
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 04 January 2023
Sec. Chemical Biology
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1085463
This article is part of the Research Topic Computational and Experimental Insights in Proton and Ion Translocating Bioenergetic Systems View all 5 articles
 Tamara N. Grund1
Tamara N. Grund1 Yoshiki Kabashima2
Yoshiki Kabashima2 Tomoichirou Kusumoto2Di Wu1
Tomoichirou Kusumoto2Di Wu1 Sonja Welsch3Junshi Sakamoto2Hartmut Michel1
Sonja Welsch3Junshi Sakamoto2Hartmut Michel1 Schara Safarian1,4,5*
Schara Safarian1,4,5*Cytochromes bd are essential for microaerobic respiration of many prokaryotes including a number of human pathogens. These enzymes catalyze the reduction of molecular oxygen to water using quinols as electron donors. Their importance for prokaryotic survival and the absence of eukaryotic homologs make these enzyme ideal targets for antimicrobial drugs. Here, we determined the cryoEM structure of the menaquinol-oxidizing cytochrome bd-type oxygen reductase of the facultative anaerobic Actinobacterium Corynebacterium glutamicum at a resolution of 2.7 Å. The obtained structure adopts the signature pseudosymmetrical heterodimeric architecture of canonical cytochrome bd oxidases formed by the core subunits CydA and CydB. No accessory subunits were identified for this cytochrome bd homolog. The two b-type hemes and the oxygen binding heme d are organized in a triangular geometry with a protein environment around these redox cofactors similar to that of the closely related cytochrome bd from M. tuberculosis. We identified oxygen and a proton conducting channels emerging from the membrane space and the cytoplasm, respectively. Compared to the prototypical enzyme homolog from the E. coli, the most apparent difference is found in the location and size of the proton channel entry site. In canonical cytochrome bd oxidases quinol oxidation occurs at the highly flexible periplasmic Q-loop located in the loop region between TMHs six and seven. An alternative quinol-binding site near heme b595 was previously identified for cytochrome bd from M. tuberculosis. We discuss the relevance of the two quinol oxidation sites in actinobacterial bd-type oxidases and highlight important differences that may explain functional and electrochemical differences between C. glutamicum and M. tuberculosis. This study expands our current understanding of the structural diversity of actinobacterial and proteobacterial cytochrome bd oxygen reductases and provides deeper insights into the unique structural and functional properties of various cytochrome bd variants from different phylae.
An estimated 2.4 billion years ago, the “Great Oxidation Event”, presumably originating from the activity of the photosynthetic ancestors of cyanobacteria, raised atmospheric O2 levels over four orders of magnitude (to about 10% of current levels) and drastically changed the evolutionary course of life while also leading to one of the greatest mass extinction events on earth (Schirrmeister et al., 2013; Lyons et al., 2014). Certain organisms managed to escape oxidative damage by inhabiting oxygen-free niches. However, the more important adaption to these extreme environmental conditions was the evolution of metalloproteins that catalyze the reduction of highly toxic dioxygen to harmless water and at the same time harness the oxidizing power of oxygen for organismal energy metabolism (Hoganson et al., 1998).
Among these enzymes, terminal oxygen reductases catalyze the reduction of molecular oxygen to water employing electrons from the oxidation of either cytochrome c (cyt. c) or membrane dissolved quinols (QH2). They are classified into i) heme-copper oxidases (HCO), which contain, as a unifying characteristic, a heme-copper binuclear center, ii) alternative oxidases (AOX), first identified in plants, and iii) cytochrome bd oxidases, which are only present in prokaryotes (May et al., 2017; Borisov and Siletsky, 2019). All so-far experimentally characterized bd-type oxidases use exclusively quinols as substrates, whereas HCOs comprise both cyt. c- and QH2-oxidizing members.
The cytochrome bd enzyme was first described nearly a century ago and since then has been identified as a membrane-integrated respiratory terminal oxidase unique to the prokaryotic domain, including a number of human pathogens (Cook and Poole, 2016; Borisov et al., 2020). While bd-type enzymes catalyze the reduction of molecular oxygen to water, they do not actively pump protons across the cytoplasmic membrane as do members of the HCO superfamily. Instead, cytochrome bd generates an electrochemical proton gradient via charge separation by consuming substrate protons from the cytoplasmic space and the release of protons upon quinol oxidation to the perisplasm. Thus, the contribution to the proton motive force (pmf) is smaller than that of proton-pumping HCO-type oxidases (Puustinen et al., 1991; Borisov et al., 2011; Borisov and Verkhovsky, 2015).
Canonical cytochrome bd oxidases share a common core architecture of two subunits, denoted CydA and CydB, which may be accompanied by up to two additional single-transmembrane helix subunits (Miller and Gennis, 1983; VanOrsdel et al., 2013; Hoeser et al., 2014; Safarian et al., 2016; Safarian et al., 2019; Theßeling et al., 2019; Grund et al., 2021; Safarian et al., 2021; Wang et al., 2021; Friedrich et al., 2022). As implied by designation, cytochromes bd contain two b-type hemes (low-spin b558, high-spin b595) and one d-type heme involved in quinol oxidation, electron transfer and oxygen reduction. Substrate-binding occurs within CydA at the quinol binding domain (Q-loop) located on the periplasmic side of the membrane. Members of the canonical cytochrome bd oxidases are further subdivided into those containing either a short or a long hydrophilic Q-loop domain (S- and L-subfamily, respectively) (Osborne and Gennis, 1999; Sakamoto et al., 1999; Kusumoto et al., 2000). The role of the N-terminal insertion of the Q-loop domain has been studied previously, yet it still remains elusive whether this extension fulfils a purely structural or additionally also a functional role within cytochrome bd oxidases (Goojani et al., 2020; Theßeling et al., 2020).
