 Yingxia Lu
Yingxia Lu Xuan Dong
Xuan Dong Xiaozhen Huang
Xiaozhen Huang De-gang Zhao
De-gang Zhao Yichen Zhao
Yichen Zhao Lei Peng
Lei Peng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 13 October 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1053227
This article is part of the Research Topic Advances in Novel Natural Product Pesticides View all 24 articles
Eucommia ulmoides Oliv. (Duzhong), a valued traditional herbal medicine in China, is rich in antibacterial proteins and is effective against a variety of plant pathogens. Fusarium oxysporum is a pathogenic fungus that infects plant roots, resulting in the death of the plant. In this study, transcriptomic and proteomic analyses were used to explore the molecular mechanism of E. ulmoides counteracts F. oxysporum infection. Transcriptomic analysis at 24, 48, 72, and 96 h after inoculation identified 17, 591, 1,205, and 625 differentially expressed genes (DEGs), while proteomics identified were 66, 138, 148, 234 differentially expressed proteins (DEPs). Meanwhile, GO and KEGG enrichment analyses of the DEGs and DEPs showed that they were mainly associated with endoplasmic reticulum (ER), fructose and mannose metabolism, protein processing in the ER, type II diabetes mellitus, the ribosome, antigen processing and presentation, and the phagosome. In addition, proteome and transcriptome association analysis and RT-qPCR showed that the response of E. ulmoides to F. oxysporum was likely related to the unfolded protein response (UPR) of the ER pathway. In conclusion, our study provided a theoretical basis for the control of F. oxysporum.
Eucommia ulmoides Oliv. (Duzhong) is a traditional Chinese herbal medicine and is recorded in the ancient pharmacy classics, Shen Nong’s Herbal Classic and Compendium of Materia Medical (Anderson, 1982). It is the only existing species of the family Eucommiaceae (Tokumoto et al., 2016). The leaves and bark of E. ulmoides have been listed in the Chinese Pharmacopoeia (Qian et al., 2010), and its medicinal components have attracted extensive attention. It has been used to treat hypertension, hyperlipidemia, obesity, diabetes, inflammation, and other conditions. In addition, it also has antiviral, antibacterial, antioxidant, antitumor, and other activities (Qian et al., 2010; Fujiwara et al., 2016; Hussain et al., 2017; Cho et al., 2018; Lee et al., 2018).
Fusarium oxysporum is a widespread plant pathogenic fungus, belonging to the imperfect fungi. It is a soil-borne parasite and produces large numbers of sickle-shaped conidia through asexual reproduction after infecting the plant (Gordon and Martyn, 1997; Michielse and Rep, 2010). It infects the plant roots and causes deterioration of the vascular bundles, damages the functioning of the ducts, and may cause the death of the plant through damage at all stages of the plant growth (Pietro et al., 2010). Fusarium infection affects a large number of crops with important economic value, such as the Musaceae, Rosaceae, Cucurbitaceae, Brassicaceae, Solanaceae, and Fabaceae (Gordon and Martyn, 1997; Edel-Hermann and Lecomte, 2019), which has brought serious losses to agricultural production. Our laboratory’s previous research found that the total protein extracted from E. ulmoides had a significant inhibitory effect on F. oxysporum (Liu et al., 2007). However, the molecular mechanism of E. ulmoides counteracts F. oxysporum infection was not clear yet.
With the development of high-throughput sequencing technologies, “omics” analyses have been widely applied to the study of plant-microbe interactions, especially in the study of plant responses to pathogen stress. The transcriptome represents the sum of all transcripts produced by a particular tissue or cell of a species at a specific time (Guo et al., 2021). Transcriptome sequencing can determine the expression of all genes during a particular period, which provides an important reference for gene transcription (Wilhelm and Landry, 2009). Transcriptome sequencing has been used extensively to study biotic and abiotic stresses in plants, and the technology can provide a comprehensive picture of gene expression in target biological tissues under specific stress conditions. Comparative transcriptome analysis can identify changes in the expression of genes associated with specific physiological or pathological conditions (Shi et al., 2020). Proteomics is used to identify and quantify the overall protein content of cells, tissues, or organisms. It is an effective complement to other omics analyses, such as genomics and transcriptomics. It provides a useful basis for elucidating the structure and function of proteins, resolving cellular signaling pathways (Wasinger et al., 1995; Mann et al., 2001; Monti et al., 2005), and understanding pathogenic mechanisms (Saleh et al., 2019).
With the development of biological information, in this study, we explore to study the molecular mechanism of E. ulmoides in response to F. oxysporum using transcriptome and proteome methods. This study will provide a theoretical basis for the control of F. oxysporum.
The seeds of E. ulmoides were collected from E. ulmoides trees that had been established for over 10 years in the Key Laboratory of Mountain Plant Resource Protection and Germplasm Innovation, Institute of Agricultural Bioengineering, Guizhou University. F. oxysporum was isolated from Passiflora edulis Sims according to the method of Sarwar et al. (2005) showing characteristic symptoms of wilt and identified by the Tea Biotechnology Laboratory of Tea College of Guizhou University. Plants of P. edulis Sims showing characteristic symptoms of wilt were collected with fibrous tertiary roots on the basis of visual observations and brought in the laboratory for isolation of F. oxysporum.
