 Hugo A. López Peña
Hugo A. López Peña Ka Un Lao
Ka Un Lao Katharine Moore Tibbetts
Katharine Moore Tibbetts- Department of Chemistry, Virginia Commonwealth University, Richmond, VA, United States
We report on pump-probe control schemes to manipulate fragmentation product yields in p-nitrotoluene (PNT) cation. Strong field ionization of PNT prepares the parent cation in the ground electronic state, with coherent vibrational excitation along two normal modes: the C–C–N–O torsional mode at 80 cm−1 and the in-plane ring-stretching mode at 650 cm−1. Both vibrational wave packets are observed as oscillations in parent and fragment ion yields in the mass spectrum upon optical excitation. Excitation with 650 nm selectively fragments the PNT cation into
1 Introduction
Chemists have sought to control molecular dissociation with lasers for decades. Tunable monochromatic laser light was believed to enable “bond-selective chemistry” through resonant energy absorption at the vibrational frequency of the targeted bond Bloembergen and Yablanovitch (1978). However, early attempts to control bond-cleavage by tuning the laser frequency failed due to rapid intramolecular vibrational energy redistribution (IVR) across coupled vibrational modes Bloembergen and Zewail (1984). Effective control of unimolecular dissociation was only achieved with the development of high-intensity ultrashort pulsed lasers and coherent control techniques that operate on timescales faster than IVR.
Coherent control over molecular dissociation has primarily been achieved by the “closed-loop” scheme of optimally designing shaped laser pulses with automated learning algorithms, initially proposed by Judson and Rabitz (1992). Gerber and co-workers reported the first experimental implementation of closed-loop control over ionization and dissociation of CpFe(CO)2Cl (Cp = cyclopentadienyl) into CpFeCOCl+ or FeCl+ Assion et al. (1998). This success spurred the application of closed-loop control to selectively dissociate various molecules including halogenated alkanes Damrauer et al. (2002), Langhojer et al. (2005), Plenge et al. (2011), Moore Tibbetts et al. (2013) and acetones Langhojer et al. (2005), Cardoza et al. (2006). However, shaped pulse control fails to enhance specific dissociation pathways in certain molecules including p-nitrotoluene Lozovoy et al. (2008). Moreover, the “black box” nature of closed-loop control makes it difficult to fully understand the physical mechanisms by which an optimal pulse shape achieves product selectivity, even using additional specialized pulse shaping procedures Xing et al. (2017).
Understanding the physical mechanisms underlying coherent control of molecular dissociation can be achieved using two-pulse “pump-probe” excitation schemes Tannor and Rice (1985), Zewail (1988). Pump-probe measurements with complementary quantum chemical calculations of the relevant electronic potential energy surfaces (PESs) have revealed bond-cleavage mechanisms facilitated by coherent vibrational motions in numerous organic cations Moore Tibbetts (2019). For instance, coherent excitation of the I–C–Br bending mode in CH2IBr+ upon strong-field ionization facilitates dissociation into CH2Br+ upon excitation of the D0→D3 transition at a specific point on the D0 PES Nichols et al. (2009). Similarly, coherent vibrational motions along the phenyl–substituent torsional coordinate in the molecular cations of acetophenone Bohinski et al. (2014), Tibbetts et al. (2015) and nitrobenzene López Peña et al. (2021) facilitate CH3 and NO2 loss, respectively, upon excitation of the vibrational wave packet at 90° phenyl–substituent dihedral angle. Although pump-probe measurements advance understanding of molecular dissociation facilitated by coherent vibrational dynamics, they enable only limited control over relative fragment yields because the probe wavelength typically excites resonantly to a single electronic excited state. As a result, one preferential fragment or a specific distribution of fragments is usually observed.
In this work, we demonstrate selective coherent excitation to three different electronic excited states from ground-state p-nitrotoluene (PNT) cation using probe pulses at 800, 650, and 400 nm. This selective excitation results in different relative yields of the
2 Materials and Methods
2.1 Experiments
Portions of the experimental setup have been described in our previous works Ampadu Boateng et al. (2018), Ampadu Boateng et al. (2019a). Briefly, a commercial Ti:sapphire regenerative amplifier (Astrella, Coherent, Inc.) producing 30 fs, 800 nm, 2.2 mJ pulses was used to pump an optical parametric amplifier (OPA, TOPAS Prime) to produce sub-20 fs 1,300 nm or 1,500 nm pump pulses. The pump wavelength was 1,300 nm for measurements with the 400 and 650 nm probes, whereas the pump wavelength was 1,500 nm for measurements with the 800 nm probe for reasons that will be discussed in the Results section. The 650 nm probe pulse was obtained from OPA output split with a 50:50 (r:t) beam splitter and frequency doubled with a β-barium borate (BBO) crystal. The 800 nm probe pulse was obtained from the transmitted portion of the incident laser beam from a 90:10 (r:t) beam splitter prior to the OPA and down-collimated using a reflective telescope with reduction factor 2. The 400 nm probe pulse was obtained by down-collimating the 800 nm beam with a telescope with reduction factor 3.33 comprised of a plano-convex lens (f = 250 mm) and a plano-concave lens (f = −75 mm) placed on a linear translation stage, followed by frequency doubling with a BBO crystal. The transmissive telescope geometry was necessary to allow for fine adjustment of the focal spot of the 400 nm probe beam to overlap with the focal spot of the 1,300 nm pump beam when both are focused with the same plano-convex lens (f = 200 mm). Both the 800 and 650 nm probe pulses have duration of ∼25 fs as measured by frequency-resolved optical gating Ampadu Boateng et al. (2018), Ampadu Boateng et al. (2019a). The 400 nm probe pulse duration was estimated at 70 fs as measured by the cross-correlation of the O2 signal from air in the mass spectrometer with 1,300 nm pump/400 nm probe excitation (Supplementary Figure S1). PNT (Sigma Aldrich, 99%) was introduced into the time-of-flight mass spectrometer (Jordan TOF) via an effusive inlet under gentle heating. Pump-probe measurements were taken over the delay range of −500 fs (probe before pump) to +2,500 fs (pump before probe) in steps of 5 fs for the 800 nm probe and 10 fs otherwise. Mass spectra were recorded at each pump-probe delay and averaged over 1,000 laser shots with a 1 GHz digital oscilloscope (LeCroy WaveRunner 610Zi).
