 Carlos Emiliano Buelna-García1,2
Carlos Emiliano Buelna-García1,2 Cesar Castillo-Quevedo3
Cesar Castillo-Quevedo3 Jesus Manuel Quiroz-Castillo1
Jesus Manuel Quiroz-Castillo1 Edgar Paredes-Sotelo1
Edgar Paredes-Sotelo1 Manuel Cortez-Valadez4
Manuel Cortez-Valadez4 Martha Fabiola Martin-del-Campo-Solis3
Martha Fabiola Martin-del-Campo-Solis3 Tzarara López-Luke5
Tzarara López-Luke5 Marycarmen Utrilla-Vázquez6
Marycarmen Utrilla-Vázquez6 Ana Maria Mendoza-Wilson7
Ana Maria Mendoza-Wilson7 Peter L. Rodríguez-Kessler8
Peter L. Rodríguez-Kessler8 Alejandro Vazquez-Espinal9
Alejandro Vazquez-Espinal9 Sudip Pan10
Sudip Pan10 Aned de Leon-Flores11
Aned de Leon-Flores11 Jhonny Robert Mis-May6
Jhonny Robert Mis-May6 Adán R. Rodríguez-Domínguez12
Adán R. Rodríguez-Domínguez12 Gerardo Martínez-Guajardo13*
Gerardo Martínez-Guajardo13* Jose Luis Cabellos6*
Jose Luis Cabellos6*- 1Departamento de Investigación en Polímeros y Materiales, Universidad de Sonora, Hermosillo, Mexico
- 2Organización Científica y Tecnológica del Desierto, Hermosillo, Mexico
- 3Departamento de Fundamentos del Conocimiento, Centro Universitario del Norte, Universidad de Guadalajara, Colotlán, Mexico
- 4CONACYT-Departamento de Investigación en Física, Universidad de Sonora, Hermosillo, Mexico
- 5Instituto de Investigación en Metalurgia y Materiales, Universidad Michoacana de San Nicolás de Hidalgo, Ciudad Universitaria, Morelia, Mexico
- 6Universidad Politécnica de Tapachula, Tapachula, Mexico
- 7Coordinación de Tecnología de Alimentos de Origen Vegetal, CIAD, A.C., Hermosillo, Mexico
- 8Laboratorio de Química Inorgánica y Materiales Moleculares, Facultad de Ingeniería, Universidad Autonoma de Chile, Santiago, Chile
- 9Comput. Theor. Chem. Group Departamento de Ciencias Químicas, Facultad de Ciencias Exactas, Universidad Andres Bello, Santiago, Chile
- 10Fachbereich Chemie, Philipps-Universität Marburg, Marburg, Germany
- 11Departamento de Ciencias Químico Biologicas, Universidad de Sonora, Hermosillo, Mexico
- 12Instituto de Física, Universidad Autónoma de San Luis Potosí, San Luis Potosí, Mexico
- 13Unidad Académica de Ciencias Químicas, Área de Ciencias de la Salud, Universidad Autónoma de Zacatecas, Zacatecas, Mexico
The relative populations of Cu38 isomers depend to a great extent on the temperature. Density functional theory and nanothermodynamics can be combined to compute the geometrical optimization of isomers and their spectroscopic properties in an approximate manner. In this article, we investigate entropy-driven isomer distributions of Cu38 clusters and the effect of temperature on their IR spectra. An extensive, systematic global search is performed on the potential and free energy surfaces of Cu38 using a two-stage strategy to identify the lowest-energy structure and its low-energy neighbors. The effects of temperature on the populations and IR spectra are considered via Boltzmann factors. The computed IR spectrum of each isomer is multiplied by its corresponding Boltzmann weight at finite temperature. Then, they are summed together to produce a final temperature-dependent, Boltzmann-weighted spectrum. Our results show that the disordered structure dominates at high temperatures and the overall Boltzmann-weighted spectrum is composed of a mixture of spectra from several individual isomers.
Introduction
Nanoclusters are of great interest since they allow us to study the transition from free atoms to bulk condensed systems (Wilcoxon and Abrams, 2006) by analyzing the size-dependent evolution of their properties (Ferrando et al., 2008). In particular, noble-metal nanoclusters (NMCs) have attracted attention in many fields of science due to their interesting plasmonic, catalytic (Mathew and Pradeep, 2014; Inwati et al., 2018), and photophysical properties at the nanoscale (Xavier et al., 2012). Specifically, Cu nanoclusters embedded in a dielectric matrix have attracted attention because of their tunable longitudinal surface plasmon resonance characteristics (Inwati et al., 2018). Copper is cheaper than gold and silver and has high photosensitivity, high thermal and electric conductivities, and optical properties (Zhang et al., 2019) that make it a good candidate for nanodevices (Liu et al., 2015) and nanoelectronics development (Jena and Castleman, 2006). Particularly, Cu38 has attracted attention because it has “magic number” structures (Tran and Johnston, 2009), which are defined in terms of geometric and energetic factors and related to the closing of electronic shells (Baletto et al., 2004), much like small sodium clusters (de Heer, 1993). The magicity of the Cu38 cluster is due only to energetic considerations (Doye and Wales, 1998; Baletto et al., 2004). In contrast, in small, packed barium clusters with magic numbers, stability is dominated by geometric rather than electronic effects (de Heer, 1993). It is believed that magic structures are the global minimum energy structures on the potential energy surface, and thus reflect the molecular properties of the system (Baletto et al., 2004).
From the experimental point of view, the Cu38 cluster has been widely studied via photoelectron spectroscopy (PES) in order to extract the electronic gap of the anionic Cu38 cluster resulting in a semiconductor with a 0.33 eV electronic gap (Pettiette et al., 1988; Zhang et al., 2019). PES has also been used to study the anionic Cu38 cluster, inferring that its putative global minimum should be an oblate structure instead of a highly symmetric structure (Kostko et al., 2005; Zhang et al., 2019), despite computational results for 38-noble metal atom clusters that frequently find a highly symmetric (cuboctahedral) structure (Fujima and Yamaguchi, 1989; Doye and Wales, 1998; Kostko et al., 2005).
From the theoretical point of view, several density functional theory (DFT)-based studies have been carried out, in which, for example, the thermodynamic properties of the Cu38 cluster have been reported (Taylor et al., 2008). The transition states and reaction energies of the water gas shift reaction on a Cu38 cluster and Cu slab have also been studied via DFT computations (Qi et al., 2020). In particular, the high-symmetry octahedral structure was reported as the lowest energy structure (Takagi et al., 2017) using the PW91 functional (Perdew et al., 1992), a plane-wave basis set, and the pseudopotential approximation (Itoh et al., 2009). The Cu38 cluster has also been investigated using a hybrid strategy (Hijazi and Park, 2010); in which the embedded atom potential method followed by DFT computations with the PBE functional and pseudopotential approximation was used. The authors reported a putative global minimum structure with octahedral (OH) symmetry. The second most stable structure was the incomplete-Mackay icosahedron (IMI) located 0.26 eV above the putative global minimum.
There has been some discussion about the lowest energy structure of the Cu38 cluster. Searches for the lowest-energy structure that employed many-body potentials identified a cuboctahedral structure (Doye and Wales, 1998; Grigoryan et al., 2005). Several authors used an empirical potential-energy function containing two-body atomic interactions and found that fivefold symmetry appears to be the putative global minimum in the Cu38 cluster (Erkoç, 1994; Erkoç and Shaltaf, 1999). In contrast, previous studies reported the cuboctahedron structure as the putative global minimum (Grigoryan et al., 2006) by employing empirical many-body Gupta and Sutton-Chen potentials; some other works also consider the Cu38 octahedron cluster to be the putative global minimum (Itoh et al., 2009; Hijazi and Park, 2010; Takagi et al., 2017), but several others found that the Cu38 cluster with truncated octahedron geometry is energetically more stable than other configurations (Darby et al., 2002; Itoh et al., 2009; Hijazi and Park, 2010; Núnez and Johnston, 2010; Park and Hijazi, 2012; Fernández et al., 2015; Zhao et al., 2017). Some of us pointed out that the energies computed via different methods such as DFT, second-order Møllet-Plesset approach (MP2), and Coupled cluster with single-double and perturbative triple (CCSDT) excitations yield different energetic ordering results (de la Puente et al., 1997; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021; Castillo-Quevedo et al., 2021). In the case of DFT, the functional and basis set employed, the zero-point energy correction (ZPE), the dispersion energy, and other parameters can change the energetic ordering of the low energy structures (Castillo-Quevedo et al., 2021). Moreover, practical molecular systems and materials must be studied at warm temperatures, so their molecular properties at finite temperature are dominated by Boltzmann distributions of isomers (Baletto and Ferrando, 2005; Li and Truhlar, 2014; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021; Castillo-Quevedo et al., 2021), and those of the associated materials are statistical averages over the ensemble of conformations (Mendoza-Wilson et al., 2020; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). Total energy computations using DFT methodology are typically carried out at absolute zero temperature, although the thermal properties of the inhomogeneous electron gas in the mid-1960s were studied (Mermin, 1965). Recently, DFT was extended to finite temperature (Pittalis et al., 2011; Gonis and Däne, 2018; Gazquezet al., 2019), but as far as we know, it has not been implemented in any software.
There are cases where the global minimum structure ceases to be the most likely at high temperatures, so other structures prevail. For instance, in small Ag clusters, the temperature leads to the transition from the initial Face-Centered Cubic (FCC) phase to other structures (Redel et al., 2015), thus temperature promotes face changes in materials. Interestingly, the molecular system minimizes the Gibbs free energy at temperatures other than zero and maximizes the entropy (Buelna-Garcia et al., 2021). Although the search for global and local minima is useful in understanding reactivities and catalytic efficiencies, such studies mostly neglect temperature-dependent entropic contributions to free energy when the temperature increases. Taking temperature into account requires dealing with nanothermodynamics (Hill, 1962; Baletto and Ferrando, 2005; Li et al., 2007; Li and Truhlar, 2014; Grigoryan and Springborg, 2019; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). The thermodynamics of clusters have been studied using various tools (Wales, 1996; Li et al., 2007; Li and Truhlar, 2014; Calvo, 2015; Buelna-Garcia et al., 2021) such as in the molecular-dynamics simulations of boron clusters (Martinez-Guajardo et al., 2015) and Cu38 clusters (Zhang et al., 2019).
