 Yan Liu
Yan Liu Peng Jiang1
Peng Jiang1 Wei Guan
Wei Guan Bing-You Yang
Bing-You Yang- 1Key Laboratory of Basic and Application Research of Beiyao (Heilongjiang University of Chinese Medicine), Ministry of Education, Harbin, China
- 2China Resources Double-Crane Pharmaceutical Co., Ltd., Peking, China
Five new oleanane-type triterpenoid saponins (1–5), together with 24 known saponins (6–29) were isolated from the fruit of Acanthopanax senticosus. Their structures were determined by extensive spectroscopic analysis, including 1D, 2D nuclear magnetic resonance (NMR), and high-resolution electrospray ionization mass spectrometry (HR-ESI-MS), in combination with chemical methods (acid hydrolysis). The neuroinflammation model was established by lipopolysaccharide (LPS)-induced BV2 microglia, and the neuroprotective effects of all compounds (1–29) were evaluated.
Introduction
Acanthopanax senticosus (Rupr. & Maxim.) Harms, commonly known as Ci Wu Jia or Siberian Ginseng, is a well-known traditional Chinese medicine widely distributed in the northeast of China. With high medicinal value, A. senticosus is popularly used as an “adaptogen” like Panax ginseng. Modern pharmacology study shows that this plant was used for antifatigue, anti-depression, anxiolytic, anti-irradiation, anticancer, anti-inflammatory, hypolipidemic, etc. (Huang et al., 2011; Li et al., 2016a), and these activities may be attributed to triterpenoid saponins. Insuperably, modern pharmacological studies have confirmed that A. senticosus fruits possess significant activities of antifatigue (Cong et al., 2010), antioxidant (Kim et al., 2015; Zhao et al., 2013), hypolipidemic (Yan et al., 2009), anti-obesity (Li et al., 2007; Saito et al., 2016), anti-inflammatory (Li et al., 2013), and so on. However, for the past few years, most of the phytochemical studies have been mainly focused on the root, stem, and leaves of A. senticosus, and limited researches have been investigated on its fruits (Ge et al., 2016; Huang et al., 2011; Li et al., 2016b; Li et al., 2017; Wang et al., 2012; Zhang et al., 2017).
In the present paper, we continue to further explore the active component triterpenoid saponins from the fruits of A. senticosus. The results found five previously undescribed triterpenoid saponins (1–5) (Figure 1), together with known 24 triterpenoid saponins (6–29). Their structures were elucidated mainly by spectroscopic methods including 1D and 2D nuclear magnetic resonance (NMR) experiments in combination with high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) and by comparison of their physical and spectral data with literature. Meanwhile, their neuroprotective effects were evaluated by lipopolysaccharide (LPS)-induced BV2 microglia.
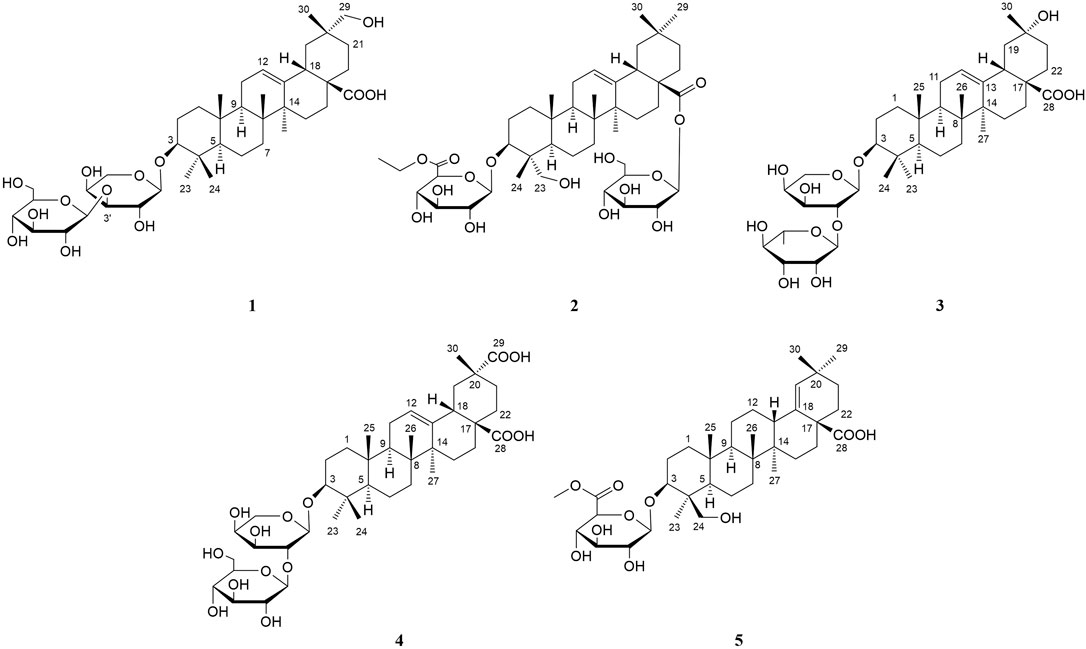
FIGURE 1. Chemical structures of compounds 1–5.
Experimental Section
General Experimental Procedures
The HR-ESI-MS data of the new triterpenoid saponins were obtained on a Thermo Orbitrap Fusion Lumos Tribrid Mass Spectrometer. The 1D and 2D NMR spectra were acquired on a Bruker DPX-600 spectrometer in Pyridine-d5 using TMS as internal standard. Preparative high-performance liquid chromatography (HPLC) (LC-20AR, Shimadzu) was performed on Waters Atlantis® Prep T3 (5 μm, 10 × 250 mm column) with a RID-20 A detector, with flow rates of 3 ml/min. Optical rotation measurements were conducted on a JASCO P-2000 instrument. Gas chromatography-mass spectrometry (GC-MS) analysis was performed on an Agilent 7890A system with a DB-5 capillary column. Absorbance (OD) value was detected on a BioTek Epoch™2 Microplate Reader. The FT-IR data of the new triterpenoid saponins was performed on Thermo Scientific Nicolet iS10. Silica gel column chromatography (CC) and octadecyl silica (ODS) chromatography were used in the separation of extracts.
Plant Material
The fruit of A. senticosus was collected in October 2018 from the Yichun, Heilongjiang Province. The plant was identified by the Professor Rui-Feng Fan of the Heilongjiang University of Chinese Medicine, and its voucher specimen (NO. 20190330) has been deposited at Heilongjiang University of Chinese Medicine.