The peels of the mature seeds of E. ulmoides were removed and the seeds surfaces were thoroughly cleaned using sterile water. Then the seeds were soaked in sterile water for 12 h and rinsed with tap water. After disinfection with 75% alcohol for 60 s, the seeds were washed several times with sterile water, followed by soaking in NaClO solution with 10% available chlorine for 15 min. After several further washes with sterile water, the seeds were dried on sterile water-absorbing paper. The seed coat was then cut using a scalpel and inoculated on solid Murashige and Skoog (MS) basal medium with vitamins (cultured in a glass incubator containing 50 ml MS medium with a diameter of 5.5 cm and a height of 12 cm) and cultured under a light/dark cycle of 16/8 h at 25°C. After 15–20 days when the cotyledons were fully expanded, the roots of E. ulmoides seedlings with 2-4 true leaves were inoculated with F. ulmoides. The inoculated seedlings were cultured in dark at 25°C. After inoculation for 0, 24, 48, 72, and 96 h, the naked eye fungal infection and necrotic part were removed and the samples for subsequent experiments were collected from the tissue 1 cm above the removed tissue. Then, the collected samples were snap-frozen in liquid nitrogen and stored at -80°C.
The collected samples (100 mg) were ground with liquid nitrogen. Total RNA was extracted using the Plant RNA Extraction Kit (ComWin Biotech Co., Ltd., Beijing, China) according to the supplied method, and three replicates were prepared for each treatment. Poly-A tailed mRNA was enriched using Oligo dT magnetic beads. The RNA was then fragmented and reverse-transcribed using random N6 primers. After synthesis of double-stranded DNA, the ends were flattened and phosphorylated at the 5′ end, while the 3′ end had a protruding “A” sticky end. The ligated product was amplified by PCR with specific primers (forward primer: 5′-GAACGACATGGCTACGA-3′ and reverse primer: 5′-TGTGAGCCAAGGAGTTG-3′), the PCR product was heat-denatured to single-stranded DNA and then cyclized with a bridge primer to obtain a single-stranded circular DNA library and sequenced using DNBSEQ platform.
The raw sequencing data contains reads with low quality, splice contamination, and high contents of unknown base N. These reads need to be removed before data analysis to ensure the reliability of the results. The filtering software SOAPnuke version 1.4.0 was used for analysis and filtering was performed using Trimmomatic v0.36 to remove the reads containing the connector, those with unknown base N contents greater than 5%, and low-quality reads with mass values of less than 10 accounting for more than 20% of the total base number of the reads. The clean reads were then used for subsequent analysis and were mapped to the Eucommia reference genome (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA357336) (Chen et al., 2022) using HISAT 2.1.0 (Kim et al., 2015). The clean reads were also compared with reference gene sequences using Bowtie 22.2.5 (Langmead and Salzberg, 2012) and RSEM 1.2.8 (Li and Dewey, 2011) was used to calculate the expression levels of the genes and transcripts. All the programs were used with default parameters. The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number of CNP0003469.
The total protein of E. ulmoides was extracted according to the method of Yu et al. (2018) and analyzed by Nano LC-1DTM plus system (Eksigent, Dublin, CA, United States) combined with Triple TOF5600 MS (Foster City, CA, United States).
Max Quant version 1.5.2.8 (Cox et al., 2011) was used for processing the LC-MS/MS data. The E. ulmoides protein database downloaded from NCBI was used for MS/MS identification. The results of the Max Quant analysis, including the initial search for the precursor mass with a tolerance of 20 μl/ml, were used for mass recalibration (Cox and Mann, 2008). The false discovery rate (FDR) for peptide and protein identification was set at 0.01. Label-free quantification with a minimum of two ratio counts was used for normalization and the sum of two ratio counts was used to determine the normalized protein levels for comparison between samples (Cox and Mann, 2008). The IBAQ algorithm was used to calculate the absolute abundance of different proteins in a single sample (Luber et al., 2010), and the proteins were filtered to eliminate common contaminants and the interference of opposite databases. The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number of CNP0003469.
To annotate unigenes which were assembled by Stringtie, sequences were searched by BLASTx against the NCBI non-redundant protein (nr) database and other databases, including the Swiss-Prot protein database, the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the Gene Ontology (GO) databases. KEGG enrichment scatter plots and GO enrichment bar plots were constructed using the OmicStudio tools at https://www.omicstudio.cn/tool.
The DESeq2 R package (https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8, accessed on November 2020) was used to analyses the differential expression between groups; it uses a model based on the negative binomial distribution to provide statistical routines for determining differential expressions in digital gene expression data. The resulting p-values were adjusted by using Benjamini and Hochberg’s approach to control the false discovery rate (FDR < 0.01). Genes with an adjusted p value < 0.01 by DESeq2 were assigned as differentially expressed genes (DEGs). The protein differences between two groups were analyzed by t-tests. Proteins with p values ≤ 0.01 were considered to be differentially expressed proteins (DEPs).
