 Mingyu Zheng
Mingyu Zheng Xiaoping Zhang
Xiaoping Zhang Yihao Cheng
Yihao Cheng Lili Sun
Lili Sun- Jiangxi Key Laboratory for Mass Spectrometry and Instrumentation, East China University of Technology, Nanchang, China
Collisional activation of protonated phenylalanine derivatives deamination products leads to hydroxyl skeletal rearrangement versus cyclization reaction, and to form hydroxylbenzyl cation via elimination of CH2CO. To better clarify this unusual fragmentation reaction, accurate mass measurements experiments, native isotope experiments, multiple-stage mass spectrometry experiments, different substituents experiments, and density functional theory (DFT) calculations were carried out to investigate the dissociation mechanistic pathways of protonated phenylalanine derivatives deamination products. In route 1, a three-membered ring-opening reaction and a 1,3-hydroxyl transfer (from the carbonyl carbon atom to the interposition carbon atom of carbonyl) occurs to form 3-hydroxy-1-oxo-3-phenylpropan-1-ylium, followed by dissociation to lose CH2CO to give hydroxy (phenyl)methylium. In route 2, a successive cyclization rearrangement reaction and proton transfer occur to form a 2-hydroxylphenylpropionyl cation or protonated 2-hydroxy-4H-benzopyran, followed by dissociation to lose CH2CO or CH≡COH to give 2-hydroxylbenzyl cation. In route 3, a successive hydroxyl transfer (from the carbonyl carbon atom to the ortho carbon atom on benzene) and two stepwise proton transfer (1,2-proton transfer to the ipso-carbon atom of the phenyl ring followed by 1,3-proton transfer to the ortho carbon atom of carbonyl) occurs to form a 2-hydroxylphenylpropionyl cation, which subsequently dissociates to form 2-hydroxylbenzyl cation by elimination of CH2CO. DFT calculations suggested that route 1 was more favorable than route 2 and route 3 from a thermodynamic point of view.
Introduction
Electrospray ionization mass spectrometry (ESI-MS) is not only a versatile technique for the analysis of a wide range of compounds but also has proven to be a powerful tool for the investigation of molecular structure, mechanism, and dynamic (Gronert, 2001; Indelicato et al., 2021; Li et al., 2021; Qiu et al., 2022). In particular, tandem mass spectrometry can offer abundant fragmentation data for structure elucidation, which is acted as a small “gas-phase chemistry laboratory”. Several specific rearrangement reactions have been reported in tandem mass spectrometry, including hydride transfer (Chai et al., 2010), benzyl cation transfer (Sun et al., 2012; Li et al., 2014; Paulose et al., 2015), sulfonyl cation transfer (Wang et al., 2016), methyl transfer (Kshirsagar and Argade, 2009; Ren et al., 2019), sulfur transfer (Zhang et al., 2019), halogen transfer (Chai et al., 2016; Zhang and Cheng, 2017), etc. Mastering these specific rearrangement transfer reactions can help to elucidate the structural information of compounds, enrich the content of gas-phase ion chemistry, and also help to discover new pathways of drug metabolism.
Phenylalanine derivatives are an important class of functionally active substances that are frequently involved in various metabolisms in the body, and their metabolites are hydroxylated products (Matthews, 2007). Melphalan, a phenylalanine derivative containing bis-β-chloroethylamine groups, plays an important role in the development of therapeutic approaches for tumors as well as cancer (Musto and D'Auria, 2007). Investigation of the fragmentation mechanism of phenylalanine derivatives can promote structure elucidation and help new metabolic pathways be discovered. The dissociation reactions of protonated amino acids such as phenylalanine have attracted considerable attention using tandem mass spectrometry (Perera et al., 2002; Lioe and O’Hair, 2005). It has been shown that protonated amino acids fragment predominantly to form their respective iminium ions by concomitant loss of H2O and CO, giving rise to very simple CID mass spectra.
In this study, we report the hydroxy transfer reaction of phenylalanine derivatives in detail. Melphalan was selected as a model to perform a detailed mechanistic investigation of the fragmentation reaction. Collisional activation of protonated melphalan occurred hydroxyl transfer, which generated hydroxybenzyl cation. Density functional theory (DFT) calculations and substituent effects were also implemented for the mechanism research.
Experimental section
Chemicals and material
Methanol HPLC grade was purchased from Sigma-Aldrich (St. Louis, MO, United States). Melphalan (compound 1) and phenylalanine (compound 2) were purchased from J&K Scientific Ltd (Shanghai, China) with a purity>99%. 3,4-Dimethoxyphenylalanine (compound 3), 4-chlorophenylalanine (compound 4), 4-methoxyphenylalanine (compound 5), 4-methylphenylalanine (compound 6), tyrosine (compound 7), 4-bromophenylalanine (compound 8) and 2-hydroxylbenzylamine were purchased from Sinopharm Chemical Reagent Co., Ltd. Their structures of them are listed in Scheme 1. Experimental ultrapure water produced by Millipore ultrapure water system.
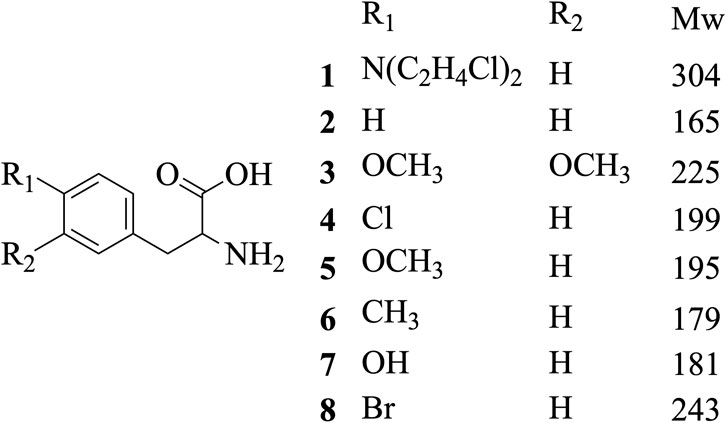
SCHEME 1. Structures of phenylalanine derivatives. (Mw: molecular weight)
Mass spectrometry
The samples were analyzed on an LTQ-XL advantage IT-MS (Thermo Scientific, San Jose, CA, United States) and an Orbitrap-XL mass spectrometer (Thermo Scientific, San Jose, CA, United States) using a homemade ESI interface in the positive ion mode. The optimized ESI source conditions were as follows: the capillary temperature at 250°C, the capillary voltage at 19 V; the tube lens at 90 V, the nebulizing gas (N2), and 25 arbitrary units (a.u.). The samples were injected by a syringe pump at a flow rate of 5 μL/min. Other LTQ-XL parameters were automatically optimized by the system. The Orbitrap-XL mass spectrometer instrument was operated at a high resolution of up to 100,000. The collision-induced dissociation (CID) MS data were obtained with helium as the collision gas. In CID-MS experiments, the desired precursor ions were isolated with an isolation window of 1.0 m/z unit. The CID-MS spectra of the protonated molecules were obtained by activation of the precursor ions at the normalized collision energy of 20–30%.
