comparable asymmetry amplitudes. In addition to that, the intensity dependence of the asymmetry amplitudes of CO2 (Endo et al., 2017) plateaus, while in OCS a monotonic decrease with increasing intensity is observed as shown in Supplementary Figure S4 of the SM. We therefore suggest another possible scenario; a population transfer between electronic states after ionization, as will be discussed later.
To summarize these results, the shoulder peak of the OC+ + S+ channel occurring around 4 eV exhibits a distinct phase dependence compared to the two channels directly dissociating from the excited states. We assign its origin to post-ionization dynamics; through this observed phase dependence, we are able to indicate that this shoulder peak arises from a population transfer between electronic excited states in doubly charged states, as supported by the computational results discussed in the following.
To further analyse the contribution of different electronic states to the observed asymmetry in both ionization and post-ionization interactions, we now discuss the computational results of the rtTDDFT calculations. Results are summarized in Table 1 and Supplementary Figure S1 of the SM, listing the total number of emitted electrons nemit, the number of electrons originating from 3π and 2π orbitals (HOMO and HOMO-1), nπ, and from 9σ and 8σ orbitals (HOMO-2 and HOMO-3), nσ, as well as the fraction of electrons originating from σ orbitals, fσ = nσ/nemit. The results show that for phase ϕ = 0°, ionization is most efficient: 1.67 electrons are removed in total. This is not surprising, as the peak of the field amplitude |Fω+2ω| is maximum for that phase. However, for a relative phase of ϕ = 180°, the ionization is less, as only 1.48 electrons, about 90% of ϕ = 0°, leave the numerical grid. Thus, as the molecule in the simulation is perfectly aligned with the laser polarization direction, at phase ϕ = 0°, ionization preferentially occurs from the sulfur atom, as the electric field vector pointing towards the oxygen atom allows the electrons to acquire momentum in the opposite direction. For phase ϕ = 180°, the situation is reversed and ionization preferentially occurs from the oxygen side. As shown by Ohmura et al. (Ohmura and Tachiya, 2008; Ohmura et al., 2014, 2019), the ionization efficiency is larger when emitting electrons from the large lobe of the 3π HOMO located at the sulfur (see Figure 6C), explaining the observed difference in nemit for the cases ϕ = 0° and ϕ = 180°. Note, that the preference of ionization from the S or the O side at specific ϕ is identical between our work and the ones of Ohmura et al. (Ohmura and Tachiya, 2008; Ohmura et al., 2014, 2019), both obtained with linear polarized light. It is however opposite to the work of Holmegaard et al. (Holmegaard et al., 2010) using circularly polarized light.
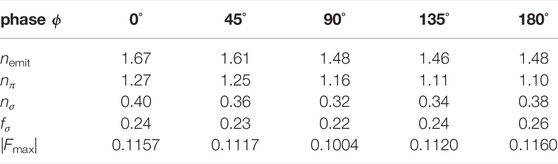
TABLE 1. Summary over the numerical results obtained from the rtTDDFT calculations. Listed are the number nemit of emitted electrons originating from 3π and 2π orbitals (HOMO and HOMO-1), nπ, and from 9σ and 8σ orbitals (HOMO-2 and HOMO-3), nσ, as well as the fraction of electrons originating from σ orbitals, fσ, for different relative phases ϕ. Additionally, the maximum field strength of the (combined) applied laser field, |Fmax|, is given in atomic units (a.u.).
Next, we consider the amount of σ electrons being emitted for different relative phases. Figure 6A,B provide an overview over the time-dependent occupations of the respective orbitals for ϕ = 0° and 180°. Overviews of other phases are provided in the SM. It should be noted that the π orbitals are doubly degenerate. We can read from Figure 6 and Table 1 that the number of electrons emitted from σ orbitals nσ is 0.40 for ϕ = 0°, while nσ = 0.38 for ϕ = 180°. Emission of σ electrons corresponds to a depopulation of the 9σ and 8σ orbitals, which leads, together with the also very pronounced depopulation of the 3π orbitals, to a final electron configuration of (9σ)−1(3π)−1 and (8σ)−1(3π)−1, and thus to the 11Π, 13Π, 21Π, and 23Π states. These states feature a small barrier towards dissociation into OC+ + S+ (Figure 2A). Thus, upon pronounced ionization of σ orbitals, the dication ultimately fragments into OC+ + S+. As a side notice, fragmentation of these electronic states into the O+ + CS+ channel is less likely, as the barrier towards dissociation is still around 2 eV (see also Figure 2B). This explains the difference of the yields between the channels observed in the experiment, and also why the main peaks at 5 eV in the two different fragment channels follow the same phase dependence (green line in Figure 3C and purple line in (f)).