2.2 Computations
Density functional theory (DFT) calculations were conducted using Gaussian 16 software Frisch et al. (2016) employing the restricted Kohn-Sham formalism for neutral species and the unrestricted formalism for cationic species. A previous work on PNT from our group Ampadu Boateng et al. (2018a) has identified the hybrid generalized gradient approximation (GGA) B3LYP functional Becke (1993), Stephens et al. (1994) in combination with the def2-TZVPP Weigend and Ahlrichs (2005) basis set as an adequate level of theory to describe this molecular system. Both the neutral and cation geometries of PNT were optimized within this level of theory. The convergence threshold for total energy was set to 10–8 eV while the force threshold was set to 10–3 eV/Å. Each geometric optimization was followed by harmonic frequency computations in order to confirm the stationary character of the state obtained.
To determine the excited-state energies of the PNT cation at different geometries, we performed single-point time-dependent DFT (TDDFT) Bauernschmitt and Ahlrichs (1996) calculations using Gaussian 16. For each cation geometry, we calculated the first 10 doublet-doublet transitions at the B3LYP/def2-TZVPP level of theory. In section 3.2 we will present TDDFT excited-state calculations for PNT radical cation at the B3LYP/def2-TZVPP level but those calculations will be further supported with selected calculations at the equation-of-motion excitation-energies coupled-cluster singles and doubles (EOM-EE-CCSD) Krylov, (2008) level. Due to the high computational cost of EOM methods, we employ the smaller 6−311+G* basis set. These EOM calculations were performed using Q-Chem 5.3 Epifanovsky et al. (2021). It is important to clarify that both TDDFT and EOM calculations on the cation are done under field-free conditions, i.e., after the strong-field pump pulse is over (see further discussion on Section 3.1).
3 Results
3.1 Pump-Probe Measurements
Pump-probe measurements were conducted with pump intensity of 6 × 1013 W cm−2. Figure 1A shows the mass spectra of PNT+ taken with only the 1,300 nm pump pulse (bottom) and with pump-probe excitation using 6 × 1012 W cm−2 probe pulses at 800, 650, and 400 nm (top). The pump-only spectrum is dominated by the intact PNT+ cation at m/z 137, with minor contribution from the
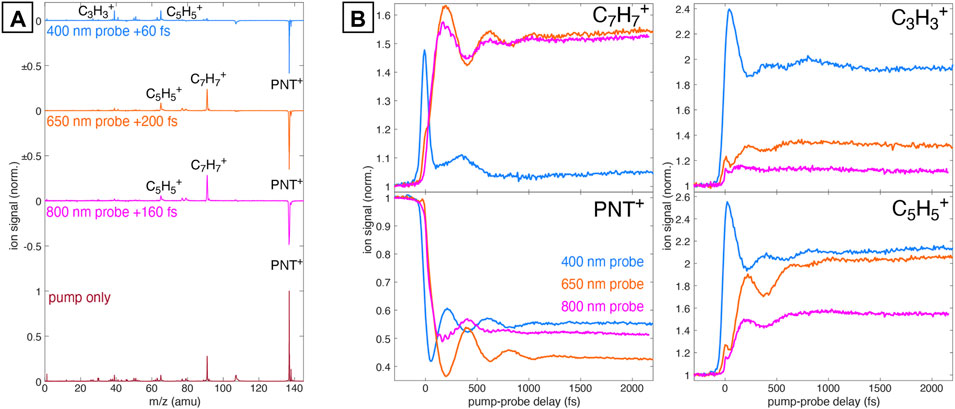
FIGURE 1. (A) Mass spectrum of PNT+ taken with 1,300 nm pump only (dark red) and the difference spectra relative to the pump-only spectrum taken with 800 nm (magenta), 650 nm (orange), and 400 nm (blue) probe pulses at the indicated delays. (B) Transient ion signals for PNT+,
Figure 1B displays the transient ion signals of the PNT+,
To further interpret the oscillatory dynamics, transient ion signals as a function of pump-probe delay, τ, were fit to the equation
where a and b are amplitude coefficients, t is the oscillation period, T1 is the coherence lifetime, T2 is a second lifetime not associated with oscillations, and c is the ion yield as τ→∞. Each transient signal at τ ≥ 70 fs (i.e., after the pump pulse is over so the instrument response function can be ignored) was fit to Eq. 1 using nonlinear least-squares curve fitting in MATLAB. A full description of the extracted coefficients can be found in the Supplementary Material, Supplementary Figure S3 and Supplementary Tables S1–S3. Although the strong-field pump pulse can populate multiple electronic states of the cation, it is reasonable to assume that, when the probe beam arrives after a delay
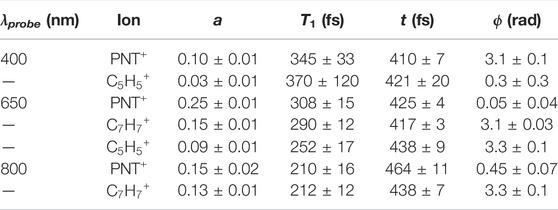
TABLE 1. Curve fitting coefficients for coherent dynamics of ion signals: oscillation amplitude (a), coherent lifetime (T1), oscillation period (t), and phase (ϕ).