Cytochromes bd are characterized by high oxygen affinity, as well as their resistance to cyanide inhibition (IC50 concentration for KCN of cytochrome bo3 from E. coli: 10 μM; IC50 concentration for KCN of cytochrome bd-I from E. coli: 2 mM) (Kita et al., 1984; Borisov and Verkhovsky, 2015). Extreme cyanide insensitivity as well as an apparent lack of a heme d signal, led to the sub-classification of cytochromes bd into the non-canonical cyanide-insensitive quinol oxidases (CIO) and canonical bd-type enzymes (Borisov et al., 2011). In CIOs, heme d is replaced by a second high-spin b-type heme (Azarkina et al., 1999; Jackson et al., 2007; Mogi et al., 2009). CIO-type terminal oxidases are encoded by the cio operon. The deduced amino acid sequences of CioA and CioB are homologous to CydA and CydB (Cunningham et al., 1997; Quesada et al., 2007). CIOs are found inter alia in Pseudomonas aeruginosa and Pseudomonas pseudoalcaligenes, and are assumed to enable aerobic respiration under cyanogenic and microaerobic growth conditions. This is of relevance for the opportunistic pathogen P. aeruginosa to attain full pathogenicity in the cyanide-mediated paralytic killing of nematodes (Cunningham et al., 1997; Quesada et al., 2007).
We previously reported X-ray and cryoEM structures of cytochrome bd oxidases from Geobacillus thermodenitrificans (3.8 Å), Escherichia coli (2.7 Å bd-I, 2.1 Å bd-II), and Mycobacterium tuberculosis (2.6 Å) (Safarian et al., 2016; Safarian et al., 2019; Grund et al., 2021; Safarian et al., 2021). While these structures show a common core architecture, cofactor arrangements, accessory subunit compositions, and Q-loop architectures indicate structural and functional diversity within the cytochrome bd family.
Here, we determined the cryoEM structure of the actinobacterial cytochrome bd from Corynebacterium glutamicum to 2.7 Å which is closely related to the mycobacterial cytochrome bd oxidases and at the same time displays spectroscopic and electrochemical properties closely matching those of cytochrome bd-I from E. coli (Nikolaev et al., 2021).
The menaquinol oxidizing cytochrome bd-type oxygen reductase of the facultative anaerobic Actinobacterium C. glutamicum (Cgbd) was produced under the activity of its native promoter without an affinity tag as described previously (Kabashima et al., 2009) (Supplementary Figure S1). The structure of the natively purified oxidase was determined via single-particle analysis cryoEM. A total of 13,390 movies were collected on a Titan Krios G3i microscope equipped with a Gatan K3 camera. The final map refined to 2.7 Å resolution (Supplementary Figures S2–S4, Supplementary Table S1).
Cytochrome bd from C. glutamicum is characterized by the canonical cytochrome bd architecture comprising the larger CydA and the slightly smaller CydB subunits. Each subunit is composed of two four-helix bundle motifs and an additional peripheral helix. The heterodimeric Cgbd forms a pseudosymmetrical complex with C2 symmetry (Figure 1). The dimerization interface is composed of the symmetry related TMHs 2, 3, and 9 of CydA and CydB, respectively. Subunit interactions are mostly characterized by van-der-Waals interactions. Unlike the E. coli and G. thermodenitrificans cytochrome bd oxidases, Cgbd does not contain small accessory subunits which resembles the composition of the closely related mycobacterial oxidases (Safarian et al., 2021; Wang et al., 2021). C. glutamicum and M. tuberculosis both belong to the phylum of Actinobacteria. The amino acid sequences of their core subunits share a sequence identity of 62% and 48% for CydA and CydB, respectively.
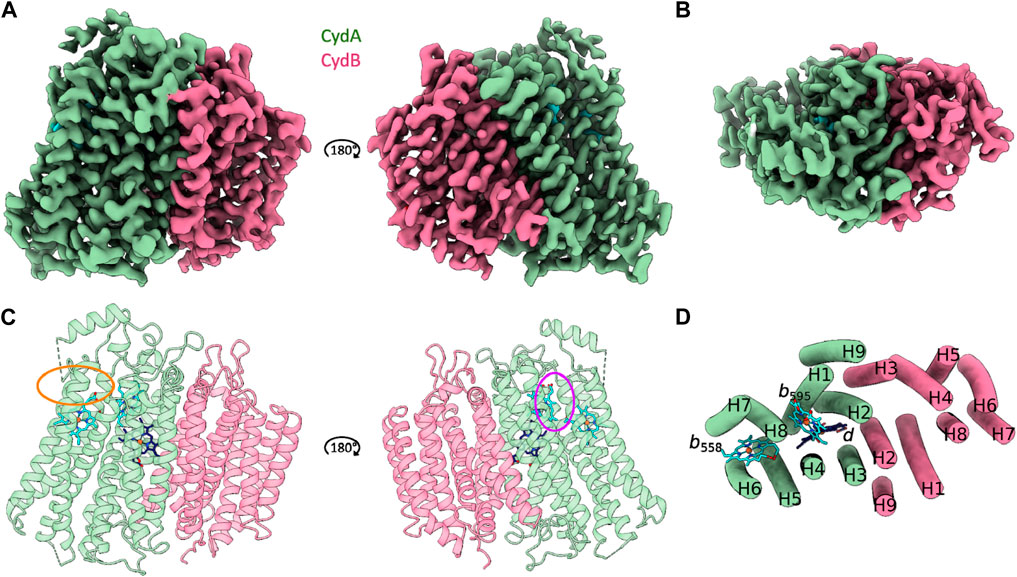
FIGURE 1. CryoEM structure of C. glutamicum cytochrome bd at 2.7 Å resolution. (A and C) Side view representations of the cryoEM map density and its corresponding ribbon model of the heterodimeric C. glutamicum cytochrome bd structure. The substrate quinol binding site 1 at the Q-loop in proximity to heme b558 is highlighted in orange. The second putative quinol binding site next to heme b595 is shown in purple. (B and D) Top view representation of the cryoEM map density and the corresponding tube model of the Cgbd structure highlighting the pseudosymmetrical arrangement of the two four helix bundles and the additional peripheral helix per subunit. Color code: CydA, green; CydB, pink; b-type hemes, cyan; heme d, dark blue.