The rtTDDFT calculations also provide another possible post-ionization origin to explain the shoulder peak in the OC+ + S+ channel: taking a closer look again on the population dynamics in Figure 6A, we observe a strong mixing between the 9σ and 8σ orbitals for ϕ = 0°. This can be traced back, because the 21Π and 23Π states (corresponding to ionization from 8σ) are, at the equilibrium structure of the neutral OCS, only about 2 eV above the 11Π and 13Π states (corresponding to ionization from 9σ, see Figure 2). As can be gathered from the time-dependent populations in Figure 6, mixing of these states starts around t = 70 fs and continues up to 100 fs, a time well after the maximum of the electric field and also after the main ionization event between t = 60–80 fs. This state mixing after the main ionization events provides another strong indication for post-ionization dynamics. Figure 2 shows that at slightly stretched geometries (R = 3.5 a. u.), the pairs of the two mixing states, 13Π and 23Π, or 11Π and 21Π, come energetically very close and the efficiency of population transfer can be expected to further increase at the wing of the pulse compared to the rtTDDFT calculations, which do not take such stretching into account. This is similar to the strong coupling of the σg and σu states in
Since the rtTDDFT calculations already strongly suggested the contribution of several excited states, we performed separate semi-classical surface-hopping calculations, in order to trace possible post-ionization dynamics beyond the fixed-nuclei approximation. As outlined in Section 2.2.2, in these calculations, the electronic states are calculated on-the-fly with the quantum chemical program package OpenMolcas including the two-color laser field, while the nuclei are treated classically.
We have investigated the following initial conditions upon double ionization: 1) all trajectories are launched in the electronic ground state of the dication, X3Σ−; 2) all trajectories start initially in the excited state 13Π ((9σ)−1(3π)−1), as suggested by rtTDDFT.
Our calculations show that for (i), double ionization occurring to the electronic ground state of the dication, none of the trajectories showed dissociation during the calculation time. These results will not be discussed any more in what follows. For (ii), ionization into the excited electronic state of OCS2+*, several pathways were identified for a relative phase of ϕ = 0°, leading to break up into OC+ + S+ (4 of 20 trajectories), O+ + CS+ (5 of 20 trajectories), and [O+ C + S]2+ (11 of 20 trajectories), the latter one being not visible experimentally. Similar values concerning the relative number of trajectories are obtained for the other relative phases of the two-color field. When looking at the population dynamics, we gather a complex behavior for all cases, including population transfer between several excited electronic states (see also Figure 7 for representative trajectories). Changes in the relative phase of the two-color field lead to changes in the ratio between the population of the electronic states. With these trajectories at hand, we focus on the OC+ + S+ channel and the question concerning the shoulder peak. In our calculations, we identify for this break-up channel trajectories which would end up at higher KER (
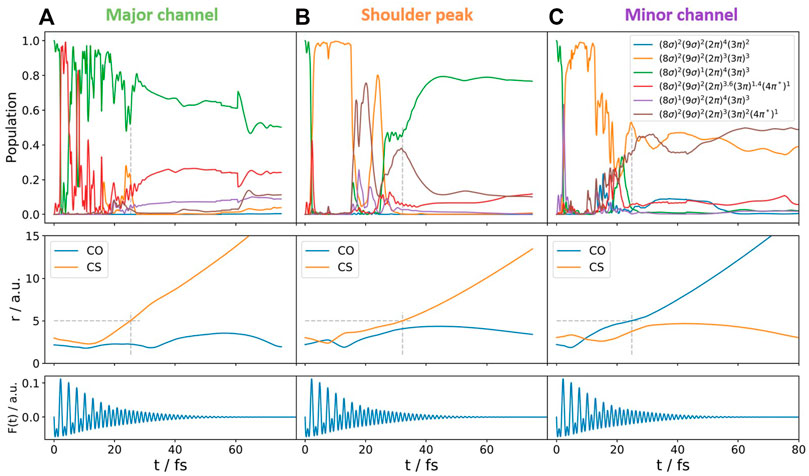