Cluster properties depend heavily on the cluster structure, size, composition, and temperature. Therefore, the first step to understanding their molecular properties is to elucidate the lowest energy structure and its isomers close in energy (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021; Baletto and Ferrando, 2005; Darby et al., 2002; Ohno and Maeda, 2006), which is a complex task due to several factors (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). The second step relies on spectroscopy, which gives insight into the structure and has been proposed as a way of detecting structural transformations within clusters. The influence of temperature on Infrared spectroscopy (IR) has been computed before for a variety of clusters (Even et al., 1989; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). The present paper uses the statistical formulation of thermodynamics and nanothermodynamics (Li et al., 2007; Li and Truhlar, 2014; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021) to compute the thermodynamic properties of the neutral Cu38 cluster, define its putative global minimum at finite temperature, compute the relative populations among the isomers, and the IR spectra as Boltzmann-weighted spectral sums of individual spectra. Our findings show that an amorphous structure strongly dominates the putative global minimum at high temperatures, whereas the truncated octahedron dominates at low temperatures. The remainder of the manuscript is organized as follows: “Free Energy Surface Exploration Method and Computational Details” gives the computational details and a brief overview of the theory and algorithms. The results and discussion are presented in “Results and Discussion”. This includes the putative global minimum at room temperature, relative populations at temperatures from 20 to 1500 K, and IR spectra as functions of the temperature. Conclusions are given in “Conclusion”.
Free Energy Surface Exploration Method and Computational Details
The putative global minimum is determined by the enthalpy (at zero temperature) or the Gibbs free energy (at temperatures other than zero). A simple analysis of the Gibbs free energy given by ∆G = ∆H − ∆ST leads to the conclusion that the entropy must be maximized in order to minimize the Gibbs free energy (Sutton and Levchenko, 2020; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). From the theoretical point of view, and in order to understand molecular properties at finite temperature, the lowest Gibbs free energy structure (or the structure with the largest entropy), as well as all structures close in energy to the lowest energy structure (or all high-entropy structures close in entropy to the structure with the highest entropy) must be known (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021), considering that experiments are performed at finite temperature.
The search for the global minimum in atomic clusters is a complex task due to the number of possible combinations, which grows exponentially with the number of atoms, leading to a combinatorial explosion problem, among others (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). Despite the difficulty of this task, several algorithms have been successfully employed in a targeted way to explore the potential and free energy surfaces. These are coupled to a local optimizer of any electronic structure package. Examples include the Ab initio Random Structure Searching approach (Pickard and Needs, 2011), simulated annealing (Kirkpatrick et al., 1983; Metropolis et al., 1953; Xiang and Gong, 2000; Xiang et al., 2013; Vlachos et al., 1993; Granville et al., 1994), the kick methodology (Pan et al., 2014; Cui et al., 2015; Vargas-Caamal et al., 2016b,a; Cui et al., 2017; Vargas-Caamal et al., 2015; Flórez et al., 2016; Ravell et al., 2018; Hadad et al., 2014; Saunders, 2004, 1987; Grande-Aztatzi et al., 2014; Mendoza-Wilson et al., 2022), and genetic algorithms (Guo et al., 2017; Dong et al., 2018; Mondal et al., 2016; Ravell et al., 2018; Grande-Aztatzi et al., 2014; Rodrı́guez-Kessler et al., 2017; Alexandrova et al., 2004; Buelna-Garcia et al., 2021). Global minimum structure searches at the DFT level are too computationally expensive to be applied to intermediate and large cluster sizes since, as mentioned earlier, the number of candidates increases exponentially with the number of atoms. In this paper, we use a two-stage procedure to explore the potential energy surface efficiently. In the primary stage, we perform a global search using an empirical methodology. The Gupta interaction potential is used to describe the Cu-Cu interactions with default parameters taken from Refs. (Cleri and Rosato, 1993; Wilson and Johnston, 2002). It is coupled to the basin hopping global optimization algorithm implemented in Python code and part of the GALGOSON global search code (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). In the second stage, all of the lowest energy structures from the primary stage are symmetrized, which is followed by geometry optimization at the DFT level, using the Gaussian-suite code (Frisch et al., 2009). The calculations employ two exchange-correlation functionals, B3PW91 and PBE, and two basis sets, def2SVP and LANL2DZ, with and without considering the D3 version of Grimme’s dispersion corrections (Grimme et al., 2010), as implemented in the Gaussian 09 code (Frisch et al., 2009). Becke’s hybrid three-parameter (Becke 1993, 1988) exchange-correlation functional in combination with the Perdew and Wang GGA functional PW91 (Perdew et al., 1992; Perdew and Wang, 1992) is known as the B3PW91 exchange-correlation functional. The B3PW91 has been employed in other studies of reactivity in copper clusters, where it has provided good performance (Fernández et al., 2015). The PBE exchange-correlation functional (Perdew et al., 1996) has shown good performance with regarding thermochemical properties (del Campo et al., 2012). The LANL2DZ basis set (Dunning and Hay, 1977) has been used in previous computational studies of copper-based molecular properties with very good agreement with experimental values (Legge et al., 2001). In a previous DFT study, the def2-SVP basis set (Weigend and Ahlrichs, 2005) provided good results in the computation of the Cu-metal ligand bond lengths (Niu et al., 2014). The true minimum energy structures are validated via vibrational analysis.
Thermochemical Properties
All of the thermodynamic properties of an ensemble of molecules can be derived from the molecular partition function (Takadaet al., 2018; Dzib et al., 2019; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021). Similarly, the wave function contains all information about a molecular system (Buelna-Garcia et al., 2021). Previous theoretical studies used the partition function to compute thermodynamic properties of Cun clusters (n = 2, 150) as a function of the temperature and showed that the magic number structures are temperature dependent (Li et al., 2007; Grigoryan and Springborg 2019). The thermodynamics of unsupported neutral Aln (2 < n < 65) clusters have also been investigated by evaluating vibrational partition functions. They reported that the dominant cluster structure is temperature dependent (Li and Truhlar, 2014). Aditionally, the atomistic thermodynamics framework has been used to predict material behaviors at realistic temperatures (Sutton and Levchenko, 2020). More recently, the partition function was used by some of us to compute the temperature-dependent relative populations and IR spectra of neutral Be4B8 and anionic Be6B11 clusters (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021), and a similar procedure was employed to compute reaction rate constants in two representative hydrogen abstraction reactions (Dzib et al., 2019). Regarding the temperature-dependent entropic contributions, the [Fe(pmea)(NCS)2] complex was studied by Brehm et al. (Brehm et al., 2006). In this study, the thermodynamic properties are computed using the partition function Q given in Eq. 1 under the rigid rotor, harmonic oscillator, Born-Oppenheimer, ideal gas, and particle-in-a-box approximations.
In Eq. 1, gi is the degeneracy factor, KB is the Boltzmann constant, T is the temperature, and −∆Ei is the total energy of a cluster (Buelna-Garcia et al., 2021; Dzib et al., 2019; McQuarrie, 1975). Within Born Oppenheimer and rigid rotor harmonic oscillator approximations, the partition function Q(T) is factorized into electronic, translational, vibrational, and rotational contributions given by Eq. 2
Table 1 shows the contributions of electronic, translational, vibrational, and rotational to the canonical partition function.

TABLE 1. Contributions to the partition function.
We considered that the energy gap between the first and higher excited states is greater than

TABLE 2. Contributions to internal energy and entropy.
Equations shows in Tables 1 and 2 are the same as those employed in previous studies (Li et al., 2007; Grimme, 2012; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021; Dzib et al., 2019) and any standard thermodynamics textbook (McQuarrie, 1975; Hill, 1986).
The vibrational frequencies are computed employing Gaussian code. The Gibbs free energy (G) and the enthalpy (H) are computed employing Eqs. 3 and 4, respectively. In these Equations R is the ideal gas constant, n is the amount of substance, and T is the absoulte temperature.
To compute the probability of the occurrence of one particular Cu38 cluster in a Boltzmann ensemble at thermal equilibrium as a function of temperature, we employed the probability of occurrence (Shortle, 2003; Li et al., 2007; Grimme, 2012; Bhattacharya et al., 2017; Schebarchov et al., 2018; Dzib et al., 2019; Goldsmith et al., 2019; Grigoryan and Springborg, 2019; Mendoza-Wilson et al., 2020; Bhumla et al., 2021; Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021) given by Eq. 5:
where
In this study, the Boltzmann-weighted IR spectrum at a finite temperature is given by Eq. 7:
where n is the total number of clusters in the ensemble, IRi is the spectrum of the ith isomer at temperature T = 0, and Pi (T) is the probability of the ith isomer, which is given by Eq. 5. We use the Boltzmann-optics-full-Ader code (BOFA) to compute the occurrence probability and the IR spectra (Buelna-Garcia et al., 2021).
