
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 06 January 2023
Sec. Medicinal and Pharmaceutical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1094841
This article is part of the Research Topic Advances in Drug discovery and Quality Evaluation View all 10 articles
 Wende Zheng1,2Borong Tu1,2Zhen Zhang1,2Jinxuan Li1,2Zhenping Yan1,2Kaize Su1,2Duanyu Deng1,2Ying Sun1,2Xu Wang1,2Bingjie Zhang3Kun Zhang1,2
Wende Zheng1,2Borong Tu1,2Zhen Zhang1,2Jinxuan Li1,2Zhenping Yan1,2Kaize Su1,2Duanyu Deng1,2Ying Sun1,2Xu Wang1,2Bingjie Zhang3Kun Zhang1,2 Wing-Leung Wong4
Wing-Leung Wong4 Panpan Wu1,2*
Panpan Wu1,2* Weiqian David Hong1,2*
Weiqian David Hong1,2* Song Ang1,2*
Song Ang1,2*Introduction: Fusidic acid (FA) has been widely applied in the clinical prevention and treatment of bacterial infections. Nonetheless, its clinical application has been limited due to its narrow antimicrobial spectrum and some side effects.
Purpose: Therefore, it is necessary to explore the structure–activity relationships of FA derivatives as antibacterial agents to develop novel ones possessing a broad antimicrobial spectrum.
Methods and result: First, a pharmacophore model was established on the nineteen FA derivatives with remarkable antibacterial activities reported in previous studies. The common structural characteristics of the pharmacophore emerging from the FA derivatives were determined as those of six hydrophobic centers, two atom centers of the hydrogen bond acceptor, and a negative electron center around the C-21 field. Then, seven FA derivatives have been designed according to the reported structure–activity relationships and the pharmacophore characteristics. The designed FA derivatives were mapped on the pharmacophore model, and the Qfit values of all FA derivatives were over 50 and FA-8 possessed the highest value of 82.66. The molecular docking studies of the partial target compounds were conducted with the elongation factor G (EF-G) of S. aureus. Furthermore, the designed FA derivatives have been prepared and their antibacterial activities were evaluated by the inhibition zone test and the minimum inhibitory concentration (MIC) test. The derivative FA-7 with a chlorine group as the substituent group at C-25 of FA displayed the best antibacterial property with an MIC of 3.125 µM. Subsequently, 3D-QSAR was carried on all the derivatives by using the CoMSIA mode of SYBYL-X 2.0.
Conclusion: Hence, a computer-aided drug design model was developed for FA, which can be further used to optimize FA derivatives as highly potent antibacterial agents.
Fusidic acid (FA), a typical antibiotic with excellent bioactivity against Staphylococcus aureus including the strain that produced cross resistance with other antibiotics, has been applied in clinical therapy since the 1960s (Collignon and Turnidge, 1999; Turnidge, 1999). The study on the antibacterial mechanism of FA showed that the elongation factor G (EF-G) of the bacteria was interfered and the production of bacterial proteins was inhibited (Tanaka et al., 1968; Bodley et al., 1969). Accordingly, the relevant protein of the EF-G has always been implemented as a target acceptor in the development of FA-type antibiotics (Borg et al., 2015; Belardinelli and Rodnina, 2017; Lu et al., 2019). However, the narrow antibacterial spectrum of FA, which merely possessed the activity against Staphylococci, limited its practical application in extensive medical treatment (Petrosillo et al., 2018). Therefore, it became increasingly important to design and synthesize new FA derivatives to explore a broad range of relationships between structures and antibacterial activity. According to the literature, the structure–activity relationships (SARs) between FA derivatives and antibacterial activity have been studied (Godtfredsen et al., 1965; Von Daehne et al., 1979; Duvold et al., 2001). The reported SAR demonstrated that the hydroxyl group at C-3 played a crucial role in drug activity. As a recent study showed blocking the metabolic sites (21-COOH and 3-OH) of FA and its derivatives could maintain the antibacterial activity with a prolonged half-life (Lu et al., 2019). Moreover, it has been reported that the hydroxylation at C-27 of FA and its derivatives could significantly cause the vanishment of the antibacterial activity (Ragab et al., 2020). Hence, the further SARs of FA should be obtained through more designed derivatives and their bioassay tests.
Nowadays, computer-aided drug design (CADD) has become an integral component involving drug discovery and development since it has enormous leverage as an auxiliary tool to raise economic efficiency and reduce time costs (Cerqueira et al., 2015). Advanced rational design techniques combined with computational methodologies have been utilized to create more effective and creative medications (Fjell et al., 2012; Cardoso et al., 2019). The rational design of innovative pharmaceuticals, with the aim of creating pharmaceutical products with more specificity by calculated simulation, has emerged as a crucial aspect of medicinal chemistry (Mouchlis et al., 2020). Pharmacophore-based and docking-based screening are two classic CADD approaches, which were usually applied in virtual screening to select the potential bioactive derivatives (Niu et al., 2012; Sangeetha et al., 2017). Recently, the discovery of a novel drug has benefited greatly from the use of pharmacophore-based virtual screening (PBVS), especially when there is a lack of information regarding the three-dimensional structure of the desired protein target (Sharma et al., 2020; Zhu et al., 2020). In addition, the investigation of the comparison showed that the result of the pharmacophore-based method had higher accuracy than the docking-based method in the experiment (Chen et al., 2009; Talambedu et al., 2017).
In this study, a pharmacophore model has been constructed to design the FA derivatives and molecular docking was used to predict the interactions between FA derivatives and the target protein EF-G. The antibacterial activities of the FA derivatives were assessed by the inhibition zone test and the MIC assay. Furthermore, the quantitative structure–activity relationships (QSARs) of FA were investigated with a thorough inquiry according to biological test data. All in all, this study has provided a novel pharmacophore model to select antibacterial FA derivatives and studied the relationship between the structures and bioactivity.
Based on a set of FA derivatives with remarkable antibacterial activity reported in previous studies (Godtfredsen et al., 1966; Riber et al., 2006; Lv et al., 2017; Kong et al., 2018; Salimova et al., 2018; Singh et al., 2020), a pharmacophore model was established to gain an insight into the necessary features for designing antibacterial agents. A total of 19 FA derivatives selected from the literature reports were aligned by using the GALAHAD module of SYBYL-X 2.0. The assessment parameters generated by two similar models are shown in Table 1, including data of specificity, N-hits, feats, energy, sterics, H-bond, and Mo-Qry. The specificity data of the model, which is a logarithmic indicator of the expected discrimination of each query, are determined by the number of features they contain and the extent of dissociation. Identical specificity values of 5.70 indicated that the two models could come to an anticipant result. Moreover, the model had nine pharmacophore features, i.e., six hydrophobic centers (HYs), two H-bond acceptors, and a negative center (NC) (Figure 1). The hydrophobic centers were distributed in the indole ring and the FA skeleton frame aromatic ring, two H-bond acceptors were found in the carbonyl group and the ester group, and the negative center was distributed in the carboxyl group of the FA derivatives at C-21, which sketched the common structural characteristics of pharmacophore emerging from the FA derivatives with antibacterial activity.
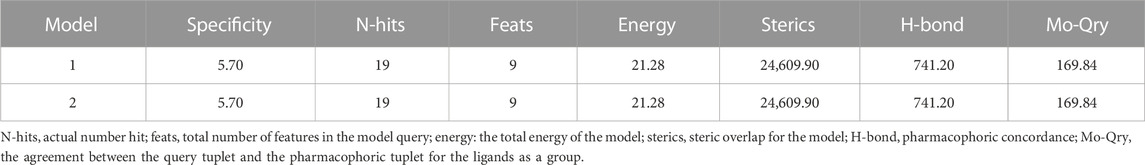
TABLE 1. Assessment parameters of the pharmacophore theory produced by the GALAHAD module.
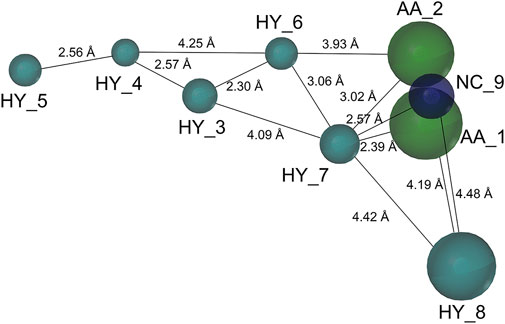
FIGURE 1. Map of a common feature pharmacophore model. AA, the atom center of the hydrogen bond acceptor; HY, hydrophobic group; and NC, negative center.
Seven FA derivatives were conceived according to the reported structure–activity relationships and the characteristics of the pharmacophore model by modifying the C-3, C-21, and C-25 positions of FA. The designed FA derivatives were validated by analyzing the matching degree with the pharmacophore model through the Qfit value. The values of all derivatives were over 50 and FA-8 possessed the highest Qfit value of 82.66, which indicated that the design of the derivative was reasonable and the designed FA derivatives possessed potential antimicrobial activity. The structures, molecular surface lipophilic potential photographs, and Qfit values of derivatives are shown in Table 2. There was still high hydrophobic potential maintained in the C-25 position when methyl was converted into an object of low-size profile, such as hydrogen, chlorine, and bromine. Additionally, strong negative electrical potential in the C-21 position field was not significantly altered by the creation of the lactonic ring. The aim of modification at C-3 was to maintain and even strengthen the lipophilic tendency within this range of the FA skeleton frame.