Subtraction of the incoherent dynamics (second and third terms in Eq. 1) allows for clearer visualization of the oscillatory dynamics and frequency analysis via fast Fourier Transform (FFT). Figure 2A displays the coherent transient ion dynamics of PNT+ and oscillatory fragment ions for each probe wavelength. A clear π phase shift in the PNT+ signals (red) between the 400 nm probe and 650 nm or 800 nm probes is visible, as indicated by the dotted lines at 200, 620, and 1,040 fs. Figure 2B displays the FFT amplitude of the signals shown in Figure 2A. For all probe wavelengths the FFT spectra exhibit a strong peak at 80 cm−1 assigned to the previously reported C–N–N–O torsional mode of PNT+ Ampadu Boateng et al. (2018). A closer inspection of the oscillations using the 800 nm probe (bottom panel of Figure 2A) reveals what seems to be the superposition of two coherent oscillations: one corresponding to the torsional mode already discussed and smaller amplitude oscillations corresponding to a faster vibrational mode. In line with these observations is the presence of two frequencies, at 80 and 650 cm−1, in the FFT of the 800 nm probe signal (bottom panel of Figure 2B). Additionally, it is worth noting that the superposition of vibrational modes is more evident around the first minimum of the oscillatory ion signal in PNT+. This fact will be further discussed in Section 3.3 with the aid of computational results.
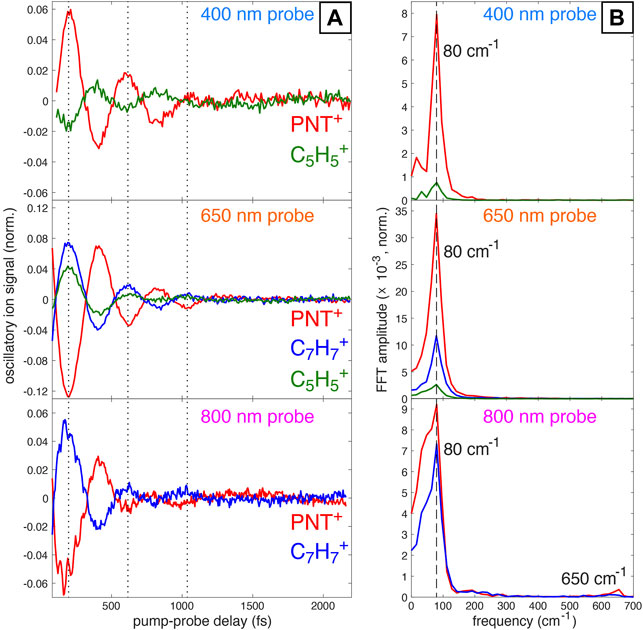
FIGURE 2. (A) Oscillatory ion signals for PNT+ and selected fragment ions obtained by subtracting off the incoherent contributions to signal fitting via Eq. 1. (B) FFT amplitudes obtained from the signals in (A) with indicated frequencies at 80 cm−1 and 650 cm−1 (for 800 nm probe).
In order to motivate forthcoming computational results, we present the optimized structures of neutral and cationic PNT at the B3LYP/def2-TZVPP level of theory in Figure 3. The corresponding coordinates are available within the Supplementary Material (Supplementary Tables S4, S5). Relevant to this work are the following changes after electron detachment: the C–C–N–O dihedral angle goes from 0.1 to 52.7° and a moderate distortion of the ring occurs. Additionally, the frequencies and corresponding intensities of the normal modes for PNT+ are presented in the Supplementary Material (Supplementary Table S6). A careful analysis of these vibrational modes shows that the C–C–N–O torsional mode has a calculated frequency of 58.96 cm−1, in reasonable agreement with the experimental frequency of 80 cm−1 (Figure 2B). Figure 3B shows the 12th normal mode calculated for the PNT cation. The relevance of this ring-stretching mode will be further discussed in Section 3.3. At this moment, it is enough to say that the calculated frequency of 604.32 cm−1 fairly matches the experimental frequency of 650 cm−1 shown in Figure 2B for the 800 nm probe.
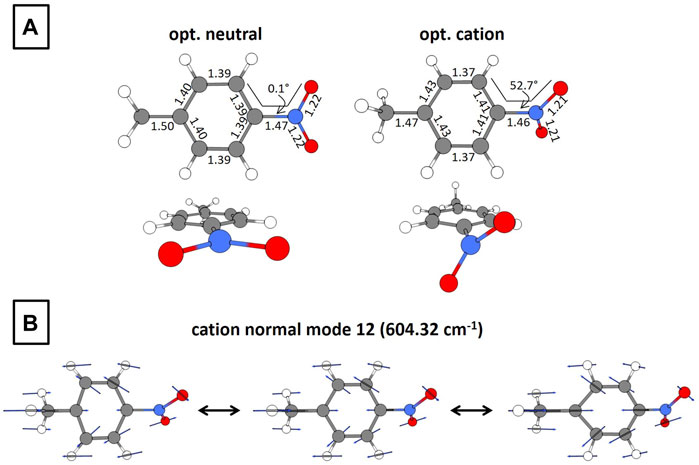
FIGURE 3. (A) Geometrical structures for optimized neutral and cationic PNT. Bond lengths are in Å and torsional angles in degrees. (B) Normal mode at 604.32 cm−1 for the optimized PNT cation. The calculations were performed at the B3LYP/def2-TZVPP level of theory.