Extraction and Isolation
The dry fruits (20 kg) of A. senticosus were extracted with 70% EtOH three times, under reflux for 2 h each time to afford a crude extract (2,216 g). The crude extract was extracted with petroleum ether, EtOAc, and n-BuOH successively, and the corresponding extract was obtained after removing the solvent, namely, PE fraction (320.0 g), EtOAc fraction (470.0 g), and n-BuOH fraction (510.0 g). The ethyl acetate layer (360.0 g) was chromatographed on a silica gel column (200–300 mesh) eluted successively with CH2Cl2/MeOH (100:1–0:1) to obtain nine fractions. Fr. VI was separated on a silica gel column (200–300 mesh) eluted successively with CH2Cl2/MeOH (50:1–0:1) to obtain five fractions (Fr. VI 1–5). Fr. VI 4 was purified by ODS chromatography to afford 48 fractions. Fr. VI 4–(46) were purified by semi-preparative HPLC (MeOH/H2O 84%) to afford compounds 14 (5.3 mg), 7 (4.9 mg), 15 (5.0 mg), 8 (37.5 mg), 18 (9.7 mg), 16 (45.9 mg), 10 (7.1 mg), and 11 (5.4 mg). Fr. VII was separated on a silica gel column (200–300 mesh), using solvent system CH2Cl2/MeOH (30:1 to 0:1) to give five fractions (Fr. VII 1–7) based on TLC analysis. Fr. VII 5 was purified by ODS chromatography to afford sixty-three fractions. Fr. VII 5–(54) were purified by semi-preparative HPLC (MeOH/H2O 78%) to afford compounds 5 (5.3 mg) and 6 (3.6 mg). Fr. VIII was separated on a silica gel column (200–300 mesh, 1 kg), using solvent system CH2Cl2/MeOH (30:1 to 0:1) to give nine fractions (Fr. VIII 1–9) based on TLC analysis. Fr. VIII 5 was purified by ODS chromatography to afford fifty fractions. Fr. VIII 5–(47) were purified by semi-preparative HPLC (MeOH/H2O 84%) to afford compounds 13 (66.4 mg) and 9 (52.8 mg). Fr. VIII 6 was purified by ODS chromatography to afford forty-two fractions. Fr. VIII 6-(24) was purified by semi-preparative HPLC (MeOH/H2O 73%) to afford compound 3 (8.8 mg). Fr. VIII 6–(25) was purified by semi-preparative HPLC (MeOH/H2O 73%) to afford compound 12 (5.3 mg). Fr. VIII 6–(27) were purified by semi-preparative HPLC (MeOH/H2O 68%) to afford compounds 1 (26.0 mg), 4 (4.2 mg), 17 (147.1 mg), and 22 (26.4 mg). Fr. VIII 6–(29) was purified by semi-preparative HPLC (MeOH/H2O 73%) to afford compound 25 (42.0 mg). Fr. VIII 6–(29D) were purified by semi-preparative HPLC (MeOH/H2O 78%) to afford compound 19 (3.6 mg). Fr. VIII 6–(30) were purified by semi-preparative HPLC (MeOH/H2O 70%) to afford compound 23 (28.1 mg). Fr. VIII 6–(30C) were purified by semi-preparative HPLC (MeOH/H2O 80%) to afford compound 2 (8.7 mg). Fr. VIII 6–(32) were purified by semi-preparative HPLC (MeOH/H2O 80%) to afford compound 26 (5.4 mg). Fr. VIII 6–(33) were purified by semi-preparative HPLC (MeOH/H2O 81%) to afford compound 24 (9.1 mg). Fr. VIII 6–(34) were purified by semi-preparative HPLC (MeOH/H2O 82%) to afford compounds 28 (8.2 mg) and 27 (7.6 mg). Fr. VIII 6–(35) were purified by semi-preparative HPLC (MeOH/H2O 84%) to afford compounds 29 (2.8 mg), 20 (7.7 mg), and 21 (23.5 mg).
Spectroscopic Data
Acasentrioid A (1): amorphous powder;
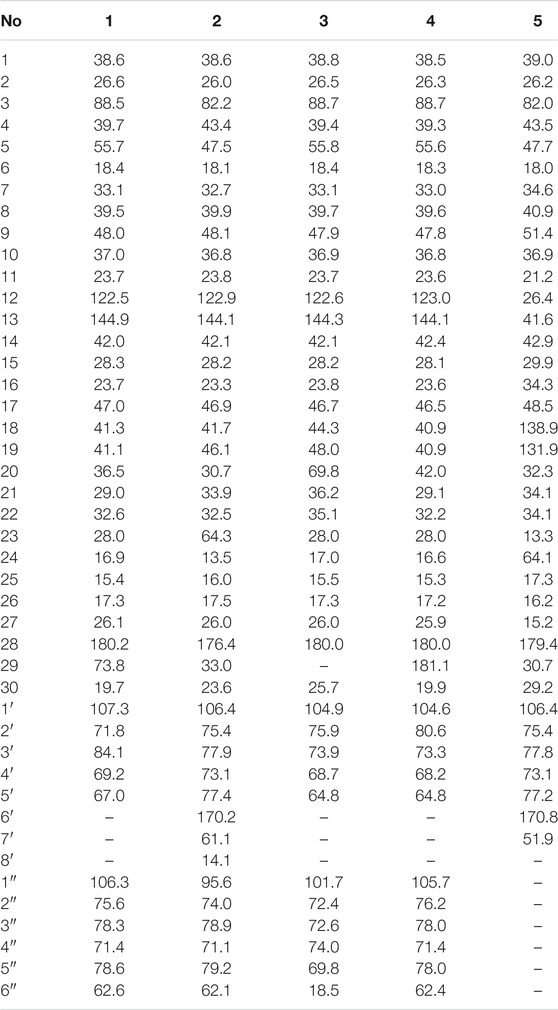
TABLE 1. 13C NMR data (δ) for compounds 1–5 (150, MHz in pyridine-d5).

TABLE 2. 1H NMR data (δ) for compounds 1–5 (600, MHz in pyridine-d5).
Acasentrioid B (2): amorphous powder;
Acasentrioid C (3): amorphous powder;
Acasentrioid D (4): amorphous powder;
Acasentrioid E (5): amorphous powder;
Hydrolysis of Compounds 1–5
Monosaccharide was determined by GC (Teng et al., 2018). Compounds 1–5 (each 1.0 mg) were dissolved in 2 ml of 2 M HCl (dioxane/H2O, 1:1, v/v), and hydrolyzed at 90°C for 3 h. After removing dioxane in a vacuum, the solution was diluted with H2O and extracted with EtOAc (3 × 1 ml). The aqueous layer was evaporated to dryness. The dried residue was dissolved in pyridine (200 μl) and treated with L-cysteine methyl ester hydrochloride (2.0 mg). After stirring the mixture for 1 h at 60°C, 100 μl of N-trimethylsilylimidazole was added, and they were kept at 60°C for 1 h. The reaction mixture was suspended in 1.0 ml H2O and extracted with n-hexane (3 × 1.0 ml). The layer of n-hexane was directly analyzed by GC with a DM-5 column (30 m × 0.25 mm, 0.25 μm) with the elution of N2 as carrier gas. Other GC conditions are as follows: column temperature: 220–270°C with the rate of 3°C/min; injector and detector temperature: 250°C; split ratio: 10:1; and injection volume:1 μl. The configurations of D-glucose, L-arabinose, D-glucuronic acid, and L-rhamnose in compounds 1–5 were determined by comparison of their retention times with those of standard samples.