Theoretical calculations
Theoretical calculations were performed using the Gaussian 09 program (Frisch et al., 2010). The geometries such as reactants, intermediates, transition states (TSs), and products were optimized by the density functional theory (DFT) method at the B3LYP/6-31+G (d,p) level. TSs were obtained by relaxed PES scans, in which a bond length was scanned to search a first-order saddle point, and subsequently optimizing the corresponding TS. Then, the relevant TS structures were searched and optimized by either TS or QST2. All TSs were confirmed by the presence of a single imaginary vibrational frequency using intrinsic reaction coordinate (IRC) method. IRC calculations at the same level of theory were performed on each transition state to further confirm that the optimized TS structures were connected to the correct reactants and products by the steepest descend path. Vibrational frequencies of all the key species were calculated at the same level of theory. Full structural details and energies of all structures involved are available in the supplementary material. The energies discussed here are the sum of electronic and thermal free energy.
Result and discussion
Fragmentation behavior of protonated melphalan
The gas phase hydroxyl transfer rearrangement reaction was explored by investigating the MS fragmentation behavior of protonated phenylalanine derivatives (Scheme 1). Melphalan (compound 1) was selected as a model to perform a detailed investigation. The tandem mass spectrum of protonated melphalan (a, m/z 305) shown in Figure 1A reveals the generation of a major fragment ion at m/z 288, corresponding to b via a neutral loss of 17 Da (NH3). Fragment ions at m/z 246 and m/z 244 are attributed to the elimination of (NH3 + CO2) and (NH3 + CH2CO) of the precursor ion, respectively, which will be discussed in detail in the following sections.
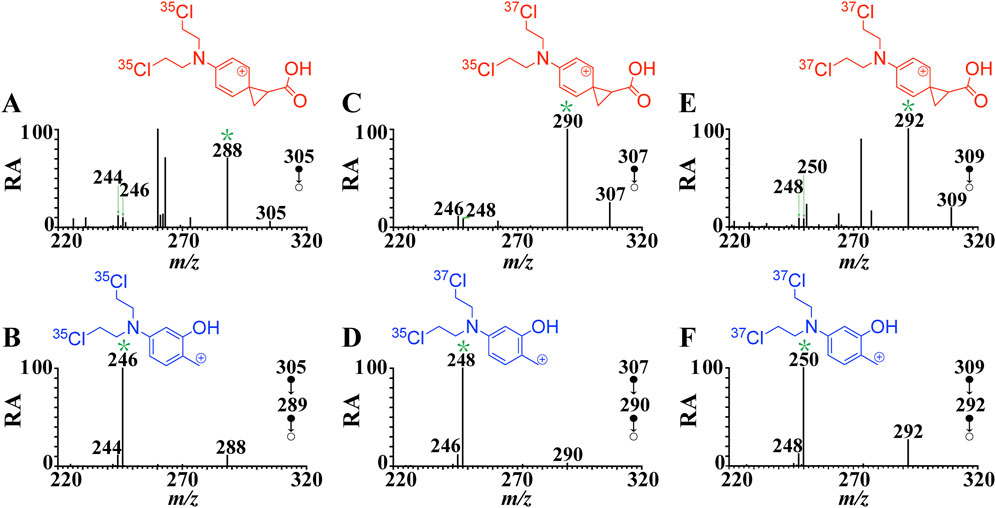
FIGURE 1. CID mass spectra of [melphalan + H]+ at the normalized collision energy of 20% (A) MS2 spectrum of [melphalan-35Cl35Cl + H ]+ (m/z 305→), (B) MS3 spectrum of [melphalan-35Cl35Cl + H]+ (m/z 305→m/z 288→), (C) MS2 spectrum of [melphalan -37Cl35Cl + H]+ (m/z 307→), (D) MS3 spectrum of [melphalan -37Cl35Cl + H]+ (m/z 307→m/z 290→) (E) MS2 spectrum of [melphalan -37Cl37Cl + H]+ (m/z 309→), (F) MS3 spectrum of [melphalan -37Cl37Cl + H]+ (m/z 309→m/z 292→). The structures in the figure correspond to the structure of the ion marked with an asterisk.
Fragmentation behavior of protonated melphalan deamination product
The characteristic fragment ion at m/z 246 can only be interpreted as a result of the (NH3 + CH2CO) elimination. To verify the formation mechanism of ion at m/z 246, MS3 experiments were performed. As shown in Figure 1B, the MS3 spectrum of protonated melphalan (m/z 305→m/z 288→) shows fragment ions at m/z 246 and m/z 244 via eliminations of CH2CO and CO2, respectively. However, there is no relevant moiety of CH2CO in the structure of b (m/z 288). Thus, the generation of the ion at m/z 246 originated from the dissociation of b (m/z 288) may via skeletal rearrangement. As reported earlier, protonated melphalan does appear to eliminate NH3 and CH2CO. This is similar to the results of the Harrison group (Tsang and Harrison, 1976; Harrison and Ichikawa, 1980) and K. W. Michael Siu et al. (Shoeib et al., 2002; El Aribi et al., 2004). O’Hair group has also done a lot of work on the gas-phase behavior of protonated phenylalanines (Lioe and O’Hair, 2005; Lioe and O’Hair, 2007), but no such a CH2CO loss has been observed, possibly due to the differences on the energy transfer to the ions which depending on the different MS instruments used. Harrison’s group has proposed a mechanism for the loss of NH3 and CH2CO from protonated phenylalanine in which the hydroxide ion migrates to form Ph-CHOH+ ion with concomitant elimination of CH2CO (Tsang and Harrison, 1976). This similar mechanism was also invoked by Ichikawa and co-workers who proposed migration of the methoxy ion in the dissociation of protonated methyl cinnamate (Harrison and Ichikawa, 1980). However, few detailed studies have been conducted on the detailed mechanism of hydroxyl transfer. Are there other mechanisms such as cyclization reaction that contribute to the formation of m/z 246?