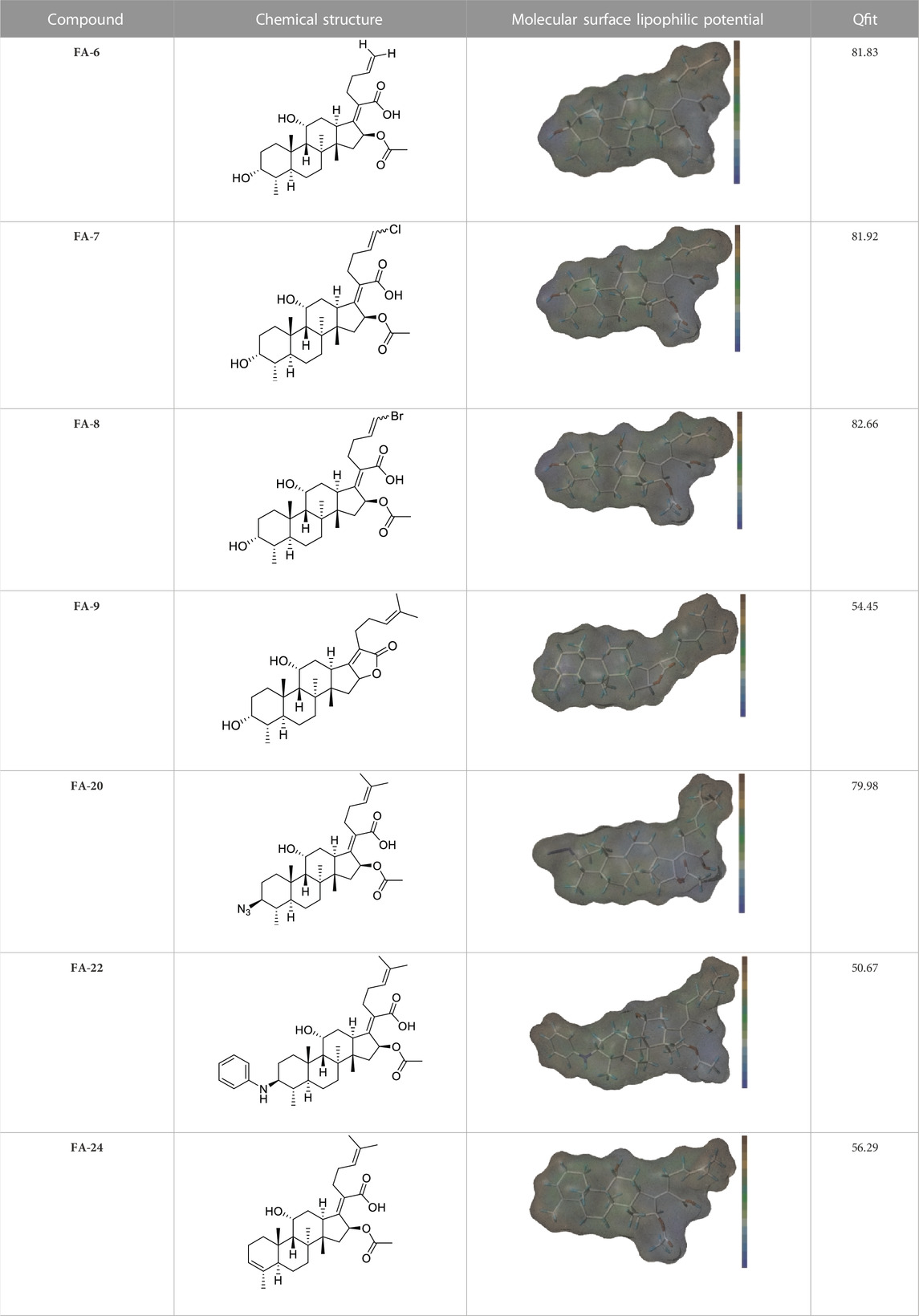
TABLE 2. Structure of the designed derivatives and the Qfit values.
The previous study reported that FA was considered as an antibiotic by interfering with the EF-G of S. aureus (Chen et al., 2010). Therefore, it was applied therapeutically to treat Gram-positive bacterial infections, such as S. aureus (Lannergård et al., 2009). As shown in Figure 2, the target derivatives FA-8, 9, 20, and 22 were docked to the EF-G to investigate the action between the molecular and the receptor protein. The results showed that the interactions between the carboxyl groups of FA derivatives and the binding pockets existed. Compound FA-8 could engender good affinities to Ala655, Tyr668, Glu455, and Phe88 in the active site by a hydrogen bond (Figure 2A). This kind of action of the hydrogen bond existed likewise in the mode of FA-20 and FA-22 fitting to the protein pocket (Figures 2C, D). However, FA-9 docking results saw a massive loss of these key interactions (Figure 2B), which corresponded to the low pharmacophore score. In addition, brominated FA-8 had a halogen bond with Asp87, and the azide group in FA-20 formed a salt bridge with Glu93, which may be positive features to obtain better activities. The results illustrated that the molecular docking model and the pharmacophore model could not be unanimous. It was not surprising that many epactal interactions, such as hydrogen bond and halogen bond interactions, predicted by the binding model may well compensate for some losses of key interactions.
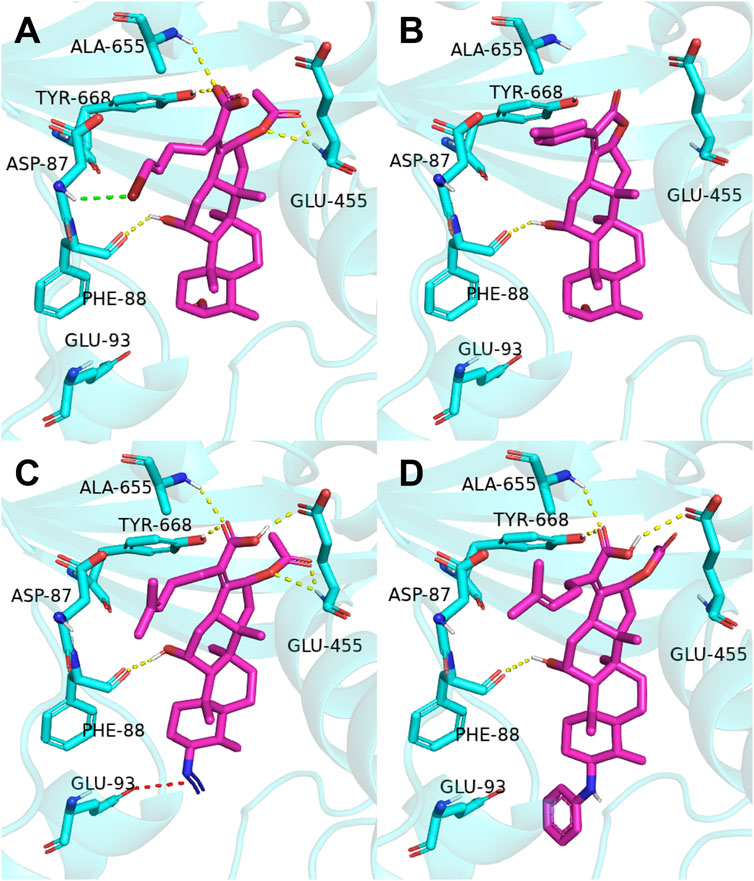
FIGURE 2. Binding mode of four derivatives in the S. aureus EF-G pocket: (A) FA-8; (B) FA-9; (C) FA-20; and (D) FA-22. The relevant ligand molecules were colored by magenta, and the vital amino acid was colored by cyan. The red color dash indicated the salt bridge force; the yellow color dash indicated the hydrogen bond; and the green color indicated the halogen bond interaction.
The previous studies put forward some enlightenment that the modification at C-25 of FA could be beneficial for maintaining the antibacterial effect (Riber et al., 2006; Zhao et al., 2013). A group of target FA derivatives (FA-6 to FA-8) was synthesized, as shown in Scheme 1. The C-25 position of FA had been modified successfully according to the literature reports; however, there were very few modifications with simple and small-sized atoms with functional characteristics at this position that have been carried out to estimate the antibacterial activity. We have commenced with this route by preparing several vital intermediate FA derivatives. The FA triethylamine, as an acid-binding agent, and chloromethyl pivalate were dissolved in DMF and stirred overnight at 50°C. Thus, the FA-1 was procured with protected carboxyl groups. The FA-2 was produced by oxidation of FA-1 with N-methyl morpholine N-oxide (NMO) in the presence of ozone at 0°C (Schwartz et al., 2006). The derivatives FA-6, FA-7, and FA-8 were obtained through the next simple steps such as Wittig’s reaction and de-esterification. To clarify the stability of the ester group at C-16 under different alkaline conditions, potassium carbonate and sodium hydroxide were used to promote the lactone reaction of FA and the intermediate FA-1, respectively. As a result, the lactone derivative FA-9 was generated by the esterification of FA with sodium hydroxide. The syntheses of the derivatives (FA-6∼9) are outlined in Scheme 1. The new FA derivatives were determined by using NMR, HRMS, and CHNS-O elemental analyzer.
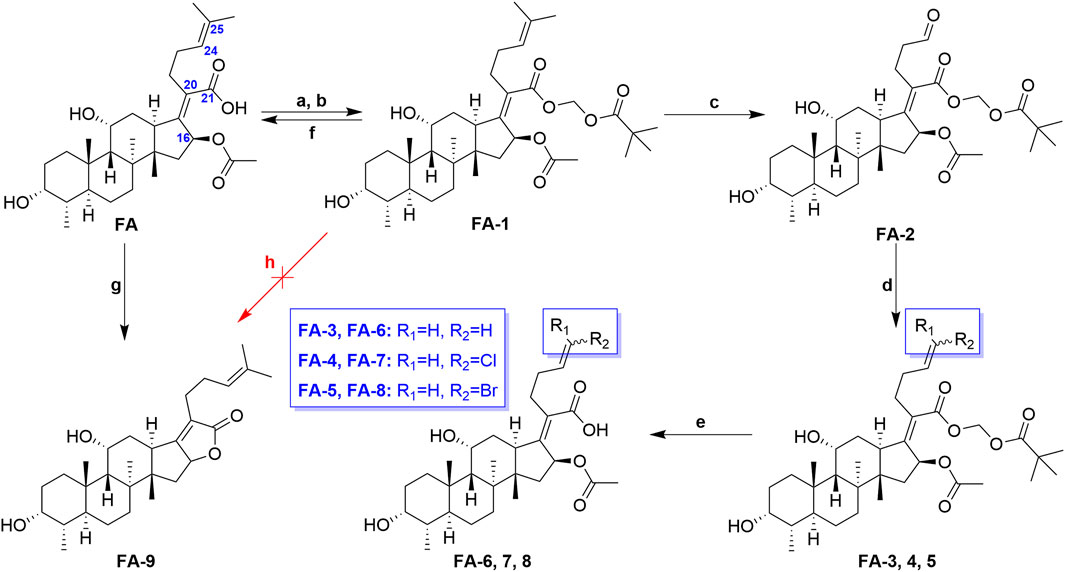
SCHEME 1. Synthetic route of FA-6∼9. Reagents and conditions: (A) DMF, Et3N (1.3 eq.), 30 min; (B) tBuCO2CH2Cl, overnight, 50°C; (C) O3, NMO (1 eq.), DCM, 0°C; (D) Wittig’s reaction; (E) K2CO3 (2 eq.), methanol, r. t., 1 h; (F) K2CO3 (2 eq.), methanol, r. t., overnight; (G) 2 N NaOH (5 eq.), ethanol, refluxed, overnight; and (H) K2CO3 (2 eq.), methanol, r. t., overnight.