Bioassay for Cytotoxicity Activities
In each well of a 96-well plate, 100 μl of logarithmic growth phase cells (density 1.5 × 105/ml) was inoculated and cultured at 37°C and 5% CO2 until the cells attached to the wall. A drug-containing medium (0, 50, 100, 200, 400, and 600 μM) was added to each well of the administration group, and an equal volume of medium was also added to the blank group, and they were incubate at 37°C and 5% CO2 for 24 h. After the culture, 10 μl of CCK-8 was added to each well and incubated for 1–4 h at 37°C and 5% CO2. The absorbance (OD) value of each well at 450 nm was detected with a microplate reader, repeating three times, and its IC50 value was calculated.
Bioassay for NO Production Inhibitory Activities
The anti-neuroinflammatory effect of compounds 1–29 was evaluated by LPS-induced BV2 microglia reported previously (Luo et al., 2020). The BV2 microglia cells were plated into a 96-well plate. After adding LPS (1 μg/ml) to each well for 12 h, it was treated with or without compounds of various concentrations (0, 100, 200, 300, 400, and 600 μM) for 12 h. The NO production in the supernatant was measured by the Griess reaction. The absorbance at 570 nm was measured using a microplate reader. The NO concentration and the inhibitory rate were calculated through a calibration curve. Quercetin was used as the positive control. Experiments were repeated three times.
Results and Discussion
Structure Elucidation of Compounds
Compound 1 was obtained as an amorphous powder. The negative HR-ESI-MS showed a deprotonated molecular ion peak at m/z 784.4845 [M + NH4]+ (calculated for C41H70NO13, 784.4842), indicating its molecular formula of C41H66O13. The 1H NMR spectrum displayed characteristic resonances of an olean-12-ene skeleton, namely, six methyls [δH 0.83, 0.96, 1.00, 1.22, 1.29, and 1.31 (3H each, all s, H-25, 24, 26, 30, 23, and 27)], one oxygenated methylene [δH 3.61 (2H, s, H-29)], one oxygenated methine [δH 3.33 (1H, dd, J = 11.5, 3.8 Hz, H-3)], one olefin [δH 5.51 (1H, br. s, H-12)], and two anomeric proton signals [δH 4.74 (1H, d, J = 7.3 Hz, H-1′) and 5.39 (1H, d, J = 7.7 Hz, H-1″)] (see Tables 1, 2). Coupled with DEPT spectrum, the 13C NMR spectrum showed the presence of 41 signals, of which 30 signals were assigned to a triterpene of oleanane skeleton, containing one carboxyl group (δC 180.2), two olefinic carbons (δC 122.5 and 144.9), two anomeric carbons (δC 107.3 and 106.3), one downfield glycosylation-shifted oxygenated methine (δC 88.5), a hydroxymethyl carbon (δC 73.8), and six methyls (δC 15.4, 16.9, 17.3, 19.7, 26.1, and 28.0) (see Tables 1, 2). These observations implied that compound 1 might be an oleanane-type triterpenoid saponin.
The HMBC cross-peaks of the anomeric proton H-1′ (δH 4.74)/C-3 showed the by ether bond location of the sugar chain at C-3. The HMBC connections of H2-22 (δH 2.16)/C-17, C-28 indicated a carboxy fragment attached at C-17 (Figure 2). Based on the above analysis, the structure of compound 1 was similar to compound 21, and the major difference was the substituent C-29 is changed from methyl to oxymethylene in compound 1, which was supported by the HMBC correlation of the anomeric protons of H2-29 (δH 3.61) with C-19, C-20, C-21, and C-30 (Figure 2). The β orientations of both pyranose sugars were deduced according to the large coupling constants of the anomeric protons (J = 7.3 Hz, H-1′; J = 7.7 Hz H-1″). To determine the absolute configuration of the arabinopyranose and glucopyranose, compound 1 was hydrolyzed by 2 mM HCl to obtain the sugar, and then, the trimethylsilyl thiazolidine derivatives of the sugar and standards, L-arabinose, and D-glucose were prepared. By comparing the retention times of these three trimethylsilyl thiazolidine derivatives obtained from GC, the absolute configuration of the arabinopyranose and glucopyranose in 1 was determined to be L and D, respectively.
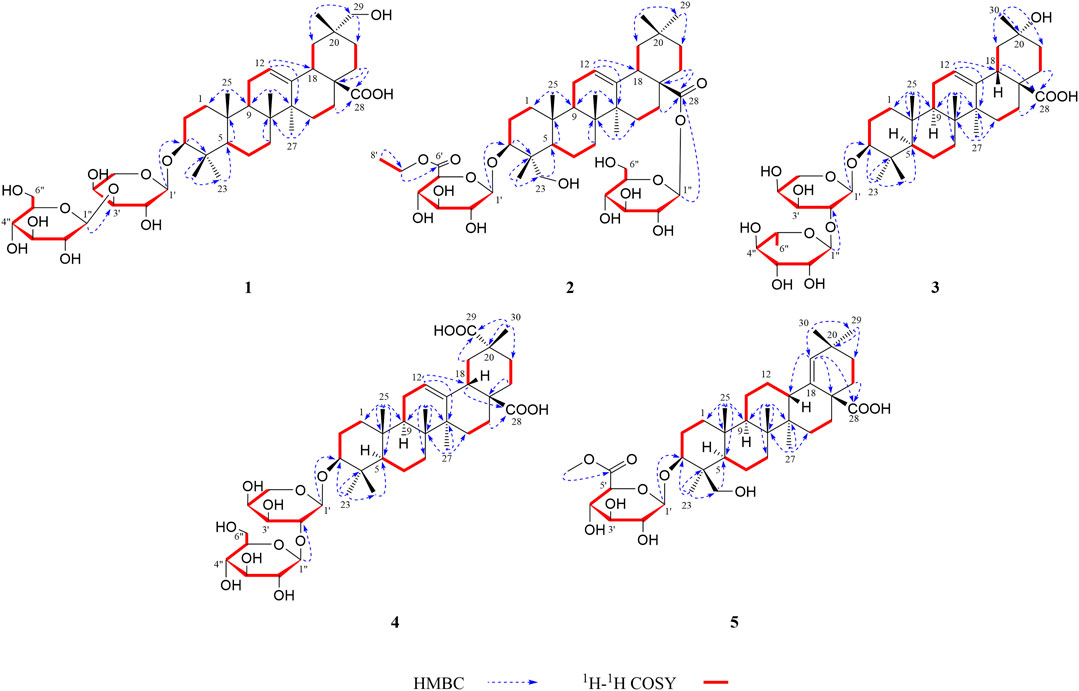
FIGURE 2. 1H–1H COSY and key HMBC correlations of compounds 1–5.