To correlate transcript and protein expression profiles, accession numbers from the proteome were extracted and compared with the annotated RNA-Seq libraries. A protein-mRNA correlation analysis was performed using the regularized-logarithm transformation (rlog) value of the spectral counts and the normalized log2 probe intensity for mRNAs. Briefly, we calculated the global Spearman correlation coefficient, rho, for protein-mRNA pairs within all DEPs and DEGs, similar regulatory patterns of DEPs and DEGs and opposite regulatory patterns of DEPs and DEGs, respectively. Adjusted p values based on the analysis of DEPs and DEGs were computed by the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). The KEGG enrichment analysis and Advanced Heatmap Plots were performed using the OmicStudio tools by R version 3.6.3 (https://www.omicstudio.cn).
Total RNA was extracted from powdered E. ulmoides samples using the Plant RNA Extraction Kit (ComWin Biotech Co., Ltd., Beijing, China). The RNA was reverse-transcribed using a Goldenstar™ RT6 cDNA Synthesis Kit (Tsingke Biotechnology, Beijing, China) according to the manufacturer’s instructions. Real-time quantitative polymerase chain reaction (RT-qPCR) was performed using the reaction sample (10 μl) with SoFast EvaGreen® Supermix (Bio-Rad, Hercules, CA, United States) and the RT-PCR assay was completed using the CFX ConnectTM Real-Time PCR System (Bio-Rad, Hercules, CA, United States), each sample was run in triplicate with appropriate negative controls, under the same condition used in previous studies: 95°C for 3 min, then 39 cycles of 95°C for 10 s, 65°C for 20 s, and 72°C for 10 s. Fluorescence data were collected at the end of the second step and, following cycling, the melting curve was determined in the range of 65–95°C with an increment of 0.05°C/s. Gene expression was normalized using elongation factor-1 alpha as an internal control. The specific primers used in RT-qPCR were designed by the RNA Folding Form (http://www.unafold.org/mfold/applications/rna-folding-form.php) and Primer Quest™ Tool (https://sg.idtdna.com/pages/tools/primerquest) and listed in Table 1. The 2−∆∆Ct method was used to calculate the relative expression levels. Each treatment was analyzed for three times.

TABLE 1. List of primers used in this study.
The roots morphology of E. ulmoides seedlings inoculated with F. oxysporum was observed. As shown in Figure 1, after 24 h of inoculation, no mycelia were observed on the MS basal medium and the roots of E. ulmoides seedlings, while small numbers of new mycelia of F. oxysporum were visible after 48 h; no obvious lesions or necrosis were seen on the roots at either time. After 72 h of inoculation, large numbers of new mycelia of F. oxysporum were apparent, although there was no evidence of lesions or necrosis. After 96 h of inoculation, the root of E. ulmoides was eroded by F. oxysporum, with clear signs of necrosis and finger sheath-like detachments.

FIGURE 1. The roots morphology of E. ulmoides seedlings after 0, 24, 48, 72, and 96 h of inoculation with F. oxysporum.
A total of 15 samples (label as ck-1, ck-2, ck-3, 24h-1, 24h-2, 24h-3, 48h-1, 48h-2, 48h-3, 72h-1, 72h-2, 72h-3, 96h-1, 96h-2, 96h-3) were tested using the DNBSEQ platform. The quality indicators of the filtered reads are shown in Supplementary Table S1. The results showed that a total of 95.99 GB high-quality reads were obtained from the RNA-Seq transcriptome sequencing. Each sample had three independent biological replicates and each sample produced an average of 6.40 GB of data. The Q20 and Q30 of each sample were over 96.04% and 87.42%, respectively. The block diagram of the FPKM values for each sample was shown in Supplementary Figure S1, which showed that the data are accurate and could be used directly for further analysis. The average comparison rate between the samples and the genome was 68.49%. A total of 5177 new genes were predicted and the expression of 28,604 genes was detected, of which 23,478 were known genes and 5,126 were possibly new genes. A total of 57,545 new transcripts were detected, of which 15,439 belonged to new variable splicing subtypes of known protein-coding genes, 5,177 were transcripts of new protein-coding genes, and the remaining 36,929 were long non-coding RNAs.
After 72 h of inoculation with F. oxysporum, the Venn diagram (Figure 2A) showed that the greatest number (1205) of DEGs in E. ulmoides were present, and the DEGs are visually represented in the volcano plot (Supplementary Figure S2) with the numbers of up- and downregulated genes being 292 and 913, respectively. Meanwhile, after 72 h of inoculation, the annotation of genes using the GO database indicated that the total number of genes (TB) and the significantly DEGs between the inoculated and uninoculated treatments (TS) with F. oxysporum were 23,802 and 1,013, respectively. The top four DEGs annotations in the biological process (BP) category were found to be cellular process, metabolic process, organic substance metabolic process, and cellular metabolic process, with the numbers of S genes (the number of significantly DEGs annotated in the designated database) were 296, 274, 231, and 211, respectively. In the cellular components (CC) category, DEGs were enriched in the membrane, membrane part, intrinsic component of membrane, and integral component of membrane categories, with S gene numbers of 403, 382, 371, and 370, respectively. The CC categories of cell, cell part, intracellular, intracellular part, intracellular organelle, and organelle were also enriched, and the numbers of S genes were 312, 300, 249, 249, 218, and 218, respectively. In the molecular functions (MF) category, the DEGs were enriched in catalytic activity, binding, ion binding, heterocyclic compound binding, organic cyclic compound binding, and transferase activity. The numbers of genes were 542, 478, 310, 309, 309, 212, and 212, respectively (Figure 2B; Supplementary Figure S3). KEGG pathway enrichment (Figure 2C; Supplementary Figure S4) showed that there were 12,550 TB genes and 600 TS genes. DEGs were significantly enriched in metabolic pathways, biosynthesis of secondary metabolites, plant-pathogen interaction, MAPK signaling pathway-plant, fructose and mannose metabolism, phosphatidylinositol signaling system, and protein processing in ER, with the gene numbers of 288, 174, 56, 55, 12, 10, and 9, respectively.