Results and Discussion
Low-Energy Structures
The ball and stick models shown in Figure 1 depict the lowest-energy structures of neutral Cu38 clusters and some competing isomers. We use the B3PW91/def2SVP level of theory and consider the Grimme (DFT-D3) dispersion pairwise correction (Grimme et al., 2010), at room temperature and at 1 atm pressure. We found that the tetrakaidecahedron is the lowest energy structure. It has fourteen faces: six equivalent square FCC(100) faces and eight equivalent hexagons. This shape is obtained when cutting the corners off a 3D diamond shape. It is an FCC-like truncated octahedron (TO). The calculated structure belongs to the C1 symmetry point group and to the 1A electronic ground state. Its lowest IR active vibration frequency is 32.57 cm−1 and it is a semiconductor with an electronic gap of 0.623 eV. Previous works regarding the exploration of the potential energy surface of Cu38 using genetic algorithms via the Gupta potential have often found highly symmetric TO structures (Kostko et al., 2005; Darby et al., 2002), which have also been reported using the Sutton-Chen potential with Monte Carlo simulations (Zhao et al., 2017). The optimized Cu-Cu bond length is 2.4670 Å, which is in good agreement with the reported bond length in the Cu-Cu dimer via DFT calculations (2.248 Å) (Kabir et al., 2004; Guvelioglu et al., 2006) and is consistent with the experimental Cu-Cu bonding distance of 2.22 Å (Kabir et al., 2004). Our computed TO structure diameter is 7.8 Å, which is in good agreement with the 8 Å reported in previous DFT calculations (Guvelioglu et al., 2006).
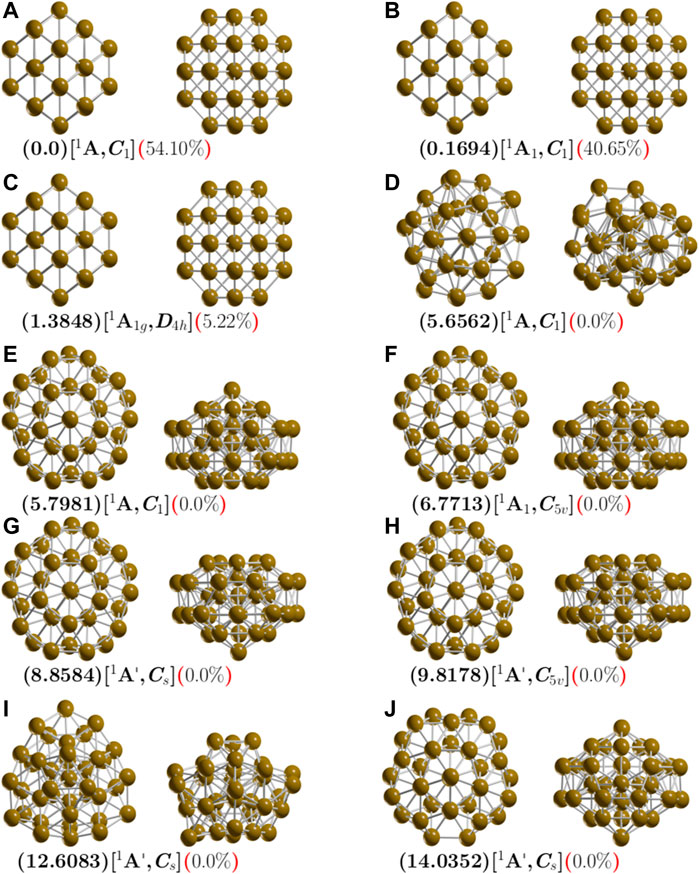
FIGURE 1. (Color online) Optimized geometries in front and side views of neutral Cu38 clusters at the PBE-D3/def2-SVP level of theory. The calculations consider the D3 version of Grimme’s dispersion corrections and the ZPE correction energy. The first letter is the isomer label, relative Gibbs free energies are in kcal/mol (in round parenthesis) at 298.15 K, electronic groups and symmetry point groups are [in square parenthesis], and the probability of occurrence is shown (in round, red parenthesis) at 298.15 K.
The structure with the second-lowest energy lies at 0.16 kcal/mol at 298.15 K above the putative global minimum and is also a TO structure with C1 symmetry and 1A electronic ground state. Its lowest IR active vibration frequency is 32.13 cm−1 and it is a semiconductor with an electronic gap of 0.623 eV, thus, it is fairly similar to the putative global minimum. The next structure is slightly higher in energy. It is located 1.38 kcal/mol above the putative global minimum and is also a TO structure. It has D4h symmetry and 1A1g electronic ground state. Its lowest IR active vibration frequency is 33.44 cm−1. We also explore TO structures, starting with highly-symmetric OH and TH. After geometry optimization without constraints, the OH and TH symmetries become C1 and D4h. The perfect OH symmetry could be deformed due to the Jahn–Teller effect (Kabir et al., 2004; Guvelioglu et al., 2006). This effect must be considered when calculating the total energy (Zlatar et al., 2010; Zhang et al., 2018) because the computed optical properties could change due to relative population at finite temperatures (Opik and Pryce, 1957). In one of our recent works, we clarified the origin of Gibbs free energy differences between two similar structures with different symmetry point group due to rotational entropy, specifically the RTln(σ) factor (Buelna-Garcı́a et al., 2021). In this work, the energy difference of 0.16 kcal/mol between the two isomers depicted in Figure 1A with C1 symmetries and a root-mean-square deviation (RMSD) of 0.08 is due to the Jahn-Teller effect. The structure located 1.38 kcal/mol above the putative global minimum with D4h symmetry exists due to rotational entropy. The next structure, shown in Figure 1D is located 5.65 kcal/mol above the putative global minimum and has point group symmetry C1 and electronic ground state 1A. Its lowest IR active vibration frequency is 24.16 cm−1. It is a distorted-structure semiconductor with an electronic gap of 1.0 eV, calculated Cu-Cu bond distance of 2.50 Å and molecular diameter of 9.1 Å. Its Cu-Cu bond distance and diameter are slightly larger than the global minimum. This structure possesses the smallest relative ZPE energy, as shown in Supplementary Figure S1, and the smallest vibrational frequency mode of all the isomers. The next two higher energy structures are shown in Figures 1E,F reach 5.8 kcal/mol. They are IMIs with C1 and C5V point group symmetries and electronic ground states 1 and 1A1, respectively. In both cases, the molecular diameter is 8.54 Å, the electronic gap is 0.97 eV, and the Cu-Cu bonding distance is 2.47 Å. Other, higher energy structures are shown in Figures 1G–J. These do not contribute to any molecular properties at zero and finite temperatures. Supplementary Figure S2 depicts lowest-energy-structure screening at the B3PW91/def2SVP level without considering the Grimme D3 atom-pairwise correction. The lowest energy structure is the IMI structure with point group symmetry C1 and electronic ground state 1A. The molecular diameter is 8.69 Å, which is slightly larger than that of the TO structure (7.8 Å). The average bond distance is 2.50 Å. We find the IMI structure to be the most stable at the PBE-D3/Def2SVP level. In contrast, at the PBE-D3/LANL2DZ level, we find the TO structure to be the putative global minimum. A complete description of the structures located at higher energies is presented in the Supplementary Material. For the Cu38 clusters, we point out that the energetic ordering of the isomers, the energy gaps among the isomers, and the putative global minimum interchange when we consider the dispersion interactions.
Energetics
Employing different methods to compute energies yields different results due to differences in the functional and basis set (Yanai et al., 2004), and the resulting energetic ordering changes (de la Puente et al., 1997; Buelna-Garcia et al., 2021). A comparison of total energy, among isomers, computed with two different exchange-correlation functionals and two basis sets, one which does and one which does not consider the Grimme D3 dispersion, is shown in Table 3. The optimizations performed at the B3PW91/PBE-def2TZVP level that consider the dispersion yield the same type of lowest-energy equilibrium geometries and similar energetic isomer ordering. From the energetic point of view, the inclusion of dispersion is more important than the type of functional or basis set. The first line of Table 3 shows the relative Gibbs free energies computed at the B3PW91-D3/def2TZVP level of theory. The isomer labeled ib in Table 3 and depicted in Figure 1B is located 0.16 kcal/mol above the putative global minimum. In contrast, the second line of Table 3 shows the relative Gibbs free energies computed at the B3PW91/def2TZVP level of theory. Here, the isomer ib in Table 3 that is depicted in Figure 1B is located 0.95 kcal/mol above the putative global minimum. For isomer with label ib, the inclusion of dispersion decreases the Gibbs free energy relative to the putative global minimum (from 0.95 to 0.16 kcal/mol). For isomer with label ic, considering dispersion decreases the Gibbs free energy relative to the putative global minimum from 2.0 to 1.38 kcal/mol. In contrast, the inclusion of dispersion for isomer with label id, the relative Gibbs free energy increases from 2.4 to 5.65 kcal/mol. In summary, considering dispersion reduces the Gibbs free energies of the lowest-energy structures where the Boltzmann factors are not zero. An overall comparison of free energies computed using functional B3PW91, the second line of Table 3, and PBE in four-line in Table 3, shows a reduction in the relative Gibbs free energies when the PBE functional is employed. The LANL2DZ basis set increases the relative Gibbs free energies of the low-energy isomers, as shown by comparing line six and one of Table 3.
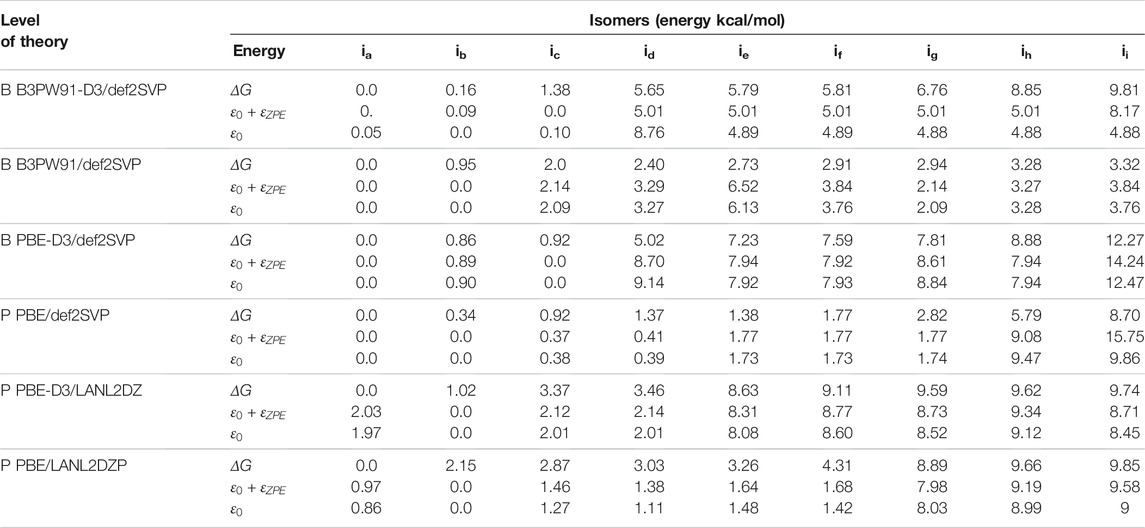
TABLE 3. A comparison of the energetic isomer ordering as determined using the B3PW91/Def2SVP, PBE/Def2SVP, and PBE/LANL2DZ levels of theory. The Gibbs free energy is computed at room temperature. The electronic energy includes the ZPE energy correction.