In the NOESY spectrum (Figure 3), the correlation peaks of H3-24/H3-25/H3-26/H-18/H3-30 suggested the β orientations of H3-24, H3-25, H3-26, H-18, and H3-30. Conversely, the correlation peaks of H-3/H3-23/H-5/H-9/H3-27/H-22α (δH 2.16)/H2-29 indicated that H-3, H-5, H-9, H3-23, H3-27, and H2-29 were α-oriented. Therefore, the structure of compound 1 was elucidated to be 3-O-β-glucopyranosyl-(1→3)-β-arabinopyranosyl-29-hydroxy-olean-12-en-28-oic acid, named acasentrioid A.
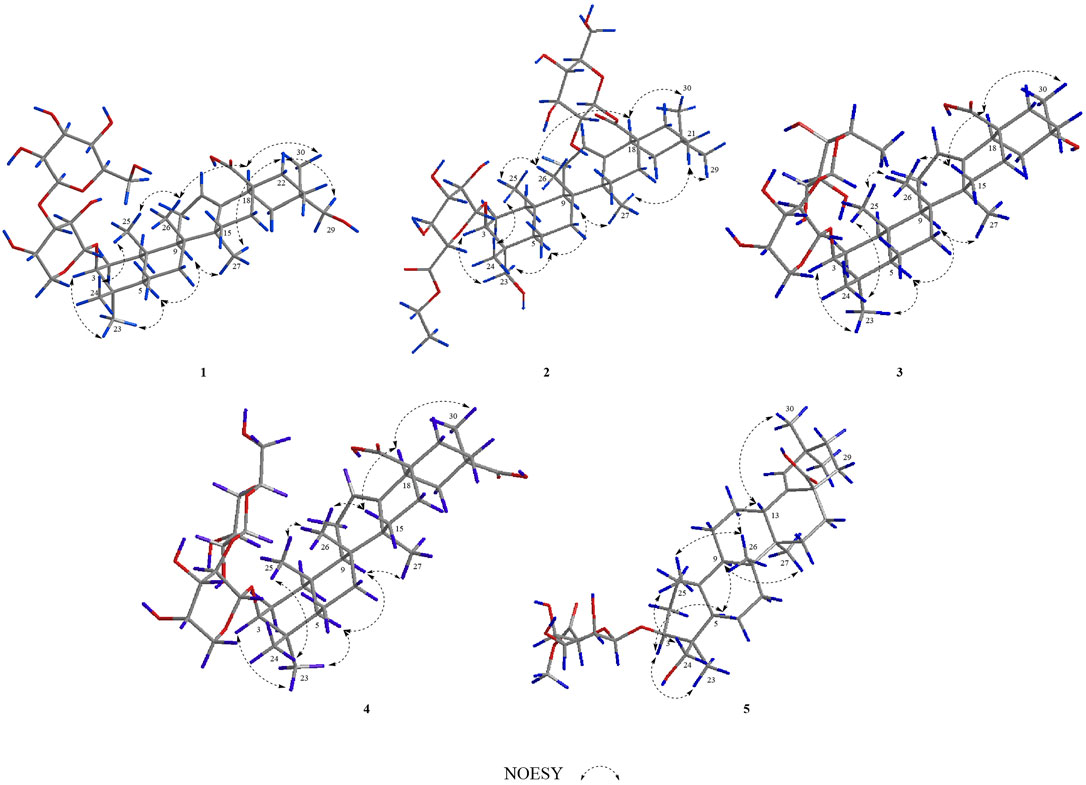
FIGURE 3. Key NOESY correlations of compounds 1–5.
Compound 2 was isolated as an amorphous powder. Its molecular formula, C44H70O15, was determined by the negative HR-ESI-MS at m/z 856.5043 [M + NH4]+ (calculated for C44H74NO15, 856.5053). The 1H NMR spectrum displayed a skeleton characteristic similar to compound 1, namely, six methyls [δH 0.87, 0.88, 0.92, 0.93, 1.11, and 1.19 (3H each, all s, H-30, 29, 25, 24, 26, and 27)], together with one methyl triplet at δH 1.14 (3H, t, J = 7.1 Hz, H-8′), one hydroxymethyl [δH 3.70 (1H, d, J = 10.9 Hz, H-23a), δH 4.34 (1H, d, J = 10.9 Hz, H-23b)], one oxygenated methine [δH 4.30 (1H, dd, J = 4.3, 12.3 Hz, H-3)], one olefin [δH 5.41 (1H, t, J = 3.1 Hz, H-12)], and two anomeric proton signals [δH 5.20 (1H, d, J = 7.7 Hz, GlcA-H-1′) and 6.33 (1H, d, J = 8.1 Hz, Glc-H-1″)] (see Tables 1, 2). There are 44 signals displayed in the 13C NMR spectrum, of which 30 signals corresponded to the triterpene of the oleanane skeleton. Combined with the DEPT spectrum, the 13C NMR spectrum showed resonances for one carboxyl group (δC 176.4), two olefinic carbons (δC 122.9 and 144.1), two anomeric carbons (δC 106.4 and 95.6), one downfield glycosylation-shifted oxygenated methine (δC 82.2), oxygenated methylene (δC 64.3), and seven methyls (δC 13.5, 14.1, 16.0, 17.5, 23.6, 26.0, and 33.0) (see Tables 1, 2). These observations implied that compound 2 might be as well an oleanane-type triterpenoid saponin.
The heteronuclear multiple bond coherence (HMBC) cross-peaks of the anomeric protons H-1′ (δH 5.20)/C-3 and H-1″ (δH 6.33)/C-28 show that sugar is attached to the ether bond of C-3 and C-28, respectively. The HMBC connections of H2-22 (δH 1.74, 1.81)/C-17, C-28 indicated a glycosylated carboxyl fragment attached at C-17 (Figure 2). Based on the above analysis, the structure of 2 was similar to ilexoside XLVIII (Amimoto et al., 1993), except for the ethyl group (δC 14.1 and 61.1) linked to C-6′ (δC 170.2) by ether bond in compound 2, which was supported by the HMBC correlation of the ethyl protons H3-8′ and H-7′ with C-7′ and C-6′, respectively (Figure 2). The β orientations of both pyranose sugars were deduced according to the large coupling constants of the anomeric protons (J = 7.7 Hz, H-1′; J = 8.1 Hz, H-1″). The absolute configurations of both glucuronopyranosyl and glucopyranosyl were determined to be D by the same chemical methods and GC analysis as 1.
In the NOESY spectrum (Figure 3), the correlation peaks of H3-24/H3-25/H3-26/H-18/H3-30 suggested the β orientations of H3-24, H3-25, H3-26, H-18, and H3-30. Conversely, the correlation peaks of H-3/H2-23/H-5/H-9/H-27/H-21β (δH 1.07)/H3-29 indicated that H-3, H-5, H-9, H2-23, H3-27, and H3-29 were α-oriented. Thus, the structure of compound 2 was defined as 3-O-β-D-6′-ethyl-glucuronopyranosyl-23-hydroxy-olean-12-en-28-β-D-glucopyranosyl, named acasentrioid B.