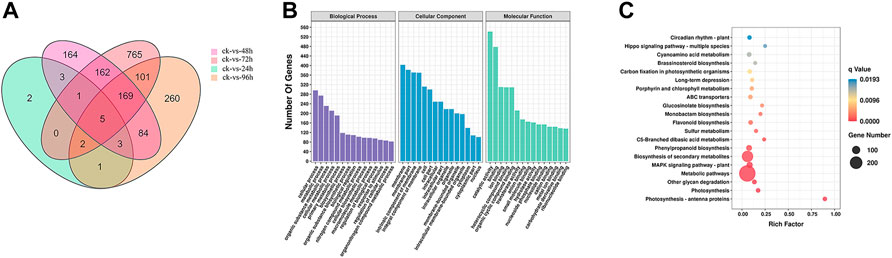
FIGURE 2. (A) Venn diagram of DEGs in E. ulmoides compared before (ck) with 24, 48, 72, and 96 h of inoculation with F. oxysporum, respectively. (B) GO analysis of DEGs in E. ulmoides between 0 and 72 h of inoculation with F. oxysporum. (C) KEGG enrichment analysis of DEGs in E. ulmoides between 0 and 72 h of inoculation with F. oxysporum.
After the component analysis (Supplementary Figure S5) and cluster analysis (Supplementary Figure S6) of the DEPs, the data from three biological replicates obtained after 0, 24, 48, 72, and 96 h of inoculation with F. oxysporum were analyzed, indicating that both the samples and the data were reliable and could be further analyzed. A total of 1,595 peptides were obtained by searching and analyzing the peptides in the total proteomic library of E. ulmoides obtained before and after inoculation with F. oxysporum. Overall, 1397, 1268, 1206, 1220, and 1166 peptides were obtained after 0, 24, 48, 72, and 96 h of inoculation, respectively.
The Venn diagram (Figure 3A) showed that the number of DEPs was highest at 96 h after inoculation with F. oxysporum. The DEPs were visualized using a volcano plot (Supplementary Figure S7), indicating that eight proteins were up-regulated and 226 were downregulated after 96 h of inoculation compared with their expression before inoculation. The GO database indicated that the significant DEPs were enriched in small-molecule metabolic process, translation, carbohydrate metabolic process, protein folding, response to stress, and other BP. The number of DEPs in these categories were 50, 30, 27, 17, and 14, respectively. In the CC category, DEPs were enriched in cytoplasm, membrane, ribosome, chloroplast, and chloroplast, with 77, 27, 26, and 15 proteins in the respective categories. In terms of MF, the numbers of DEPs enriched in ion binding, oxidoreductase activity, hydrolase activity, structural molecule activity were 84, 45, 35, 28, and 27, respectively (Figure 3B; Supplementary Figure S8). KEGG pathway analysis (Figure 3C; Supplementary Figure S9) showed the most significant enrichment in the ribosome, carbon metabolism, biosynthesis of amino acids, glycolysis/gluconeogenesis, pyruvate metabolism, protein processing in ER, phagosome, antigen processing and presentation, and type II diabetes categories. The number of DEPs in each of these pathways was 18, 15, 10, 9, 7, 6, 5, 4, and 2, respectively.
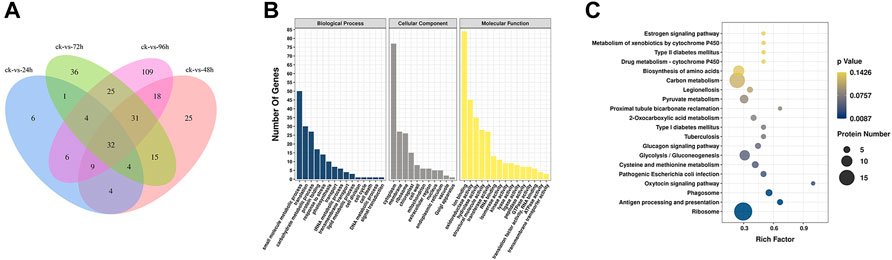
FIGURE 3. (A) Venn diagram of DEPs in E. ulmoides compared before (ck) with 24, 48, 72, and 96 h of inoculation with F. oxysporum, respectively. (B) GO analysis of DEPs in E. ulmoides between 0 and 72 h of inoculation with F. oxysporum. (C) KEGG enrichment analysis of DEPs in E. ulmoides between 0 and 72 h of inoculation with F. oxysporum.
To correlate the transcript and protein expression profiles, the DEPs from the proteomic analysis were compared with the annotated RNA-Seq library. The results showed that 153 of the 326 proteins were expressed in the same pattern as their mRNAs, indicating that the expression of almost half of the proteins was regulated directly at the transcriptional level. Cluster analysis on these 326 DEGs and DEPs (Supplementary Figure S10) indicated that both DEGs and DEPs were reliable and could be further analyzed. Figure 4 showed 153 and 158 genes with similar and opposite regulatory patterns at the transcriptional and protein levels were analyzed for correlations, obtaining the pearson correlation coefficients (r) of 0.718 and -0.677, respectively. The red boxes in the KEGG pathways (Figure 5) highlight the pathways in which both DEPs and DEGs were common enriched in transcription, translation, and folding, sorting and degradation.