3.2 Coherent Control of Torsional Wave Packet Excitation
As mentioned in the previous section, a former work from our group has assigned the main coherent oscillations of PNT+ to a torsional wave packet involving the C–C–N–O dihedral angle Ampadu Boateng et al. (2018). In order to explain the observation of different fragment distributions and ion yield dynamics depending on the wavelength of the probe beam, we used the following approach: starting from the optimized cation geometry with a C–C–N–O dihedral angle of 52.7° we performed a relaxed scan of this torsional mode using the ModRedundant keyword in Gaussian 16. This procedure generated a collection of geometries that span all the torsional mode, which were used to perform single-point TDDFT calculations. The resulting potential energy surfaces (PESs) can be seen in Figure 4A. In line with previous findings on the closely related nitrobenzene cation López Peña et al. (2021), we found that the PESs can be classified into two groups: the first one comprising from D1 to D4 with all the surfaces showing a marked dependence on the dihedral angle, and a second group comprising from D5 to D7 with a less marked dependence on the dihedral angle. The influence of the dihedral angle on the excitation probability, quantified by means of a harmonic estimate of the oscillator strength, can be observed in Figure 4B. It is noteworthy that the oscillator strength for the D0→D4 transition (f04) is particularly high and dependent on the geometry, reaching its maximum value at 90°. Moreover, this transition is the only one that has a substantial oscillator strength at 90°. Also notable is the parabolic shape of f06 centered at 90° dihedral angle. As a consequence, there is higher probability for the D0→D6 transition at geometries where PNT+ is nearly planar, i.e., near the neutral PNT geometry. Figure 4A also shows the energies corresponding to 400, 650, and 800 nm photons for comparison (3.1, 1.91, and 1.55 eV respectively). A careful evaluation of all the information contained in Figure 4 as a whole reveals that the 400 nm probe can selectively access an allowed transition from D0 to D6 at dihedral angles close to 0°, whereas the 650 nm probe can selectively access a strongly allowed transition from D0 to D4 at a dihedral angle of 90°. This ability to access different electronic excited states at different locations along the potential energy surface accounts for the selective fragmentation observed with different probe wavelengths at different time delays. Additionally, the 800 nm probe should be capable of promoting the D0→Dn, n = 1, 2 transitions at any dihedral angle.
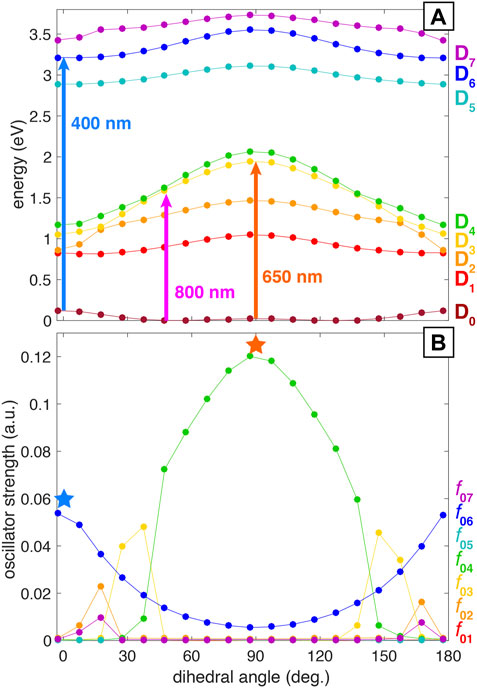
FIGURE 4. Computed potential energy surfaces (A) and oscillator strengths (B) for PNT+ along the C–C–N–O dihedral angle.
In order to support our TDDFT calculations we also performed single-point computations at three selected geometries calculated at the B3LYP/def2-TZVPP level using the EOM-EE-CCSD method. The geometries considered were the optimized neutral and cationic species (Figure 3A; Supplementary Tables S4, S5) and the geometry with a dihedral angle of 87.4° obtained by means of the relaxed scan previously described (Supplementary Table S7). Table 2 shows the excitation energies and harmonic estimates of oscillator strengths calculated at both levels of theory for PNT cation with a C–C–N–O dihedral angle of 87.4°. These methodologies produced qualitatively similar results but there are some differences that are worth noting: EOM calculations introduce a larger energetic gap between D1 and D2 states of ∼1.2 eV. Consequently, states D2, D3, and D4 from EOM computations are shifted upwards when compared with TDDFT results. Also, according to the oscillator strength values, EOM method points towards D3 as the bright state while TDDFT locates the D4 state as the bright one. Despite this disagreement in the ordering of states, the relevant fact is that both methodologies confirm the presence of an excited state with substantial oscillator strength at 90° dihedral angle and with excitation energy around 2 eV. Possible sources for the disagreement between both levels of theory will be briefly discussed on Section 4. Supplementary Tables S8, S9 of Supplementary Material show analogous comparisons between the two methodologies for PNT+ at the optimized neutral and cation geometries respectively.
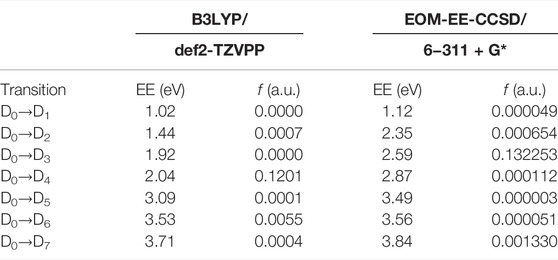
TABLE 2. Excitation energies (EE) and oscillator strengths (f) for PNT+ with a C–C–N–O dihedral angle of 87.4° at the B3LYP/def2-TZVPP and EOM-EE-CCSD/6-311+G* levels of theory.