Scheme 2 shows the syntheses of FA-17∼22 and FA-24. Briefly, FA-1 was treated with the methane sulfonyl chloride and the pyridine in dichloromethane and afforded product FA-10. Subsequently, on one hand, the methane sulfonyloxy in FA-10 was replaced by the azide group, phenylamino group, and halogens to afford FA-11∼16, respectively. On the other hand, methane sulfonyloxy was reduced into a double bond in the positions of C-3 and C-4 to give FA-23. Finally, all the culminating products (FA-17∼22 and FA-24) were obtained by deblocking the protected ester at the C-21 position with potassium carbonate as the base reagent according to the ester stability experiment of FA derivatives. In this procedure, the related derivatives were identified mainly by HRMS.
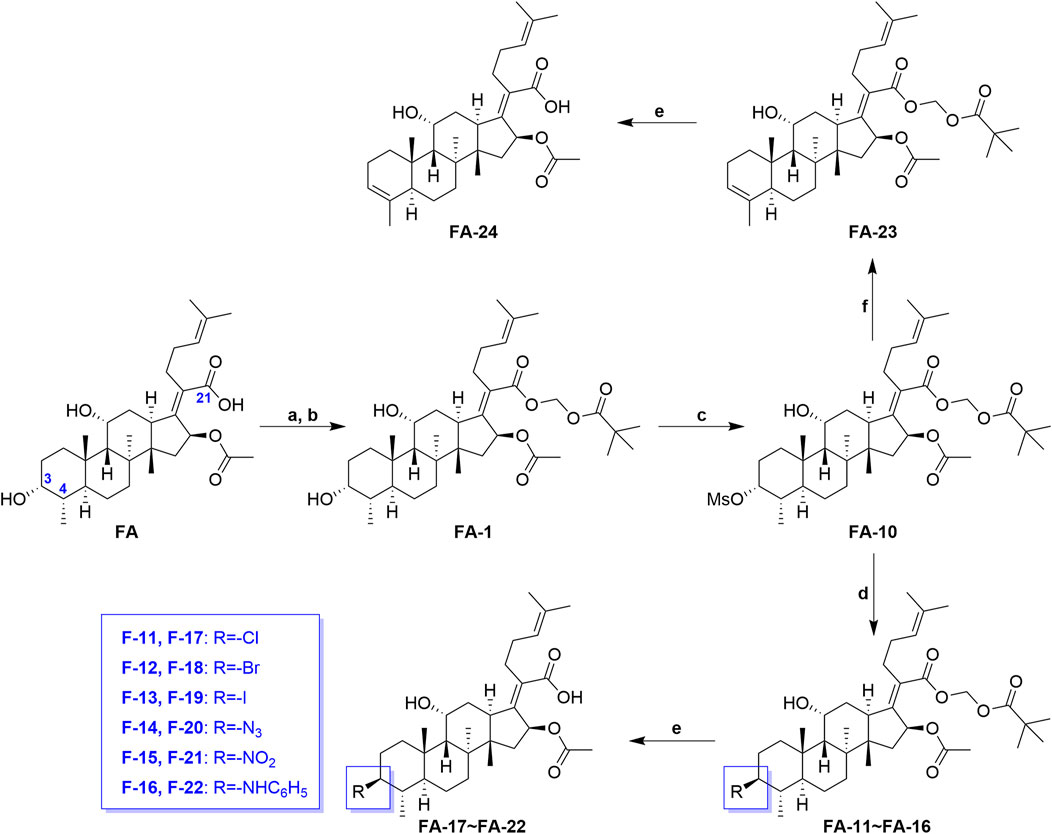
SCHEME 2. Synthetic route of FA-17∼22 and FA-24. Reagents and conditions: (A) DMF, Et3N (1.3 seq.), 30 min; (B) tBuCO2CH2Cl, overnight, 50°C; (C) pyridine (2 eq.), methane sulfonyl chloride (5 eq.), DCM, overnight; (D) tetrabutylammonium chloride/bromide/iodide/nitrite/sodium azide (2 eq.), THF, reflux, overnight; (E) K2CO3 (2 eq.), methanol, r. t., 1 h; (F) 2,6-lutidine, 130°C, 2 h.
As shown in Table 3, the antibacterial activities of the FA derivatives were assessed using the inhibition zone test. Compound FA-6 possessed remarkable activity against Gram-positive germs with the corresponding inhibition zone diameters of 18.89 ± 0.03, 19.72 ± 0.12, and 14.96 ± 1.21 mm in a relatively low dosage (0.83 nmol). As the dosage increased, FA-17∼22 and FA-24 displayed obvious inhibition zones against Gram-positive bacteria. However, there was no sign for all target derivatives to inhibit Gram-negative germs in this test.
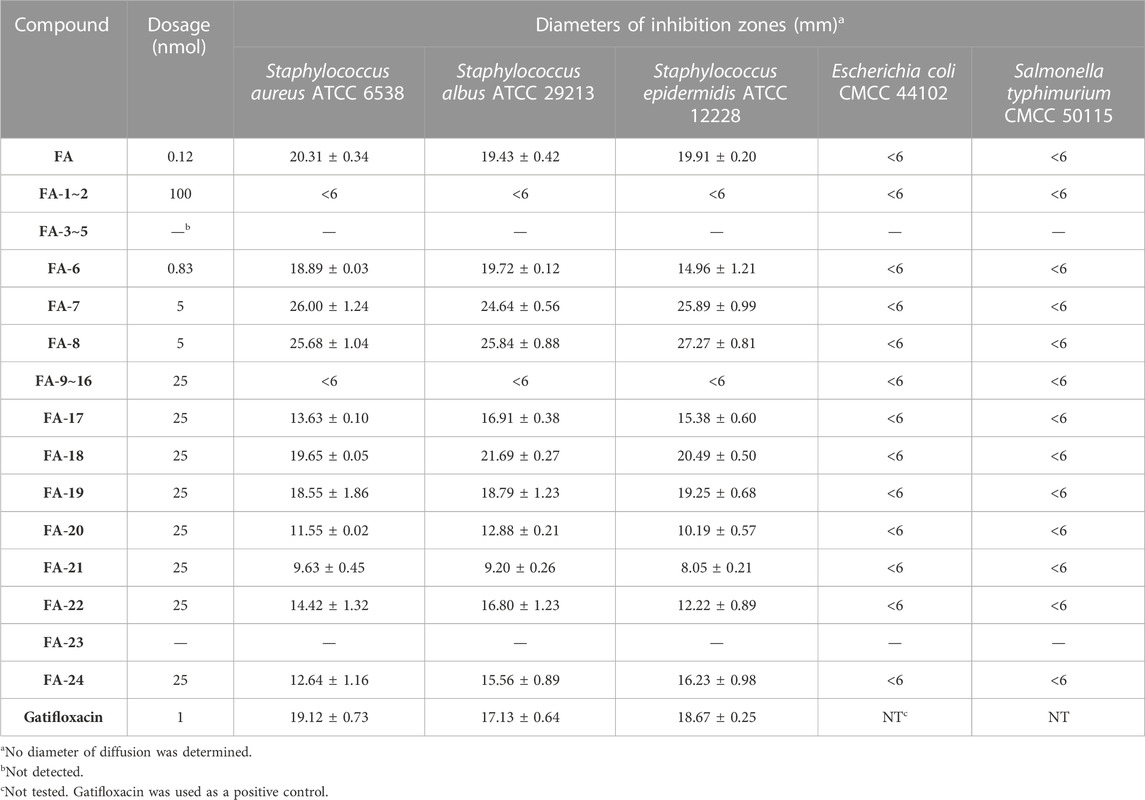
TABLE 3. The inhibition zone test of FA and its derivatives against bacterial strains.
Thenceforward, the MIC test was carried out to evaluate the antibacterial effect of the FA derivatives. As shown in Table 4, the C-25 positions of the FA derivatives were altered chemically with the halogen and hydrogen groups and the derivatives maintained the bioactivity against Gram-positive bacteria. FA-7 with a chlorine group as the substituent group at C-25 displayed the best medicinal property with a MIC of 3.125 µM. None of the intermediates showed any antibacterial activity in this assay. Simultaneously, it was noteworthy that esterification at C-21 resulted in the complete loss of activity. Therefore, the integrity of carboxyl at C-21 was indispensable for the preservation of activity. Additionally, the results of the bioassay showed that FA-20, FA-21, and FA-22 possessed weaker antimicrobial activity than those owned by the halogen groups. Therefore, the existence of the halogen groups was much more conducive to antibacterial activity.