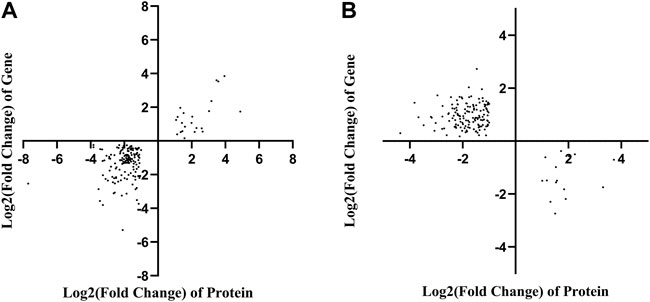
FIGURE 4. Correlation analysis based on the proteomics and transcriptomics. (A) Correlation analysis of differentially expressed proteins and genes with the same tendency of change. (B) The correlation analysis of significant differential expression of proteins and genes with opposite trends.
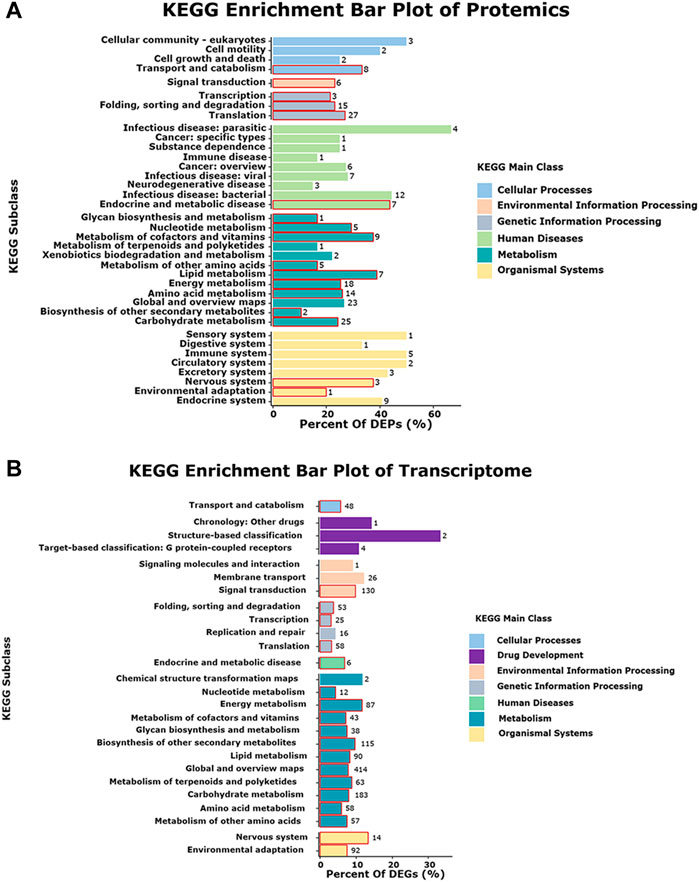
FIGURE 5. Pathway enriched in both the proteome and transcriptome. The red boxes represent in the pathways that are enriched in both protein and mRNA. (A) KEGG enrichment bar plot of Protemics. (B) KEGG enrichment bar plot of Transcriptome.
The relative expression level of the DEPs and DEGs enriched in the ER pathway (ID: ko04141) were further analyzed using RT-qPCR method. The results showed that the relative expression level of translation initiation factor eIF-2B subunit alpha after 24, 48, 72, and 96 h of inoculation with F. oxysporum was 0.05, 0.15, 0.61, and 0.24 times lower than that of ck group (Figure 6A). The relative expression level of BCL2-associated athanogene 1 was 5.75 and 16.53 times higher at 48 and 72 h after inoculation, respectively, than ck group (Figure 6B). The ER contains at least three types of molecular chaperones of protein folding. The first of these includes the heat shock molecular chaperones such as HSP20, HSP70, and HSP90, the second includes the lectin molecular chaperones calcium-linked protein and calcium network protein, while the last category comprises enzymes related to protein processing and Ca2+ homeostasis, such as protein disulfide isomerase (PDI). The results of this study (Figures 6C–J) showed that, except for calreticulin, whose expression was suppressed after inoculation with F. oxysporum, the expression of all other molecular chaperones, including ER chaperone BiP, HSP20 family protein, heat shock 70 kDa protein 1/2/6/8, heat shock protein hsp70-hsp90, PDI A6, and calnexin, increased significantly at 72 h after inoculation, with the relative expression of hypoxia upregulated 1 increased 65.53 times more than before inoculation. During ER stress, the ER-associated degradation (ERAD) is activated at the transcriptional level to remove misfolded proteins, increasing their degradation by the ubiquitin-proteasome system. Figures 6K-L showed that the expression of the ubiquitin-binding enzyme E2 and ubiquitin-ligase F-box increased significantly at 72 h after inoculation, which were 10.07 and 75.26 times higher than those before inoculation, respectively.