The catalytically active subunit CydA of Cgbd harbors three prosthetic heme groups (b558, b595, and d) and contains the quinol oxidizing domain (Q-loop). The Q-loop is located between TMHs six and seven and protrudes into the periplasm. While the QC region is fully resolved, residues 268 to 317 which constitute the QN region could not be traced with high confidence. The Q-loop starts with the short helix Qh1 comprising the conserved residues Lys263.ACgbd and Glu268.ACgbd. These residues have been implicated in quinol binding and oxidation as well as coordination of propionate A from heme b558 in cytochromes bd from Proteobacteria (Matsumoto et al., 2006; Mogi et al., 2006). The QN region of Cgbd appears to be characterized by increased movement dynamics similar to what has been observed for cytochrome bd from Mycobacterium smegmatis (Msbd) (Wang et al., 2021). The QC region of Cgbd is fully resolved and consists of the periplasm exposed helix Qh3 that is followed by a 180° turn and leads back to TMH7. In the cytochrome bd structure of Mycobacterium tuberculosis (Mtbd), a hydrophobic cluster stabilizing the interaction of Qh3 with the periplasmic loop connecting TMHs eight and nine (PL8) was identified (Safarian et al., 2021). The residues Pro409.ACgbd, Trp410.ACgbd, and Pro414.ACgbd of PL8 and Tyr335.ACgbd of Qh3 are conserved in Cgbd, while the residues Tyr321.AMtbd and Phe325.AMtbd of Qh3 are replaced by Ala326.ACgbd and Tyr330.ACgbd. Since the residues Tyr335.ACgbd and Tyr330.ACgbd, which are relevant for the activity of the mycobacterial enzyme are conserved, a common role of Qh3 and periplasmic loop 8 (PL8) in stabilization can be inferred (Sviriaeva et al., 2020).
The two b-type hemes b558 and b595 as well as the chlorin-type heme d are organized in the well-characterized triangular cytochrome bd-specific geometry and are located in proximity of the periplasmic surface (Figure 2). The coordinating and vicinal residues, as well as the arrangement of the b-type hemes and heme d are conserved between cytochromes bd from Proteobacteria and Actinobacteria (Safarian et al., 2019; Theßeling et al., 2019; Grauel et al., 2021; Grund et al., 2021; Safarian et al., 2021; Wang et al., 2021). A noteable variation in the triangular heme arrangement is however found in the structure of cytochrome bd from Geobacillus thermodenitrificans (Gtbd), in which the high-spin heme b and heme d have “switched” positions.
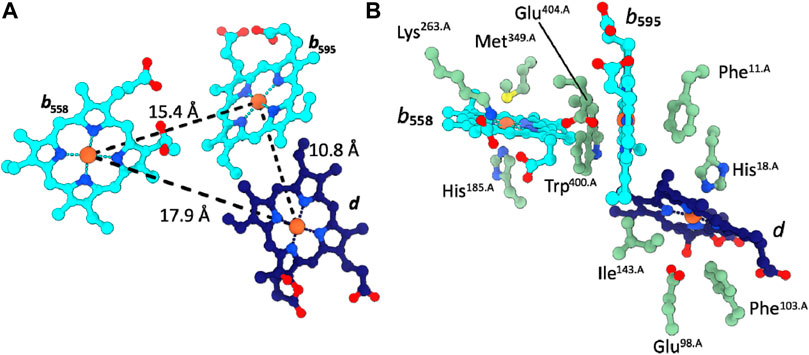
FIGURE 2. Cofactor arrangement in Cgbd. (A) Triangular heme arrangement in CydA. (B) Axial ligands and proximal side chains of the prosthetic heme groups in Cgbd. Color code: CydA, green; b-type hemes, cyan; heme d, dark blue.
The structural subunit CydB does not harbor cofactors or structural quinone molecules as observed in the proteobacterial cytochromes bd-I and bd-II (Safarian et al., 2019; Theßeling et al., 2019; Grauel et al., 2021; Grund et al., 2021). This is in agreement with the structures of the closely related Mtbd and Msbd that also lack structural quinones in the smaller subunit CydB (Safarian et al., 2021; Wang et al., 2021). Analogous to Mtbd, TMHs six and seven in CydB are connected by a short linker of three amino acids (CydBCgbd: amino acids 220–222), whereas the proteobacterial enzymes contain an elongated β-sheet extending along the periplasmic surface of CydB and a short α-helix oriented in a roughly 90° angle to TMH6 (Supplementary Figure S5).
The structural framework of a cytochrome bd-type enzyme requires, in addition to domains and cofactors for substrate oxidation and electron transfer, specific channels and cavities for allowing the access of protons and dioxygen to the reduction site at heme d, which is buried deeply within the core of the enzyme at the interface of CydA and CydB. In the Cgbd structure, both an oxygen and a proton pathway were identified (Figures 3A, B) (Sehnal et al., 2013; Pravda et al., 2018). The proton channel (H-channel) runs perpendicular to the membrane plane and connects the cytoplasm to the active site at heme d. The H-channel is predicted to start as a shallow cavity at the cytoplasmic site in proximity to TMHs two and three of CydB and subsequently constricts and elongates at the interface of CydA and CydB towards the reduction site. The entrance of the H-channel is shifted towards CydB compared to the proteobacterial channels (Figure 3C). The typical opening in the proteobacterial structures is obstructed by the extended C-terminal stretch of CydA in Cgbd, while the entrance of the H-channel of Cgbd is blocked by the short cytoplasmic C-terminal helix of CydB/AppB in cytochromes bd of E. coli. On the other hand, the location of the hydrophobic oxygen channel (O-channel) is matching well with the pathways observed in the proteobacterial and mycobacterial enzymes. The O-channel is predominantly lined by hydrophobic and uncharged residues and starts at the lipid interface between TMHs one and nine of CydB and extends parallel to the membrane plane, connecting the lipid bilayer to the reduction site at the chlorin-type heme d.