FIGURE 7. Quantum amplitudes (in the limit of infinite trajectories, this would correspond to populations) of different excited states (top), bond length C-O and C-S (middle), and the temporal shapes of the applied electric fields (bottom) during the dissociation of selected trajectories. These represent typical trajectories following (A,B) the OC+ + S+ channel and (C) the O+ + CS+ channel. A KER
To discuss the effects of post-ionization interactions from another point of view, the inter-channel branching ratio between the OC+ and the O+ fragments is defined as
where
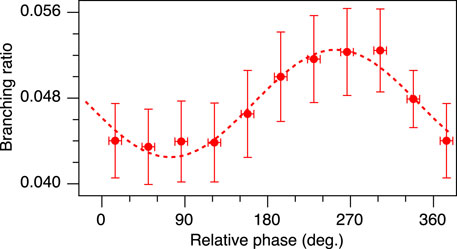
FIGURE 8. Inter-channel branching ratio between the OC+ and the O+ fragments ejected to the same direction as a function of the relative phase at a total peak intensity of 2×1014 W/cm2 and a ratio α = 0.14. Red circles correspond to experimental data points, the dotted line is a sine fit.
We have thus demonstrated the selective bond scission of polar molecules by changing the relative phase of phase-locked two-color intense laser fields and attributed the origin of such selectivity to post-ionization interactions.
To summarize, we have investigated the Coulomb explosion of the polar molecule OCS in phase-locked two-color intense laser fields. A clear 2π-oscillation was observed in both the OC+ + S+ and O+ + CS+ channels. We have succeeded in the observation of a shoulder peak that has not been discovered previously, because it is a dark channel for synchrotron radiation that opens however up due to strong light-matter interactions in intense laser fields. In the OC+ + S+ channel, a clear dependency of the asymmetry on the kinetic energy was found due to this dissociation pathway. The phase shift of 270° between both sub-channels of OC+ + S+ is attributed to a population transfer between the involved electronic states in the applied intense laser fields as demonstrated by rtTDDFT calculations. In addition, the inter-channel branching ratio (when breaking the C–O and C–S bonds oriented to the same direction with respect to the laser polarization direction) also oscillated with 2π, depending on the relative phase. The selective scission of chemical bonds in polar molecules has been demonstrated by using a simple pulse shaping technique. Here presented results indicate that not only the ionization step, but also post-ionization interactions play important roles even for heavy polar molecules, where the anisotropy of the fragments is mainly dominated by tunneling ionization. Two-color phase-locked laser fields provide a variety of controllable parameters, such as the total intensity, the intensity ratio, the relative phase and the polarization, while not involving a complex experimental implementation. This approach is therefore useful to investigate the mechanisms of coherent reaction control for a wide variety of molecular systems by tailored intense laser fields.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
TE performed all experiments and data analysis. FL was responsible for the ALLS facility at INRS in Canada. KMZ, MR, FF, and SG performed all simulations. AH, FL, SG, TE, and HI discussed results. HI conceived and directed the project, supervised experiments, and analyzed data. TE, SG, and HI wrote the original draft. All authors contributed to the discussions of the results and to the production of the manuscript.