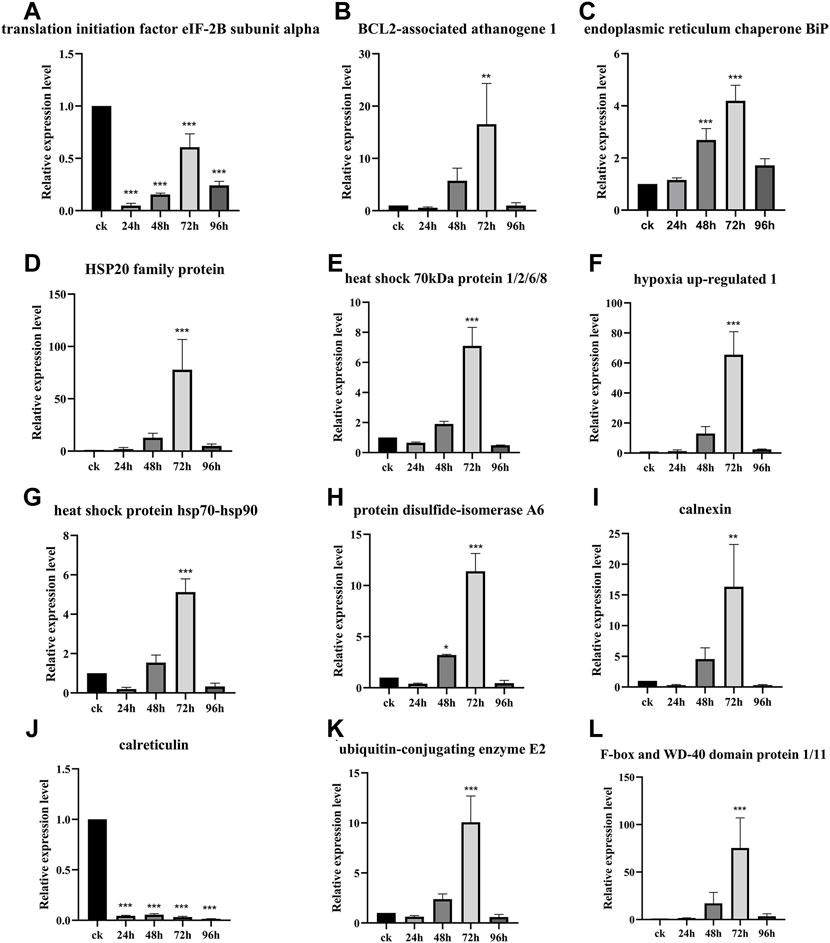
FIGURE 6. Gene relative expression analysis of the ER pathway-related genes. Data are shown as the mean of three biological replicates ± SD. The * represents significant difference (**p < 0.01, ***p < 0.001, Student’s t-test). (A) Translation initiation factor eIF-2B subunit alpha. (B) BCL2-associated athanogene 1. (C) Endoplasmic reticulum chaperone BiP. (D) HSP20 family protein. (E) Heat shock 70 kDa protein 1/2/6/8. (F) Hypoxia upregulated 1. (G) Heat shock protein hsp70-hsp-90. (H) Protein disulfide-isomerase A6. (I) Calnexin. (J) Calreticulin. (K) Ubiquitin-conjugating enzyme E2. (L) F-box and WD-40 domain protein 1/11.
F. oxysporum is an important pathogenic fungus, which can cause skin and eye lesions in mammals through infection of breaks in the skin, and also causes fusarium wilt in many crops through soil transmission, leading to serious economic losses in agricultural production. The control of F. oxysporum in agricultural production relies mainly on chemical fungicides which pose a major risk to the environment and food safety (Mukhtar, 2007; Jahanshir and Dzhalilov, 2010; Swarupa et al., 2014). It is, thus, important to investigate the antimicrobial mechanisms of traditional Chinese herbal medicine for the development of plant immune inducer (Dewen et al., 2017) and antimicrobial drugs with high efficacy and low toxicity. E. ulmoides is a traditional Chinese medicinal plant, recorded in The Classic of Herbal Medicine, that has proven antifungal property against Botrytis cinerea in our previous study (Liu et al., 2008).
In our present study, transcriptomic and proteomic analyses results showed that the KEGG pathway mainly concentrated in protein processing in ER, fructose and mannose metabolism, phosphatidylinositol signaling system, ribosome, protein processing in ER, phagosome, and antigen processing and presentation. Some previous studies (Malhotra and Kaufman, 2007; Wen and Wei, 2015; Ma, 2019; Zhang and Wei, 1997) had demonstrated that these pathways were related to the ER and ER misfolding. Therefore, our results demonstrated that E. ulmoides responds to F. oxysporum infection by mechanisms related to ER. The ER is an important organelle in the secretion pathway of eukaryotes and participates in the coordination of a variety of cellular activities such as protein processing and calcium homeostasis. It is also one of the important hubs for plant responses to various stresses (Martinez and Chrispeels, 2003; Helenius and Aebi, 2004; Kamauchi et al., 2005; Chen and Brandizzi, 2013; Doblas et al., 2013; Howell, 2013; Mishiba et al., 2013; Pollier et al., 2013; Cai et al., 2014; Williams et al., 2014). When plants are stressed by pathogenic infections, there is an accumulation of large numbers of unfolded or misfolded proteins in the ER lumen, leading to ER stress and adversely affecting the normal functioning of the organelle (Howell, 2013). If the protein accumulation exceeds the ER capacity, the cell responds by upregulating specific ER-associated genes and enzymes to restore homeostasis. This process is termed the ER stress response (Howell, 2013; Jing and Wang, 2020). Plant ER stress sensors can recognize ER stress and trigger the ER stress response, also called the unfolded protein response (UPR). The UPR has been shown to play an important role in interactions between plants and microorganisms (Wan and Jiang, 2016). The ER stress signaling pathway is closely related to plant immunity and represents an important part of the plant’s resistance to pathogen infection (Zhang et al., 2016; Qiang et al., 2021; Zhou et al., 2021). However, there is a lack of information on how plants respond to pathogen infection and activate ER stress-mediated immunity (ERSI).