Occurrence Probabilities at Finite Temperature
Figure 2 shows the relative populations of neutral Cu38 clusters computed at different levels of theory for temperatures ranging from 20 to 1500 K. In order to gain insight into the effects of dispersion on the relative population, this is computed with and without the D3 Grimme dispersion. For ease of comparison, the results are displayed in side-by-side plots in Figure 2.
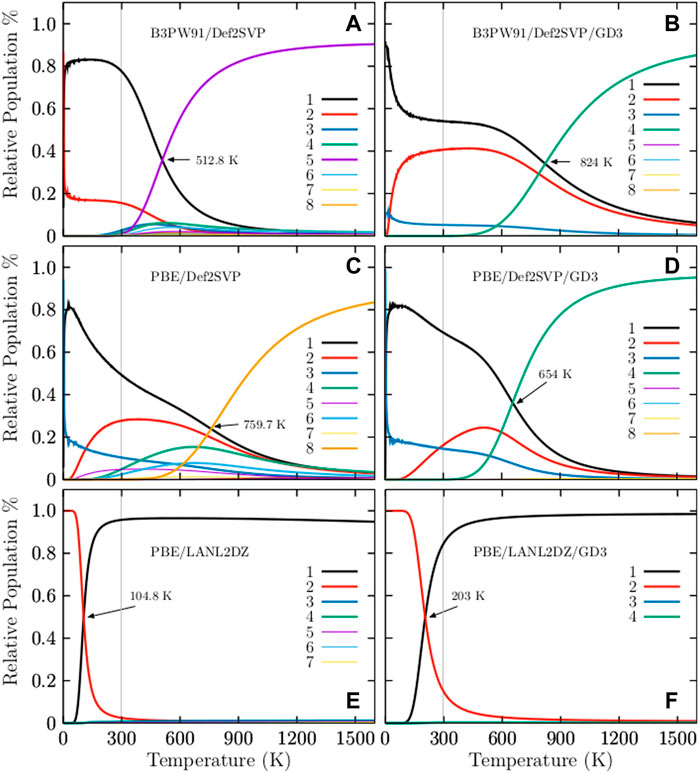
FIGURE 2. (Color online) The occurrence probabilities for temperatures ranging from 20 to 1500 K at six different levels of theory: (A) B3PW91-D3/def2-SVP, (B) B3PW91-D3/def2-SVP, (C) PBE/def2-SVP, (D) PBE-D3/def2-SVP, (E) PBE/LANL2DZ, and (F) PBE-D3/LANL2DZ. The cases (B,D), and (F) are computed considering the D3 Grimme dispersion. In all cases, the effect of the dispersion on the solid-solid transformation point in the temperature scale is large for the Cu38 cluster. At hot temperatures the dominant structure is an amorphous geometry depicted in Figure 1D, whereas, the TO structure depicted in Figure 1A is the strongly dominant structure at cold temperatures and at the B3PW91-D3/def2-SVP level of theory.
The occurrence probability of the TO structure with C1 symmetry at the B3PW91/def2-SVP level of theory is depicted in the solid, black line in Figure 2A. This occurrence probability of the TO structure strongly dominates from 0 to 300 K; thus, all of the molecular properties in this range of temperature are due only to this structure, and the occurrence probability starts to decay exponentially just before 300 K and nearly disappears at 900 K. The probability of finding the amorphous structure with point group symmetry C1 is depicted in the solid, violet line in Figure 2A. This probability starts to grow exponentially and becomes dominant at a temperature between 512.8 and 900 K. The TO and the amorphous structure co-exist at a solid-solid transition temperature of 512.8 K. The effect of dispersion can be seen Figure 2B. The relative population is computed at the B3PW91-D3/def2-SVP level of theory. The dispersion effect is dramatic; the solid-solid transformation point shifts from 512.8 to 824 K, an increase of 160%. In Figure 2B, one can see that the molecular properties below 600 K are due to only the TO structure. The probability of finding the amorphous structure, depicted via the solid, green line in Figure 2B, starts to increase exponentially just before 600 K. The TO and the amorphous structure co-exist at 824 K. The probability of finding the TO structure starts to decay exponentially at 600 K and is still around 20% at 900 K. The occurrence probabilities of various Cu38 isomers at the PBE/def2-SVP level of theory are displayed in Figure 2C. The dominant putative global minimum structure at T = 0 is the inverted incomplete-Mackay icosahedron (IIMI) structure depicted in Figure 3A with C1 symmetry.
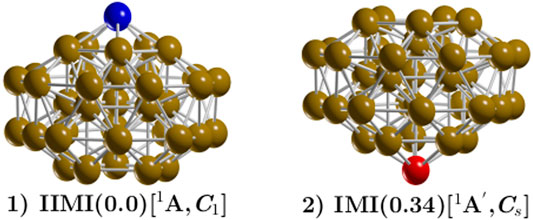
FIGURE 3. (Color online) The inverted incomplete-Mackay icosahedron (IIMI) is labeled 1 and has C1 symmetry. The incomplete-Mackay icosahedron (IMI) is labeled 2, with symmetry Cs, and is located 0.34 kcal/mol energy above the putative minimum global at 298.15 K. The yellow, red, and blue colored spheres represent copper atoms. The IIMI structure is the result of interchanging the red Cu atom depicted in the IMI structure to the position of the blue atom in the IIMI structure. The IMI structure has been reported in reference Ref Zhang et al. (2019). as the low-energy structure. The Highest Occupied Molecular Orbital (HOMO)—Lowest Unoccupied Molecular Orbital (LUMO) gap of the IMI structure is 0.24 eV (0.356 eV reported in previous DFT studies (Zhang et al., 2019). In contrast, the HOMO-LUMO gap for the IIMI structure is 0.30 eV. This, provides a plausible explanation for the higher energetic stability of why the IIMI structure is energetically more stable.
The probability of finding the IIMI structure is shown via the black, solid line in Figure 2C. This probability decays almost linearly until a temperature of 1000 K, where the probability of occurrence almost disappears. At the solid-solid transformation point (759.7 K), the IIMI structure co-exists with an amorphous structure. The probability of finding the amorphous structure starts to increase at 600 K and starts to dominate heavily as the putative global minimum above the solid-solid transformation point. The probability of finding the IMI structure is depicted via the red, solid line in Figure 2C. The probability is maximized (30%) at room temperature. Interestingly, the probabilities of the IIMI and IMI structures do not cross at low temperatures. Previous work reported that the IMI structure can be highly competitive at finite temperature (Zhang et al., 2019). Still, our findings show that the amorphous structure with C1 symmetry is quite dominant at high temperatures, whereas the IIMI structure is strongly dominant at low temperatures.
For ease of comparison, Figure 3 displays the IIMI and the IMI structures side by side. The IIMI structure dominates at low temperatures. The dispersion effect shifts the solid-solid transformation point down from 759.7 to 654 K, as shown in Figure 2D. The probability of finding the IIMI structure is depicted via the black, solid line as a function of temperature. The probability decays approximately linearly from 50 to 500 K; after that, it decays exponentially until 900 K, where it disappears. At around 400 K, the probability of finding the amorphous structure (depicted via the green, solid line Figure 2D starts to grow exponentially. At 654 K, it co-exists with the IIMI structure. Above 654 K, the amorphous structure becomes energetically favorable.