We sincerely thank the Canada Foundation for Innovation, NSERC and FRQNT. TE and AH acknowledges support from the JSPS Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (S2602). FF and SG highly acknowledge support by the German Science Foundation (DFG) provided via the International Research Training Group (IRTG 2101) “Guided Light, Tightly Packed”. MR and SG highly acknowledge support from the European Research Council via the Consolidator Grant QUEM-CHEM (772676). KMZ and SG are part of the Max Planck School of Photonics supported by BMBF, Max Planck Society, and Fraunhofer Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Philippe Lassonde for technical support, as well as Dr. Benji Wales and Prof. Joseph Sanderson for providing the analysis software for the CEI measurement files.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.859750/full#supplementary-material
Alnaser, A. S., Kübel, M., Siemering, R., Bergues, B., Kling, N. G., Betsch, K. J., et al. (2014). Subfemtosecond Steering of Hydrocarbon Deprotonation through Superposition of Vibrational Modes. Nat. Commun. 5, 3800. doi:10.1038/ncomms4800
Alnaser, A. S., and Litvinyuk, I. V. (2017). Subfemtosecond Directional Control of Chemical Processes in Molecules. J. Phys. B: Mol. Opt. Phys. 50, 032002. doi:10.1088/1361-6455/50/3/032002
Andrade, X., Alberdi-Rodriguez, J., Strubbe, D. A., Oliveira, M. J. T., Nogueira, F., Castro, A., et al. (2012). Time-dependent Density-Functional Theory in Massively Parallel Computer Architectures: the octopus Project. J. Phys. Condens. Matter 24, 233202. doi:10.1088/0953-8984/24/23/233202
Aquilante, F., Autschbach, J., Carlson, R. K., Chibotaru, L. F., Delcey, M. G., De Vico, L., et al. (2016). Molcas 8: New Capabilities for Multiconfigurational Quantum Chemical Calculations across the Periodic Table. J. Comput. Chem. 37, 506–541. doi:10.1002/jcc.24221
Assion, A., Baumert, T., Bergt, M., Brixner, T., Kiefer, B., Seyfried, V., et al. (1998). Control of Chemical Reactions by Feedback-Optimized Phase-Shaped Femtosecond Laser Pulses. Science 282, 919–922. doi:10.1126/science.282.5390.919
Bergt, M., Brixner, T., Kiefer, B., Strehle, M., and Gerber, G. (1999). Controlling the Femtochemistry of Fe(CO)5. J. Phys. Chem. A. 103, 10381–10387. doi:10.1021/jp992541k
Brites, V., Eland, J. H. D., and Hochlaf, M. (2008). OCS2+ Dication Spectroscopy and Electronic States. Chem. Phys. 346, 23–33. doi:10.1016/j.chemphys.2007.11.009
Brixner, T., Kiefer, B., and Gerber, G. (2001). Problem Complexity in Femtosecond Quantum Control. Chem. Phys. 267, 241–246. doi:10.1016/S0301-0104(01)00223-3
Brumer, P., and Shapiro, M. (1986). Control of Unimolecular Reactions Using Coherent Light. Chem. Phys. Lett. 126, 541–546. doi:10.1016/S0009-2614(86)80171-3
Bryan, W. A., Newell, W. R., Sanderson, J. H., and Langley, A. J. (2006). Observation of Multiple Ionization Pathways for OCS in an Intense Laser Field Resolved by Three-Dimensional Covariance Mapping and Visualized by Hierarchical Ionization Topology. Phys. Rev. A. 74, 053409. doi:10.1103/physreva.74.053409
Cardoza, D., Baertschy, M., and Weinacht, T. (2005). Understanding Learning Control of Molecular Fragmentation. Chem. Phys. Lett. 411, 311–315. doi:10.1016/j.cplett.2005.06.044