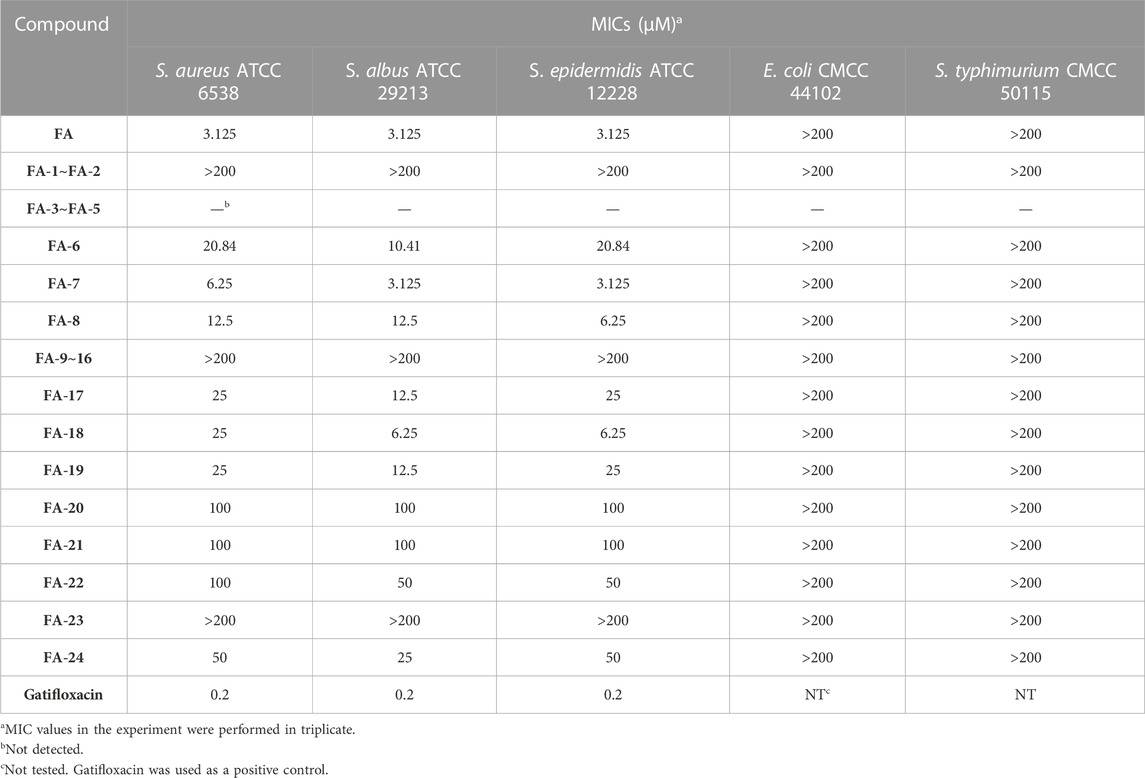
TABLE 4. MICs of FA and its derivatives against the bacterial strains.
Based on the pMIC (negative logarithm of the MIC) values of the synthesized FA derivatives, a comparative molecular similarity index analysis (CoMSIA) model was constructed to explore the structure–activity relationship of the constructed FA derivatives against S. aureus. Cross-validated coefficients (q2), non-cross-validated correlation coefficients (r2), standard error of estimates, and F-test values F) were 0.55, 0.921, 0.167, and 110.547 in the constructed CoMSIA model, respectively. The obtained q2 and r2 values were in the range of the internal validations (q2 > 0.5 and r2 > 0.8), which indicated that the predictive accuracy of the constructed 3D-QSAR models was credible. The results displayed a linear relationship between the experimental and predicted values as shown in the scatter plot (Figure 3A). As shown in Figure 3B, the aligned compounds were imported into Phase to make partial least-squares (PLS). The model was then used to correlate the activities of these compounds with the Phase field data calculated from their 3D structures. The steric contour map of CoMSIA is given in Figure 3C, and the result suggested that the larger the size of substituents at C-3, C-21, and C-25 positions, the stronger will be the antibacterial activity of derivatives. The electrostatic contour map in Figure 3D shows that the introduction of the atom with high electrostatic potential at C-21 and C-25 would be beneficial to improve the antibacterial activity of derivatives. In addition, the hydrophobic group and the group of hydrogen bond acceptors at C-3 and C-21 positions would contribute to enhanced antibacterial activities, as shown in Figures 3E, F. Overall, the presences of halogen groups at the positions C-3 and C-25 were more advantageous for maintaining antibacterial activity than FA.
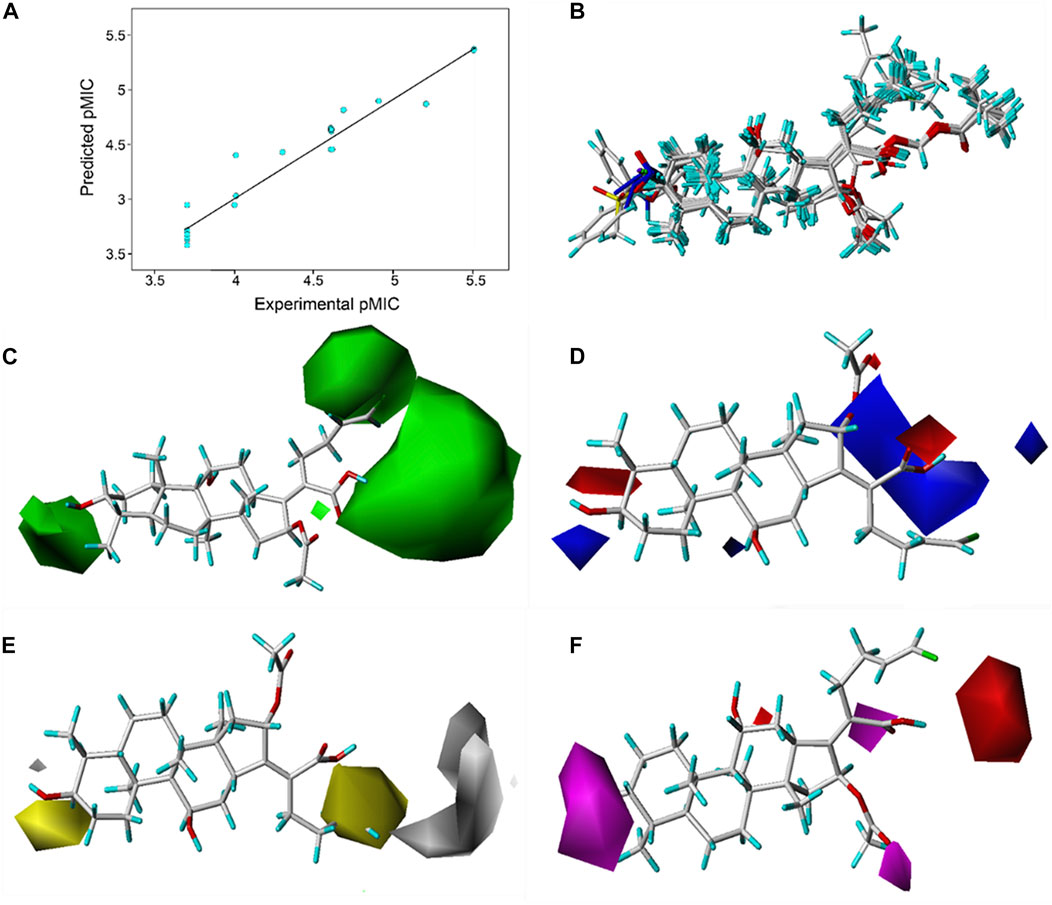
FIGURE 3. (A) Relationship between the experimental and predicted antibacterial activities by the QSAR model, pMIC = -lg MIC, showing R2 = 0.921; (B) flexible alignment of derivatives shown in the stick model; (C) steric fields, green favorable and yellow disfavored; (D) Phase electrostatic fields, blue favorable and red disfavored; (E) hydrophobic fields, yellow favorable and white disfavored; and (F) H-bond acceptor fields, magenta favorable and red disfavored.
The pharmacophore model was constructed using the Genetic Algorithm with Linear Assignment of Hypermolecular Alignment of Database (GALAHAD) module of SYBYL-X 2.1 software (Tripos Inc., St. Louis, MO, United States), and two similar models with varied parameters including specificity, N-hits, feats, and energy were first generated by setting 2, 5, and 4 for the parameters of population size, maximum generation, and mols, respectively. The pharmacophore model that was suitable for screening should basically meet the following requirements: specificity >4, N-hits (the number of compounds used for the construction), and relatively low energy that indicated stability. A decoy set method was then applied to evaluate the quality of the model. The decoy set in this study was composed of 19 FA derivatives with notable antibacterial activities taken from the published literature reports, as shown in Table 5. Following the creation of the pharmacophore models, the most effective model was carried out and a 3D search query was applied for the designed derivatives. Then, a column of Qfit parameters was loaded with the FA derivatives. Qfit is a value between 0 and 100, where 100 is the best. It represents how close the ligand atoms of the compounds match the query target coordinates. Meanwhile, the Qfit values for derivatives were shown to assess the degree of correlation with antibacterial activity. In this study, the minimum standard value of Qfit was first set to 50, and seven compounds with Qfit values of more than 50 were obtained.
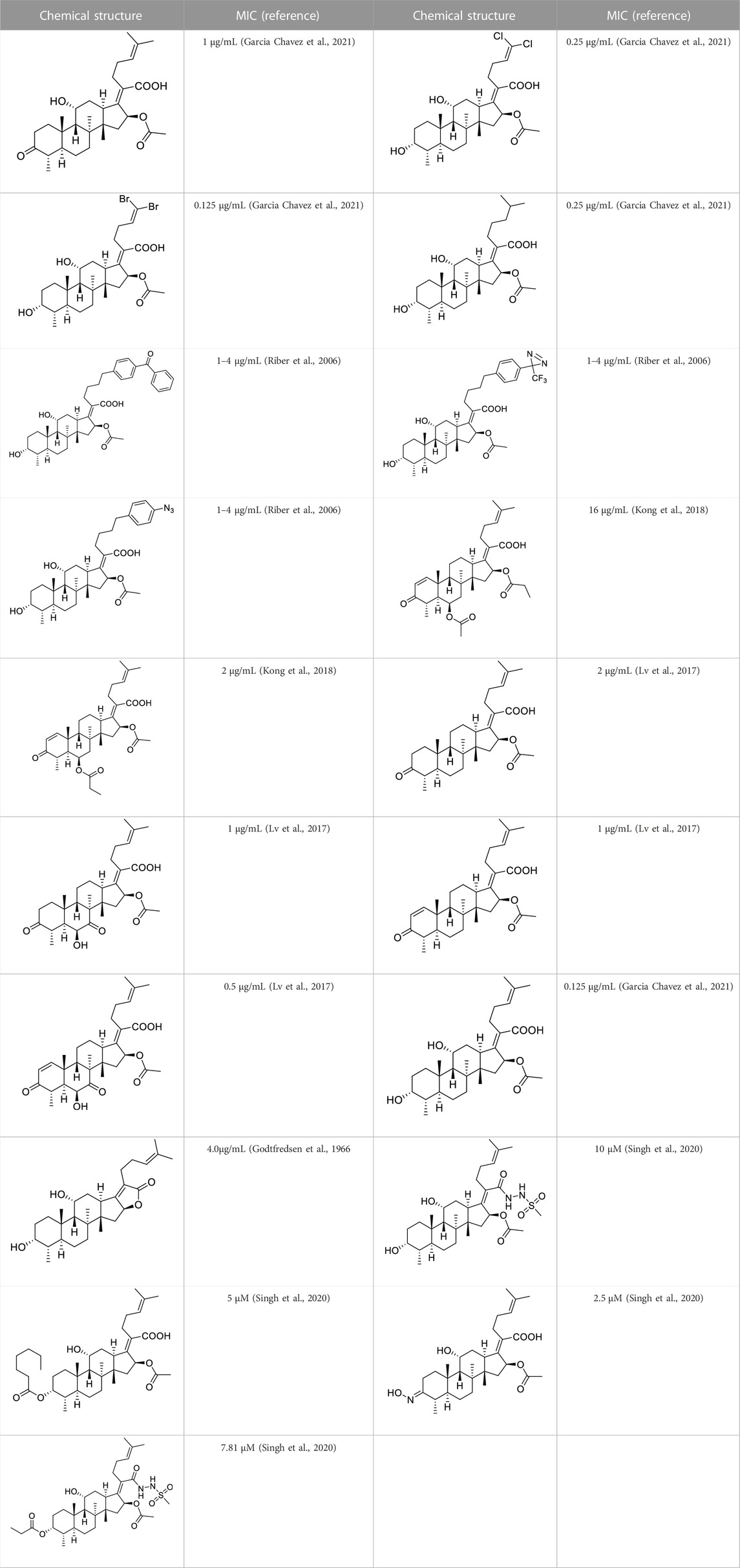
TABLE 5. Known FA derivatives with notable antibacterial activities used to generate a pharmacophore model.