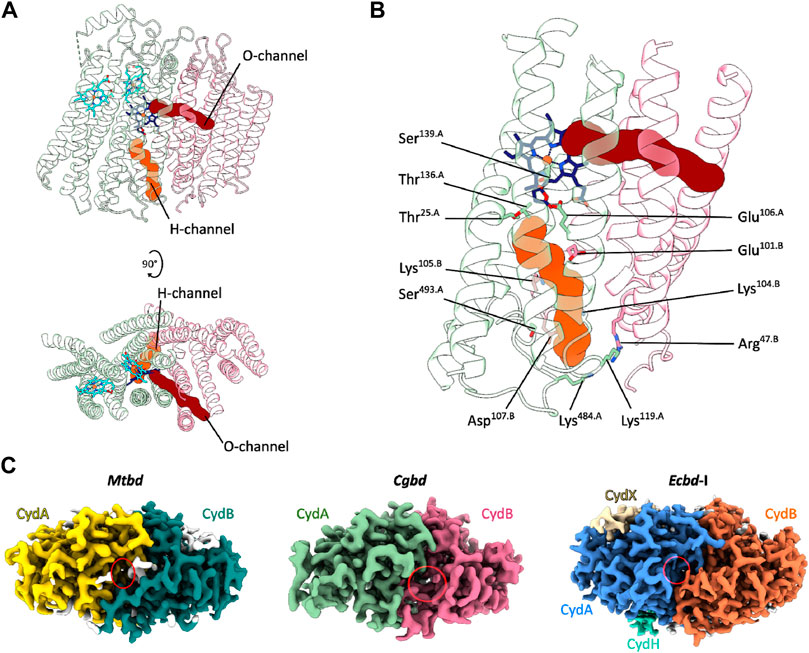
FIGURE 3. Oxygen and proton pathways in Cgbd. (A) Oxygen (O) and proton (H) channels predicted by MOLE2. The O-channel starts at the membrane interface between TMHs one, nine and two of CydB and TMH three of CydA and continues towards heme d. The predicted H-channel allows proton transfer from the cytoplasm to the reduction site. (B) Hydrophilic residues lining the H-channel. (C) Variation in location and size of the cytoplasmic proton channel entry sites in cytochromes bd. Depicted are the cryoEM maps of Mtbd (EMD-12451) (Safarian et al., 2021), Cgbd and Ecbd-I (EMD-4908) (Safarian et al., 2019) viewed from the cytoplasmic side of the membrane. The cavity elongating into the H-channel is highlighted by a red ellipse.
Two substrate quinone binding sites have been characterized for bd-type oxidases (for a comparison of all quinone sites please see Supplementary Figure S6). One at the Q-loop next to heme b558 which is best characterised in Ecbds (site 1). A second quinol binding-site composed of TMH1, TMH9 and heme b595 has been identified exclusively in Mtbd (site 2) (Safarian et al., 2019; Grauel et al., 2021; Safarian et al., 2021). At first glance, the overall structure of Cgbd is showing strong resemblance to that of Mtbd, however the structure of Qh3-PL8 region near site 1 is slightly different.
In Mtbd, the Qh3-PL8 region is mostly stabilized by a hydrophobic cluster composed of aromatic side chains as described previously (Safarian et al., 2021). In addition, a hydrogen bond between the side chain carbonyl group of Gln412.AMtbd and Arg324.AMtbd seems to strengthen the interaction between PL8 and Qh3 (Figure 4). Furthermore, electrostatic interactions between Arg324.A, Glu320.A, and Gln312.A seem to rigidify the conformation of the Mtbd Qc region even more. As Glu320.AMtbd, Tyr321.AMtbd and Arg324.AMtbd located within Qh3 are all substituted to alanine residues in homologous positions of Cgbd (Ala325.ACgbd, Ala326.ACgbd and Ala329.ACgbd), it is conceivable that the Qh3-PL8 interaction in Cgbd might be weaker than in Mtbd. In consequence, the local charge and steric environment in close proximity of the well-conserved Gln260.ACgbd and Lys263.ACgbd residues of Qh1, which have been suggested to be important for quinol oxidation in Ecbd-I, is significantly different (Mogi et al., 2006). These structural dissimilarities may be first indications that site 1 could represent the quinol binding and oxidation region in Cgbd. The increased movement of the Cgbd QN region reflected by unclearly-resolved densities of residues 268 to 317 (CydA) may support a model in which site 1 represents the electrochemically active substrate-binding domain. In fact, quinol oxidase activity of Cgbd is about 200–400 s−1 (Kusumoto et al., 2000), considerably higher than that of Msbd (22 s−1) (Wang et al., 2021).
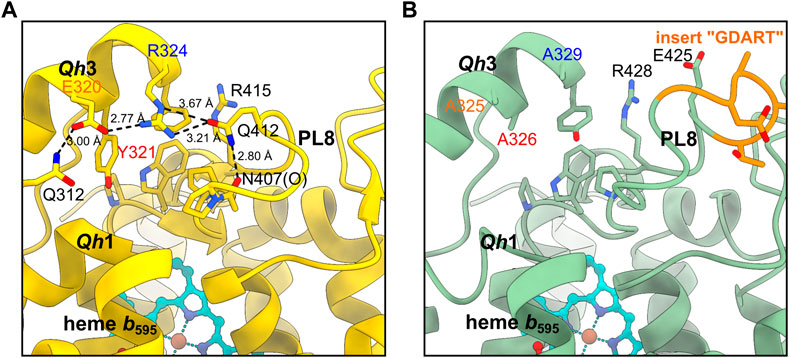
FIGURE 4. Comparison of the Qh3-PL8 region in (A) Mtbd and (B) Cgbd. The stabilizing residues E320, Y321 and R324 of Qh3 in Mtbd (7NKZ) (Safarian et al., 2021) are replaced by alanines in Cgbd, presumably leading to a weaker interaction between Qh3 and PL8.
In contrast to the structures of Cgbd and Ecbd-I, the Q-loop domain is fully resolved in Mtbd (Safarian et al., 2021). This may be because the QN region of Mtbd contains an internal disulfide bond which contributes to an overall increased stability and rigidity. Based on this unique finding, we previously suggested that the existence or absence of a disulfide bond within the Q-loop domain may correlate with utilization of either site 1 or site 2 as the electron entry route from substrate quinols (Safarian et al., 2021). Interestingly, these two cysteine residues (Cys271.ACgbd and Cys290.ACgbd) are conserved in Cgbd, yet our structure suggests that they are not forming a covalent bond, furthermore raising the question about the mechanism of disulfide bond generation and breakage within Q-loops of cytochromes bd in a physiological and metabolic context.