Here we are interested in the characteristic fragment ion m/z 246 marked as p2 (100%). P2 is 42 Da (CH2 = C=O or CH≡COH) less than the precursor ion b. The elemental compositions of fragment ions at m/z 246 and m/z 244 were determined by accurate mass measurements performed on a high-resolution Orbitrap-XL mass spectrometer (Supplementary Figure S1 and Supplementary Table S1). The high-resolution MS results further confirmed that fragment ions at m/z 246 and m/z 244 were generated via eliminations of CH2CO and CO2, respectively. Thus, the formation of p2 may be interpreted as a result of both rearrangement (hydroxyl transfer or cyclization reaction) and dissociation (elimination of CH2 = C=O or CH≡COH) of b (m/z 288).
Many literatures have reported that the loss of NH3 form protonated phenylalanine is predicted to occur via a neighbouring group attack by the aryl group to form a phenonium cation rather than by 1,2-hydride migration from the perspective of experiment or calculation (Shoeib et al., 2002; El Aribi et al., 2004; Lioe and O’Hair, 2005; Lioe and O’Hair, 2007; Lam and O'Hair, 2010). In addition, Liam and O'Hair have fully considered several structures for the [MH-NH3]+ fragment and showed that the most stable one is not the phenonium cation but that the most favorable NH3 loss pathway is however leadint to this structure (Dookeran et al., 1996; Lam and O'Hair, 2010). Thus, the structure of deamination product (ion b, m/z 288) was also considered as a ternary ring structure (see Scheme 2) in this work. The potential pathways for the generation of product ions at m/z 246 and m/z 244 were displayed in Scheme 2. For the generation of m/z 244, the hydrogen atom on carboxyl undergoes 1,3-H transfer to form an intermediate isomer, which undergoes the dissociation to form p1 (spiro [2.5]octa-5,7-dien-6-N(C2H4Cl)2-4-ylium) via elimination of CO2 (Scheme 2A). Three potential routes for the generation of m/z 246 (p2) from protonated melphalan deamination product b) were proposed in Scheme 2B. In route 1, firstly, under collision activation of structure b, a three-membered ring opening reaction occurred to form intermediate c, then the hydroxyl is transferred from the carbonyl carbon atom (C3) to the interposition carbon atom (C6) of carbonyl leading to intermediate d, followed by the elimination of CH2CO to give Ph-CHOH+ ion (p2). In route 2, the oxygen atom on the carbonyl group first conducts a nucleophilic attack on the positively charged ortho-position carbon atom of the phenyl ring group, thereby generating the bicyclic intermediate e. Then, the proton at carbon atom (C8) of intermediate e undergoes a 1,2-transfer to form intermediate f, followed by dissociation to lose CH≡COH to give HO-Ph-CH2+ ion (p2′, route 2-A). Alternatively, the formed intermediate e undergoes enol interconversion to form a keto structure g, and the proton at carbon atom (C8) of intermediate g directly undergoes further 1,2-transfer to form intermediate g1, followed by dissociation to lose CH2CO to give p2’ (route 2-B). In route 3, the hydroxyl is transferred from the carbonyl carbon atom (C3) to the ortho carbon atom (C8) of the benzene ring, leading to the intermediate h, which further undergoes a successive proton transfer to form the intermediate g1 and dissociates to form p2’.
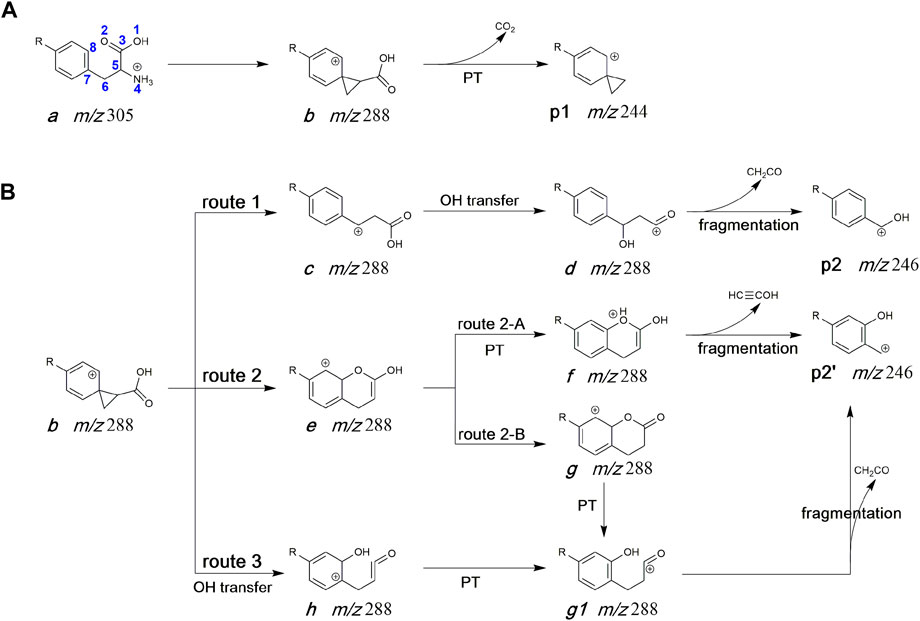
SCHEME 2. Proposed fragmentation mechanism of protonated melphalan. (A) The formation process of m/z 244. (B) The formation process of m/z 246. R: N(C2H4Cl)2, PT: proton transfer.