Compound 3 was also obtained as an amorphous powder with the molecular formula of C40H64O12 as indicated by the molecular ion peak m/z 737.4490 [M + H]+ (calculated 737.4471 for C40H65O12) in the HR-ESI-MS spectra. The 1H NMR spectra gave seven angular methyl signals at δH 0.82 (3H, s, H-25), 0.99 (3H, s, H-26), 1.06 (3H, s, H-24), 1.17 (3H, s, H-23), 1.26 (3H, s, H-27), 1.58 (3H, s, H-30), and 1.63 (3H, d, J = 5.9 Hz, Ara-H-6″); one oxygenated methine at δH 3.25 (1H, dd, J = 4.1, 11.5 Hz, H-3); one olefin at δH 5.53 (1H, br.s, H-12); and two anomeric proton signals at δH 4.91 (1H, d, J = 5.1 Hz, Ara-H-1′) and 6.16 (1H, s, Rha-H-1″) (see Tables 1, 2). Accordingly, the 13C NMR spectra also revealed seven methyl signals at δC 15.5 (C-25), 17.0 (C-24), 17.3 (C-26), 18.5 (C-6″), 26.0 (C-27), 28.0 (C-23), and 25.7 (C-30); an oxygen-substituted methine signal at δC 88.7 (C-3); oxygen-substituted quaternary carbon signal at δC 69.8 (C-20); two olefinic carbon signals at δC 122.6 (C-12) and 144.3 (C-13); and one carboxyl carbon signal at δC 180.0 (C-28), along with two anomeric carbon signals at δC 101.7 (C-1″) and 104.9 (C-1′) as determined by the HSQC and DEPT spectra. All these NMR data were characteristic resonances of olean-12-ene skeleton triterpenes. The NMR data of 3 resembled those of 17, and the major difference was the substituent C-29 is changed from methyl to hydroxyl in compound 3, which was supported by the chemical shift δC 69.8 (C-20) and the HMBC correlation of the anomeric protons of H3-30 (δH 1.58) with C-19, C-20, and C-21 (Figure 2). The HMBC connections of H2-22 (δH 2.07) and H-18 (δH 3.35)/C-28 indicated a carboxy fragment attached at C-17 (Figure 2). The absolute configurations of both arabinopyranose and rhamnopyranose were determined to be L by the same chemical methods and GC analysis as 1. The coupling constant of the anomeric hydrogen J = 5.1 Hz (δH 4.91, d, H-1′) established the α-arabinopyranosyl linkage in 3. In the NOESY spectrum (Figure 3), the correlation peaks of H3-24/H3-25/H3-26/H-15β/H-18/H3-30 indicated that the H3-24, H3-25, H3-26, H-18, and H3-30 were β-oriented. Conversely, the correlation peaks of H-3/H3-23/H-5/H-9/H3-27 suggested the α orientations of H-3, H-5, H-9, H3-23, H3-27, and OH-20. Thus, compound 3 was defined as 3β-[(α-L-rhamnopyranosyl-(1→2)-α-L-arabinopyranosyl)oxy]-20α,30-dihydroxy-norolean-12-en-28-oic acid, named acasentrioid C.
Compound 4 was obtained as an amorphous powder. Its molecular formula C41H64O14 was established by the HR-ESI-MS spectrum m/z 798.4648 [M + NH4]+ (calculated for C41H68NO14, 798.4634) and was supported by the 13C NMR spectroscopic data. The 13C NMR spectrum of 4 displayed 41 carbons, of which 30 were assigned to the aglycone part and the remaining 11 were assigned to two sugar units comprising one pentose and one hexose. The NMR spectra showed signals for six angular methyl at δH 0.80, 0.96, 1.00, 1.18, 1.25, and 1.55 (3H each, all s, H-25, 26, 24, 23, 27, and 30), and their corresponding carbons at δC 15.3 (C-25), 17.2 (C-26), 16.6 (C-24), 28.0 (C-23), 25.9 (C-27), and 19.9 (C-30); an olefinic group at δH 5.51 (1H, t, J = 2.9 Hz, H-12) and δC 123.0 (C-12) and 144.1 (C-13); one oxygenated methine at δH 3.17 (1H, dd, J = 4.2 and 11.8 Hz, H-3) and δC 88.7 (C-3); two anomeric proton signals at δH 4.93 (1H, d, J = 5.7 Hz, Ara-H-1′) and 5.17 (1H, d, J = 7.8 Hz, Glc-H-1″) and their corresponding carbons at δC 104.6 (C-1′) and 105.7 (C-1″); and two carboxy groups at δC 180.0 (C-28) and 181.1 (C-29), which were characteristic for the triterpenoid saponin with oleanane skeleton. Two-dimensional NMR data of 4 resembled those of 20, and the major difference was the substituent C-29 is changed from methyl to carboxyl in compound 3, which was supported by the chemical shift δC 181.1 (C-29) and the HMBC correlation of H-19 (δH 2.57)/C-29 and H3-30 (δH 1.55)/C-20, C-21, and C-29 (Figure 2). The absolute configurations of the arabinopyranose and glucopyranose were determined to be L and D, respectively, by the same chemical methods and GC analysis as 1, and the coupling constant of anomeric protons J = 5.7 Hz (δH 4.93, d, H-1′) and J = 7.8 Hz (δH 5.17, d, H-1″) established the α-arabinopyranosyl and β-glucopyranosyl linkage in 4. In the NOESY spectrum (Figure 3), the correlation peaks of H3-24/H3-25/H3-26/H-15β/H-18/H3-30 indicated that the H3-24, H3-25, H3-26, H-18, and H3-30 were β-oriented. Conversely, the correlation peaks of H-3/H3-23/H-5/H-9/H3-27 suggested the α orientations of H-3, H-5, H-9, H3-23, H3-27, and COOH-20. Thus, compound 4 was defined as 3β-[(O-β-D-glucopyranosyl-(1→2)-α-L-arabinopyranosyl)oxy]olean12-ene-28,29-dioic acid, named acasentrioid D.