IR Spectra at Finite Temperature
The properties observed in a molecule are statistical averages over the ensemble of geometrical conformations or isomers accessible to the cluster. Thus, the molecular properties are governed by the Boltzmann distributions of the isomers, which can change significantly with the temperature, primarily due to entropic effects (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021; Li et al., 2007). The many soft vibrational modes that the clusters possess are the major contributions to the entropy. The IR spectrum is related to vibrations or rotations that alter the dipole moment and it is observed in molecules with a dipole moment. The IR spectrum is also related to the curvature of the relationship between the potential and the interatomic distance. Complete information regarding molecular vibrations allows us to analyze catalytic chemical reactions (Tinnemans et al., 2006; Brandhorst et al., 2006; Hashimoto et al., 2019). IR spectra are used to identify functional groups and chemical bond information. However, assigning IR bands to vibrational molecular modes in measured spectra can be difficult and requires DFT calculations; as mentioned earlier, the temperature is not considered in these computations and discrepancies between experimental and computed IR spectra can result from finite temperatures, anharmonic effects, and the multi-photon nature of experiments. IR computations assume single-photon processes (Buelna-Garcia et al., 2021). The IR spectra of isolated metal clusters in the gas phase were measured for vanadium cluster cations and neutral and cationic niobium clusters (Fielicke et al., 2005). Even though Cu clusters are important in catalysis and were the first clusters produced experimentally (Powers et al., 1982), the available structural information is limited to photoelectron spectroscopy studies of anions, mass spectrometry, and visible-range photodissociation spectra (Lushchikova et al., 2019). Previous work determined the structures of small cationic copper clusters based on a combination of IR spectroscopy of Cun+-Arm complexes and DFT calculations (Lushchikova et al., 2019). In this work, the IR spectra of the isomers were computed using the Gaussian package under harmonic approximation at the PBPW91-D3 [106]/def2TZVP level and a full width at half maximum of 8 cm−1. The Grimme D3 dispersion was considered as implemented in the Gaussian code (Frisch et al., 2009). Imaginary frequencies were checked in all calculations to ensure that the resulting structures were not transition states. The computed frequencies were scaled by a factor of 0.98 to estimate the observed frequencies. Here, the total IR spectrum is computed as a weighted Boltzmann sum of the IR spectrum of each isomer in the distribution at a finite temperature (Buelna-Garcia et al., 2021; Buelna-Garcia et al., 2021; Lecoultre et al., 2011; Sieber et al., 2004). The spectrum is calculated using Eq. 7 and employing the occurrence probabilities displayed in Figure 2. We know of a few theoretical studies on the computation of IR spectra of metal clusters as weighted sums of the IR/UV spectra of the isomers (Sieber et al., 2004). The weighted Boltzmann IR spectra of Cu38 clusters at various temperatures are shown in Figure 4. The transition metal clusters are quite stable and their vibrational frequencies are found to be below 400 cm−1 (Lapoutre et al., 2013). This is in good agreement with our computed spectra displayed in Figure 4. In particular, the IR spectrum at a low temperature is displayed in Figure 4A. There are three dominant peaks at 125, 225, and 250 cm−1. The vibrational mode located at 125 cm−1 is a breathing mode that moves the atoms at the surface, whereas the mode at 250 cm−1 is a breathing mode where the core atoms move. The IR spectra in Figures 4A,B are similar in the 0–600 K temperature range because the relative populations in this temperature range are dominated strongly by TO structures with C1 and D4h structures, as shown in Figure 2B. The IR spectra starts to become small at 700 K. Large changes in the IR spectra happen at a temperature of approximately 800 K, where the solid-solid transition point is located, and where the amorphous structure depicted in Figure 2D and the TO structure depicted in Figure 2A coexist and contribute similarly to the overall IR Boltzmann weighted spectra. Figure 4C shows the IR Boltzmann weighted spectra at 800–1200 K. For temperatures up to 1300 K, the IR Boltzmann weighted spectra are displayed in Figure 4D. At 1200 K, the IR Boltzmann weighted spectra are similar to the spectrum of the amorphous structure depicted in Figure 2D. In contrast, the IR Boltzmann weighted spectra displayed in Figure 4A are similar to the individual IR spectrum of a TO structure. In general, the effect of temperature on the IR spectra is to extenuate the IR spectrum as the temperature increases.
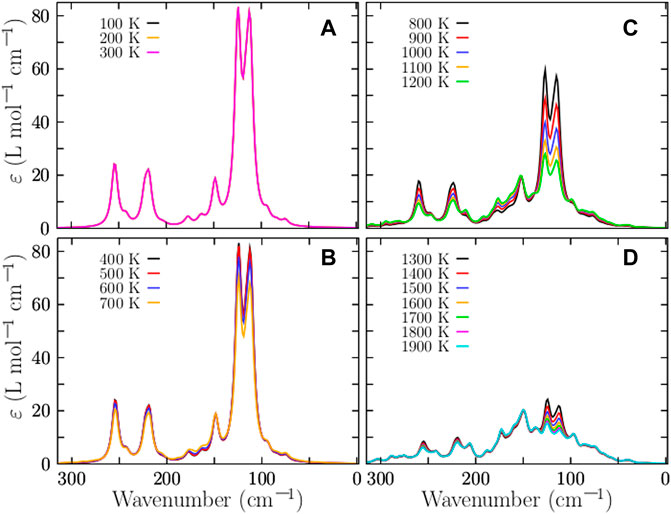
FIGURE 4. (Color online) Temperature-dependent IR Boltzmann -spectra -weighted at room temperature of the neutral Cu38 cluster are shown in panels (A)–(D) for various temperatures. The computed IR spectrum of each isomer is multiplied by its corresponding Boltzmann weight at finite temperature; Then, they are summed together to produce a final Boltzmann-weighted IR spectrum. Each spectrum of each isomer is computed using density functional theory as implemented in Gaussian code at the B3PW91-D3/def2-TZVP level of theory. The large change in the IR spectra occurs at a temperature of 824 K, as we can see in panel (D), and this agrees with the relative occurrence displayed in Figure 2B. The bulk melting temperature of copper is 1358 K (Grigoryan and Springborg, 2019), considering this, our result below of this temperature are well-behaved.
Conclusion
The temperature and entropic effects produce several competing structures because energy separation between isomers on the free energy surface is small and changes the dominant structure. Thus, it is likely that various isomers interconvert at finite temperature. Our findings show that the amorphous structure with C1 symmetry is quite dominant at hot temperatures. These energetically competing structures provide various portions of the overall IR spectrum. In contrast, higher-energy structures with significant energy separation between isomers on the potential/free energy surface do not contribute to the overall IR spectrum. The main contribution to the molecular properties comes from low-energy structures that are close to the global minimum, where the temperature-dependent Boltzmann factor weights are not equal to zero. The Boltzmann-weights depend strongly on the energy separation; the IR spectrum is constant if the energy separation is significant. One motif is dominant in cold conditions and the other in hot conditions. In addition, the level of theory and dispersion influences the location of the TSS point in the temperature scale. Our computations at the six levels of theory clearly show (relative population) that low-symmetry isomers become more stable at high temperatures due to the entropic effect on a Boltzmann ensemble at thermal equilibrium. Our unbiased global search of the free energy surface shows that there is an amorphous structure that dominates at high temperatures. As far as we know, this is a novel high-temperature structure putative global minimum. Computations of the relative populations at a high level of theory is recommended as immediate future work.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
CB-G, worked on the methodology, software, and validation; performed calculations; and drafted and wrote the manuscript. CC-Q performed calculations and analysis; provided resources; and drafted and wrote the manuscript. JQ-C worked on the methodology, validation; performed calculations; and drafted the manuscript; EP-S worked on the methodology, software, and validation; performed calculations; and drafted and wrote the manuscript; MC-V worked on the methodology, software, and validation; performed calculations and drafted the manuscript; MM-d-C-S performed calculations and drafted the manuscript; TL-L worked on the methodology, software, and validation; performed calculations and drafted the manuscript; MU-V worked on the methodology, software, and validation; performed calculations and drafted the manuscript; AM-W worked on the methodology, software, and validation; performed calculations and drafted the manuscript; PR-K worked on the methodology, software, and validation; performed calculations and drafted the manuscript; AV-E performed software development, design, and validation; performed calculations and analysis; drafted the manuscript; performed data analyses and investigations; wrote the manuscript; SP performed software development, design, and validation; performed calculations and analysis; drafted the manuscript; AdL-F performed software development, design, and validation; performed calculations and analysis; drafted the manuscript; JM-M performed software development, design, and validation; performed calculations and analysis; drafted the manuscript; AR-D performed calculations and analysis, and drafted the manuscript; GM-G performed calculations and analysis, and drafted the manuscript; JC conceived the study; performed software development, design, and validation; performed calculations and analysis; drafted the manuscript, performed data analyses and investigations; provided resources; and revised and wrote the manuscript. All authors have read and agreed to the submitted version of the manuscript.