Native 37Cl isotope experiments
The postulated fragmentation mechanism in Scheme 2 was supported by the MS/MS analysis on the native 37Cl isotopic ion (Figures 1C–F). The chlorine element has two isotopes, 35Cl and 37Cl in nature, with a relative abundance at 100% and 33%, respectively. Abundant native 37Cl isotope protonated molecular ions of melphalan at m/z 307 and m/z 309 were generated in the positive mode, and ions at m/z 305, m/z 307, and m/z 309 with characteristic abundance ratio of 9:6:1 (Supplementary Figure S1A). As shown in Figure 1C, the product ion b at m/z 290 was observed in the fragmentation spectra of the native 37Cl isotopologue of [1 + H]+ at m/z 307 (one 37Cl atom and one 35Cl atom). This indicates that b contains two Cl atoms. Similarly, as shown in Figure 1D, the mass shift of p2 (from m/z 246 to m/z 248) and p1 (from m/z 244 to m/z 246) increase by 2 Da in the MS3 spectrum of the protonated 37Cl isotopologue (m/z 307→m/z 290→), which provides evidence that p1 and p2 both contain two Cl atoms. In addition, the product ion b at m/z 292 was observed (Figure 1E) in the fragmentation spectrum of the native 37Cl isotope of [1 + H]+ at m/z 309 (two 37Cl atoms). And the mass shift of p2 (from m/z 246 to m/z 250) and p1 (from m/z 244 to m/z 248) increase by 4 Da in the MS3 spectrum of the protonated 37Cl isotope (m/z 309→m/z 292→) (Figure 1F, which further proves that p1 and p2 both contain two Cl atoms. These native isotope experiment results further confirm that fragment ion p1 (m/z 244) and p2 (m/z 246) are generated due to the elimination of CO2 and CH2 = C=O/CH≡COH from b, respectively.
Density functional theory calculations
To further investigate the mechanism displayed in Scheme 2, density functional theory (DFT) calculations were carried out at the B3LYP/6-31+G (d,p) level of theory. There are multiple potential protonation sites for melphalan, including O1 of the hydroxyl, O2 of the carbonyl, N4 of the amino, N9 of the nitrogen mustard, and Cl10 of the nitrogen mustard. The structures with different protonation sites of melphalan were optimized at the same level B3LYP/6-31+G (d,p) and the relative energies of these structures are summarized in Table 1. The free energy of the protonation site on the N4 atom is 40.9 kJ/mol, 132.0 kJ/mol, 133.0 kJ/mol, and 95.4 kJ/mol lower than those of protonation sites on the N9 atom, O1 atom, O2 atom, and Cl10 atom, respectively. Overall, the calculation results indicate that the N4 atom is the most thermodynamically favorable protonation site, which gives rise to ion a (m/z 305) as described in Scheme 2. Subsequently, the breakage of the C5–N4 bond in ion a occurs easily, resulting in the formation of ion b (m/z 288) with a three-membered ring structure and NH3. The detailed process was displayed in Supplementary Figure S2. The same mechanism to form a deamination product with a three-membered was also invoked by O’Hair and co-workers (Lioe et al., 2004; Lioe and O’Hair, 2007).
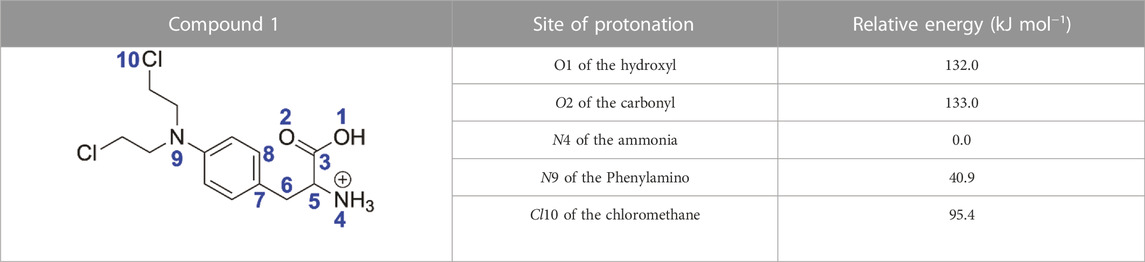
TABLE 1. Relative energies of [1 + H]+ with different protonation sites calculated at B3LYP/6-31+G (d,p) level.
The potential routes to the ion m/z 246 in the subsequent fragmentations of b (m/z 288) were compared by theoretical calculations (Figure 2), and details of the corresponding structures are available in the Supplementary Material. In route 1, firstly, under collision activation, structure b can undergo rotation of -COOH to generate intermediate b1 via overcoming a small energy barrier. Then, the three-membered ring in intermediate b1 undergoes an opening reaction to form intermediate c via a transition state (TS1 in Figure 3). This process needs to surmount an energy barrier of 161 kJ/mol. The formed intermediate c continues to undergo rotation of -COOH to generate a more stable intermediate c1. The hydroxyl in c1 is transferred from the carbonyl carbon atom (C3) to the interposition carbon atom (C6) of carbonyl through a four-membered ring transition state (TS2 in Figure 3), leading to the formation of ion d with a new carbon-oxygen bond. This process needs to surmount an energy barrier of 178 kJ/mol. As shown in Figure 3, the distance between the C3 atom and the O1 atom is increased to 1.874 Å in TS2, which is appreciably longer than a covalent C3–O1 bond (1.353 Å) in b. And the distance between the C6 atom and O1 atom is decreased to 1.520 Å in TS2. The reason for the breakage of the C3–O1 bond and the formation of the C6–O1 bond in c is due to the nucleophilic attack of the hydroxyl on the positively charged carbon atom (C6) via the formation of the four-membered ring transition state. The formed d continues to undergo the cleavage of the C5–C6 bond induced by the positive charge in the C3 atom and gives rise to p2 and CH2CO with an energy barrier of 104 kJ mol−1 (TS3). The sum free energy of the separated ion p2 and CH2CO is higher than that of b by 18 kJ/mol.
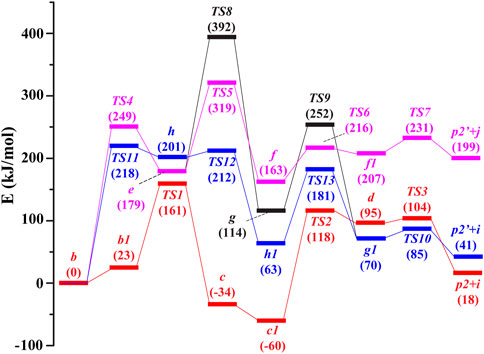
FIGURE 2. Potential energy diagram for the generation of m/z 246 in the dissociation of [melphalan + H–NH3]+ at the B3LYP/6-31+G (d,p) level. Red line: route 1, rose red line: route 2-A, black line: route 2-B, blue line: route 3. i: CH2CO, j: CH≡COH.