In Mtbd, site 2 is occupied by a MK-9 group, while it is appearing empty in Cgbd (Figure 5) (Safarian et al., 2021). At higher contour levels a weak density might be present, yet as judged by its size and shape at the current resolution, it more likely represents a bound lipid molecule rather than a quinone group (Supplementary Figure S3B). Nevertheless, the local environment formed by TMH1, TMH9 and heme b595 in Cgbd is almost identical to that of Mtbd. The residues forming the binding pocket for the naphthoquinone head group of MK-9 in Mtbd are conserved in Cgbd (Arg8.ACgbd, Trp9.ACgbd and Met405.ACgbd), and thus site 2 may still represent a presumable secondary MK-9 biding site. In Ecbd-I, site 2 is occupied by the accessory subunit CydH, which forms a different surface at this peripheral site of the cytochrome bd complex and consequently does not form any cavities which would allow for quinol binding close to the heme b595 group. The absence of the CydH subunit and the resulting access to site 2 might furthermore be the reason why we could previously show that the addition of menaquinones modulates the activity of Ecbd-II in a concentration dependent manner (Grund et al., 2021).
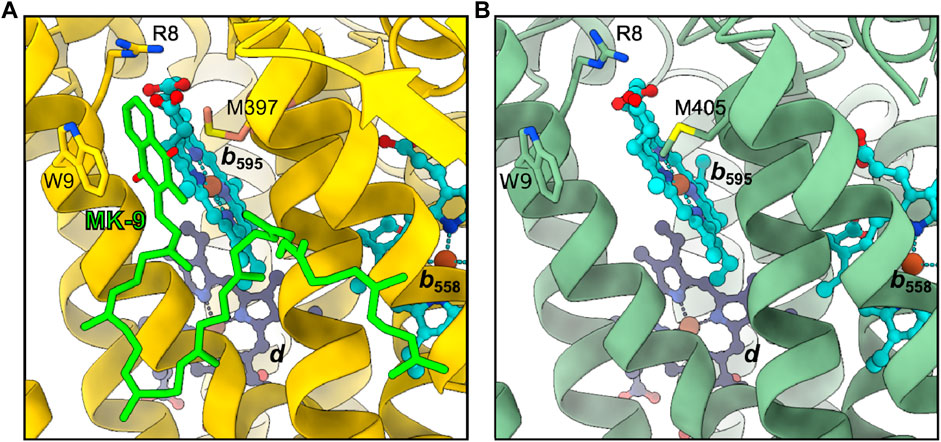
FIGURE 5. Second quinol binding site. (A) In Mtbd, site 2 harbors a bound MK-9 molecule stabilized by W9, R8 and M397. (B) Site 2 appears unoccupied in Cgbd, however the residues interacting with MK-9 in Mtbd are conserved in Cgbd.
We have previously reported that preincubation of Cgbd and Gtbd with quinones prior to induction of catalytic oxygen reduction activity enhances the turnover of the enzyme (Sakamoto et al., 1999; Kusumoto et al., 2000). The activity enhancement observed in these cases may be explained by the binding of quinones to site 2, the putative regulatory or acceleratory quinone binding site. It appears that the access of quinones to site 2 is possible in Gtbd and Cgbd since no accessory subunits are found at this position in the X-ray and cryoEM structures, respectively. However, Trp9.ACgbd/Mtbd, which seems important for forming stacking interactions with the naphthoquinone head group of bound MK-9, is not conserved in Gtbd. The absence of the Trp residue could explain the different preference of preincubated quinone species observed for Cgbd and Gtbd (Sakamoto et al., 1999; Kusumoto et al., 2000). The MK-9 that binds to site 2 of Mtbd is derived from endogenous sources, and its amount may vary depending on cultivation conditions and sample preparation procedures such as purification methods and the choice of detergents. Future studies are required to characterize site 2 interactions between the protein environment, the heme b595 group, and exogenously added quinones in greater detail.
In conclusion, the atomic structure of the cytochrome bd oxidase from C. glutamicum confirms common features among actinobacterial cytochrome bd oxidases: (i) the arrangement of three prosthetic heme cofactors in a triangular manner, (ii) the absence of accessory subunits, (iii) the location of the H- and the O-channels, (iv) structural features of a second quinone binding site (site 2). On the other hand, it is also reaffirmed that there are considerable differences between cytochrome bd oxidases of Actinobacteria and those of Firmicutes and Proteobacteria. To gain a deeper and more precise understanding of the catalytic mechanisms of the so-far characterized cytochrome bd homologs of Actinobacteria, follow-up structural studies of cytochromes bd in the presence of substrate quinols or substrate-derived inhibitors are indispensable.
Chemicals were purchased from Sigma unless otherwise stated. Cytochrome bd from C. glutamicum was produced in C. glutamicum subsp. lactofermentum (ΔctaD/pPC4-cydABCD). This production strain was established as described previously (Kabashima et al., 2009). In brief, the entire cydABDC genes including its authentic promoter region were amplified from genomic DNA of C. glutamicum by polymerase chain reaction (PCR) and inserted into the E. coli-C. glutamicum shuttle vector, pPC4. The cytochrome aa3-deficient mutant of C. glutamicum (ΔctaD) was transformed with the resulting plasmid, pPC4-cydABDC. Transformants were selected on solid medium containing 50 μg/ml kanamycin and 10 μg/ml chloramphenicol. Individual colonies were cultured in liquid medium for 30 h and the clone with the highest heme d content in membrane fractions was selected. Expression of cytochrome bd coding genes is induced at late stationary phase, in response to the low oxygen level due to its native promoter. After harvesting, cells were resuspended in 10 mM NaPi buffer at pH 7.0 containing 0.5% (w/v) NaCl. Cell disruption was performed via vigorous mixing with glass beads (diameter: 0.18 mm) in a cell disrupting mixer (Bead-Beater, Biospec). Subsequently, low-speed centrifugation (5,000 g, 10 min, 4°C) was carried out to remove undisrupted cells and cell debris. The supernatant was then subjected to ultracentrifugation (100,000 g, 30 min, 4°C). Pelleted membranes were resuspended in 10 mM NaPi buffer at pH 7.0 and used for further protein purification.
The membrane fraction was resuspended at 10 mg/ml in a first washing buffer [10 mM NaPi (pH 7.4), 0.5 M NaCl, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine] and stirred for 30 min at 4°C. The suspension was subjected to ultracentrifugation (100,000 g, 20 min, 4°C) and the pelleted membranes were resuspended at 10 mg/ml in a second washing buffer [10 mM NaPi (pH 7.4), 1.5% Na-cholate, 0.5% Na-deoxycholate and 0.1 M NaCl] and stirred for 30 min at 4°C. The membranes were sedimented at 100,000 g for 20 min and the pellet was subsequently resuspended at a concentration of 5 mg/ml in a solubilization buffer [10 mM NaPi (pH 7.4), 10% glycerol, 1% n-dodecyl-β-D-maltoside (DDM), 0.1 mM PMSF and 1 mM benzamidine]. Solubilization was carried out for 90 min at 4°C. The solubilized membrane fraction was separated from remaining crude membranes by centrifugation at 100,000 g for 20 min at 4°C.