Funding
CEB-G thanks Conacyt for the scholarship that they provided (860052). EP-S thanks Conacyt for the scholarship that they provided (1008864).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Dra. Carmen Heras and L.C.C. Daniel Mendoza for granting us access to their clusters and computational support. Computational resources for this work were provided partially by the high-performance computing Area of the University of Sonora. We are also grateful to the Computational and Experimental Research Group for Materials and Energy at UPTap for providing access to the high-performance supercomputer LYNX of the Polytechnic University of Tapachula (UPTap), including access to the ELBAKYAN and PAKAL supercomputers. Powered@NLHPC: this research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.841964/full#supplementary-material
References
Alexandrova, A. N., Boldyrev, A. I., Fu, Y.-J., Yang, X., Wang, X.-B., and Wang, L.-S. (2004). Structure of the NaxClx+1− (x=1-4) Clusters Viaab Initiogenetic Algorithm and Photoelectron Spectroscopy. J. Chem. Phys. 121, 5709–5719. doi:10.1063/1.1783276
Baletto, F., and Ferrando, R. (2005). Structural Properties of Nanoclusters: Energetic, Thermodynamic, and Kinetic Effects. Rev. Mod. Phys. 77, 371–423. doi:10.1103/RevModPhys.77.371
Baletto, F., Rapallo, A., Rossi, G., and Ferrando, R. (2004). Dynamical Effects in the Formation of Magic Cluster Structures. Phys. Rev. B 69, 235421. doi:10.1103/PhysRevB.69.235421
Becke, A. D. (1988). Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Bhattacharya, S., Berger, D., Reuter, K., Ghiringhelli, L. M., and Levchenko, S. V. (2017). Theoretical Evidence for Unexpected O-Rich Phases at Corners of MgO Surfaces. Phys. Rev. Mater. 1, 071601. doi:10.1103/PhysRevMaterials.1.071601
Bhumla, P., Kumar, M., and Bhattacharya, S. (2021). Theoretical Insights into C-H Bond Activation of Methane by Transition Metal Clusters: the Role of Anharmonic Effects. Nanoscale Adv. 3, 575–583. doi:10.1039/D0NA00669F
Brandhorst, M., Cristol, S., Capron, M., Dujardin, C., Vezin, H., Le bourdon, G., et al. (2006). Catalytic Oxidation of Methanol on Mo/Al2O3 Catalyst: An EPR and Raman/infrared Operando Spectroscopies Study. Catal. Today 113, 34–39. doi:10.1016/j.cattod.2005.11.008Recent Advances in In situ and Operando Studies of Catalytic Reactions
Brehm, G., Reiher, M., Le Guennic, B., Leibold, M., Schindler, S., Heinemann, F. W., et al. (2006). Investigation of the Low-Spin to High-Spin Transition in a Novel [Fe(pmea)(NCS)2] Complex by IR and Raman Spectroscopy and DFT Calculations. J. Raman Spectrosc. 37, 108–122. doi:10.1002/jrs.1437
Buelna-Garcia, C. E., Cabellos, J. L., Quiroz-Castillo, J. M., Martinez-Guajardo, G., Castillo-Quevedo, C., de Leon-Flores, A., et al. (2021). Exploration of Free Energy Surface and thermal Effects on Relative Population and Infrared Spectrum of the Be6B11− Fluxional Cluster. Materials 14. doi:10.3390/ma14010112
Buelna-García, C. E., Robles-Chaparro, E., Parra-Arellano, T., Quiroz-Castillo, J. M., del-Castillo-Castro, T., Martínez-Guajardo, G., et al. (2021). Theoretical Prediction of Structures, Vibrational Circular Dichroism, and Infrared Spectra of Chiral Be4B8 Cluster at Different Temperatures. Molecules 26, 3953. doi:10.3390/molecules26133953
Calvo, F. (2015). Thermodynamics of Nanoalloys. Phys. Chem. Chem. Phys. 17, 27922–27939. doi:10.1039/C5CP00274E
Castillo-Quevedo, C., Buelna-Garcia, C. E., Paredes-Sotelo, E., Robles-Chaparro, E., Zamora-Gonzalez, E., Martin-del-Campo-Solis, M. F., et al. (2021). Effects of Temperature on Enantiomerization Energy and Distribution of Isomers in the Chiral Cu13 Cluster. Molecules 26, 5710. doi:10.3390/molecules26185710
Cleri, F., and Rosato, V. (1993). Tight-binding Potentials for Transition Metals and Alloys. Phys. Rev. B 48, 22–33. doi:10.1103/PhysRevB.48.22
Cui, Z.-h., Ding, Y.-h., Cabellos, J. L., Osorio, E., Islas, R., Restrepo, A., et al. (2015). Planar Tetracoordinate Carbons with a Double Bond in CAl3E Clusters. Phys. Chem. Chem. Phys. 17, 8769–8775. doi:10.1039/C4CP05707D
Cui, Z.-h., Vassilev-Galindo, V., Luis Cabellos, J., Osorio, E., Orozco, M., Pan, S., et al. (2017). Planar Pentacoordinate Carbon Atoms Embedded in a Metallocene Framework. Chem. Commun. 53, 138–141. doi:10.1039/C6CC08273D
Darby, S., Mortimer-Jones, T. V., Johnston, R. L., and Roberts, C. (2002). Theoretical Study of Cu-Au Nanoalloy Clusters Using a Genetic Algorithm. J. Chem. Phys. 116, 1536–1550. doi:10.1063/1.1429658
de Heer, W. A. (1993). The Physics of Simple Metal Clusters: Experimental Aspects and Simple Models. Rev. Mod. Phys. 65, 611–676. doi:10.1103/RevModPhys.65.611
de la Puente, E., Aguado, A., Ayuela, A., and López, J. M. (1997). Structural and Electronic Properties of Small Neutral (MgO)n Clusers. Phys. Rev. B 56, 7607–7614. doi:10.1103/PhysRevB.56.7607
del Campo, J. M., Gázquez, J. L., Trickey, S. B., and Vela, A. (2012). Non-empirical Improvement of Pbe and its Hybrid PBE0 for General Description of Molecular Properties. J. Chem. Phys. 136, 104108. doi:10.1063/1.3691197
Dong, X., Jalife, S., Vásquez‐Espinal, A., Ravell, E., Pan, S., Cabellos, J. L., et al. (2018). Li2B12 and Li3B12: Prediction of the Smallest Tubular and Cage‐like Boron Structures. Angew. Chem. Int. Ed. 57, 4627–4631. doi:10.1002/anie.201800976
Doye, J. P. K., and Wales, D. J. (1998). Global Minima for Transition Metal Clusters Described by Sutton-Chen Potentials. New J. Chem. 22, 733–744. doi:10.1039/A709249K
Dunning, T. H., and Hay, P. J. (1977). Gaussian Basis Sets for Molecular Calculations. In Methods of Electronic Structure Theory, ed. H. F. Schaefer- (Plenum, New York: Springer US), chap. 1. 1-27. doi:10.1007/978-1-4757-0887-5_1
Dzib, E., Cabellos, J. L., Ortíz‐Chi, F., Pan, S., Galano, A., and Merino, G. (2019). Eyringpy : A Program for Computing Rate Constants in the Gas Phase and in Solution. Int. J. Quan. Chem 119, e25686. doi:10.1002/qua.25686
Erkoç, S. (1994). An Empirical many-body Potential Energy Function Constructed from Pair-Interactions. Z. Phys. D - Atoms, Mol. Clusters 32, 257–260. doi:10.1007/BF01437156
Erkoç, Ş., and Shaltaf, R. (1999). Monte Carlo Computer Simulation of Copper Clusters. Phys. Rev. A. 60, 3053–3057. doi:10.1103/PhysRevA.60.3053
Even, U., Ben-Horin, N., and Jortner, J. (1989). Multistate Isomerization of Size-Selected Clusters. Phys. Rev. Lett. 62, 140–143. doi:10.1103/PhysRevLett.62.140
Fernández, E., Boronat, M., and Corma, A. (2015). Trends in the Reactivity of Molecular O2 with Copper Clusters: Influence of Size and Shape. J. Phys. Chem. C 119, 19832–19846. doi:10.1021/acs.jpcc.5b05023
Ferrando, R., Jellinek, J., and Johnston, R. L. (2008). Nanoalloys: From Theory to Applications of alloy Clusters and Nanoparticles. Chem. Rev. 108, 845–910. doi:10.1021/cr040090g
Fielicke, A., von Helden, G., and Meijer, G. (2005). Far-infrared Spectroscopy of Isolated Transition Metal Clusters. Eur. Phys. J. D 34, 83–88. doi:10.1140/epjd/e2005-00124-7
Flórez, E., Acelas, N., Ibargüen, C., Mondal, S., Cabellos, J. L., Merino, G., et al. (2016). Microsolvation of NO3−: Structural Exploration and Bonding Analysis. RSC Adv. 6, 71913–71923. doi:10.1039/C6RA15059D
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09, Revision B.01. Wallingford: Gaussian, Inc.,.
Fujima, N., and Yamaguchi, T. (1989). Magnetic Anomaly and Shell Structure of Electronic States of Nickel Microclusters. J. Phys. Soc. Jpn. 58, 3290–3297. doi:10.1143/JPSJ.58.3290
Gázquez, J. L., Franco‐Pérez, M., Ayers, P. W., and Vela, A. (2019). Temperature‐dependent Approach to Chemical Reactivity Concepts in Density Functional Theory. Int. J. Quan. Chem 119, e25797. doi:10.1002/qua.25797
Goldsmith, B. R., Florian, J., Liu, J.-X., Gruene, P., Lyon, J. T., Rayner, D. M., et al. (2019). Two-to-three dimensional transition in neutral gold clusters: The crucial role of van der Waals interactions and temperature. Phys. Rev. Mater. 3, 016002. doi:10.1103/PhysRevMaterials.3.016002
Gonis, A., and Däne, M. (2018). Extension of the Kohn-Sham Formulation of Density Functional Theory to Finite Temperature. J. Phys. Chem. Sol. 116, 86–99. doi:10.1016/j.jpcs.2017.12.021
Grande-Aztatzi, R., Martínez-Alanis, P. R., Cabellos, J. L., Osorio, E., Martínez, A., and Merino, G. (2014). Structural Evolution of Small Gold Clusters Doped by One and Two boron Atoms. J. Comput. Chem. 35, 2288–2296. doi:10.1002/jcc.23748
Granville, V., Krivanek, M., and Rasson, J.-P. (1994). Simulated Annealing: a Proof of Convergence. IEEE Trans. Pattern Anal. Machine Intell. 16, 652–656. doi:10.1109/34.295910
Grigoryan, V. G., Alamanova, D., and Springborg, M. (2005). Structure and Energetics of Nickel, Copper, and Gold Clusters. Eur. Phys. J. D 34, 187–190. doi:10.1140/epjd/e2005-00141-6
Grigoryan, V. G., Alamanova, D., and Springborg, M. (2006). Structure and Energetics of CuN clusters with (2⩽N⩽150): An Embedded-Atom-Method Study. Phys. Rev. B 73, 115415. doi:10.1103/PhysRevB.73.115415
Grigoryan, V. G., and Springborg, M. (2019). Temperature and Isomeric Effects in Nanoclusters. Phys. Chem. Chem. Phys. 21, 5646–5654. doi:10.1039/C9CP00123A
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Grimme, S. (2012). Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. Eur. J. 18, 9955–9964. doi:10.1002/chem.201200497
Guo, J.-C., Feng, L.-Y., Wang, Y.-J., Jalife, S., Vásquez-Espinal, A., Cabellos, J. L., et al. (2017). Coaxial Triple-Layered versus Helical Be6B11− Clusters: Dual Structural Fluxionality and Multifold Aromaticity. Angew. Chem. Int. Ed. 56, 10174–10177. doi:10.1002/anie.201703979
Guvelioglu, G. H., Ma, P., He, X., Forrey, R. C., and Cheng, H. (2006). First Principles Studies on the Growth of Small Cu Clusters and the Dissociative Chemisorption ofH2. Phys. Rev. B 73, 155436. doi:10.1103/PhysRevB.73.155436
Hadad, C. Z., Florez, E., Merino, G., Cabellos, J. L., Ferraro, F., and Restrepo, A. (2014). Potential Energy Surfaces of WC6 Clusters in Different Spin States. J. Phys. Chem. A. 118, 5762–5768. doi:10.1021/jp4099045
Hashimoto, K., Badarla, V. R., Kawai, A., and Ideguchi, T. (2019). Complementary Vibrational Spectroscopy. Nat. Commun. 10, 4411. doi:10.1038/s41467-019-12442-9
Hijazi, I. A., and Park, Y. H. (2010). Structure of Pure Metallic Nanoclusters: Monte Carlo Simulation and Ab Initio Study. Eur. Phys. J. D 59, 215–221. doi:10.1140/epjd/e2010-00133-5
Hill, T. (1986). An Introduction to Statistical Thermodynamics. Addison-Wesley series in chemistry. Dover Publications.