3.3 Assignment of 650 cm−1 Wave Packet
In Section 3.1 we showed experimental evidence of the superposition of two vibrational wave packets and identified the associated frequencies as 80 and 650 cm−1. The 80 cm−1 has previously been assigned to the C–C–N–O torsional mode but the identity of the vibrational mode associated with the 650 cm−1 frequency remains to be fully elucidated. The match between the experimental frequency of 650 cm−1 (Figure 2B) and the computationally calculated frequency of 604.32 cm−1 (Figure 3B and Supplementary Table S6) lays the foundation for the hypothesis that this ring-stretching mode is the one supporting the additional nuclear wave packet found in this work.
In order to test this hypothesis we simulated the superposition of the two vibrational modes by considering three geometries within the torsional mode with 0.1, 52.7, and 87.4 C–C–N–O dihedral angles. Then, from these three geometries we performed frequency calculations to identify the ring-stretching mode in each case. The geometry with 0.1° torsional angle corresponds to the vertical cation, i.e., the cation under the optimized neutral geometry, while the structure with 52.7° dihedral angle corresponds to the optimized cation with a ring-stretching mode at 604.32 cm−1 (Supplementary Table S6). Having identified the ring-stretching modes for each of the three geometries we made the additional hypothesis that the D0→D2 transition is the one allowing the observation of the ring-stretching wave packet. This hypothesis is supported by our TDDFT calculations showing that 800 nm is nearly resonant with the D0→D2 transition at any dihedral angle (Figure 4A) and by the experimental fact that the superposition of vibrational wave packets is only observable with 800 nm excitation. With these ideas in mind we took five “snapshots” within each of the three ring-stretching modes and retrieve the corresponding geometries. Those 15 geometries, five per each ring-stretching mode, served as the basis for single-point TDDFT calculations to obtain the D0→D2 excitation energies and the corresponding harmonic estimates for the oscillator strengths.
The results of these calculations are shown in Figure 5. It is important to offer some clarification regarding the abscissa axis of this figure: since the ring-stretching mode involves the collective motion of many atoms as can be seen in Figure 3B, the description of the mode in terms of a single parameter is not an easy task. Due to this complication we show the C–N bond distance as a signature of the mode, but it should be kept in mind that the abscissa axis represents the whole ring-stretching mode, as highlighted in the structures shown in Figure 5. This being said, Figure 5A shows the excitation energy for the D0→D2 transition as a function of the C–N bond distance for three different torsional geometries with 0.1, 52.7, and 87.4 dihedral angles. The figure also shows that 800 nm (1.55 eV) provides enough energy to promote the D0→D2 transition for all the C–N bond lengths under all torsional geometries. Additionally, 800 nm is nearly resonant with the excitation energy corresponding to the torsional geometry of 87.4° at all C–N bond distances. This last observation might explain why the superposition of vibrational modes is more evident around the first minimum of PNT+ and the first maximum of
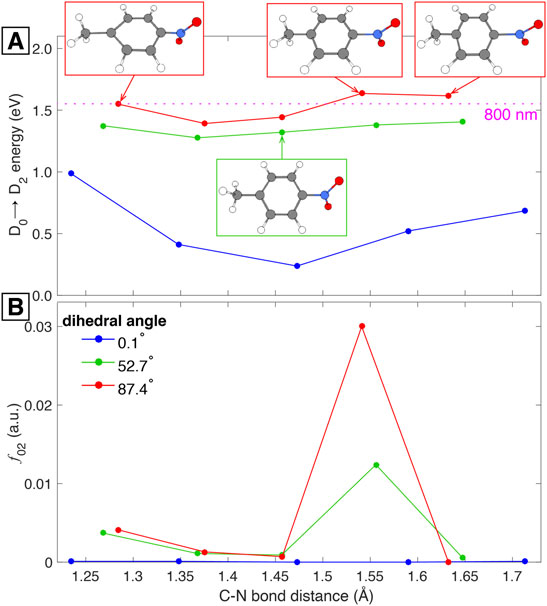
FIGURE 5. Computed potential energy surfaces (A) and oscillator strengths (B) for PNT+ along C–N bond distance under different C–C–N–O dihedral angles.
4 Discussion
This study represents the first demonstration of selective excitation to different electronic excited states upon coherent vibrational motion in a large organic cation. The motion of the two nuclear wave packets in PNT+ produces three distinct transient electronic transitions amenable to optical excitation: 1) D0→D6 transition with energy 3.1 eV at C–C–N–O dihedral angle of 0°, 2) D0→D4 (or D3) transition with energy 2.0 eV at a 90° dihedral angle, and 3) D0→D2 transition with energy 1.6 eV at 90° dihedral angle with elongated C–N bond length of 1.54 Å. The electronic structure of PNT+ along both the torsional and ring-stretching coordinates enables selective excitation to different excited states at specific geometries. The electronic structure was mainly explored by means of TDDFT calculations and further supported at the EOM-CCSD level. The upwards energetic shift observed for the D2, D3, and D4 states at the EOM level when compared with TDDFT results can be rationalized by the small 6−311+G* basis set being used for EOM computations (in contrast with the def2-TZVPP basis set for TDDFT calculations). Different groups had found that, as the basis set is expanded by adding diffuse and polarization functions, the excitation energies computed at the EOM-CCSD level decrease Cristian and Krylov (2003), Laurent et al. (2015). Therefore, a closer match between the excitation energies predicted by both levels of theory would be expected if a bigger basis set is employed for EOM calculations. Additionally, the discrepancy in which EOM identifies D3 as the bright state while TDDFT points to D4 has been previously observed for various systems Grimme and Parac (2003), Lopata et al. (2011), Prlj et al. (2015), Prlj et al. (2016), Acharya et al. (2018), López Peña et al. (2021) Nevertheless, both levels of theory confirm the presence of an excited state with substantial oscillator strength at 90° dihedral angle and with excitation energy around 2 eV. Overall, we conclude that the computationally inexpensive TDDFT level of theory is adequate enough for aiding and rationalizing the design of pump-probe control schemes.