The RT-qPCR results showed that the gene relative expression of ER chaperone protein BiP was significantly up-regulated after 48 and 72 h of inoculation with F. oxysporum. Upregulated expression of BiP is often considered to be a marker for ERS and UPR activation (Pobre et al., 2019). Meanwhile, the gene relative expression of translation initiation factor eIF-2B subunit alpha was inhibited after inoculation with F. oxysporum. It is possible that this could be the result of incorrect folding or wrong assembly of the protein, leading to binding by BiP to retain its unfolded state and prevent further misfolding. These reactions can lead to the phosphorylation of protein kinase R (PKR)-like ER kinase (PERK) protein dimer and cytoplasmic domain and eIF2. Phosphorylation of the latter at serine 51 blocks the binding of eIF2 to ATP and reduces the translation of its downstream mRNA (Harding et al., 2002; Ni et al., 2009). These reactions reduce the translation and synthesis of proteins, reduce the influx of new proteins to the ER, and thus prevent further increases in the numbers of unfolded proteins, thus lowering ERS (Harding et al., 2002; Ni et al., 2009). Combining the results of the analysis of BiP and eIF2α in this study with reported results, we speculate that the activation of the UPR in the E. ulmoides response to F. oxysporum is likely to be via the PERK/ATF4 of the ER pathway. This would prolong ERS and prevent the restoration of intracellular homeostasis and apoptosis would be induced by activation of the apoptosis regulator B lymphocyte 2 gene family (Marciniak et al., 2004). In this study, we also observed a significant increase in the expression of the BCL2-associated athanogene, further suggesting that the PERK/ATF4 of the ER pathway may play an important role in E. ulmoides response to F. oxysporum. At present, most of the studies on the UPR in plants have focused on IRE1-X box binding protein, transcriptional activator 6, and site-2 proteases (S2P) (Celli and Tsolis, 2015). However, the PERK/ATF4 of the ER pathway has only been studied in animals and has not been reported in plants (Zhang et al., 2021). Both the proteomic and transcriptomic association analysis and RT-qPCR verification indicated that the response of E. ulmoides to F. oxysporum is likely to be associated with the PERK/ATF4 of the ER pathway.
The UPR can coordinate transcriptional and translational changes in cells, up-regulate the expression of molecular chaperones involved in folding and stabilizing the proteins in the ER lumen, and induce the expression of stress proteins in the ER. In this study, the expression of ER molecular chaperones such as HSP20, HSP70, HSP90, protein disulfide isomerase (PDI), and calnexin (CNX) was significantly increased at 72 h after inoculation. It has been reported that HSP chaperones such as BIP and HSP90 can recognize misfolded proteins (Park and Seo, 2015; Usman et al., 2017; Ul Haq et al., 2019). The lectin molecular chaperones calnexin (CNX), calreticulin (CRT), and CRT are proteins related to protein processing and Ca2+ homeostasis, including PDI, CNX, and CRT, which could help localize unfolded proteins in the ER lumen to prevent unfolded/misfolded proteins from being exported. In ERS, the ERAD is activated at the transcriptional level to remove and degrade misfolded proteins. This process restores protein homeostasis (Harding et al., 1999; Balch et al., 2008). In addition, CNX links misfolded proteins through transmembrane domain interactions, activating protein degradation by the ubiquitin-proteasome system and signal transduction associated with phagocytosis, thus accomplishing the degradation of unfolded or misfolded proteins. The role of protein ubiquitination in plant growth and development and stress response has been widely recognized (Belknap and Garbarino, 1996). In this study E2 and F-box protein were significantly increased at 72 h after inoculation. These results suggest that molecular chaperones in the ER pathway (ko04141) are also involved in the response of E. ulmoides to F. oxysporum.