Compound 5 was obtained as an amorphous powder. The HR-ESI-MS indicated a precise [M + H]+ ion at m/z 663.4121 (calculated for C37H59O10, 663.4103), indicating an empirical molecular formula of C41H64O14. In the 1H NMR spectrum, six quaternary methyl group protons at δH 0.83, 0.89, 0.92, 1.02, 1.04, and 1.11 (3H each, all s, H-25, 27, 23, 26, 30, and 29); a methoxy group proton at δH 3.69 (3H, s, H-7′); an olefinic proton at δH 5.27 (1H, s, H-19); an oxygenated methine proton at δH 4.32 (1H, dd, J = 4.3, 12.1 Hz, H-3), hydroxymethyl protons at δH 3.70 (1H, d, J = 11.0 Hz, H-24a) and δH 4.34 (1H, d, J = 11.0 Hz, H-24b); and an anomeric proton at δH 5.22 (1H, d, J = 7.7 Hz, GlcA-H-1′) along with six quaternary methyl group carbons at δC 17.3 (C-25), 15.2 (C-27), 13.3 (C-23), 16.2 (C-26), 29.2 (C-30), and 30.7 (C-29); methoxy group carbons at δC 51.9 (C-7′); an oxygen-bearing methine carbon at δC 82.0 (C-3); a set of olefinic carbons at δC 138.9 (C-18) and 131.9 (C-19); a hydroxymethyl carbon at δC 64.1 (C-24); a carboxyl carbon at δC 179.4 (C-28); an ester group carbon at δC 170.8 (C-6′); and an anomeric carbon at δC 106.4 (C-1′) in its 13C NMR suggested the aglycone belongs to oleanane-type triterpene (see Tables 1, 2). A detailed analysis of HSQC, HMBC, COSY, and NOESY spectra of 5 assisted the complete assignment of its 1H and 13C NMR data, which were similar to those of 3β,23-dihydroxyolean-18-en-28-oic acid (Cai and Geng, 2016). The only difference is the presence of glucuronopyranoside-6′-O-methyl ester at C-3 in 5, while there is no glycoside at C-3 in 3β,23-dihydroxyolean-18-en-28-oic acid, which was further confirmed by the HMBC correlations of H-1′ with C-3 and of H3-7′ with C-6′ (Figure 2). The absolute configurations of the glucuronopyranosyl were determined to be D by the same chemical methods and GC analysis as 1, and the coupling constant of anomeric proton J = 7.7 Hz (δH 5.22, d, H-1′) established the β-glucuronopyranoside in 5. In the NOESY spectrum (Figure 3), the correlation peaks of H2-24/H3-25/H3-26/H-13/H3-30 indicated that the H2-24, H3-25, H3-26, H-13, and H3-30 were β-oriented. Conversely, the correlation peaks of H-3/H3-23/H-5/H-9/H3-27 suggested the α orientations of H-3, H-5, H-9, H3-23, and H3-27. Thus, compound 5 was defined as 3β,23-dihydroxyolean-18-en-28-oic acid 3-O-β-D-glucuronopyranoside-6′-O-methyl ester, named acasentrioid E.
The structures of known compounds 6–29 were determined as HN-saponin D1 (6) (Kizu et al., 1985), hederagenin glycosides 3-O-α-L-arabinopyranoside (7) (Grishkovets et al., 2005), oleanolic acid 3-O-β-D-glucuronopyranoside (8) (Li et al., 2012), HN-saponin K (9) (Kizu et al., 1985), 3-O-β-D-glucuronopyranosyl-3β,16α-dihydroxyolean-12-en-28-oic acid (10) (Ushijima et al., 2008), gypsogenin 3-O-glucuronide (11) (Bouguet-Bonnet et al., 2002), elatoside G (12) (Yoshikawa et al., 1995), hederagenin-3-O-β-D- glucuronopyranoside 6′-O-methyl ester (13) (Cao et al., 2011), tragopogonsaponin A methyl ester (14) (Warashina et al., 1991), 3-O-6′-O-methyl-β-D-glucuronopy-ranoside of gypsogenin (15) (Iwamoto et al., 1985), 3-O-β-D-(6′-O-methyl-glucuronopyranosyl) oleanolic acid (16) (Melek et al., 1996), 3-O-α-rhamnopyranose- (1→2)-α-arabinopyranosyl-29-hydroxy-olean-12-en-28-oic acid (17) (Shao et al., 1989), 3-O-[α-L-rhamnopyranosyl-(1→2)-α-L-arabinopyranosyl] oleanolic acid (18) (Nakanishi et al., 1993), HN-saponin F (19) (Mizui et al., 1988), saponin PE (20) (Zhong et al., 2001), oleanolic acid 3-O-β-D-glucopyranosyl (1→3)-α-L-arabinopyranoside (21) (Satoh et al., 1994), lucyoside F (22), lucyoside H (23) (Takemoto et al., 1984), 3-O-β-D-glucopyranosyl-(1→2)-β-D-glucopyranosyl oleanolic acid (24) (Reginatto et al., 2001), 3-O-β-D-glucuronopyranosyl methyl ester-28-O-β-D-glucopyranoside (25) (Li et al., 2007), oleanolic acid 3-O-α-L-rhamnopyranosyl(1→2)-α-L-arabinopyranosyl-28-O-β-D-glucopyranosyl ester (26) (Fan et al., 2013), paritriside E (27) (Wu et al., 2012), 3-O-α-arabinopyranosyl-(1→2)-β-glucopyranoside-30-norolean-12,20(29)-dien-28-oic acid (28) (Shao et al., 1989), and 3-[(O-β-D-glucopyranosyl-(1→3)-α-L-arabinopyranosyl)oxy]-30- noroleana-12,20(29)-dien-28-oic acid (29) (Jitsuno and Mimaki, 2010) by comparison with literature data (Supplementary Table S1). Compounds 7 and 20 were isolated from A. senticosus for the first time. Compounds 6, 9, 12, 16, 19, 21, 23, and 24, were isolated from Acanthoganax Miq. species for the first time. Compounds 10, 11, 14, 15, 22, 26, 27, and 29 were isolated from the family Araliaceae for the first time.
Bioactive Activity
The cytotoxicity of compounds 1–29 on BV2 microglia was determined by the CCK-8 assay, and the results are listed in Table 3. The neuroinflammation model was established by LPS-induced BV2 microglia, and the neuroprotective effect of compounds (1–29) in vitro was evaluated. Unfortunately, the results of the evaluation were not ideal. Compounds 5, 10, 12, 13, and 16 had moderate inhibitory effects on neuroinflammation, as indicated in Table 4, and other compounds had no anti-neuroinflammatory activity. Based on the existing results and analyzing its structure–activity relationship, we speculate in the structure of oleanane-type triterpene saponins; when the C-16 hydroxyl group is substituted or the structure contains only one methyl glucuronate, the compound has moderate anti-neuroinflammatory effects.

TABLE 3. Cytotoxic activities of compounds 1–29 on BV2 Cells (IC50, μM).
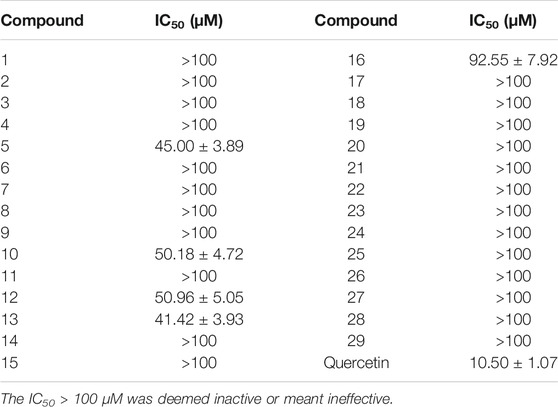
TABLE 4. Inhibitory effects of compounds 1–29 on NO in LPS-induced BV-2 Cells (n = 3, x ± s).