To anticipate the ligand–receptor interactions, molecular docking experiments were conducted using SYBYL-X 2.0 software. The target protein EF-G (PDB: 2XEX) of S. aureus was selected as a receptor to bind the derivatives, and several significant residues were identified as the active protein pocket. The FA-binding site was bordered by Arg464, His457, Leu456, Thr436, Asp434, and Phe88, which created a specific cavity. All the hydrogens were added to EF-G in the structural model to improve the quality of the model. The Tripos force field and Gasteiger–Huckel charges were assigned for the EF-G and FA derivatives, respectively. After docking, the ligand–receptor complexes were opened in PyMol software for the visualizer to analyze the interaction.
All reagents were purchased from commercial suppliers of Adamas Reagent Ltd. (Shanghai, China) in analytical reagent grade and were used directly without further purification. Flash chromatography was carried out with silica gel (200–300 mesh) which was supplied by Innochem Co., Ltd. (Beijing, China). Analytical TLC was performed on pre-coated silica gel F254 plates (0.25 mm; E. Merck), and the products were visualized by UV detection or treated with an ethanolic solution of p-anisaldehyde spray followed by heating. The derivatives of FA were characterized by 1H NMR, 13C NMR, HRMS, and elemental analysis. The antibacterial activity was assayed by using a multi-model plate reader (Infinite 200).
First, chloromethyl pivalate (1.52 ml; 9.062 mmol) was added to the solution of FA (2 g; 4.513 mmol) in dry N, N-dimethylformamide (30 ml) at room temperature for 10 min. This was followed by drop-wise addition of triethylamine (0.7 mL; 5.890 mmol). The resulting reaction mixture was stirred at 50°C overnight. After completion of the reaction (TLC), the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by column chromatography using n-hexane: ethyl acetate = 1: 4 as the eluent, affording the target compound as a white solid (1.62 g; 81%). Mp: 76°C–78°C. 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 1H), 5.86 (d, J = 8.3 Hz, and 1H), 5.74 (dd, 2H), 5.08 (t, J = 7.0 Hz, and 1H), 4.34 (s, 1H), 3.74 (d, J = 8.3 Hz, and 1H), 3.05 (d, J = 11.1 Hz, and 1H), 2.54–2.36 (m, 2H), 2.31 (d, J = 14.1 Hz, and 1H), 2.25–2.06 (m, 4H), 1.98 (s, 3H), 1.93–1.79 (m, 2H), 1.79–1.70 (m, 2H), 1.67 (s, 3H), 1.61–1.47 (m, 8H), 1.37 (s, 3H), 1.29 (t, J = 16.0, 7.6 Hz, and 2H), 1.21 (s, 9H), 1.18–1.02 (m, 2H), 0.97 (s, 3H), 0.90 (d, J = 16.7, 7.5 Hz, and 6H). 13C NMR (100 MHz, CDCl3) δ 176.0, 171.3, 169.1, 151.9, 131.6, 128.3, 123.9, 79.8, 74.3, 71.4, 68.2, 49.2, 48.8, 44.3, 39.4, 39.0, 38.8, 37.1, 36.2, 36.2, 35.6, 32.4, 30.3, 30.0, 28.8, 28.2, 26.9, 25.7, 24.2, 22.8, 20.8, 20.8, 17.9, 17.8, 15.9. Anal. calcd. for C37H58O8: C 70.44, H 9.27; found: C 69.81, H 9.32. HRMS (ESI): C37H58O8Na (653.4029) [M + Na]+ = 653.4027.
In a 50-mL round-bottom flask, FA-1 (100 mg; 0.166 mmol) was dissolved in dichloromethane (5 mL). Then NMO (18.56 mg; 0.249 mmol) and OsO4 (0.9 μL, 0.0166 mmol, and 0.1 eq.) in MeCN were added, respectively. The solution of 2% O3/O2 (nominal output of 1 mmol O3/min) was introduced directly above the solution via a glass pipet for 6.6 min (nominally 2.2 equiv ozone relative to alkene) at 0°C for 1 h. The reaction was then quenched by the addition of 10 mL of saturated sodium thiosulfate. The reaction was stirred for an additional 45 min and then concentrated in vacuo to remove dichloromethane. Subsequently, EtOAc and brine were added, and the layers were separated. The aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using an eluent (n-hexane: ethyl acetate = 3: 2, V: V). FA-2 as a white solid was obtained. Yield: 90%. 1H NMR (400 MHz, CDCl3) δ 9.76 (s, 1H), 5.96 (d, J = 8.4 Hz, 1H), 5.74 (dd, J = 28.0, 5.4 Hz, 2H), 4.35 (s, 1H), 3.71 (d, J = 2.0 Hz, 1H), 3.10 (d, J = 10.6 Hz, 1H), 2.75–2.46 (m, 5H), 2.39–2.24 (m, 2H), 2.18 (d, J = 3.9 Hz, 2H), 1.98 (s, 3H), 1.90–1.40 (m, 8H), 1.36 (s, 3H), 1.28 (dd, J = 12.2, 11.0 Hz, 2H), 1.21 (s, 9H), 1.17–1.03 (m, 2H), 0.97 (s, 3H), 0.90 (d, J = 6.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 201.2, 177.1, 170.1, 167.4, 153.2, 127.1, 79.9, 74.2, 71.4, 67.9, 49.4, 48.8, 44.6, 43.8, 39.5, 38.8, 38.7, 36.8, 36.6, 35.5, 35.3, 31.3, 30.9, 29.8, 26.8, 23.6, 23.4, 21.3, 21.1, 20.8, 17.7, and 16.0. HRMS (ESI): C34H52O9Na (627.3509) [M + Na]+ = 627.3503.
A solution of methyltriphenylphosphonium bromide (389.04 mg; 1.09 mmol) and potassium tert-butoxide (122.2 mg, 1.09 mol) was added to the mixture of FA-2 (441.49 mg, 0.73 mol) in 20 mL of toluene under nitrogen. The reaction was kept refluxed overnight and monitored by TLC. After completion of the reaction (TLC), the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using an eluent (n-hexane: ethyl acetate = 3: 2, V: V). FA-3 as a white solid was obtained. Yield: 75%. Anal. calcd. for C35H54O8: C 69.74, H 9.03; found: C 68.93, H 8.96. HRMS (ESI): C35H54O8Na (625.3716) [M + Na]+ = 625.3709 (Zhao et al., 2016).
A solution of FA-2 (200 mg, 0.34 mmol) and (chloromethyl) triphenylphosphonium chloride (212 mg, 0.68 mmol) in anhydrous tetrahydrofuran (15 mL) was cooled to 0°C under nitrogen for 15 min, and then n-butyllithium (416.7 μL) dissolved in n-hexane (0.68 mmol, 1.6 mol/L) was added drop-wise above the solution and stirred at 0°C for 30 min. After completion of the reaction (TLC), the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using an eluent (n-hexane: ethyl acetate = 3: 2, V: V). FA-4 as a white solid was obtained. Yield: 43% (Zhao et al., 2016).
A solution of FA-2 (200 mg, 0.34 mmol) and (bromomethyl) triphenylphosphonium bromide (296.7 mg, 0.68 mmol) in anhydrous tetrahydrofuran (15 mL) was cooled to 0°C under nitrogen for 15 min, and then, n-butyllithium (416.7 μL) dissolved in n-hexane (0.68 mmol, 1.6 mol/L) was added drop-wise above the solution and stirred at 0°C for 30 min. After completion of the reaction (TLC), the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using an eluent (n-hexane: ethyl acetate = 3: 2, V: V). FA-5 as a white solid was obtained. Yield: 32%. 1H NMR (400 MHz, CDCl3) δ 6.22–6.11 (m, 1H), 6.11–6.00 (m, 1H), 5.93–5.84 (m, 1H), 5.80 (dd, J = 8.9, 5.4 Hz, 1H), 5.70 (d, J = 5.4 Hz, 1H), 4.34 (s, 1H), 3.75 (s, 1H), 3.07 (t, J = 12.2 Hz, 1H), 2.64–2.44 (m, 2H), 2.42–2.05 (m, 6H), 1.98 (d, J = 1.6 Hz, 3H), 1.93–1.69 (m, 4H), 1.65–1.48 (m, 4H), 1.37 (s, 3H), 1.35–1.23 (m, 3H), 1.21 (s, 9H), 1.27–1.03 (m, 2H), 0.99 (s, 3H), and 0.92 (d, J = 6.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 177.1, 170.2, 167.7, 152.5, 136.4, 133.2, 128.2, 108.7, 105.5, 79.8, 74.3, 71.4, 68.2, 49.2, 48.9, 44.5, 39.5, 39.0, 38.8, 37.1, 36.2, 36.2, 35.7, 32.5, 30.3, 30.0, 29.8, 27.0, 26.9, 24.2, 22.7, 20.8, 20.7, 18.0, and 15.9. HRMS (ESI): C35H53BrNaO8 (703.2822) [M + Na]+ = 703.2606.