Dimitrovski, D., Abu-samha, M., Madsen, L. B., Filsinger, F., Meijer, G., Küpper, J., et al. (2011). Ionization of Oriented Carbonyl Sulfide Molecules by Intense Circularly Polarized Laser Pulses. Phys. Rev. A. 83, 023405. doi:10.1103/physreva.83.023405
Doblhoff-Dier, K., Kitzler, M., and Gräfe, S. (2016). Theoretical Investigation of Alignment-dependent Intense-Field Fragmentation of Acetylene. Phys. Rev. A. 94, 013405. doi:10.1103/PhysRevA.94.013405
Endo, T., Fujise, H., Hasegawa, H., Matsuda, A., Fushitani, M., Tolstikhin, O. I., et al. (2019). Angle Dependence of Dissociative Tunneling Ionization of NO in Asymmetric Two-Color Intense Laser fields. Phys. Rev. A. 100, 053422. doi:10.1103/PhysRevA.100.053422
Endo, T., Fujise, H., Kawachi, Y., Ishihara, A., Matsuda, A., Fushitani, M., et al. (2017). Selective Bond Breaking of CO2 in Phase-Locked Two-Color Intense Laser fields: Laser Field Intensity Dependence. Phys. Chem. Chem. Phys. 19, 3550–3556. doi:10.1039/C6CP07471E
Endo, T., Fujise, H., Matsuda, A., Fushitani, M., Kono, H., and Hishikawa, A. (2016). Coincidence Momentum Imaging of Asymmetric Coulomb Explosion of CO2 in Phase-Locked Two-Color Intense Laser fields. J. Electron Spectrosc. Relat. Phenomena 207, 50–54. doi:10.1016/j.elspec.2015.12.010
Endo, T., Neville, S. P., Wanie, V., Beaulieu, S., Qu, C., Deschamps, J., et al. (2020). Capturing Roaming Molecular Fragments in Real Time. Science 370, 1072–1077. doi:10.1126/science.abc2960
Godbout, N., Salahub, D. R., Andzelm, J., and Wimmer, E. (1992). Optimization of Gaussian-type Basis Sets for Local Spin Density Functional Calculations. Part I. Boron through Neon, Optimization Technique and Validation. Can. J. Chem. 70, 560–571. doi:10.1139/v92-079
Gräfe, S., Meier, C., and Engel, V. (2005). Instantaneous Dynamics and Quantum Control fields: Principle and Numerical Applications. J. Chem. Phys. 122, 184103. doi:10.1063/1.1891728
Hasegawa, H., Walmsley, T., Matsuda, A., Morishita, T., Madsen, L. B., Jensen, F., et al. (2022). Asymmetric Dissociative Ionization of Tetrafluoromethane in ω-2ω Intense Laser fields. Front. Chem.. doi:10.3389/fchem.2022.857863
Holmegaard, L., Hansen, J. L., Kalhøj, L., Louise Kragh, S., Stapelfeldt, H., Filsinger, F., et al. (2010). Photoelectron Angular Distributions from strong-field Ionization of Oriented Molecules. Nat. Phys 6, 428–432. doi:10.1038/nphys1666
Ibrahim, H., Héjjas, M., Fushitani, M., and Schwentner, N. (2009a). Phase Sensitive Control of Vibronic Guest−Host Interaction: Br2 in Ar Matrix. J. Phys. Chem. A. 113, 7439–7450. doi:10.1021/jp900287m
Ibrahim, H., Héjjas, M., and Schwentner, N. (2009b). Tracing, Amplifying, and Steering Chromophore-Bath Coherences by Ultrashort Pulse Trains. Phys. Rev. Lett. 102, 088301. doi:10.1103/PhysRevLett.102.088301
Ibrahim, H., Lefebvre, C., Bandrauk, A. D., Staudte, A., and Légaré, F. (2018). H2: the Benchmark Molecule for Ultrafast Science and Technologies. J. Phys. B: Mol. Opt. Phys. 51, 042002. doi:10.1088/1361-6455/aaa192
Kane, D. J., and Trebino, R. (1993). Characterization of Arbitrary Femtosecond Pulses Using Frequency-Resolved Optical Gating. IEEE J. Quan. Electron. 29, 571–579. doi:10.1109/3.199311