Conclusion
In summary, five previously undescribed oleanane-type triterpenoid saponins (1–5), together with twenty-four known saponins (6–29), were isolated from the fruit of A. senticosus. The structures of all compounds were elucidated by extensive spectroscopic analysis, including 1D, 2D NMR, and HR-ESI-MS, in combination with chemical methods (acid hydrolysis). The neuroinflammation model was established by LPS-induced BV2 microglia, and the neuroprotective effects of all compounds (1–29) were evaluated. Unfortunately, the results of the evaluation were not ideal. Compounds 5, 10, 12, 13, and 16 had moderate inhibitory effects on neuroinflammation, while other compounds have no anti-neuroinflammatory activity.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was financially supported by Heilongjiang Touyan Innovation Team Program, Heilongjiang University of Chinese Medicine Funds (2018pt01 and 2018bs03).
Conflict of Interest
Author M-LZ is employed by China Resources Double-Crane Pharmaceutical Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.825763/full#supplementary-material
References
Amimoto, K., Yoshikawa, K., and Arihara, S. (1993). Triterpenoid Saponins of Aquifoliaceous Plants. XII. Ilexosides XLVI-LI from the Leaves of Ilex Rotunda Thunb. Chem. Pharm. Bull. 41, 77–80. doi:10.1248/cpb.41.77
Bouguet-Bonnet, S., Rochd, M., Mutzenhardt, P., and Henry, M. (2002). Total Assignment of1H and13C NMR Spectra of Three Triterpene Saponins from Roots ofSilene Vulgaris (Moench) Garcke. Magn. Reson. Chem. 40, 618–621. doi:10.1002/mrc.1069
Cai, Y. T., and Geng, H. W. (2016). Chemical Constituents from the Roots of Rhodomyrtus Tomentosa. Zhong Yao Cai 39, 1303–1307. doi:10.13863/j.issn1001-4454.2016.06.025
Cao, B. Y., Liu, R. X., Wang, J., Jia, L. Y., Zheng, Q. W., and Lu, J. C. (2011). Isolation and Identification of Chemical Constituents of Roots from Gardenia Jasminoides Ellis. J. Shenyang Pharm. Univ. 28, 784–817. doi:10.14066/j.cnki.cn21-1349/r.2011.10.013
Cong, D. L., Wang, H. T., Gao, X. Y., Zheng, Y. L., and Zhang, C. X. (2010). Anti-fatigue Effect of Acanthopanax Senticosus Fruits. J. Jilin Univ. (Medicine Edition) 36, 891–894. doi:10.13481/j.1671-587x.2010.05.008
Fan, W. H., Liu, J. Y., Gong, Y. X., Ma, J., Zhou, N., and Xu, Y. N. (2013). A New Triterpenoid Saponin from Pulsatilla Cernua. Nat. Product. Sci. 19, 150–154. www.koreascience.or.kr/article/JAKO201319953221993.page
Ge, Y.-W., Tohda, C., Zhu, S., He, Y.-M., Yoshimatsu, K., and Komatsu, K. (2016). Effects of Oleanane-type Triterpene Saponins from the Leaves of Eleutherococcus Senticosus in an Axonal Outgrowth Assay. J. Nat. Prod. 79, 1834–1841. doi:10.1021/acs.jnatprod.6b00329
Grishkovets, V. I., Panov, D. A., Kachala, V. V., and Shashkov, A. S. (2005). Triterpene Glycosides from Kalopanax Septemlobum. 1. Glycosides A, B, C, F, G1, G2, I2, H, and J from Leaves of Kalopanax Septemlobum Var. Maximowichii Introduced to Crimea. Chem. Nat. Compd. 41, 194–199. doi:10.1007/s10600-005-0110-2
Huang, L., Zhao, H., Huang, B., Zheng, C., Peng, W., and Qin, L. (2011). Acanthopanax Senticosus: Review of Botany, Chemistry and Pharmacology. Pharmazie 66, 83–97. doi:10.1691/ph.2011.0744
Iwamoto, M., Okabe, H., Yamauchi, T., Tanaka, M., Rokutani, Y., Hara, S., et al. (1985). Studies on the Constituents of Momordica Cochinchinensis Spreng. I. Isolation and Characterization of the Seed Saponins, Momordica Saponins I and II. Chem. Pharm. Bull. 33, 464–478. doi:10.1248/cpb.33.464
Jitsuno, M., and Mimaki, Y. (2010). Triterpene Glycosides from the Aerial Parts of Larrea Tridentata. Phytochemistry 71 (17-18), 2157–2167. doi:10.1016/j.phytochem.2010.09.012
Kim, Y.-H., Cho, M., Kim, D.-B., Shin, G.-H., Lee, J.-H., Lee, J., et al. (2015). The Antioxidant Activity and Their Major Antioxidant Compounds from Acanthopanax Senticosus and A. Koreanum. Molecules 20, 13281–13295. doi:10.3390/molecules200713281
Kizu, H., Kitayama, S., Nakatani, F., Tomimori, T., and Namba, T. (1985). Studies on Nepalese Crude Drugs. III. On the Saponins of Hedera Nepalensis K. KOCH. Chem. Pharm. Bull. 33, 3324–3329. doi:10.1248/cpb.33.3324
Li, F., Li, W., Fu, H., Zhang, Q., and Koike, K. (2007). Pancreatic Lipase-Inhibiting Triterpenoid Saponins from Fruits of Acanthopanax Senticosus. Chem. Pharm. Bull. 55, 1087–1089. doi:10.1248/cpb.55.1087
Li, C. S., Yu, H. W., Chen, X. Z., Wu, X. Q., Fang, D. M., and Li, G. Y. (2012). Chemical Constituents of Deutzia Setchuenensis. Nat. Product. Res. Dev. 24, 465–468. doi:10.16333/j.1001-6880.2012.04.031
Li, W., Luo, Q., and Jin, L. H. (2013). Acanthopanax Senticosus Extracts Have a Protective Effect on Drosophila Gut Immunity. J. Ethnopharmacology 146, 257–263. doi:10.1016/j.jep.2012.12.040
Li, J.-L., Li, N., Lee, H.-S., Xing, S.-S., Qi, S.-Z., Tuo, Z.-D., et al. (2016a). Four New Sesqui-Lignans Isolated from Acanthopanax Senticosus and Their Diacylglycerol Acyltransferase (DGAT) Inhibitory Activity. Fitoterapia 109, 185–189. doi:10.1016/j.fitote.2016.01.009
Li, T., Ferns, K., Yan, Z.-Q., Yin, S.-Y., Kou, J.-J., Li, D., et al. (2016b). Acanthopanax Senticosus: Photochemistry and Anticancer Potential. Am. J. Chin. Med. 44, 1543–1558. doi:10.1142/S0192415X16500865
Li, J.-L., Li, N., Xing, S.-S., Zhang, N., Li, B.-B., Chen, J.-G., et al. (2017). New Neo-Lignan from Acanthopanax Senticosus with Protein Tyrosine Phosphatase 1B Inhibitory Activity. Arch. Pharm. Res. 40, 1265–1270. doi:10.1007/s12272-015-0659-7