The finding that 400 nm excitation of PNT+ selectively produces
for each probe wavelength at the pump-probe delay producing the greatest PNT+ signal depletion identified in Figure 1. The fractional yields for each ion at a series of probe intensities from 2 to 15 TW cm−2 are shown in Figure 6. Although greater enhancement of fragment ion yields is observed at higher probe intensities, 15 TW cm−2 represents a practical upper limit because at higher intensity the probe pulse creates ions even in the absence of the pump pulse. Examination of Figure 6 shows that
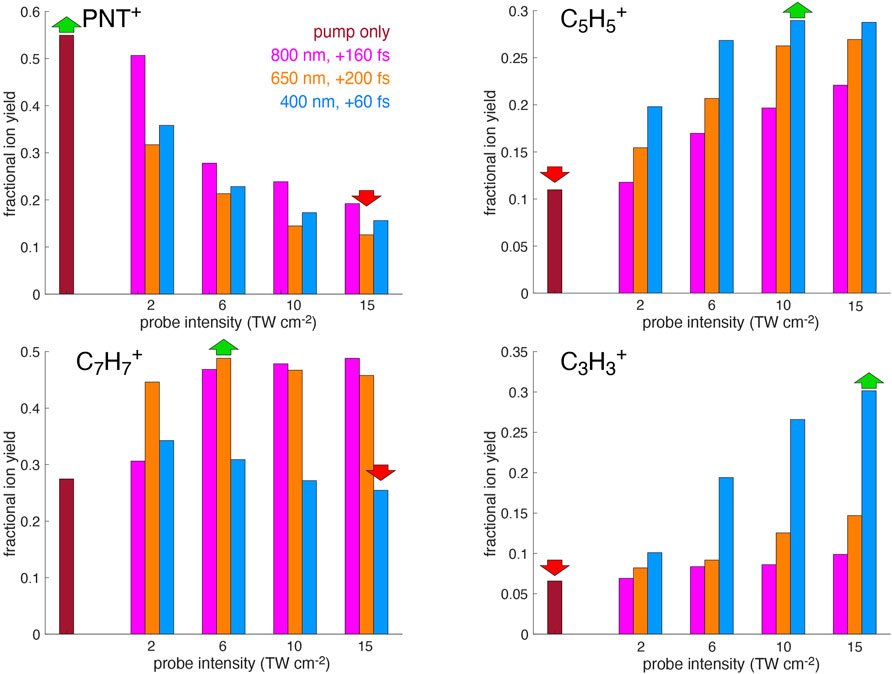
FIGURE 6. Fractional yields of PNT+,
Finally, the observation that strong field ionization of PNT launches nuclear wave packets along two distinct vibrational modes is of particular interest. Numerous previous pump-probe studies of other substituted benzenes including nitrobenzene López Peña et al. (2021), o-nitrotoluene Ampadu Boateng et al. (2019a), azobenzene Munkerup et al. (2017) and alkyl phenyl ketones Konar et al. (2014), Bohinski et al. (2014), Tibbetts et al. (2015) have observed only torsional wave packet motion upon strong-field ionization. To the best of our knowledge, the only previous study to definitively observe multimode coherent vibrational motion in a cation prepared by strong field ionization found a superposition of the C–I stretch and I–CH3 umbrella modes in CH3I+ Wei et al. (2017). Although in a previous work we had proposed multimode coherent excitation in diisopropyl methylphosphonate ion to explain observed ion yield oscillations at two different frequencies Ampadu Boateng et al. (2019b), no PES calculations were performed to confirm distinct excitation pathways. Hence, we recommend the combined strategy of pump-probe measurements with multiple probe wavelength and PES computations along possible coherently excited coordinates as performed in this work to identify possible multimode coherent excitation in other organic cations prepared by strong-field ionization.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
KT designed and directed the research. HL, JS, and DA conducted the experiments. HL performed the calculations with input from KL. KL contributed calculation codes. HL, JS, and KT analyzed the data and wrote the manuscript with input from all authors.
Funding
This work was supported by the U.S. Army Research Office through Contract W911NF-19-1-0099. DA and HL acknowledge generous financial support from Altria Graduate Research Fellowships.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
High Performance Computing resources provided by the High Performance Research Computing (HPRC) Core Facility at Virginia Commonwealth University (https://chipc.vcu.edu) were used for conducting portions of the research reported in this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.859095/full#supplementary-material
Abbreviations
BBO, β-barium borate; DFT, density functional theory; EOM-EE-CCSD, equation-of-motion excitation-energies coupled cluster singles and doubles; OPA, optical parametric amplifier; PES, potential energy surface; PNT, p-nitrotoluene; TDDFT, time-dependent density functional theory; IVR, intramolecular vibrational energy redistribution.