Kaneyasu, T., Ito, M., Soejima, K., Hikosaka, Y., and Shigemasa, E. (2015). Site-specific Formation of Metastable OCS2+studied by Auger-Electron-Ion Coincidence Method. J. Phys. B: Mol. Opt. Phys. 48, 125101. doi:10.1088/0953-4075/48/12/125101
Karamatskos, E. T., Raabe, S., Mullins, T., Trabattoni, A., Stammer, P., Goldsztejn, G., et al. (2019). Molecular Movie of Ultrafast Coherent Rotational Dynamics of Ocs. Nat. Commun. 10, 3364. doi:10.1038/s41467-019-11122-y
Kling, M. F., Siedschlag, C., Verhoef, A. J., Khan, J. I., Schultze, M., Uphues, T., et al. (2006). Control of Electron Localization in Molecular Dissociation. Science 312, 246–248. doi:10.1126/science.1126259
Kotur, M., Weinacht, T., Pearson, B. J., and Matsika, S. (2009). Closed-loop Learning Control of Isomerization Using Shaped Ultrafast Laser Pulses in the Deep Ultraviolet. J. Chem. Phys. 130, 134311. doi:10.1063/1.3103486
Kowalewski, M., and Seeber, P. (2022). Sustainable Packaging of Quantum Chemistry Software with the Nix Package Manager. Int. J. Quan. Chem. doi:10.1002/qua.26872
Langford, M. L., Harris, F. M., Reid, C. J., Ballantine, J. A., and Parry, D. E. (1991). Triplet-state Energy Levels of CO2+2, COS2+ and CS2+. Chem. Phys. 149, 445–457. doi:10.1016/0301-0104(91)90044-T
Levis, R. J., Menkir, G. M., and Rabitz, H. (2001). Selective Bond Dissociation and Rearrangement with Optimally Tailored, strong-field Laser Pulses. Science 292, 709–713. doi:10.1126/science.1059133
Li, H., Ray, D., De, S., Znakovskaya, I., Cao, W., Laurent, G., et al. (2011). Orientation Dependence of the Ionization of CO and NO in an Intense Femtosecond Two-Color Laser Field. Phys. Rev. A. 84, 043429. doi:10.1103/PhysRevA.84.043429
Li, H., Tong, X. M., Schirmel, N., Urbasch, G., Betsch, K. J., Zherebtsov, S., et al. (2016). Intensity Dependence of the Dissociative Ionization of DCl in Few-Cycle Laser fields. J. Phys. B: Mol. Opt. Phys. 49, 015601. doi:10.1088/0953-4075/49/1/015601
Ma, P., Wang, C., Luo, S., Li, X., Hu, W., Yu, J., et al. (2019). Bond-breakage-dependent Dissociative Ionization of an Asymmetric Molecule in an Intense Femtosecond Laser Field. Phys. Rev. A. 99, 023423. doi:10.1103/physreva.99.023423
Mai, S., Marquetand, P., and González, L. (2018). Nonadiabatic Dynamics: The SHARC Approach. Wires Comput. Mol. Sci. 8, e1370. doi:10.1002/wcms.1370
Malinovsky, V. S., Meier, C., and Tannor, D. J. (1997). Optical Paralysis in Electronically Congested Systems: Application to Large-Amplitude Vibrational Motion of Ground State Na2. Chem. Phys. 221, 67–76. doi:10.1016/s0301-0104(97)00126-2
Masuoka, T., and Koyano, I. (1991). Dissociative Single, Double, and Triple Photoionization of OCS in the Region hν=20-100 eV Studied by Mass Spectrometry and the Photoion-Photoion Coincidence Method. J. Chem. Phys. 95, 909–917. doi:10.1063/1.461099
Masuoka, T., Koyano, I., and Saito, N. (1992). Kinetic‐energy Release in the Dissociative Double Photoionization of OCS. J. Chem. Phys. 97, 2392–2399. doi:10.1063/1.463078
Miura, S., Ando, T., Ootaka, K., Iwasaki, A., Xu, H., Okino, T., et al. (2014). Carrier-envelope-phase Dependence of Asymmetric C D Bond Breaking in C2D2 in an Intense Few-Cycle Laser Field. Chem. Phys. Lett. 595-596, 61–66. doi:10.1016/j.cplett.2014.01.045
NixOS (2021). Nixpkgs 21.05 Manual. Available at: https://nixos.org/manual/nixpkgs/stable/(Accessed December 29, 2021).