The supernatant was applied to a Q-Sepharose column, washed with a buffer containing 10 mM NaPi (pH 7.4), 10% glycerol, 0.05% DDM, 0.1 mM PMSF and 1 mM benzamidine and eluted with a NaCl step gradient (150, 280 and 500 mM NaCl). After desalting of the oxidase containing fractions with an Amicon Ultra-100 K concentrator, the oxidase sample was applied to a DEAE-Toyopearl (Tosoh Bioscience) column, washed with a buffer containing 10 mM NaPi, (pH 7.4), 10% glycerol, 0.05% 2,2-didecylpropane-1,3-bis-β-d-maltopyranoside (LMNG), 0.1 mM PMSF and 1 mM benzamidine and eluted by a NaCl step gradient (150, 280 and 500 mM NaCl). Cytochrome bd elutes at a NaCl concentration of 280 mM from the DEAE-Toyopearl column. The oxidase containing fractions were pooled, flash-frozen in liquid nitrogen and stored at −80°C until further use.
For sample polishing, the sample was thawed on ice, subjected to centrifugation (21,130 g, 10 min, 4°C) and applied to a Superdex™ 200 Increase 10/300 GL column (GE Healthcare), which had been pre-equilibrated with 3 CV of a buffer containing 20 mM Tris/HCl (pH 7), 100 mM NaCl and 0.002% (w/v) LMNG. The chromatography was performed on an Äkta™ pure system (GE Healthcare) with a flow rate of 0.5 ml/min and a fraction volume of 0.2 ml. Peak fractions were pooled and concentrated to 2.5 mg/ml for downstream cryoEM sample preparation.
Quantifoil R1.2/1.3 300 mesh Au-carbon grids were washed in chloroform and subsequently glow discharged with a PELCO easiGlow device at 15 mA for 90 s. A volume of 4 μL sample was applied on a grid immediately before plunge freezing. Samples were vitrified at 4°C, 100% humidity, and a blot force of 20 using a Vitrobot IV device (Thermo Scientific). Blotting was carried out for 4 s before plunge-freezing in liquid ethane.
Electron micrographs were recorded using a Titan Krios G3i microscope operated at 300 kV (Thermo Scientific). Data were acquired automatically using EPU software (Thermo Scientifc) with aberration free image shift (AFIS) in faster acquisition mode using a K3 direct electron detector in combination with a post-column energy-imaging filter (Gatan) at a nominal magnification of ×105,000, corresponding to a calibrated pixel size of 0.837 Å. Dose-fractionated movies were recorded for 4.99 s at an electron flux of 15 e− x pixel−1 x s−1, corresponding to a total dose of about 107 e− x A−2. A focus range of −1.1 to −2.1 μm was applied throughout data collection.
Movies were motion corrected and dose weighted with MotionCor2 using a 5 × 5 patch (Zheng et al., 2017). Initial CTF (contrast transfer function) parameters for each movie were determined with Gctf (Zhang, 2016). At this step images with estimated poor resolution (>3.2 Å) and high astigmatism (>250) were removed. Autopicking was performed using crYOLO (Wagner et al., 2019) and all further data processing including 2D classification, 3D classification, 3D refinement and Bayesian polishing was performed using Relion 3.1 (Zivanov et al., 2018). A total of 10,586,211 particles were picked and cleaned up with four rounds of two-dimensional (2D) classification. One round of 3D classification provided the final particle stack of 410,010 particles which was used for all subsequent rounds of auto-refinement, CTF refinement and Bayesian polishing. Final maps were generated after subtraction of the detergent micelle followed by CTF refinement. An overview of the processing workflow is given in Supplementary Figure S2.
All model-building steps were performed using COOT (version 0.9.6) (Emsley et al., 2010). The cryoEM structure of cytochrome bd from Mycobacterium tuberculosis (pdb:7NKZ) was used as starting model (Safarian et al., 2021). After backbone fitting and side chain docking, we performed real-space refinement in Phenix (version 1.18.2). Refinement results were manually inspected and corrected if necessary. Map-to-model cross validation was performed in Phenix (version 1.18.2). FSC0.5 was used as cut-off to define resolution. Model parameters and corresponding cryoEM map statistics are summarized in Supplementary Table S1. The finalized model was visualized using ChimeraX (Goddard et al., 2018). Tunnels and interior cavities were mapped with MOLE2.5 (bottleneck radius: 1.1 Å, bottleneck tolerance: 3 Å, origin radius 5 Å, surface radius 8 Å, probe radius 5 Å, interior threshold 1.1 Å) (Sehnal et al., 2013; Pravda et al., 2018).
The cryoEM map file of cytochrome bd from C. glutamicum was deposited in the EMD database and can be found under accession number EMD-15851. The model file of cytochrome bd from C. glutamicum was deposited to the PDB under accession number 8B4O.
TG optimized sample conditions, prepared grids, collected cryoEM data, processed cryoEM data, refined the structure, built the model, co-drafted the manuscript, and prepared figures. YK and TK produced enzyme, established purification, and co-drafted the manuscript. DW collected cryoEM data. SW calibrated and aligned the microscope. JS, HM, and SS initiated, designed and supervised the project. JS, HM, and SS evaluated data, and drafted the manuscript.