A solution of derivatives (FA-3, FA-4, and FA-5, respectively; 0.0924 mmol) and potassium carbonate (25.55 mg, 0.185 mmol) in methanol was stirred at room temperature for 1 h and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using an eluent (n-hexane: ethyl acetate = 3: 2, V: V). FA-6, FA-7, and FA-8 were obtained, respectively.
24-ene-Fusidic acid (FA-6). White solid. Yield: 55%. 1H NMR (400 MHz, CDCl3) δ 5.88 (d, J = 8.3 Hz, 1H), 5.86–5.73 (m, 1H), 5.04 (d, J = 17.1 Hz, 1H), 4.97 (d, J = 10.1 Hz, 1H), 4.34 (s, 1H), 3.73 (s, 1H), 3.05 (d, J = 12.0 Hz, 1H), 2.45–2.41 (m, 2H), 2.29 (d, J = 13.8 Hz, 1H), 2.24–2.03 (m, 5H), 1.99 (s, 3H), 1.84 (t, J = 13.2 Hz, 2H), 1.78–1.66 (m, 2H), 1.66–1.53 (m, 3H), 1.49 (d, J = 12.8 Hz, 1H), 1.43 (s, 1H), 1.38 (s, 3H), 1.29 (d, J = 5.6 Hz, 1H), 1.26 (s, 1H), 1.18–1.05 (m, 2H), 0.98 (s, 3H), and 0.92 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 172.4, 171.4, 148.6, 137.6, 130.1, 115.1, 74.4, 71.4, 68.2, 49.4, 48.8, 43.9, 39.6, 38.9, 36.8, 36.6, 35.8, 35.5, 33.8, 31.8, 30.0, 29.8, 28.0, 23.6, 23.3, 21.1, 20.6, 17.6, and 15.9. Anal. calcd. for C29H44O6: C 71.28, H 9.08; found: C 70.42, H 9.12. HRMS (TOF): C29H43O6 (487.3060) [M-H]- = 487.3058.
(E)-25-chlorohexa-24-ene-fusidic acid (FA-7). White solid. Yield: 55%. 1H NMR (400 MHz, CDCl3) δ 6.02 (dd, J = 19.0, 10.2 Hz, 1H), 5.95–5.70 (m, 2H), 4.36 (s, 1H), 3.76 (s, 1H), 3.08 (t, J = 10.3 Hz, 1H), 2.65–2.48 (m, 2H), 2.48–2.21 (m, 3H), 2.21–2.06 (m, 3H), 1.97 (s, 3H), 1.92–1.67 (m, 4H), 1.67–1.46 (m, 4H), 1.38 (s, 3H), 1.31 (d, J = 14.3 Hz, 1H), 1.28–1.18 (m, 2H), 1.18–1.03 (m, 2H), 0.98 (s, 3H), and 0.92 (d, J = 5.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 173.9 [173.7 (for the second diastereoisomer)], 170.7, 152.44 [152.39 (for the second diastereoisomer)], 132.5 [130.2 (for the second diastereoisomer)], 128.6 [128.3 (for the second diastereoisomer)], 118.9 [118.0 (for the second diastereoisomer)], 74.44 [74.40 (for the second diastereoisomer)], 71.6, 68.2 [68.1 (for the second diastereoisomer)], 49.27 [49.26 (for the second diastereoisomer)], 48.78 [48.77 (for the second diastereoisomer)], 44.50 [44.45 (for the second diastereoisomer)], 39.5, 38.90 [38.88 (for the second diastereoisomer)], 36.9, 36.4, 36.0 [35.9 (for the second diastereoisomer)], 35.6 [35.5 (for the second diastereoisomer)], 32.08 [32.06 (for the second diastereoisomer)], 31.2 [27.9 (for the second diastereoisomer)], 30.3 [30.1 (for the second diastereoisomer)], 29.8 [27.4 (for the second diastereoisomer)], 27.2 [27.0 (for the second diastereoisomer)], 24.0, 23.06 [23.03 (for the second diastereoisomer)], 20.9, 20.6, 17.9, and 16.0. Anal. calcd. for C29H43ClO6: C 66.59, H 8.29; found: C 65.21, H 8.34. HRMS (ESI): C29H42ClO6 (521.2670) [M-H]- = 521.2656.
(E)-25-bromohexa-24-ene-fusidic acid (FA-8). White solid. Yield: 80%. 1H NMR (400 MHz, CDCl3) δ 6.22–6.15 (m, 1H), 6.15–6.03 (m, 1H), 5.92 (d, J = 8.3 Hz, 1H), 4.35 (s, 1H), 3.76 (s, 1H), 3.08 (t, J = 12.0 Hz, 1H), 2.62–2.46 (m, 2H), 2.46–2.06 (m, 6H), 1.98 (s, 3H), 1.93–1.67 (m, 4H), 1.67–1.46 (m, 4H), 1.38 (s, 3H), 1.35–1.23 (m, 2H), 1.21 (t, J = 7.0 Hz, 1H), 1.18–1.03 (m, 2H), 0.98 (s, 3H), and 0.95–0.88 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 173.7, 170.9, 152.3, 136.8 [133.6 (for the second diastereoisomer)], 128.9, 108.7 [105.6 (for the second diastereoisomer)], 74.5, 71.7, 68.4, 49.4, 49.0, 44.6, 39.6, 39.1, 37.1, 36.4, 36.2, 35.8, 33.2, 32.4 [30.3 (for the second diastereoisomer)], 30.2, 30.0, 27.2, 24.2, 23.0, 21.0, 20.8, 18.1, and 16.1. Anal. calcd. for C29H43BrO6: C 61.37, H 7.64; found: C 60.41, H 7.66. HRMS (ESI): C29H4279BrO6 (565.2165) [M-H]- = 565.2147; C29H4281BrO6 (567.2144) [M-H]- = 567.2146.
A solution of FA (200 mg, 0.32 mmol) and sodium hydroxide (1.95 mmol, 1.2 mL) in methanol was refluxed overnight and monitored by TLC. After completion of the reaction, 1 N hydrochloric acid was added to adjust the pH to 2–3. The mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using an eluent (n-hexane: ethyl acetate = 1: 1, V: V). FA-9 as a white solid was obtained. Yield: 74%. 1H NMR (400 MHz, CDCl3) δ 5.11 (t, J = 6.0 Hz, 1H), 4.95 (dd, J = 10.9, 4.2 Hz, 1H), 4.39 (s, 1H), 3.75 (s, 1H), 3.53 (d, J = 11.7 Hz, 1H), 2.41–2.15 (m, 6H), 2.15–1.96 (m, 4H), 1.90–1.78 (m, 2H), 1.75 (d, J = 13.3 Hz, 1H), 1.68 (s, 3H), 1.66–1.62 (m, 1H), 1.60 (s, 3H), 1.57–1.46 (m, 6H), 1.32–1.07 (m, 4H), 0.97 (s, 3H), 0.94 (d, J = 6.7 Hz, 3H), and 0.82 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 176.8, 169.1, 132.9, 123.6, 123.4, 82.0, 71.5, 68.0, 55.3, 50.6, 40.9, 38.3, 37.2, 36.9, 36.0, 34.2, 31.8, 31.6, 30.2, 30.0, 27.6, 25.8, 24.2, 23.5, 23.2, 21.2, 20.1, 17.9, and 16.1. Anal. calcd. for C29H44O4: C 76.27, H 9.71; found: C 74.91, H 9.57. HRMS (TOF): C29H44O4Na (479.3137) [M + Na]+ = 479.3136.
A solution of FA-1 (615 mg, 0.98 mmol) and pyridine (236.8 μL, 2.4 mmol) in anhydrous dichloromethane (20 mL) was stirred at 0°C for 15 min and methane sulfonyl chloride (1.95 mmol, 151.23 μL) was added drop-wise. The reaction was stirred overnight and monitored by TLC. After completion of the reaction, 1 N hydrochloric acid was added to adjust pH to 2–3. The mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. FA-10 as a white solid was obtained. Yield: 60%. HRMS (TOF): C38H60O10NaS (731.3805) [M + Na]+ = 731.3812.
A solution of FA-10 (100 mg, 0.141 mmol) and tetrabutylammonium chloride (78.5 mg, 0.282 mmol) in tetrahydrofuran (5 mL) was stirred at 80°C for 1 h and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. FA-11 as a white solid was obtained. Yield: 49.1%. HRMS (TOF): C37H57O7NaCl (671.3691) [M + Na]+ = 671.3678.
A solution of FA-10 (100 mg, 0.141 mmol) and tetrabutylammonium bromine (91.0 mg, 0.282 mmol) in dimethyl sulfoxide (5 mL) was stirred at room temperature overnight and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. FA-12 as a white solid was obtained. Yield: 51.0%. HRMS (ESI): C37H57O7Na79Br (715.3171) [M + Na]+ = 715.3185; C37H57O7Na81Br (717.3177) [M + Na]+ = 717.3165.
A solution of FA-10 (100 mg, 0.141 mmol) and tetrabutylammonium iodide (104.3 mg, 0.282 mmol) in tetrahydrofuran (5 mL) was stirred at room temperature for 6 h and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. FA-13 as a white solid was obtained. Yield: 40.5%. HRMS (ESI): C37H57O7NaI (763.3047) [M + Na]+ = 763.3053.
A solution of FA-10 (100 mg, 0.141 mmol) and sodium azide (18.3 mg, 0.282 mmol) in dimethyl sulfoxide (5 mL) was stirred at 90°C overnight and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. FA-14 as a white solid was obtained. Yield: 15.7%. HRMS (ESI): C37H57N3O7Na (678.4094) [M + Na]+ = 678.4101.