Luo, D., Xie, J.-Z., Zou, L.-H., Qiu, L., Huang, D.-P., Xie, Y.-F., et al. (2020). Lanostane-type Triterpenoids from Ganoderma Applanatum and Their Inhibitory Activities on NO Production in LPS-Induced BV-2 Cells. Phytochemistry 177, 112453. doi:10.1016/j.phytochem.2020.112453
Melek, F. R., Miyase, T., El-Gindi, O. D., Abdel-Khalik, S. M., and Haggag, M. Y. (1996). Saponins from Fagonia Mollis. Phytochemistry 42, 1405–1407. doi:10.1016/0031-9422(95)00087-9
Mizui, F., Kasai, R., Ohtani, K., and Tanaka, O. (1988). Saponins from Brans of Quinoa, Chenopodium Quinoa WILLD. I. Chem. Pharm. Bull. 36, 1415–1418. doi:10.1248/cpb.36.1415
Nakanishi, T., Tanaka, K., Murata, H., Somekawa, M., and Inada, A. (1993). Phytochemical Studies of Seeds of Medicinal Plants. III. Ursolic Acid and Oleanolic Acid Glycosides from Seeds of Patrinia Scabiosaefolia Fischer. Chem. Pharm. Bull. 41, 183–186. doi:10.1248/cpb.41.183
Reginatto, F. H., Kauffmann, C., Schripsema, J., Guillaume, D., Gosmann, G., and Schenkel, E. P. (2001). Steroidal and Triterpenoidal Glucosides from Passiflora Alata. J. Braz. Chem. Soc. 12, 32–36. doi:10.1590/S0103-50532001000100003
Saito, T., Nishida, M., Saito, M., Tanabe, A., Eitsuka, T., Yuan, S.-H., et al. (2016). The fruit of Acanthopanax senticosus (Rupr. et Maxim.) Harms improves insulin resistance and hepatic lipid accumulation by modulation of liver adenosine monophosphate-activated protein kinase activity and lipogenic gene expression in high-fat diet-fed obese mice. Nutr. Res. 36, 1090–1097. doi:10.1016/j.nutres.2016.09.004
Satoh, Y., Sakai, S., Katsumata, M., Nagasao, M., Miyakoshi, M., Ida, Y., et al. (1994). Oleanolic Acid Saponins from Root-Bark of Aralia Elata. Phytochemistry 36, 147–152. doi:10.1016/S0031-9422(00)97028-6
Shao, C.-J., Kasai, R., Xu, J.-D., and Tanaka, O. (1989). Saponins from Leaves of Acanthopanax Senticoshs Harms., Ciwujia. II. Structures of Ciwujianosides A1, A2, A3, A4 and D3. Chem. Pharm. Bull. 37, 42–45. doi:10.1248/cpb.37.42
Takemoto, T., Arihara, S., Yoshikawa, K., Kusumoto, K., Yano, I., and Hayashi, T. (1984). Studies on the Constituents of Cucurbitaceae Plants. VI. On the Saponin Constituents of Luffa Cylindrica Roem. (1). Yakugaku Zasshi 104, 246–255. doi:10.1248/yakushi1947.104.3_246
Teng, Y., Zhang, H., Zhou, J., Zhan, G., and Yao, G. (2018). Hebecarposides A−K, Antiproliferative Lanostane-type Triterpene Glycosides from the Leaves of Lyonia Ovalifolia Var. Hebecarpa. Phytochemistry 151, 32–41. doi:10.1016/j.phytochem.2018.03.012
Ushijima, M., Komoto, N., Sugizono, Y., Mizuno, I., Sumihiro, M., Ichikawa, M., et al. (2008). Triterpene Glycosides from the Roots of Codonopsis Lanceolata. Chem. Pharm. Bull. 56, 308–314. doi:10.1248/cpb.56.308
Wang, Z.-B., Jiang, H., Xia, Y.-G., Yang, B.-Y., and Kuang, H.-X. (2012). α-Glucosidase Inhibitory Constituents from Acanthopanax Senticosus Harm Leaves. Molecules 17, 6269–6276. doi:10.3390/molecules17066269
Warashina, T., Miyase, T., and Ueno, A. (1991). Novel Acylated Saponins from Tragopogon Porrifolius L. Isolation and the Structures of Tragopogonsaponins A-R. Chem. Pharm. Bull. 39, 388–396. doi:10.1248/cpb.39.388
Wu, X., Wang, L., Wang, G.-C., Wang, H., Dai, Y., Yang, X.-X., et al. (2013). Triterpenoid Saponins from Rhizomes of Paris Polyphylla Var. Yunnanensis. Carbohydr. Res. 368, 1–7. doi:10.1016/j.carres.2012.11.027
Yan, Z. W., Zhou, M. J., Liu, J. P., Lu, D., and Li, P. Y. (2009). Effects of Total Saponins from the Pulp of Acanthopanax senticosus (Rupr. et Maxim) Harms on Blood Lipidin Mice with Hyperlipidemia. Spec. Wild Econ. Anim. Plant Res. 4, 13. doi:10.16720/j.cnki.tcyj.2009.04.005
Yoshikawa, M., Yoshizumi, S., Ueno, T., Matsuda, H., Murakami, T., Yamahara, J., et al. (1995). Medicinal Foodstuffs. I. Hypoglycemic Constituents from a Garnish Foodstuff "Taranome," the Young Shoot of Aralia Elata SEEM.: Elatosides G, H, I, J, and K. Chem. Pharm. Bull. 43, 1878–1882. doi:10.1248/cpb.43.1878
Zhang, L., Li, B.-B., Li, H.-Z., Meng, X., Lin, X., Jiang, Y.-Y., et al. (2017). Four New Neolignans Isolated from Eleutherococcus Senticosus and Their Protein Tyrosine Phosphatase 1B Inhibitory Activity (PTP1B). Fitoterapia 121, 58–63. doi:10.1016/j.fitote.2017.06.025
Zhao, Z., Xu, X., Ye, Q., and Dong, L. (2013). Ultrasound Extraction Optimization of Acanthopanax Senticosus Polysaccharides and its Antioxidant Activity. Int. J. Biol. Macromolecules 59, 290–294. doi:10.1016/j.ijbiomac.2013.04.067
Keywords: triterpenoid saponins, Acanthopanax senticosus (Rupr. & Maxim.) Harms, fruit, cytotoxicity, neuroprotective
Citation: Liu Y, Jiang P, Zhang M-L, Pan J, Guan W, Li X-M, Yang B-Y and Kuang H-X (2022) Triterpenoid Saponins From the Fruit of Acanthopanax senticosus (Rupr. & Maxim.) Harms. Front. Chem. 10:825763. doi: 10.3389/fchem.2022.825763
Received: 30 November 2021; Accepted: 07 January 2022;
Published: 21 February 2022.
Edited by:
Xiaoxiao Huang, Shenyang Pharmaceutical University, ChinaReviewed by:
Le Zhou, South China Sea Institute of Oceanology (CAS), ChinaMing Bai, Shenyang Pharmaceutical University, China
Copyright © 2022 Liu, Jiang, Zhang, Pan, Guan, Li, Yang and Kuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing-You Yang, eWJ5d2F0ZXJAMTYzLmNvbQ==; Hai-Xue Kuang, aHhrdWFuZzU2QDE2My5jb20=