This work was supported by the Max Planck Society, the Nobel Laureate Fellowship of the Max Planck Society, and by a Grant-in-Aid for Scientific Research (C) (16K07299 to JS) from the Japan Society for the Promotion of Science.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1085463/full#supplementary-material
Azarkina, N., Siletsky, S., Borisov, V., von Wachenfeldt, C., Hederstedt, L., and Konstantinov, A. A. (1999). A cytochrome bb’-type quinol oxidase in Bacillus subtilis strain 168. J. Biol. Chem. 274, 32810–32817. doi:10.1074/jbc.274.46.32810
Borisov, V. B., Gennis, R. B., Hemp, J., and Verkhovsky, M. I. (2011). The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 1807, 1398–1413. doi:10.1016/j.bbabio.2011.06.016
Borisov, V. B., and Siletsky, S. A. (2019). Features of organization and mechanism of catalysis of two families of terminal oxidases: Heme-copper and bd-type. Biochem. Mosc 84, 1390–1402. doi:10.1134/S0006297919110130
Borisov, V. B., Siletsky, S. A., Paiardini, A., Hoogewijs, D., Forte, E., Giuffrè, A., et al. (2020). Bacterial oxidases of the cytochrome bd family: Redox enzymes of unique structure, function, and utility as drug targets. Antioxid. Redox Signal 34, 1280–1318. doi:10.1089/ars.2020.8039
Borisov, V. B., and Verkhovsky, M. I. (2015). Oxygen as acceptor. Ecosal Plus 6. doi:10.1128/ecosalplus.ESP-0012-2015
Cook, G. M., and Poole, R. K. (2016). BIOCHEMISTRY. A bacterial oxidase like no other? Science 352, 518–519. doi:10.1126/science.aaf5514
Cunningham, L., Pitt, M., and Williams, H. D. (1997). The cioAB genes from Pseudomonas aeruginosa code for a novel cyanide-insensitive terminal oxidase related to the cytochrome bd quinol oxidases. Mol. Microbiol. 24, 579–591. doi:10.1046/j.1365-2958.1997.3561728.x
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501. doi:10.1107/S0907444910007493
Friedrich, T., Wohlwend, D., and Borisov, V. B. (2022). Recent advances in structural studies of cytochrome bd and its potential application as a drug target. Int. J. Mol. Sci. 23, 3166. doi:10.3390/ijms23063166
Goddard, T. D., Huang, C. C., Meng, E. C., Pettersen, E. F., Couch, G. S., Morris, J. H., et al. (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. doi:10.1002/pro.3235
Goojani, H. G., Konings, J., Hakvoort, H., Hong, S., Gennis, R. B., Sakamoto, J., et al. (2020). The carboxy-terminal insert in the Q-loop is needed for functionality of Escherichia coli cytochrome bd-I. Biochim. Biophys. Acta Bioenerg. 1861, 148175. doi:10.1016/j.bbabio.2020.148175
Grauel, A., Kägi, J., Rasmussen, T., Makarchuk, I., Oppermann, S., Moumbock, A. F. A., et al. (2021). Structure of Escherichia coli cytochrome bd-II type oxidase with bound aurachin D. Nat. Commun. 12, 6498. doi:10.1038/s41467-021-26835-2
Grund, T. N., Radloff, M., Wu, D., Goojani, H. G., Witte, L. F., Jösting, W., et al. (2021). Mechanistic and structural diversity between cytochrome bd isoforms of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 118, e2114013118. doi:10.1073/pnas.2114013118
Hoeser, J., Hong, S., Gehmann, G., Gennis, R. B., and Friedrich, T. (2014). Subunit CydX of Escherichia coli cytochrome bd ubiquinol oxidase is essential for assembly and stability of the di-heme active site. FEBS Lett. 588, 1537–1541. doi:10.1016/j.febslet.2014.03.036
Hoganson, C. W., Pressler, M. A., Proshlyakov, D. A., and Babcock, G. T. (1998). From water to oxygen and back again: Mechanistic similarities in the enzymatic redox conversion between water and dioxygen. Biochimica Biophysica Acta (BBA) - Bioenergetics 1365, 170–174. doi:10.1016/S0005-2728(98)00057-7
Jackson, R. J., Elvers, K. T., Lee, L. J., Gidley, M. D., Wainwright, L. M., Lightfoot, J., et al. (2007). Oxygen reactivity of both respiratory oxidases in Campylobacter jejuni: The cydAB genes encode a cyanide-resistant, low-affinity oxidase that is not of the cytochrome bd type. J. Bacteriol. 189, 1604–1615. doi:10.1128/JB.00897-06
Kabashima, Y., Kishikawa, J.-I., Kurokawa, T., and Sakamoto, J. (2009). Correlation between proton translocation and growth: Genetic analysis of the respiratory chain of Corynebacterium glutamicum. J. Biochem. 146, 845–855. doi:10.1093/jb/mvp140
Kita, K., Konishi, K., and Anraku, Y. (1984). Terminal oxidases of Escherichia coli aerobic respiratory chain. II. Purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol. Chem. 259, 3375–3381. doi:10.1016/s0021-9258(17)43305-9
Kusumoto, K., Sakiyama, M., Sakamoto, J., Noguchi, S., and Sone, N. (2000). Menaquinol oxidase activity and primary structure of cytochrome bd from the amino-acid fermenting bacterium Corynebacterium glutamicum. Arch. Microbiol. 173, 390–397. doi:10.1007/s002030000161
Lyons, T. W., Reinhard, C. T., and Planavsky, N. J. (2014). The rise of oxygen in Earth’s early ocean and atmosphere. Nature 506, 307–315. doi:10.1038/nature13068
Matsumoto, Y., Murai, M., Fujita, D., Sakamoto, K., Miyoshi, H., Yoshida, M., et al. (2006). Mass spectrometric analysis of the ubiquinol-binding site in cytochrome bd from Escherichia coli. J. Biol. Chem. 281, 1905–1912. doi:10.1074/jbc.M508206200
May, B., Young, L., and Moore, A. L. (2017). Structural insights into the alternative oxidases: Are all oxidases made equal? Biochem. Soc. Trans. 45, 731–740. doi:10.1042/BST20160178
Miller, M. J., and Gennis, R. B. (1983). The purification and characterization of the cytochrome d terminal oxidase complex of the Escherichia coli aerobic respiratory chain. J. Biol. Chem. 258, 9159–9165. doi:10.1016/S0021-9258(17)44645-X