A solution of FA-10 (100 mg, 0.141 mmol) and tetrabutylammonium nitrate (85.9 mg, 0.282 mmol) in dimethyl sulfoxide (5 mL) was stirred at 70°C overnight and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. FA-15 as a white solid was obtained. HRMS (ESI): C37H57NO9Na (682.3931) [M + Na]+ = 682.3917.
A solution of FA-10 (100 mg, 0.141 mmol), triethylamine (28.6 mg, 0.282 mmol), and aniline (26.3 mg, 0.282 mmol) in tetrahydrofuran (5 mL) was stirred at 90°C overnight and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. FA-16 as a white solid was obtained. Yield: 45.2%. HRMS (ESI): C43H64NO7 (706.4683) [M + H]+ = 706.4667.
A solution of FA-10 (0.759 g, 1.32 mmol, 1.0 equiv) in 2,6-lutidine (5.00 mL) was heated to 130°C and stirred at the same temperature for 2 h and monitored by TLC. Upon completion, the reaction mixture was cooled to 23°C and then concentrated directly. A white solid was obtained. Yield: 95.5%. HRMS (ESI): C37H56O7Na (635.3924) [M + Na]+ = 635.3918.
A solution of the derivatives (FA-11∼FA-16 and FA-23, respectively; 0.185 mmol) and potassium carbonate (25.55 mg, 0.185 mmol) in methanol (5 mL) was stirred at room temperature for 1 h and monitored by TLC. After completion of the reaction, the mixture was diluted with EtOAc and washed with water. The EtOAc layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography using an eluent (n-hexane: ethyl acetate = 1: 1, V: V). The derivatives (FA-17∼24) were obtained, respectively.
3α-Chloro-21-fusidic acid (FA-17, C31H47O5Cl). White solid, Yield: 97%. HRMS (ESI): C31H47O5NaCl (557.3010) [M + Na]+ = 557.2997.
3α-Bromo-21-fusidic acid (FA-18, C31H47O5Br). White solid. Yield: 85.5%. HRMS (ESI): C31H46O579Br (577.2529) [M-H]- = 577.2534; C31H46O581Br (579.2508) [M-H]- = 579.2522.
3α-Iodo-21-fusidic acid (FA-19, C31H47O5I). White solid. Yield: 97.6%. HRMS (ESI): C31H46O5I (625.2390) [M-H]- = 625.2380.
3α-Azido-21-fusidic acid (FA-20, C31H47N3O5). White solid. Yield: 58.0%. HRMS (ESI): C31H46N3O5 (540.3437) [M-H]- = 540.3428.
3α-Nitrohexadecahydro-21-fusidic acid (FA-21, C31H48NO7). White solid. Yield: 58.0%.
3α-Phenylamino-21-fusidic acid (FA-22, C37H53NO5). White solid. Yield: 65.5%. HRMS (ESI): C37H52NO5 (590.3845) [M-H]- = 590.3838.
3-Ene-21-fusidic acid (FA-24, C31H46O5). White solid. Yield: 98.3%. HRMS (ESI): C31H46O5Na (521.3243) [M + Na]+ = 521.3235.
The standard agar diffusion method with a slight modification was used for the determination of the antibacterial efficacy of the FA derivatives (Luangtongkum et al., 2007; Gaudreau et al., 2008; Benamrouche et al., 2014). S. aureus (ATCC 6538), S. albus (ATCC 29213), S. epidermidis (ATCC 12228), S. typhimurium (CMCC 50115), and E. coli (CMCC 44102) were cultured in a liquid medium, Mueller–Hinton Agar (MHA), at 37°C. Bacterial suspensions of 1.5 × 106 CFU/mL with 400 μL prepared were uniformly inoculated onto MHA solidified in 60-mm Petri dishes. Sterile filter paper disks of 6 mm diameter containing 5 μL different concentrations of compounds were pressed gently against the surface of the agar. A disk containing Gatifloxacin was used as a positive control, while DMSO was used as the negative control. Then the disks were incubated in a constant temperature incubator at 37°C for 24 h and the bacteriostatic circles were observed. The inhibition zone (IZ) diameter was measured using a vernier caliper. All the experiments were performed in triplicate.
The MIC was determined by a microdilution method in 96-well plates according to the Clinical and Laboratory Standards Institute (CLSI) with a slight modification. (Sader et al., 2006). Liquid media were used to cultivate the test bacteria at 37°C. Then, 195-μL bacterial suspensions containing 1.5 × 105 CFU/mL with 5 μL different derivative concentrations were added to 96-cell plates and the plates were incubated at 37°C for 24 h. In each well, OD values of derivatives were measured at 600 nm and compared with blank controls without bacteria and negative controls with bacteria. The lowest concentration of compounds, which did not show any visible growth of the test organisms after macroscopic evaluation, was determined as the MIC. Gatifloxacin served as the positive control and DMSO served as the negative control.
The MIC values (μM) of the constructed 22 derivatives (FA, FA-1, FA-2, and FA-6∼24) were converted into their corresponding negative logarithms (pMIC) for the 3D-QSAR model analysis by SYBYL-X 2.0 software (Shanghai Tri-I. Biotech. Inc., China) (Wang et al., 2019; Chen et al., 2020). Three-dimensional molecular conformations were successively optimized using the Gasteiger−Huckel charge, Tripos force field, and Powell conjugate gradient algorithm until the obtained convergence criteria were minimized in molecular energies. Three-dimensional structures of derivatives were aligned on the common scaffold of the template molecule FA-7 that exhibited the best in vitro antibacterial activity against Gram-positive bacteria among the 22 synthesized derivatives. A partial least-squares (PLS) technique was applied for optimizing the obtained 3D-QSAR model. Subsequently, the obtained PLS coefficients and standard descriptor values were used to generate their corresponding contour maps including steric, electrostatic, hydrophobic, and hydrogen bond acceptors.
In this study, a ligand-based pharmacophore model was constructed and seven FA derivatives were designed according to the reported structure–activity relationship and the pharmacophore characteristics. The designed FA derivatives were applied to analyze the matching degree with the pharmacophore model through Qfit values, and partially designed FA derivatives were docked onto the EF-G of S. aureus to study the bonding with the target protein. Finally, the designed FA derivatives were synthesized and their antibacterial activities were evaluated by the inhibition zone test and the MIC test. Afterward, 3D-QSAR was carried out on all the derivatives, and the results indicated that the substituents at the C-3, C-21, and C-25 positions would exert an influence on the antibacterial activity of derivatives. In summary, this study provides a promising computational approach to design FA derivatives with highly potent antibacterial activity.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Writing—original draft, software, formal analysis, and methodology: WZ and BT; writing—original draft, resources, and methodology: ZZ; software, formal analysis, and investigation: JL; methodology and investigation: ZY; validation and investigation: KS; formal analysis and investigation: DD; methodology and investigation: YS; validation and investigation: XW; project administration and resources: BZ; funding acquisition, supervision, and project administration: KZ and W-LW; writing—review and editing, supervision, project administration, and data curation: PW and WH; and writing—review and editing, methodology, project administration, and funding acquisition: SA. All authors read and agreed to the published version of the manuscript.
This study was supported by the National Natural Science Foundation of China (Nos. 81803390 and 22077020), Natural Science Foundation of Guangdong Province (No. 2021A1515010221), Hong Kong and Macao Joint Research and Development Foundation of 2021 (No. 2021WGALH09), Special Funds for the Cultivation of Guangdong College Students’ Scientific and Technological Innovation (“Climbing Program” Special Funds, No. pdjh 2022a0523), and Special Fund Project of Science and Technology Innovation Strategy of Guangdong Province 2018 and 2020 [Nos. Jiangke (2018)352 and Jiangke (2020)182]. The authors acknowledge the Foundation of the Department of Education of Guangdong Province (Nos. 2020KZDZX1202 and 2022KTSCX144).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with the author BZ at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1094841/full#supplementary-material
Belardinelli, R., and Rodnina, M. V. (2017). Effect of fusidic acid on the kinetics of molecular motions during EF-G-induced translocation on the ribosome. Sci. Rep. 7, 10536. doi:10.1038/s41598-017-10916-8
Benamrouche, N., Lazri, M., Hassiba, T. M., and Rahal, K. (2014). Comparison of Corynebacterium diphtheriae susceptibility testing to antibiotics by the broth dilution and diffusion (E-test and disk) methods. Med. Maladies Infect. 44, 392–393. doi:10.1016/j.medmal.2014.07.007
Bodley, J. W., Zieve, F. J., Lin, L., and Zieve, S. T. (1969). Formation of the ribosome-G factor-GDP complex in the presence of fusidic acid. Biochem. Biophysical Res. Commun. 37, 437–443. doi:10.1016/0006-291X(69)90934-6
Borg, A., Holm, M., Shiroyama, I., Hauryliuk, V., Pavlov, M., Sanyal, S., et al. (2015). Fusidic acid targets elongation factor G in several stages of translocation on the bacterial ribosome. J. Biol. Chem. 290, 3440–3454. doi:10.1074/jbc.M114.611608
Cardoso, M. H., Orozco, R. Q., Rezende, S. B., Rodrigues, G., Oshiro, K. G. N., Candido, E. S., et al. (2019). Computer-aided design of antimicrobial peptides: Are we generating effective drug candidates? Front. Microbiol. 10, 3097. doi:10.3389/fmicb.2019.03097