Hill, T. L. (1962). Thermodynamics of Small Systems. J. Chem. Phys. 36, 3182–3197. doi:10.1063/1.1732447
Inwati, G. K., Rao, Y., and Singh, M. (2018). Thermodynamically Induced In Situ and Tunable Cu Plasmonic Behaviour. Sci. Rep. 8, 3006. doi:10.1038/s41598-018-20478-y
Itoh, M., Kumar, V., Adschiri, T., and Kawazoe, Y. (2009). Comprehensive Study of Sodium, Copper, and Silver Clusters over a Wide Range of Sizes 2≤N≤75. J. Chem. Phys. 131, 174510. doi:10.1063/1.3187934
Jena, P., and Castleman, A. W. (2006). Clusters: A Bridge across the Disciplines of Physics and Chemistry. Proc. Natl. Acad. Sci. 103, 10560–10569. doi:10.1073/pnas.0601782103
Kabir, M., Mookerjee, A., and Bhattacharya, A. K. (2004). Structure and Stability of Copper Clusters: A Tight-Binding Molecular Dynamics Study. Phys. Rev. A. 69, 043203. doi:10.1103/PhysRevA.69.043203
Kirkpatrick, S., Gelatt, C. D., and Vecchi, M. P. (1983). Optimization by Simulated Annealing. Science 220, 671–680. doi:10.1126/science.220.4598.671
Kostko, O., Morgner, N., Astruc Hoffmann, M., and von Issendorff, B. (2005). Photoelectron Spectra of Nan- and Cun- with N = 20-40: Observation of Surprising Similarities. Eur. Phys. J. D 34, 133–137. doi:10.1140/epjd/e2005-00099-3
Lapoutre, V. J. F., Haertelt, M., Meijer, G., Fielicke, A., and Bakker, J. M. (2013). Communication: Ir Spectroscopy of Neutral Transition Metal Clusters through Thermionic Emission. J. Chem. Phys. 139, 121101. doi:10.1063/1.4822324
Lecoultre, S., Rydlo, A., Félix, C., Buttet, J., Gilb, S., and Harbich, W. (2011). Optical Absorption of Small Copper Clusters in Neon: Cun, (N = 1-9). J. Chem. Phys. 134, 074303. doi:10.1063/1.3552077
Legge, F. S., Nyberg, G. L., and Peel, J. B. (2001). DFT Calculations for Cu-, Ag-, and Au-Containing Molecules. J. Phys. Chem. A. 105, 7905–7916. doi:10.1021/jp0101918
Li, Z. H., Jasper, A. W., and Truhlar, D. G. (2007). Structures, Rugged Energetic Landscapes, and Nanothermodynamics of Aln (2 ≤ N ≤ 65) Particles. J. Am. Chem. Soc. 129, 14899–14910. doi:10.1021/ja073129i
Li, Z. H., and Truhlar, D. G. (2014). Nanothermodynamics of Metal Nanoparticles. Chem. Sci. 5, 2605–2624. doi:10.1039/C4SC00052H
Liu, P., Wang, H., Li, X., Rui, M., and Zeng, H. (2015). Localized Surface Plasmon Resonance of Cu Nanoparticles by Laser Ablation in Liquid media. RSC Adv. 5, 79738–79745. doi:10.1039/C5RA14933A
Lushchikova, O. V., Huitema, D. M. M., López-Tarifa, P., Visscher, L., Jamshidi, Z., and Bakker, J. M. (2019). Structures of Cun+ (N = 3-10) Clusters Obtained by Infrared Action Spectroscopy. J. Phys. Chem. Lett. 10, 2151–2155. doi:10.1021/acs.jpclett.9b00539
Martínez-Guajardo, G., Luis Cabellos, J., Díaz-Celaya, A., Pan, S., Islas, R., Chattaraj, P. K., et al. (2015). Dynamical Behavior of Borospherene: A Nanobubble. Sci. Rep. 5, 11287–11297. doi:10.1038/srep11287
Mathew, A., and Pradeep, T. (2014). Noble Metal Clusters: Applications in Energy, Environment, and Biology. Part. Part. Syst. Charact. 31, 1017–1053. doi:10.1002/ppsc.201400033
Mendoza-Wilson, A. M., Balandrán-Quintana, R. R., and Cabellos, J. L. (2020). Thermochemical Behavior of Sorghum Procyanidin Trimers with C4-C8 and C4-C6 Interflavan Bonds in the Reaction with Superoxide Anion Radical and H2O2-Forming NADH-Oxidase Flavoenzyme. Comput. Theor. Chem. 1186, 112912. doi:10.1016/j.comptc.2020.112912
Mendoza-Wilson, A. M., Balandrán-Quintana, R. R., Valdés-Covarrubias, M. Á., and Cabellos, J. L. (2022). Potential of Quercetin in Combination with Antioxidants of Different Polarity Incorporated in Oil-In-Water Nanoemulsions to Control Enzymatic browning of Apples. J. Mol. Struct. 1254, 132372. doi:10.1016/j.molstruc.2022.132372
Mermin, N. D. (1965). Thermal Properties of the Inhomogeneous Electron Gas. Phys. Rev. 137, A1441–A1443. doi:10.1103/PhysRev.137.A1441
Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H., and Teller, E. (1953). Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 21, 1087–1092. doi:10.1063/1.1699114
Mondal, S., Cabellos, J. L., Pan, S., Osorio, E., Torres-Vega, J. J., Tiznado, W., et al. (2016). 10-π-Electron arenes à la carte: structure and bonding of the [E-(CnHn)-E]n−6 (E = Ca, Sr, Ba; n = 6-8) complexes. Phys. Chem. Chem. Phys. 18, 11909–11918. doi:10.1039/C6CP00671J
Niu, S., Huang, D.-L., Dau, P. D., Liu, H.-T., Wang, L.-S., and Ichiye, T. (2014). Assessment of Quantum Mechanical Methods for Copper and Iron Complexes by Photoelectron Spectroscopy. J. Chem. Theor. Comput. 10, 1283–1291. doi:10.1021/ct400842p
Núñez, S., and Johnston, R. L. (2010). Structures and Chemical Ordering of Small Cu−Ag Clusters. J. Phys. Chem. C 114, 13255–13266. doi:10.1021/jp1048088
Ohno, K., and Maeda, S. (2006). Global Reaction Route Mapping on Potential Energy Surfaces of Formaldehyde, Formic Acid, and Their Metal-Substituted Analogues. J. Phys. Chem. A. 110, 8933–8941. doi:10.1021/jp061149l
Opik, U., and Pryce, M. H. L. (1957). Studies of the Jahn-Teller Effect. I. A Survey of the Static Problem. Proc. R. Soc. Lond. A. 238, 425–447. doi:10.1098/rspa.1957.0010
Pan, S., Moreno, D., Cabellos, J. L., Romero, J., Reyes, A., Merino, G., et al. (2014). In Quest of strong Be-Ng Bonds Among the Neutral Ng-Be Complexes. J. Phys. Chem. A. 118, 487–494. doi:10.1021/jp409941v
Park, Y. H., and Hijazi, I. A. (2012). Critical Size of Transitional Copper Clusters for Ground State Structure Determination: Empirical Andab Initiostudy. Mol. Simulation 38, 241–247. doi:10.1080/08927022.2011.616502
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Perdew, J. P., Chevary, J. A., Vosko, S. H., Jackson, K. A., Pederson, M. R., Singh, D. J., et al. (1992). Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Phys. Rev. B 46, 6671–6687. doi:10.1103/PhysRevB.46.6671
Perdew, J. P., and Wang, Y. (1992). Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 45, 13244–13249. doi:10.1103/PhysRevB.45.13244
Pettiette, C. L., Yang, S. H., Craycraft, M. J., Conceicao, J., Laaksonen, R. T., Cheshnovsky, O., et al. (1988). Ultraviolet Photoelectron Spectroscopy of Copper Clusters. J. Chem. Phys. 88, 5377–5382. doi:10.1063/1.454575
Pickard, C. J., and Needs, R. J. (2011). Ab Initiorandom Structure Searching. J. Phys. Condens. Matter 23, 053201. doi:10.1088/0953-8984/23/5/053201
Pittalis, S., Proetto, C. R., Floris, A., Sanna, A., Bersier, C., Burke, K., et al. (2011). Exact Conditions in Finite-Temperature Density-Functional Theory. Phys. Rev. Lett. 107, 163001. doi:10.1103/physrevlett.107.163001
Powers, D. E., Hansen, S. G., Geusic, M. E., Puiu, A. C., Hopkins, J. B., Dietz, T. G., et al. (1982). Supersonic Metal Cluster Beams: Laser Photoionization Studies of Copper Cluster (Cu2). J. Phys. Chem. 86, 2556–2560. doi:10.1021/j100211a002
Qi, D., Luo, X., Yao, J., Lu, X., and Zhang, Z. (2020). Computational Study of Reverse Water Gas Shift Reaction on Cu38 Cluster Model and Cu Slab Model. J. Theor. Comput. Chem. 19, 2050008. doi:10.1142/S021963362050008X
Ravell, E., Jalife, S., Barroso, J., Orozco-Ic, M., Hernández-Juárez, G., Ortiz-Chi, F., et al. (2018). Structure and Bonding in CE5 − (E=Al-Tl) Clusters: Planar Tetracoordinate Carbon versus Pentacoordinate Carbon. Chem. Asian J. 13, 1467–1473. doi:10.1002/asia.201800261