Ohmura, H., Saito, N., and Morishita, T. (2014). Molecular Tunneling Ionization of the Carbonyl Sulfide Molecule by Double-Frequency Phase-Controlled Laser fields. Phys. Rev. A. 89, 013405. doi:10.1103/PhysRevA.89.013405
Ohmura, H., and Tachiya, M. (2008). Robust Quantum Control of Molecular Tunneling Ionization in the Space Domain by Phase-Controlled Laser fields. Phys. Rev. A. 77, 023408. doi:10.1103/PhysRevA.77.023408
Ohmura, H., Yoshida, T., and Saito, N. (2019). Four-mode Multi-Selection in the Dual Phase Control of a Molecular Ionization Induced by Fourier-Synthesized Laser fields. Appl. Phys. Lett. 114, 054101. doi:10.1063/1.5082578
Rabitz, H. (2006). Strong-arming Molecular Dynamics. Science 314, 264–265. doi:10.1126/science.1134083
Ray, D., He, F., De, S., Cao, W., Mashiko, H., Ranitovic, P., et al. (2009). Ion-Energy Dependence of Asymmetric Dissociation of D2 by a Two-Color Laser Field. Phys. Rev. Lett. 103, 223201. doi:10.1103/PhysRevLett.103.223201
Richter, M., Marquetand, P., González-Vázquez, J., Sola, I., and González, L. (2011). SHARC: Ab Initio Molecular Dynamics with Surface Hopping in the Adiabatic Representation Including Arbitrary Couplings. J. Chem. Theor. Comput. 7, 1253–1258. doi:10.1021/ct1007394
Ridard, J., Levy, B., and Millié, P. (1988). Theoretical Investigation of the Dissociation of OCS2+ in the Process OCS(X1Σ+) + hv → OCS2+ → CO+ (X2Σ+) + S+ (4S). Chem. Phys. 122, 403–412. doi:10.1016/0301-0104(88)80022-3
Roos, B. O., Taylor, P. R., and Sigbahn, P. E. M. (1980). A Complete Active Space SCF Method (CASSCF) Using a Density Matrix Formulated Super-CI Approach. Chem. Phys. 48, 157–173. doi:10.1016/0301-0104(80)80045-0
Roudnev, V., and Esry, B. D. (2007). General Theory of Carrier-Envelope Phase Effects. Phys. Rev. Lett. 99, 220406. doi:10.1103/PhysRevLett.99.220406
Sato, Y., Kono, H., Koseki, S., and Fujimura, Y. (2003). Description of Molecular Dynamics in Intense Laser Fields by the Time-dependent Adiabatic State Approach: Application to Simultaneous Two-Bond Dissociation of CO2 and its Control. J. Am. Chem. Soc. 125, 8019–8031. doi:10.1021/ja0344819
Schmidt, B. E., Unrau, W., Mirabal, A., Li, S., Krenz, M., Wöste, L., et al. (2008). Poor Man's Source for Sub 7 Fs: a Simple Route to Ultrashort Laser Pulses and Their Full Characterization. Opt. Express 16, 18910–18921. doi:10.1364/OE.16.018910
Shen, Z., Yan, Y., Cheng, J., Shuang, F., Zhao, Y., and He, G. (1999). Pump-dump Control and the Related Transient Absorption Spectroscopies. J. Chem. Phys. 110, 7192–7201. doi:10.1063/1.478623
Song, Q., Gong, X., Ji, Q., Lin, K., Pan, H., Ding, J., et al. (2015). Directional Deprotonation Ionization of Acetylene in Asymmetric Two-Color Laser fields. J. Phys. B: Mol. Opt. Phys. 48, 094007. doi:10.1088/0953-4075/48/9/094007
Tancogne-Dejean, N., Oliveira, M. J. T., Andrade, X., Appel, H., Borca, C. H., Le Breton, G., et al. (2020). Octopus, a Computational Framework for Exploring Light-Driven Phenomena and Quantum Dynamics in Extended and Finite Systems. J. Chem. Phys. 152, 124119. doi:10.1063/1.5142502
Tannor, D. J., and Rice, S. A. (1985). Control of Selectivity of Chemical Reaction via Control of Wave Packet Evolution. J. Chem. Phys. 83, 5013–5018. doi:10.1063/1.449767
Wales, B., Bisson, É., Karimi, R., Beaulieu, S., Ramadhan, A., Giguère, M., et al. (2014). Coulomb Imaging of the Concerted and Stepwise Break up Processes of Ocs Ions in Intense Femtosecond Laser Radiation. J. Electron Spectrosc. Relat. Phenomena 195, 332–336. doi:10.1016/j.elspec.2014.05.003