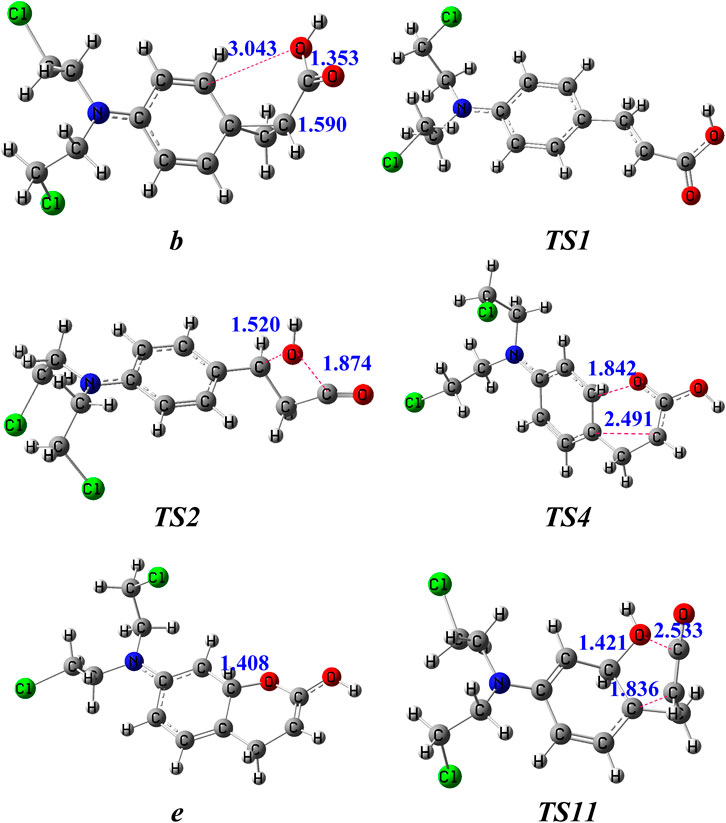
FIGURE 3. The optimized structures of key intermediates for the generation of m/z 246 in the dissociation of [melphalan + H–NH3]+ at the B3LYP/6-31+G (d,p) level. The bond lengths are given in Å.
In route 2, firstly, the oxygen atom (O2) on the carbonyl group conducts a nucleophilic attack on the positively charged ortho-position carbon atom (C8) of the phenyl ring group through a six-membered ring transition state (TS4 in Figure 3), leading to the formation of intermediate ion e with a new carbon-oxygen bond. This process needs to surmount an energy barrier of 249 kJ/mol. The ring expansion rearrangement reaction occurs with a concerted process with cleavage of the C5–C7 bond and formation of the C8–O2 bond, which can be viewed as an electrophilic substitution of the phenyl ring (Figure 3). As shown in Figure 3, the distance between the C5 atom and C7 atom is increased to 2.491 Å in TS4, which is appreciably longer than a covalent C5–C7 bond (1.590 Å) in b. And the distance between the C8 atom and O2 atom is ranged from 4.135 Å in b to 1.842 Å in TS4 and ranged from 1.842 Å in TS4 to 1.408 Å in e. The reason for the breakage of the C5–C7 bond and the formation of the C8–O2 bond in b is due to the nucleophilic attack of the carbonyl oxygen atom on the positively charged carbon atom on the phenyl ring group via the formation of the six-membered ring transition state. The free energy of ion e is 179 kJ/mol higher than that of ion b.
Then, the proton at the ortho-position carbon atom (C8) of the phenyl ring in e undergoes a 1,2-transfer (TS5) to the O2 atom to form intermediate f. This process needs to surmount an energy barrier of 319 kJ/mol. The formed f continues to undergo the cleavage of the O2–C3 bond induced by the positive charge in the O2 atom (TS6) and the cleavage of the C5–C6 bond induced by the positive charge in C3 atom (TS7), with an energy barrier of 216 kJ/mol (TS6) and 231 kJ/mol (TS7), respectively, and gives rise to 4-N(C2H4Cl)2-2-hydroxylbenzyl cation (p2′) and CH≡COH (route 2-A). The sum free energy of the separated ion p2′ and CH≡COH is higher than that of b by 199 kJ/mol. Alternatively, the formed intermediate e undergoes enol interconversion to form a keto structure g, this process needs to surmount an energy barrier of 392 kJ/mol (TS8). Then, the formed g continues to undergo a 1,2-proton transfer (from the ortho-position carbon atom (C8) of the phenyl ring to the oxygen atom (O2) with an energy barrier of 252 kJ/mol (TS9) to form intermediate g1. The formed g1 further undergoes the cleavage of the C5–C6 bond induced by the positive charge in the C3 atom and gives rise to 4-N(C2H4Cl)2-2-hydroxylbenzyl cation (p2’) and CH2CO (route 2-B).
In route 3, firstly, the hydroxyl in b is transferred from the carbonyl carbon atom (C3) to the ortho carbon atom on benzene (C8) through a five-membered ring transition state (TS11 in Figure 3), leading to the formation of ion h with a new carbon-oxygen bond. This process needs to surmount an energy barrier of 218 kJ/mol. The rearrangement occurs with a concerted process with cleavage of the C3–O1 bond and formation of the C8–O1 bond, which can be viewed as an electrophilic substitution of the phenyl ring (Figure 3). As shown in Figure 3, the distance between the C3 atom and the O1 atom is increased to 2.533 Å in TS11, which is appreciably longer than a covalent C3–O1 bond (1.353 Å) in b. And the distance between the C8 atom and the O1 atom is ranged from 3.043 Å in b to 1.421 Å in TS11. The reason for the breakage of the C3–O1 bond in b and the formation of the C8–O1 bond in h is due to the nucleophilic attack of the hydroxyl on the positively charged carbon atom on the phenyl ring group via the formation of the five-membered ring transition state. In h, the hydroxyl was connected to the C8 atom and the three-membered ring was opened (Figure 3). The free energy of ion h is 201 kJ/mol higher than that of ion b. Then, the formed h continues to undergo a two-stepwise proton transfer (1,2-proton transfer from the ortho-carbon atom (C8) of the phenyl ring to the ipso-carbon atom (C7) of the phenyl ring followed by 1,3-proton transfer to the ortho carbon atom (C5) of carbonyl) to form g1. The formed g1 continues to undergo the cleavage of the C5–C6 bond induced by the positive charge in the C3 atom and gives rise to 4-N(C2H4Cl)2-2-hydroxylbenzyl cation (p2′) and CH2CO with a small energy barrier of 85 kJ mol−1 (TS10). The sum free energy of the separated ion p2’ and CH2CO is higher than that of b by 41.0 kJ/mol.