References
Acharya, A., Chaudhuri, S., and Batista, V. S. (2018). Can Tddft Describe Excited Electronic States of Naphthol Photoacids? a Closer Look with EOM-CCSD. J. Chem. Theor. Comput. 14, 867–876. doi:10.1021/acs.jctc.7b01101
Ampadu Boateng, D., Gutsev, G. L., Jena, P., and Tibbetts, K. M. (2018). Dissociation Dynamics of 3- and 4-nitrotoluene Radical Cations: Coherently Driven C-NO2 bond Homolysis. J. Chem. Phys. 148, 134305. doi:10.1063/1.5024892
Ampadu Boateng, D., Word, M. K. D., Gutsev, L. G., Jena, P., and Tibbetts, K. M. (2019a). Conserved Vibrational Coherence in the Ultrafast Rearrangement of 2-nitrotoluene Radical Cation. J. Phys. Chem. A. 123, 1140–1152. doi:10.1021/acs.jpca.8b11723
Ampadu Boateng, D., Word, M. K., and Tibbetts, K. (2019b). Probing Coherent Vibrations of Organic Phosphonate Radical Cations with Femtosecond Time-Resolved Mass Spectrometry. Molecules 24, 509. doi:10.3390/molecules24030509
Assion, A., Baumert, T., Bergt, M., Brixner, T., Kiefer, B., Seyfried, V., et al. (1998). Control of Chemical Reactions by Feedback-Optimized Phase-Shaped Femtosecond Laser Pulses. Science 282, 919–922. doi:10.1126/science.282.5390.919
Bauernschmitt, R., and Ahlrichs, R. (1996). Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 256, 454–464. doi:10.1016/0009-2614(96)00440-X
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Bloembergen, N., and Yablonovitch, E. (1978). Infrared‐laser‐induced Unimolecular Reactions. Phys. Today 31, 23–30. doi:10.1063/1.2995035
Bloembergen, N., and Zewail, A. H. (1984). Energy Redistribution in Isolated Molecules and the Question of Mode-Selective Laser Chemistry Revisited. J. Phys. Chem. 88, 5459–5465. doi:10.1021/j150667a004
Bohinski, T., Moore Tibbetts, K., Tarazkar, M., Romanov, D. A., Matsika, S., and Levis, R. J. (2014). Strong Field Adiabatic Ionization Prepares a Launch State for Coherent Control. J. Phys. Chem. Lett. 5, 4305–4309. doi:10.1021/jz502313f
Cardoza, D., Pearson, B. J., Baertschy, M., and Weinacht, T. (2006). Charge-transfer as a Mechanism for Controlling Molecular Fragmentation. J. Photochem. Photobiol. A: Chem. 180, 277–281. doi:10.1016/j.jphotochem.2006.02.007
Cristian, A.-M. C., and Krylov, A. I. (2003). Electronic Structure of the π-bonded Al-C2H4 Complex: Characterization of the Ground and Low-Lying Excited States. J. Chem. Phys. 118, 10912–10918. doi:10.1063/1.1576212
Damrauer, N. H., Dietl, C., Krampert, G., Lee, S.-H., Jung, K.-H., and Gerber, G. (2002). Control of Bond-Selective Photochemistry in CH2 BrCl Using Adaptive Femtosecond Pulse Shaping. Eur. Phys. J. D - At. Mol. Opt. Phys. 20, 71–76. doi:10.1140/epjd/e2002-00101-8
Epifanovsky, E., Gilbert, A. T. B., Feng, X., Lee, J., Mao, Y., Mardirossian, N., et al. (2021). Software for the Frontiers of Quantum Chemistry: An Overview of Developments in the Q-Chem 5 Package. J. Chem. Phys. 155, 084801. doi:10.1063/5.0055522
[Dataset] Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 revision c.01. Wallingford CT: Gaussian Inc.
Grimme, S., and Parac, M. (2003). Substantial Errors from Time-dependent Density Functional Theory for the Calculation of Excited States of Large π Systems. ChemPhysChem 4, 292–295. doi:10.1002/cphc.200390047
Judson, R. S., and Rabitz, H. (1992). Teaching Lasers to Control Molecules. Phys. Rev. Lett. 68, 1500–1503. doi:10.1103/PhysRevLett.68.1500
Konar, A., Shu, Y., Lozovoy, V. V., Jackson, J. E., Levine, B. G., and Dantus, M. (2014). Polyatomic Molecules under Intense Femtosecond Laser Irradiation. J. Phys. Chem. A. 118, 11433–11450. doi:10.1021/jp505498t
Kraus, P. M., Mignolet, B., Baykusheva, D., Rupenyan, A., Horný, L., Penka, E. F., et al. (2015). Measurement and Laser Control of Attosecond Charge Migration in Ionized Iodoacetylene. Science 350, 790–795. doi:10.1126/science.aab2160
Krylov, A. I. (2008). Equation-of-Motion Coupled-Cluster Methods for Open-Shell and Electronically Excited Species: The Hitchhiker's Guide to Fock Space. Annu. Rev. Phys. Chem. 59, 433–462. doi:10.1146/annurev.physchem.59.032607.093602
Langhojer, F., Cardoza, D., Baertschy, M., and Weinacht, T. (2005). Gaining Mechanistic Insight from Closed Loop Learning Control: The Importance of Basis in Searching the Phase Space. J. Chem. Phys. 122, 014102. doi:10.1063/1.1826011
Laurent, A. D., Blondel, A., and Jacquemin, D. (2015). Choosing an Atomic Basis Set for TD-DFT, SOPPA, ADC(2), CIS(D), CC2 and EOM-CCSD Calculations of Low-Lying Excited States of Organic Dyes. Theor. Chem. Acc. 134, 76. doi:10.1007/s00214-015-1676-9