Redel’, L. V., Gafner, Y. Y., and Gafner, S. L. (2015). Role of "magic" Numbers in Structure Formation in Small Silver Nanoclusters. Phys. Solid State. 57, 2117–2125. doi:10.1134/S106378341510025X
Rodríguez-Kessler, P. L., Pan, S., Florez, E., Cabellos, J. L., and Merino, G. (2017). Structural Evolution of the Rhodium-Doped Silver Clusters AgnRh (N ≤ 15) and Their Reactivity toward NO. J. Phys. Chem. C 121, 19420–19427. doi:10.1021/acs.jpcc.7b05048
Saunders, M. (1987). Stochastic Exploration of Molecular Mechanics Energy Surfaces. Hunting for the Global Minimum. J. Am. Chem. Soc. 109, 3150–3152. doi:10.1021/ja00244a051
Saunders, M. (2004). Stochastic Search for Isomers on a Quantum Mechanical Surface. J. Comput. Chem. 25, 621–626. doi:10.1002/jcc.10407
Schebarchov, D., Baletto, F., and Wales, D. J. (2018). Structure, Thermodynamics, and Rearrangement Mechanisms in Gold Clusters-Insights from the Energy Landscapes Framework. Nanoscale 10, 2004–2016. doi:10.1039/C7NR07123J
Shortle, D. (2003). Propensities, Probabilities, and the Boltzmann Hypothesis. Protein Sci. 12, 1298–1302. doi:10.1110/ps.0306903
Sieber, C., Buttet, J., Harbich, W., Félix, C., Mitrić, R., and Bonači ć Koutecký, V. (2004). Isomer-specific Spectroscopy of Metal Clusters Trapped in a Matrix: Ag9. Phys. Rev. A. 70, 041201. doi:10.1103/physreva.70.041201
Sutton, C., and Levchenko, S. V. (2020). First-principles Atomistic Thermodynamics and Configurational Entropy. Front. Chem. 8, 757. doi:10.3389/fchem.2020.00757
Takada, A., Conradt, R., and Richet, P. (2018). Partition Function and Configurational Entropy in Non-equilibrium States: A New Theoretical Model. Entropy 20, 218. doi:10.3390/e20040218
Takagi, N., Ishimura, K., Matsui, M., Fukuda, R., Ehara, M., and Sakaki, S. (2017). Core-Shell versus Other Structures in Binary Cu38-nMn Nanoclusters (M = Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au; N = 1, 2, and 6): Theoretical Insight into Determining Factors. J. Phys. Chem. C 121, 10514–10528. doi:10.1021/acs.jpcc.6b13086
Taylor, C. D., Neurock, M., and Scully, J. R. (2008). First-principles Investigation of the Fundamental Corrosion Properties of a Model Cu[sub 38] Nanoparticle and the (111), (113) Surfaces. J. Electrochem. Soc. 155, C407. doi:10.1149/1.2926598
Tinnemans, S. J., Mesu, J. G., Kervinen, K., Visser, T., Nijhuis, T. A., Beale, A. M., et al. (2006). Combining Operando Techniques in One Spectroscopic-Reaction Cell: New Opportunities for Elucidating the Active Site and Related Reaction Mechanism in Catalysis. Catal. Today 113, 3–15. doi:10.1016/j.cattod.2005.11.076Recent Advances in In situ and Operando Studies of Catalytic Reactions
Tran, D. T., and Johnston, R. L. (2009). Theoretical Study of Cu38−nAun Clusters Using a Combined Empirical Potential-Density Functional Approach. Phys. Chem. Chem. Phys. 11, 10340–10349. doi:10.1039/b912501a
Vargas-Caamal, A., Cabellos, J. L., Ortiz-Chi, F., Rzepa, H. S., Restrepo, A., and Merino, G. (2016a). How many Water Molecules Does it Take to Dissociate HCl? Chem. Eur. J. 22, 2812–2818. doi:10.1002/chem.201504016
Vargas-Caamal, A., Ortiz-Chi, F., Moreno, D., Restrepo, A., Merino, G., and Cabellos, J. L. (2015). The Rich and Complex Potential Energy Surface of the Ethanol Dimer. Theor. Chem. Acc. 134, 16. doi:10.1007/s00214-015-1615-9
Vargas-Caamal, A., Pan, S., Ortiz-Chi, F., Cabellos, J. L., Boto, R. A., Contreras-Garcia, J., et al. (2016b). How strong Are the Metallocene-Metallocene Interactions? Cases of Ferrocene, Ruthenocene, and Osmocene. Phys. Chem. Chem. Phys. 18, 550–556. doi:10.1039/C5CP05956A
Vlachos, D. G., Schmidt, L. D., and Aris, R. (1993). Comparison of Small Metal Clusters: Ni, Pd, Pt, Cu, Ag, Au. Z. Phys. D - Atoms, Mol. Clusters 26, 156–158. doi:10.1007/BF01425649
Wales, D. J. (1996). Structure, Dynamics, and Thermodynamics of Clusters: Tales from Topographic Potential Surfaces. Science 271, 925–929. doi:10.1126/science.271.5251.925
Weigend, F., and Ahlrichs, R. (2005). Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/B508541A
Wilcoxon, J. P., and Abrams, B. L. (2006). Synthesis, Structure and Properties of Metal Nanoclusters. Chem. Soc. Rev. 35, 1162–1194. doi:10.1039/B517312B
Wilson, N. T., and Johnston, R. L. (2002). A Theoretical Study of Atom Ordering in Copper-Gold Nanoalloy Clusters. J. Mater. Chem. 12, 2913–2922. doi:10.1039/B204069G
Xavier, P. L., Chaudhari, K., Baksi, A., and Pradeep, T. (2012). Protein-protected Luminescent noble Metal Quantum Clusters: an Emerging Trend in Atomic Cluster Nanoscience. Nano Rev. 3, 14767. doi:10.3402/nano.v3i0.1476710.3402/nano.v3i0.14767
Xiang, Y., and Gong, X. G. (2000). Efficiency of Generalized Simulated Annealing. Phys. Rev. E 62, 4473–4476. doi:10.1103/PhysRevE.62.4473
Xiang, Y., Gubian, S., Suomela, B., and Hoeng, J. (2013). Generalized Simulated Annealing for Global Optimization: The GenSA Package. R. J. 5, 13–29. doi:10.32614/rj-2013-002
Yanai, T., Tew, D. P., and Handy, N. C. (2004). A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3lyp). Chem. Phys. Lett. 393, 51–57. doi:10.1016/j.cplett.2004.06.011
Zhang, C., Duan, H., Lv, X., Cao, B., Abliz, A., Wu, Z., et al. (2019). Static and Dynamical Isomerization of Cu38 Cluster. Sci. Rep. 9, 7564. doi:10.1038/s41598-019-44055-z
Zhang, J., Wang, C.-Z., Zhu, Z., Liu, Q. H., and Ho, K.-M. (2018). Multimode Jahn-Teller Effect in Bulk Systems: A Case of the NV0 center in diamond. Phys. Rev. B 97, 165204. doi:10.1103/physrevb.97.165204
Zhao, B., Zhang, R., Huang, Z., and Wang, B. (2017). Effect of the Size of Cu Clusters on Selectivity and Activity of Acetylene Selective Hydrogenation. Appl. Catal. A: Gen. 546, 111–121. doi:10.1016/j.apcata.2017.08.001
Zlatar, M., Gruden-Pavlović, M., Schläpfer, C.-W., and Daul, C. (2010). Intrinsic Distortion Path in the Analysis of the Jahn-Teller Effect. J. Mol. Struct. THEOCHEM 954, 86–93. doi:10.1016/j.theochem.2010.04.020DFT 09, 13th International Conference on the Applications of Density Functional Theory in Chemistry and Physics
Keywords: nanothermodynamics, IR, DFT, Cu-nanoclusters, genetic-algorithm, relative populations, temperature
Citation: Buelna-García CE, Castillo-Quevedo C, Quiroz-Castillo JM, Paredes-Sotelo E, Cortez-Valadez M, Martin-del-Campo-Solis MF, López-Luke T, Utrilla-Vázquez M, Mendoza-Wilson AM, Rodríguez-Kessler PL, Vazquez-Espinal A, Pan S, de Leon-Flores A, Mis-May JR, Rodríguez-Domínguez AR, Martínez-Guajardo G and Cabellos JL (2022) Relative Populations and IR Spectra of Cu38 Cluster at Finite Temperature Based on DFT and Statistical Thermodynamics Calculations. Front. Chem. 10:841964. doi: 10.3389/fchem.2022.841964
Received: 23 December 2021; Accepted: 24 January 2022;
Published: 01 March 2022.
Edited by:
Albert Poater, University of Girona, SpainReviewed by:
Jinasena Wathogala Hewage, University of Ruhuna, Sri LankaMichael Springborg, Saarland University, Germany
Copyright © 2022 Buelna-García, Castillo-Quevedo, Quiroz-Castillo, Paredes-Sotelo, Cortez-Valadez, Martin-del-Campo-Solis, López-Luke, Utrilla-Vázquez, Mendoza-Wilson, Rodríguez-Kessler, Vazquez-Espinal, Pan, de Leon-Flores, Mis-May, Rodríguez-Domínguez, Martínez-Guajardo and Cabellos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerardo Martinez-Guajardo, Z2VybXR6Z3VhamFyZG9AdWF6LmVkdS5teA==; Jose Luis Cabellos, am9zZS5jYWJlbGxvc0B1cHRhcGFjaHVsYS5lZHUubXg=