From the calculation results in Figure 2 for the formation of product ion m/z 246, it can be found that the sum free energy of the separated p2′ and CH2CO is 158 kJ/mol lower than that of the separated p2′ and CH≡COH, indicating that generation of (p2’ + CH2CO) is easier than that of (p2’ + CH≡COH) from a thermodynamic point of view. The maximum energy barrier in route 2-B (black line) for the formation of (p2’ + CH2CO) is 174 kJ/mol higher than that in route 3 (blue line) for the formation of (p2’ + CH2CO), indicating route 3 is easier to occur. Moreover, the maximum energy barrier in route 3 (blue line) for the formation of (p2’ +CH2CO) is 57 kJ/mol higher than that in route 1 (red line) for the formation of (p2 + CH2CO), indicating route 1 is the most favorable. Thus, DFT calculations suggested that route 1 was more favorable than route 2 and route 3 from a thermodynamic point of view. That is to say, the collisional activation of protonated phenylalanine derivatives deamination products was more favorable to form p2 (m/z 246) via hydroxyl transfer rather than cyclization reaction. The same hydroxyl transfer mechanism was also invoked by El Aribi et al. (El Aribi et al., 2004).
For the elimination of the CO2 route (Figure S3), the H atom on the carboxyl of b directly undergoes 1,3-H transfer to the ortho carbon atom of carbonyl C5 via a four-membered ring transition state (TS14), which was further separated into spiro product p1 (spiro [2.5]octa-5,7-dien-6-N(C2H4Cl)2-4-ylium) and CO2. This process needs to surmount the energy barrier of 288 kJ/mol. The sum free energy of the separated p1 and CO2 is 114 kJ/mol lower than that of the separated p2 and CH2CO, indicating that p1 is a more stable structure. However, the maximum energy barrier in the route for the formation of (p1 + CO2) is 127 kJ/mol higher than that in route 1 (red line) for the formation of (p2+ CH2CO). Thus, DFT calculations suggested that route 1 was more favorable than the route for the elimination of CO2 from a thermodynamic point of view.
The universality of the gas-phase hydroxyl transfer
Substituent effects are very useful in probing reaction mechanisms (Guo et al., 2012; Wang et al., 2015; Zhang et al., 2015; Wang et al., 2016). To better delineate the universality of the gas-phase hydroxyl transfer reaction, a series of compounds bearing different substituents (H-substituted, OCH3-substituted, Cl-substituted, CH3-substituted, OH-substituted, Br-substituted) at the para position of the phenyl ring were also investigated by tandem MS experiments (Supplementary Figure S4-10), and the tandem MS data were summarized in Table 2. Noteworthily, corresponding fragment ions of p2 (m/z 107, m/z 167, m/z 141, m/z 137, m/z 121, m/z 123 and m/z 185 for compounds 2-8) and fragment ions of p1 (m/z 105, m/z 165, m/z 139, m/z 135, m/z 119, m/z 121 and m/z 183 for compounds 2-8) were observed for all compounds. All of these compounds show similar fragmentation behaviors in the CID-MS experiments, whereas the relative intensities of the two competing product ions varied as the substituent changed (Table 2). Mainly, the presence of the electron-donating groups on the phenyl ring favors both the H transfer to give p1 and the hydroxyl transfer to give p2, whereas the presence of the electron-withdrawing groups suppresses both p1 and p2. The reason was that the electron of the electron-donating group is transferred to the positive charge site on p2, which effectively reduces the positivity of the carbon atom of p2, thus increasing its stability. A plot of the abundance ratio of these two ions, ln [(p2)/(p1)] versus the Hammett substituent constants (Hansch et al., 1991), σp+, was obtained in Figure 4. The logarithmic values of the abundance ratios of these two ions are in line with σp+.
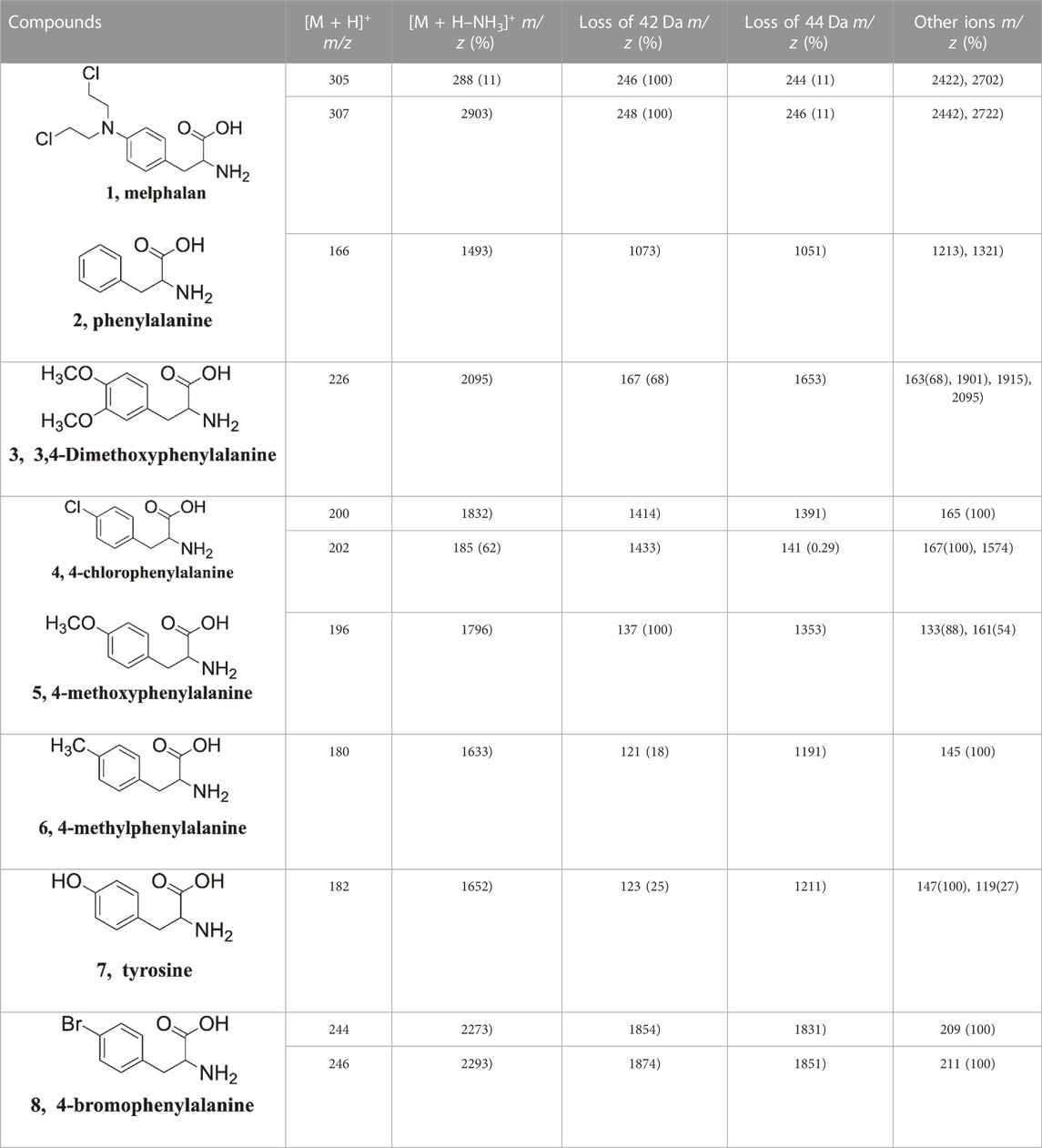
TABLE 2. The CID mass spectra data of [M + H–NH3]+ from protonated phenylalanine derivatives (1–8).