Lopata, K., Reslan, R., Kowalska, M., Neuhauser, D., Govind, N., and Kowalski, K. (2011). Excited-state Studies of Polyacenes: A Comparative Picture Using EOMCCSD, CR-EOMCCSD(T), Range-Separated (LR/RT)-TDDFT, TD-PM3, and TD-ZINDO. J. Chem. Theor. Comput. 7, 3686–3693. doi:10.1021/ct2005165
López Peña, H. A., Ampadu Boateng, D., McPherson, S. L., and Tibbetts, K. M. (2021). Using Computational Chemistry to Design Pump-Probe Schemes for Measuring Nitrobenzene Radical Cation Dynamics. Phys. Chem. Chem. Phys. 23, 13338–13348. doi:10.1039/D1CP00360G
Lozovoy, V. V., Zhu, X., Gunaratne, T. C., Harris, D. A., Shane, J. C., and Dantus, M. (2008). Control of Molecular Fragmentation Using Shaped Femtosecond Pulses. J. Phys. Chem. A. 112, 3789–3812. doi:10.1021/jp071691p
Moore Tibbetts, K., Xing, X., and Rabitz, H. (2013). Optimal Control of Molecular Fragmentation with Homologous Families of Photonic Reagents and Chemical Substrates. Phys. Chem. Chem. Phys. 15, 18012–18022. doi:10.1039/C3CP52664J
Moore Tibbetts, K. (2019). Coherent Vibrational and Dissociation Dynamics of Polyatomic Radical Cations. Chem. Eur. J. 25, 8431–8439. doi:10.1002/chem.201900363
Munkerup, K., Romanov, D., Bohinski, T., Stephansen, A. B., Levis, R. J., and Sølling, T. I. (2017). Conserving Coherence and Storing Energy during Internal Conversion: Photoinduced Dynamics of Cis- and Trans-azobenzene Radical Cations. J. Phys. Chem. A. 121, 8642–8651. doi:10.1021/acs.jpca.7b09185
Nichols, S., Weinacht, T., Rozgonyi, T., and Pearson, B. (2009). Strong-field Phase-dependent Molecular Dissociation. Phys. Rev. A. 79, 043407. doi:10.1103/physreva.79.043407
Plenge, J., Wirsing, A., Wagner-Drebenstedt, I., Halfpap, I., Kieling, B., Wassermann, B., et al. (2011). Coherent Control of the Ultrafast Dissociative Ionization Dynamics of Bromochloroalkanes. Phys. Chem. Chem. Phys. 13, 8705–8714. doi:10.1039/C0CP02742A
Prlj, A., Curchod, B. F. E., Fabrizio, A., Floryan, L., and Corminboeuf, C. (2015). Qualitatively Incorrect Features in the TDDFT Spectrum of Thiophene-Based Compounds. J. Phys. Chem. Lett. 6, 13–21. doi:10.1021/jz5022087
Prlj, A., Sandoval-Salinas, M. E., Casanova, D., Jacquemin, D., and Corminboeuf, C. (2016). Low-Lying ππ* States of Heteroaromatic Molecules: A Challenge for Excited State Methods. J. Chem. Theor. Comput. 12, 2652–2660. doi:10.1021/acs.jctc.6b00245
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab Initio calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force fields. J. Phys. Chem. 98, 11623–11627. doi:10.1021/j100096a001
Tannor, D. J., and Rice, S. A. (1985). Control of Selectivity of Chemical Reaction via Control of Wave Packet Evolution. J. Chem. Phys. 83, 5013–5018. doi:10.1063/1.449767
Tibbetts, K. M., Tarazkar, M., Bohinski, T., Romanov, D. A., Matsika, S., and Levis, R. J. (2015). Controlling the Dissociation Dynamics of Acetophenone Radical Cation through Excitation of Ground and Excited State Wavepackets. J. Phys. B: Mol. Opt. Phys. 48, 164002. doi:10.1088/0953-4075/48/16/164002
Wei, Z., Li, J., Wang, L., See, S. T., Jhon, M. H., Zhang, Y., et al. (2017). Elucidating the Origins of Multimode Vibrational Coherences of Polyatomic Molecules Induced by Intense Laser fields. Nat. Commun. 8, 735. doi:10.1038/s41467-017-00848-2
Weigend, F., and Ahlrichs, R. (2005). Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/B508541A
Xing, X., Rey-de-Castro, R., and Rabitz, H. (2017). Gaining Mechanistic Insight with Control Pulse Slicing: Application to the Dissociative Ionization of CH2BrI. J. Phys. Chem. A. 121, 8632–8641. doi:10.1021/acs.jpca.7b08835
Zewail, A. H. (1988). Laser Femtochemistry. Science 242, 1645–1653. doi:10.1126/science.242.4886.1645
Keywords: coherent control, strong field ionization, mass spectrometry, pump-probe, nuclear wave packet
Citation: López Peña HA, Shusterman JM, Ampadu Boateng D, Lao KU and Tibbetts KM (2022) Coherent Control of Molecular Dissociation by Selective Excitation of Nuclear Wave Packets. Front. Chem. 10:859095. doi: 10.3389/fchem.2022.859095
Received: 20 January 2022; Accepted: 18 March 2022;
Published: 05 April 2022.
Edited by:
Andre Bandrauk, Université de Sherbrooke, CanadaReviewed by:
Riccardo Conte, University of Milan, ItalyChaoyuan Zhu, National Chiao Tung University, Taiwan
Copyright © 2022 López Peña, Shusterman, Ampadu Boateng, Lao and Tibbetts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katharine Moore Tibbetts, a210aWJiZXR0c0B2Y3UuZWR1