Wales, B., Bisson, E., Karimi, R., Kieffer, J.-C., Légaré, F., and Sanderson, J. (2012). A Coincidence Detection Algorithm for Improving Detection Rates in Coulomb Explosion Imaging. Nucl. Instr. Methods Phys. Res. Section A: Acc. Spectrometers, Detectors Associated Equipment 667, 11–15. doi:10.1016/j.nima.2011.11.082
Wang, X., Bersohn, R., Takahashi, K., Kawasaki, M., and Kim, H. L. (1996). Phase Control of Absorption in Large Polyatomic Molecules. J. Chem. Phys. 105, 2992–2997. doi:10.1063/1.472172
Wanie, V., Ibrahim, H., Beaulieu, S., Thiré, N., Schmidt, B. E., Deng, Y., et al. (2016). Coherent Control of D2/H2 dissociative Ionization by a Mid-infrared Two-Color Laser Field. J. Phys. B: Mol. Opt. Phys. 49, 025601. doi:10.1088/0953-4075/49/2/025601
Wells, E., Todt, M., Jochim, B., Gregerson, N., Averin, R., Wells, N. G., et al. (2009). Examining the Feedback Signals Used in Closed-Loop Control of Intense Laser Fragmentation of CO+. Phys. Rev. A. 80, 063402. doi:10.1103/PhysRevA.80.063402
Wu, J., Schmidt, L. P. H., Kunitski, M., Meckel, M., Voss, S., Sann, H., et al. (2012). Multiorbital Tunneling Ionization of the CO Molecule. Phys. Rev. Lett. 108, 183001. doi:10.1103/PhysRevLett.108.183001
Wustelt, P., Oppermann, F., Yue, L., Möller, M., Stöhlker, T., Lein, M., et al. (2018). Heteronuclear Limit of Strong-Field Ionization: Fragmentation of HeH+ by Intense Ultrashort Laser Pulses. Phys. Rev. Lett. 121, 073203. doi:10.1103/PhysRevLett.121.073203
Xie, X., Doblhoff-Dier, K., Roither, S., Schöffler, M. S., Kartashov, D., Xu, H., et al. (2012). Attosecond-recollision-controlled Selective Fragmentation of Polyatomic Molecules. Phys. Rev. Lett. 109, 243001–243005. doi:10.1103/PhysRevLett.109.243001
Xie, X., Doblhoff-Dier, K., Xu, H., Roither, S., Schöffler, M. S., Kartashov, D., et al. (2014). Selective Control over Fragmentation Reactions in Polyatomic Molecules Using Impulsive Laser Alignment. Phys. Rev. Lett. 112, 163003. doi:10.1103/PhysRevLett.112.163003
Xu, Q.-y., Ben, S., Sun, Y., Xu, H.-f., Liu, X.-s., Lv, H., et al. (2020). Non-sequential Double Ionization of Triatomic Molecules OCS in Intense Laser fields. Chem. Phys. Lett. 747, 137326. doi:10.1016/j.cplett.2020.137326
Yue, L., Wustelt, P., Sayler, A. M., Oppermann, F., Lein, M., Paulus, G. G., et al. (2018). Strong-field Polarizability-Enhanced Dissociative Ionization. Phys. Rev. A. 98, 043418. doi:10.1103/PhysRevA.98.043418
Zhao, X., Xu, T., Yu, X., Ren, D., Zhang, X., Li, X., et al. (2021). Tracking the Nuclear Movement of the Carbonyl Sulfide Cation after strong-field Ionization by Time-Resolved Coulomb-Explosion Imaging. Phys. Rev. A. 103, 053103. doi:10.1103/PhysRevA.103.053103
Keywords: two-color laser field, coherent control experiment, post-ionization dynamics, real-time real-space time-dependent density functional theory, Coulomb explosion imaging
Citation: Endo T, Ziems KM, Richter M, Fröbel FG, Hishikawa A, Gräfe S, Légaré F and Ibrahim H (2022) Post-Ionization Dynamics of the Polar Molecule OCS in Asymmetric Laser Fields. Front. Chem. 10:859750. doi: 10.3389/fchem.2022.859750
Received: 21 January 2022; Accepted: 07 March 2022;
Published: 08 April 2022.
Edited by:
Andre Bandrauk, Université de Sherbrooke, CanadaReviewed by:
Bernd Von Issendorff, University of Freiburg, GermanyCopyright © 2022 Endo, Ziems, Richter, Fröbel, Hishikawa, Gräfe, Légaré and Ibrahim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: François Légaré, ZnJhbmNvaXMubGVnYXJlQGlucnMuY2E=; Heide Ibrahim, aGVpZGUuaWJyYWhpbUBpbnJzLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.