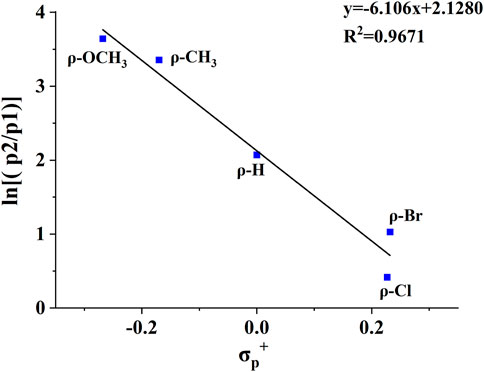
FIGURE 4. The plot of ln [(p2)/(p1)] vs the σp+ substituent constants for the CID reactions of compounds 2, 4, 5, 6, 8.
Conclusion
In summary, an intriguing intramolecular hydroxyl transfer reaction occurred in the gas-phase dissociation of protonated phenylalanine derivatives. Accurate mass measurements, native isotope experiments, multiple-stage mass spectrometry experiments, different substituents experiments, and DFT calculations indicate convincing evidence that the hydroxyl transfer was achieved to form hydroxylbenzyl cation via eliminations of NH3 and CH2CO. This study reports hydroxyl transfer reaction in the gas phase chemistry, which can not only aid us to elucidate the structural information of compounds but also help to discover new pathways of drug metabolism.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
XZ and XZ designed and supported the research. MZ and YC conducted the research. MZ, YC, LS, and XZ analyzed the data and wrote the manuscript. XZ and XZ had primary responsibility for the final content.
Funding
This work was supported by the Natural Science Foundation of China (No. 22104014, 22164002) and the Natural Science Foundation of Jiangxi Province (No. 20212BAB20213003).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1094329/full#supplementary-material
References
Chai, Y., Jiang, K., and Pan, Y. (2010). Hydride transfer reactions via ion-neutral complex: Fragmentation of protonated N-benzylpiperidines and protonated N-benzylpiperazines in mass spectrometry. J. Mass Spectrom. 45 (5), 496–503. doi:10.1002/jms.1731
Chai, Y., Xiong, X., Yue, L., Jiang, Y., Pan, Y., and Fang, X. (2016). Intramolecular halogen transfer via halonium ion intermediates in the gas phase. J. Am. Soc. Mass Spectrom. 27 (1), 161–167. doi:10.1007/s13361-015-1261-4
Dookeran, N. N., Yalcin, T., and Harrison, A. G. (1996). Fragmentation reactions of protonated α-amino acids. J. Mass Spectrom. 31 (5), 500–508. doi:10.1002/(sici)1096-9888(199605)31:5<500::aid-jms327>3.0.co;2-q(199605)31:5
El Aribi, H., Orlova, G., Hopkinson, A. C., and Siu, K. W. M. (2004). Gas-phase fragmentation reactions of protonated aromatic amino acids: Concomitant and consecutive neutral eliminations and radical cation formations. J. Phys. Chem. A 108 (17), 3844–3853. doi:10.1021/jp0374915
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2010). Gaussian 09, revision B.01. Wallingford CT: Gaussian, Inc.