Mogi, T., Akimoto, S., Endou, S., Watanabe-Nakayama, T., Mizuochi-Asai, E., and Miyoshi, H. (2006). Probing the ubiquinol-binding site in cytochrome bd by site-directed mutagenesis. Biochemistry 45, 7924–7930. doi:10.1021/bi060192w
Mogi, T., Ano, Y., Nakatsuka, T., Toyama, H., Muroi, A., Miyoshi, H., et al. (2009). Biochemical and spectroscopic properties of cyanide-insensitive quinol oxidase from Gluconobacter oxydans. J. Biochem. 146, 263–271. doi:10.1093/jb/mvp067
Nikolaev, A., Safarian, S., Thesseling, A., Wohlwend, D., Friedrich, T., Michel, H., et al. (2021). Electrocatalytic evidence of the diversity of the oxygen reaction in the bacterial bd oxidase from different organisms. Biochim. Biophys. Acta Bioenerg. 1862, 148436. doi:10.1016/j.bbabio.2021.148436
Osborne, J. P., and Gennis, R. B. (1999). Sequence analysis of cytochrome bd oxidase suggests a revised topology for subunit I. Biochim. Biophys. Acta 1410, 32–50. doi:10.1016/s0005-2728(98)00171-6
Pravda, L., Sehnal, D., Toušek, D., Navrátilová, V., Bazgier, V., Berka, K., et al. (2018). MOLEonline: A web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 46, W368–W373. doi:10.1093/nar/gky309
Puustinen, A., Finel, M., Haltia, T., Gennis, R. B., and Wikström, M. (1991). Properties of the two terminal oxidases of Escherichia coli. Biochemistry 30, 3936–3942. doi:10.1021/bi00230a019
Quesada, A., Guijo, M. I., Merchán, F., Blázquez, B., Igeño, M. I., and Blasco, R. (2007). Essential role of cytochrome bd-related oxidase in cyanide resistance of Pseudomonas pseudoalcaligenes CECT5344. Appl. Environ. Microbiol. 73, 5118–5124. doi:10.1128/AEM.00503-07
Safarian, S., Hahn, A., Mills, D. J., Radloff, M., Eisinger, M. L., Nikolaev, A., et al. (2019). Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science 366, 100–104. doi:10.1126/science.aay0967
Safarian, S., Opel-Reading, H. K., Wu, D., Mehdipour, A. R., Hards, K., Harold, L. K., et al. (2021). The cryo-EM structure of the bd oxidase from M. tuberculosis reveals a unique structural framework and enables rational drug design to combat TB. Nat. Commun. 12, 5236. doi:10.1038/s41467-021-25537-z
Safarian, S., Rajendran, C., Müller, H., Preu, J., Langer, J. D., Ovchinnikov, S., et al. (2016). Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 352, 583–586. doi:10.1126/science.aaf2477
Sakamoto, J., Koga, E., Mizuta, T., Sato, C., Noguchi, S., and Sone, N. (1999). Gene structure and quinol oxidase activity of a cytochrome bd-type oxidase from Bacillus stearothermophilus. Biochim. Biophys. Acta 1411, 147–158. doi:10.1016/s0005-2728(99)00012-2
Schirrmeister, B. E., de Vos, J. M., Antonelli, A., and Bagheri, H. C. (2013). Evolution of multicellularity coincided with increased diversification of cyanobacteria and the Great Oxidation Event. Proc. Natl. Acad. Sci. U. S. A. 110, 1791–1796. doi:10.1073/pnas.1209927110
Sehnal, D., Svobodová Vařeková, R., Berka, K., Pravda, L., Navrátilová, V., Banáš, P., et al. (2013). MOLE 2.0: Advanced approach for analysis of biomacromolecular channels. J. Cheminform. 5, 39. doi:10.1186/1758-2946-5-39
Sviriaeva, E., Subramanian Manimekalai, M. S., Grüber, G., and Pethe, K. (2020). Features and functional importance of key residues of the Mycobacterium tuberculosis cytochrome bd oxidase. ACS Infect. Dis. 6, 1697–1707. doi:10.1021/acsinfecdis.9b00449
Theßeling, A., Burschel, S., Wohlwend, D., and Friedrich, T. (2020). The long Q-loop of Escherichia coli cytochrome bd oxidase is required for assembly and structural integrity. FEBS Lett. 594, 1577–1585. doi:10.1002/1873-3468.13749
Theßeling, A., Rasmussen, T., Burschel, S., Wohlwend, D., Kägi, J., Müller, R., et al. (2019). Homologous bd oxidases share the same architecture but differ in mechanism. Nat. Commun. 10, 5138. doi:10.1038/s41467-019-13122-4
VanOrsdel, C. E., Bhatt, S., Allen, R. J., Brenner, E. P., Hobson, J. J., Jamil, A., et al. (2013). The Escherichia coli CydX protein is a member of the CydAB cytochrome bd oxidase complex and is required for cytochrome bd oxidase activity. J. Bacteriol. 195, 3640–3650. doi:10.1128/JB.00324-13
Wagner, T., Merino, F., Stabrin, M., Moriya, T., Antoni, C., Apelbaum, A., et al. (2019). SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 218. doi:10.1038/s42003-019-0437-z
Wang, W., Gao, Y., Tang, Y., Zhou, X., Lai, Y., Zhou, S., et al. (2021). Cryo-EM structure of mycobacterial cytochrome bd reveals two oxygen access channels. Nat. Commun. 12, 4621. doi:10.1038/s41467-021-24924-w
Zhang, K. (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12. doi:10.1016/j.jsb.2015.11.003
Zheng, S. Q., Palovcak, E., Armache, J.-P., Verba, K. A., Cheng, Y., and Agard, D. A. (2017). MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. doi:10.1038/nmeth.4193
Keywords: CryoEM, microbiology, oxidases, proton channel, electrochemistry
Citation: Grund TN, Kabashima Y, Kusumoto T, Wu D, Welsch S, Sakamoto J, Michel H and Safarian S (2023) The cryoEM structure of cytochrome bd from C. glutamicum provides novel insights into structural properties of actinobacterial terminal oxidases. Front. Chem. 10:1085463. doi: 10.3389/fchem.2022.1085463
Received: 01 November 2022; Accepted: 12 December 2022;
Published: 04 January 2023.
Edited by:
Petra Hellwig, Université de Strasbourg, FranceReviewed by:
Thorsten Friedrich, University of Freiburg, GermanyCopyright © 2023 Grund, Kabashima, Kusumoto, Wu, Welsch, Sakamoto, Michel and Safarian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Schara Safarian, c2NoYXJhLnNhZmFyaWFuQGJpb3BoeXMubXBnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.