Cerqueira, N. M. F. S. A., Gesto, D., Oliveira, E. F., Santos-Martins, D., Brás, N. F., Sousa, S. F., et al. (2015). Receptor-based virtual screening protocol for drug discovery. Archives Biochem. Biophysics 582, 56–67. doi:10.1016/j.abb.2015.05.011
Chen, J., Luo, Y., Wei, C., Wu, S., Wu, R., Wang, S., et al. (2020). Novel sulfone derivatives containing a 1, 3, 4-oxadiazole moiety: Design and synthesis based on the 3D-QSAR model as potential antibacterial agent. Pest Manag. Sci. 76, 3188–3198. doi:10.1002/ps.5873
Chen, Y., Koripella, R. K., Sanyal, S., and Selmer, M. (2010). Staphylococcus aureus elongation factor G – structure and analysis of a target for fusidic acid. FEBS J. 277, 3789–3803. doi:10.1111/j.1742-4658.2010.07780.x
Chen, Z., Li, H., Zhang, Q., Bao, X., Yu, K., Luo, X., et al. (2009). Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 30, 1694–1708. doi:10.1038/aps.2009.159
Collignon, P., and Turnidge, J. (1999). Fusidic acid in vitro activity. Int. J. Antimicrob. Agents 12, S45–S58. doi:10.1016/S0924-8579(98)00073-9
Duvold, T., Sørensen, M. D., Björkling, F., Henriksen, A. S., and Rastrup-Andersen, N. (2001). Synthesis and conformational analysis of fusidic acid side chain derivatives in relation to antibacterial activity. J. Med. Chem. 44, 3125–3131. doi:10.1021/jm010899a
Fjell, C. D., Hiss, J. A., Hancock, R. E. W., and Schneider, G. (2012). Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 11, 37–51. doi:10.1038/nrd3591
Garcia Chavez, M., Garcia, A., Lee, H. Y., Lau, G. W., Parker, E. N., Komnick, K. E., et al. (2021). Synthesis of fusidic acid derivatives yields a potent antibiotic with an improved resistance profile. ACS Infect. Dis. 7, 493–505. doi:10.1021/acsinfecdis.0c00869
Gaudreau, C., Girouard, Y., Gilbert, H., Gagnon, J., and Bekal, S. (2008). Comparison of disk diffusion and agar dilution methods for erythromycin, ciprofloxacin, and tetracycline susceptibility testing of campylobacter coli and for tetracycline susceptibility testing of campylobacter jejuni subsp jejuni. Antimicrob. Agents Chemother. 52, 4475–4477. doi:10.1128/AAC.00767-08
Godtfredsen, W. O., Albrethsen, C., Daehne, W. V., Tybring, L., and Vangedal, S. (1965). Transformations of fusidic acid and the relationship between structure and antibacterial activity. Antimicrob. Agents Chemother. (Bethesda) 5, 132–137. doi:10.1021/jm00319a004
Godtfredsen, W. O., Von Daehne, W., Tybring, L., and Vangedal, S. (1966). Fusidic acid derivatives. I. Relationship between structure and antibacterial activity. J. Med. Chem. 9, 15–22. doi:10.1021/jm00319a004
Kong, F., Huang, X., Ma, Q., Xie, Q., Wang, P., Chen, P., et al. (2018). Helvolic acid derivatives with antibacterial activities against Streptococcus agalactiae from the marine-derived fungus Aspergillus fumigatus HNMF0047. J. Nat. Prod. 81, 1869–1876. doi:10.1021/acs.jnatprod.8b00382
Lannergård, J., Norström, T., and Hughes, D. (2009). Genetic determinants of resistance to fusidic acid among clinical bacteremia isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 53, 2059–2065. doi:10.1128/AAC.00871-08
Lu, J., Ni, J., Wang, J., Liu, Z., Shang, K., and Bi, Y. (2019). Integration of multiscale molecular modeling approaches with the design and discovery of fusidic acid derivatives. Future Med. Chem. 11, 1427–1442. doi:10.4155/fmc-2018-0567
Luangtongkum, T., Morishita, T. Y., El-Tayeb, A. B., Ison, A. J., and Zhang, Q. (2007). Comparison of antimicrobial susceptibility testing of Campylobacter spp. by the agar dilution and the agar disk diffusion methods. J. Clin. Microbiol. 45, 590–594. doi:10.1128/JCM.00986-06
Lv, J.-M., Hu, D., Gao, H., Kushiro, T., Awakawa, T., Chen, G.-D., et al. (2017). Biosynthesis of helvolic acid and identification of an unusual C-4-demethylation process distinct from sterol biosynthesis. Nat. Commun. 8, 1644. doi:10.1038/s41467-017-01813-9
Mouchlis, V. D., Melagraki, G., Zacharia, L. C., and Afantitis, A. (2020). Computer-aided drug design of β-secretase, γ-secretase and anti-tau inhibitors for the discovery of novel alzheimer's therapeutics. Int. J. Mol. Sci. 21, 703. doi:10.3390/ijms21030703
Niu, Y., Ma, C., Jin, H., Xu, F., Gao, H., Liu, P., et al. (2012). The discovery of novel beta-secretase inhibitors: Pharmacophore modeling, virtual screening, and docking studies. Chem. Biol. Drug Des. 79, 972–980. doi:10.1111/j.1747-0285.2012.01367.x
Petrosillo, N., Granata, G., and Cataldo, M. A. (2018). Novel antimicrobials for the treatment of clostridium difficile infection. Front. Med. 5, 96. doi:10.3389/fmed.2018.00096
Ragab, A. E., Ibrahim, A.-R. S., and Leon, F. (2020). 3-O-formyl -27-hydroxyfusidic acid: A new metabolite of fusidic acid by cunninghamella echinulata. Rec. Nat. Prod. 14, 292–296. doi:10.25135/rnp.168.19.12.1505
Riber, D., Venkataramana, M., Sanyal, S., and Duvold, T. (2006). Synthesis and biological evaluation of photoaffinity labeled fusidic acid analogues. J. Med. Chem. 49, 1503–1505. doi:10.1021/jm050583t
Sader, H. S., Fritsche, T. R., and Jones, R. N. (2006). Daptomycin bactericidal activity and correlation between disk and broth microdilution method results in testing of Staphylococcus aureus strains with decreased susceptibility to vancomycin. Antimicrob. Agents Chemother. 50, 2330–2336. doi:10.1128/AAC.01491-05
Salimova, E. V., Mamaev, A. G., Tret’yakova, E. V., Kukovinets, O. S., Mavzyutov, A. R., Shvets, K. Y., et al. (2018). Synthesis and biological activity of cyanoethyl derivatives of fusidic acid. Russ. J. Org. Chem. 54, 1411–1418. doi:10.1134/S1070428018090245
Sangeetha, K., Sasikala, R. P., and Meena, K. S. (2017). Pharmacophore modeling, virtual screening and molecular docking of ATPase inhibitors of HSP70. Comput. Biol. Chem. 70, 164–174. doi:10.1016/j.compbiolchem.2017.05.011
Schwartz, C., Raible, J., Mott, K., and Dussault, P. H. (2006). Fragmentation of carbonyl oxides by N-oxides: An improved approach to alkene ozonolysis. Org. Lett. 8, 3199–3201. doi:10.1021/ol061001k
Sharma, T., Harioudh, M. K., Kuldeep, J., Kumar, S., Banerjee, D., Ghosh, J. K., et al. (2020). Identification of potential inhibitors of cathepsin-B using shape & pharmacophore-based virtual screening, molecular docking and explicit water thermodynamics. Mol. Inf. 39, 1900023. doi:10.1002/minf.201900023
Singh, K., Kaur, G., Shanika, P. S., Dziwornu, G. A., Okombo, J., and Chibale, K. (2020). Structure-activity relationship analyses of fusidic acid derivatives highlight crucial role of the C-21 carboxylic acid moiety to its anti-mycobacterial activity. Bioorg. Med. Chem. 28, 115530. doi:10.1016/j.bmc.2020.115530
Talambedu, U., Dhivya, S., Arvind Kumar, G., Chinaga Suresh, K., and Sushil Kumar, M. (2017). Recent updates on computer-aided drug discovery: Time for a paradigm shift. Curr. Top. Med. Chem. 17, 3296–3307. doi:10.2174/1568026618666180101163651
Tanaka, N., Kinoshita, T., and Masukawa, H. (1968). Mechanism of protein synthesis inhibition by fusidic acid and related antibiotics. Biochem. Biophysical Res. Commun. 30, 278–283. doi:10.1016/0006-291X(68)90447-6
Turnidge, J. (1999). Fusidic acid pharmacology, pharmacokinetics and pharmacodynamics. Int. J. Antimicrob. Agents 12, S23–S34. doi:10.1016/S0924-8579(98)00071-5
Von Daehne, W., Godtfredsen, W., and Rasmussen, P. (1979). Structure-activity relationships in fusidic acid-type antibiotics. Adv. Appl. Microbiol. 25, 95–146. doi:10.1016/s0065-2164(08)70148-5
Wang, M., Zhu, H., Wang, P., Zeng, D., Wu, Y., Liu, L., et al. (2019). Synthesis of thiazolium-labeled 1, 3, 4-oxadiazole thioethers as prospective antimicrobials: In vitro and in vivo bioactivity and mechanism of action. J. Agric. Food Chem. 67, 12696–12708. doi:10.1021/acs.jafc.9b03952
Zhao, M., Godecke, T., Gunn, J., Duan, J. A., and Che, C. T. (2013). Protostane and fusidane triterpenes: A mini-review. Molecules 18, 4054–4080. doi:10.3390/molecules18044054
Zhao, S., Wu, P., Zhang, K., Hong, W., Jiang, Z., Cui, X., et al. (2016). Chemically modified fusidic acid, preparation method and application. Guangdong: China National Intellectual Property Administration, C. N. Patent No 105,924,488.
Keywords: fusidic acid, derivatives, pharmacophore model, antibacterial, structure–activity relationships
Citation: Zheng W, Tu B, Zhang Z, Li J, Yan Z, Su K, Deng D, Sun Y, Wang X, Zhang B, Zhang K, Wong W-L, Wu P, Hong WD and Ang S (2023) Ligand and structure-based approaches for the exploration of structure–activity relationships of fusidic acid derivatives as antibacterial agents. Front. Chem. 10:1094841. doi: 10.3389/fchem.2022.1094841
Received: 10 November 2022; Accepted: 19 December 2022;
Published: 06 January 2023.
Edited by:
Xiping Cui, Guangdong University of Technology, ChinaReviewed by:
Mohammad Abrar Alam, Arkansas State University, United StatesCopyright © 2023 Zheng, Tu, Zhang, Li, Yan, Su, Deng, Sun, Wang, Zhang, Zhang, Wong, Wu, Hong and Ang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panpan Wu, d3l1Y2hlbXdwcEAxMjYuY29t; Weiqian David Hong, ZGF2aWRod3FAbGl2ZXJwb29sLmFjLnVr; Song Ang, am51YW5nc29uZ0AxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.