Gronert, S. (2001). Mass spectrometric studies of organic ion/molecule reactions. Chem. Rev. 101 (2), 329–360. doi:10.1021/cr9900836
Guo, C., Jiang, K., Yue, L., Xia, Z., Wang, X., and Pan, Y. (2012). Intriguing roles of reactive intermediates in dissociation chemistry of N-phenylcinnamides. Org. Biomol. Chem. 10 (35), 7070–7077. doi:10.1039/c2ob26011e
Hansch, C., Leo, A., and Taft, R. W. (1991). A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91 (2), 165–195. doi:10.1021/cr00002a004
Harrison, A. X. G., and Ichikawa, H. (1980). Site of protonation in the chemical ionization mass spectra of olefinic methyl esters. Org. Mass Spectrom. 15 (5), 244–248. doi:10.1002/oms.1210150507
Indelicato, S., Bongiorno, D., Ceraulo, L., Mancin, F., and Madder, A. (2021). Exploiting double exchange diels-alder cycloadditions for immobilization of peptide nucleic acids on gold nanoparticles. Front. Chem. 8, 4. doi:10.3389/fchem.2020.00004
Kshirsagar, U. A., and Argade, N. P. (2009). Facile approach to diverse range of 1, 3-diaza-heterocycles: Angular/linear selectivity paradigm and a remarkable intramolecular methyl migration. Tetrahedron 65 (27), 5244–5250. doi:10.1016/j.tet.2009.04.088
Lam, A. K. Y., and O'Hair, R. A. J. (2010). Isomer differentiation via collision-induced dissociation: The case of protonated α-β2- and β3-phenylalanines and their derivatives. Rapid Commun. Mass Spectrom. 24 (12), 1779–1790. doi:10.1002/rcm.4576
Li, F., Wu, Y., Zhang, N., Jiang, J., and Jiang, K. (2014). Competing benzyl cation transfers in the gas-phase fragmentation of the protonated benzyl phenylalaninates. Int. J. Mass Spectrom. 369, 23–29. doi:10.1016/j.ijms.2014.05.011
Li, C., Chu, S., Tan, S., Yin, X., Jiang, Y., Dai, X., et al. (2021). Towards higher sensitivity of mass spectrometry: A perspective from the mass analyzers. Front. Chem. 9, 813359. doi:10.3389/fchem.2021.813359
Lioe, H., and O’Hair, R. A. J. (2005). Neighbouring group processes in the deamination of protonated phenylalanine derivatives. Org. Biomol. Chem. 3 (20), 3618–3628. doi:10.1039/B503355A
Lioe, H., and O’Hair, R. A. J. (2007). Comparison of collision-induced dissociation and electron-induced dissociation of singly protonated aromatic amino acids, cystine and related simple peptides using a hybrid linear ion trap–FT-ICR mass spectrometer. Anal. Bioanal. Chem. 389 (5), 1429–1437. doi:10.1007/s00216-007-1535-1
Lioe, H., O’Hair, R. A. J., and Reid, G. E. (2004). Gas-phase reactions of protonated tryptophan. J. Am. Soc. Mass Spectrom. 15 (1), 65–76. doi:10.1016/j.jasms.2003.09.011
Matthews, D. E. (2007). An overview of phenylalanine and tyrosine kinetics in humans. J. Nutr. 137 (6), 1549S–1555S. doi:10.1093/jn/137.6.1549s
Musto, P., and D'Auria, F. (2007). Melphalan: Old and new uses of a still master drug for multiple myeloma. Expert Opin. inv. Drug. 16 (9), 1467–1487. doi:10.1517/13543784.16.9.1467
Paulose, J., Achuthan, R. P., Linsha, M. P., Mathai, G., Prasanth, B., Kumar Talluri, M. V., et al. (2015). Protonated N-benzyl- and N-(1-phenylethyl)tyrosine amides dissociate via ion/neutral complexes. Rapid Commun. Mass Spectrom. 29 (17), 1577–1584. doi:10.1002/rcm.7256
Perera, B. A., Gallardo, A. L., Barr, J. M., Tekarli, S. M., Anbalagan, V., Talaty, E. R., et al. (2002). Influence of a ring substituent on the tendency to form H2O adducts to Ag+ complexes with phenylalanine analogues in an ion trap mass spectrometer. J. Mass Spectrom. 37 (4), 401–413. doi:10.1002/jms.296
Qiu, L., Morato, N. M., Huang, K. H., and Cooks, R. G. (2022). Spontaneous water radical cation oxidation at double bonds in microdroplets. Front. Chem. 10, 903774. doi:10.3389/fchem.2022.903774
Ren, X., Zhang, X. P., Wang, Y. F., Cao, J. Y., Cheng, Y. Y., Feng, S. H., et al. (2019). Intramolecular and intermolecular methyl migration of fenthion studied by electrospray ionization mass spectrometry. Acta Chim. Sin. 77 (4), 358–364. doi:10.6023/a18120505
Shoeib, T., Cunje, A., Hopkinson, A. C., and Siu, K. W. M. (2002). Gas-phase fragmentation of the Ag+—Phenylalanine complex: Cation—π interactions and radical cation formation. J. Am. Soc. Mass Spectrom. 13 (4), 408–416. doi:10.1016/S1044-0305(02)00353-7
Sun, H., Chai, Y., and Pan, Y. (2012). Dissociative benzyl cation transfer versus proton transfer: Loss of benzene from protonated N-benzylaniline. J. Org. Chem. 77 (16), 7098–7102. doi:10.1021/jo301011e
Tsang, C. W., and Harrison, A. G. (1976). Chemical ionization of amino acids. J. Am. Chem. Soc. 98 (6), 1301–1308. doi:10.1021/ja00422a001
Wang, S., Yu, L., Wu, Y., Guo, C., Zhang, N., and Jiang, K. (2015). Gas-phase fragmentation of protonated N, 2-diphenyl-N'-(p-toluenesulfonyl) ethanimidamides: Tosyl cation transfer versus proton transfer. J. Am. Soc. Mass Spectrom. 26 (8), 1428–1431. doi:10.1007/s13361-015-1156-4
Wang, S., Dong, C., Yu, L., Guo, C., and Jiang, K. (2016). Dissociation of protonated N-(3-phenyl-2H-chromen-2-ylidene)- benzenesulfonamide in the gas phase: Cyclization via sulfonyl cation transfer. Rapid Commun. Mass Spectrom. 30 (1), 95–100. doi:10.1002/rcm.7420
Zhang, X., and Cheng, S. (2017). Intramolecular halogen atom coordinated H transfer via ion-neutral complex in the gas phase dissociation of protonated dichlorvos derivatives. J. Am. Soc. Mass Spectrom. 28 (10), 2246–2254. doi:10.1007/s13361-017-1736-6
Zhang, X., Zhu, P., Zhang, H., Li, Z., Jiang, K., and Lee, M. R. (2015). The competing radical eliminations in the tandem mass spectrometry of the OH-deprotonated benzyl vanillate. J. Mass Spectrom. 50 (2), 432–436. doi:10.1002/jms.3534
Keywords: hydroxyl transfer, electrospray ionization mass spectrometry, gas-phase reaction, phenylalanine, melphalan
Citation: Zheng M, Zhang X, Cheng Y, Sun L and Zhang X (2023) Hydroxyl transfer versus cyclization reaction in the gas phase: Sequential loss of NH3 and CH2CO from protonated phenylalanine derivatives. Front. Chem. 10:1094329. doi: 10.3389/fchem.2022.1094329
Received: 10 November 2022; Accepted: 23 December 2022;
Published: 09 January 2023.
Edited by:
Zheng Han, Shanghai Academy of Agricultural Sciences, ChinaCopyright © 2023 Zheng, Zhang, Cheng, Sun and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoping Zhang, emhhbmd4cHN1bnNoaW5lQDE2My5jb20=; Xinglei Zhang, bGVpemk4NTg2QDEyNi5jb20=