ER stress response plays an important role in plant response to pathogen infection and other stresses, extensive evidence shows that the ER pathway contributes to plant immunity in various ways (Li et al., 2009; Nekrasov et al., 2009; Saijo et al., 2009). There is also evidence that the ER pathway is manipulated by microbes to lead to successful colonization. For example, the symbiotic fungus Piriformospora indica establishes successful symbiosis by inducing plant ER stress but inhibiting the adaptive UPR, which eventually results in ER-PCD mediated by vacuolar processing enzymes (VPEs) (Qiang et al., 2012). In our study, we found the response of E. ulmoides to F. oxysporum was likely related to the unfolded protein response of the ER pathway. Some studies have shown that the process of apoptosis is contrary to the expression of CRT (Xu et al., 2014). Our study showed that when E. ulmoides was infected by F. oxysporum, the increased expression of ER chaperone proteins makes the protein fold correctly and causes ubiquitination and ERAD reaction, while the inhibition of CRT and EIF2 expression increases the expression of BCL2 and causes apoptosis, so as to protect plants (Figure 7). In addition, we also observed that the relative expression of the hypoxia upregulated 1 gene of the HSP70 family and the F-box and WD-40 domain protein 1/11 gene of the ERAD were highest at 72 h after inoculation, being 65.53 and 75.26 times greater, respectively, than before inoculation. It is speculated that these two genes may be key to the E. ulmoides infection response in the activation of ER pathway.
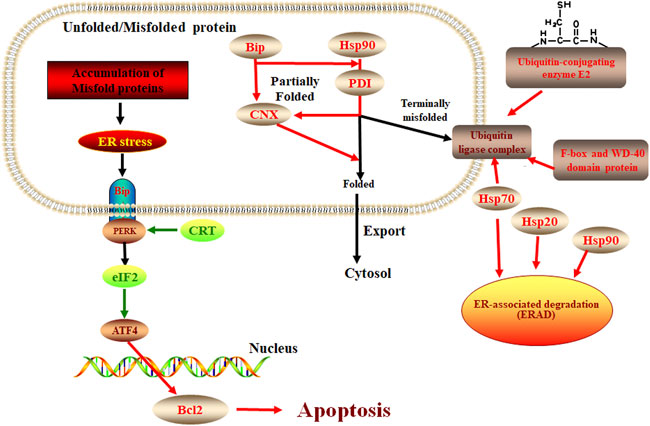
FIGURE 7. The protein processing in the ER pathway in E. ulmoide response to F. oxysporum. Red color represents up-accumulated genes or proteins in this pathway, while the green color represents down-accumulated.
In this study, proteome and transcriptome association analysis and RT-qPCR results showed that the response of E. ulmoides to F. oxysporum was likely related to the ER pathway. In the future, we will study the genes functions in the ER pathway through model plant transgenic technology to further explore the role of these genes in the response of E. ulmoides to F. oxysporum.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://db.cngb.org/- CNP0003469.
D-gZ and YZ designed the study. YL analyzed the data and wrote the article. XD performed the research. LP and XH edited the manuscript. All authors have read and agreed to the published version of the manuscript.
This research was supported by the project of Science and Technology Support Program (Agriculture) of Guizhou, China [2020]1Y001, Cloning and Polymorphism Analysis of Antibacterial Gene in Guizhou Tea [Guizhou University Renji Hezi (2018) No. 10], the Open Fund of the Key Laboratory of Mountain Plant Resources Protection and Germplasm Innovation of the Ministry of Education MOELP-201803 and National Natural Science Foundation of China (31660076).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1053227/full#supplementary-material
Anderson, W. R., and Cronquist, A. (1982). An integrated system of classification of flowering plants. Brittonia 34, 268–270. doi:10.2307/2806386
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi:10.1126/science.1141448
Belknap, W. R., and Garbarino, J. E. (1996). The role of ubiquitin in plant senescence and stress responses. Trends Plant Sci. 1, 331–335. doi:10.1016/s1360-1385(96)82593-0
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Cai, Y. M., Yu, J., and Gallois, P. (2014). Endoplasmic reticulum stress-induced PCD and caspase-like activities involved. Front. Plant Sci. 5, 41. doi:10.3389/fpls.2014.00041
Celli, J., and Tsolis, R. M. (2015). Bacteria, the endoplasmic reticulum and the unfolded protein response: Friends or foes? Nat. Rev. Microbiol. 13, 71–82. doi:10.1038/nrmicro3393
Chen, M. J., Zhai, J. H., Zhang, J. J., Li, H., Niu, X. J., Liu, Y. X., et al. (2022). Transcriptomic and physiological analyses of pigment accumulation in Eucommia ulmoides 'hongye. Phyt. B. Aires. 91, 1027–1044. doi:10.32604/phyton.2022.019106
Chen, Y., and Brandizzi, F. (2013). IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23, 547–555. doi:10.1016/j.tcb.2013.06.005
Cho, S., Hong, R., Yim, P., Yeom, M., Lee, B., Yang, W. M., et al. (2018). An herbal formula consisting of Schisandra chinensis (Turcz.) Baill, Lycium chinense Mill and Eucommia ulmoides Oliv alleviates disuse muscle atrophy in rats. J. Ethnopharmacol. 213, 328–339. doi:10.1016/j.jep.2017.10.008
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi:10.1038/nbt.1511
Cox, J., Neuhauser, N., Michalski, A., Scheltema, R. A., Olsen, J. V., and Mann, M. (2011). Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805. doi:10.1021/pr101065j
Dewen, Q., Yijie, D., Yi, Z., Shupeng, L., and Fachao, S. (2017). Plant immunity inducer development and application. Mol. Plant. Microbe. Interact. 30, 355–360. doi:10.1094/MPMI-11-16-0231-CR
Doblas, V. G., Amorim-Silva, V., Pose, D., Rosado, A., Esteban, A., Arro, M., et al. (2013). The SUD1 gene encodes a putative E3 ubiquitin ligase and is a positive regulator of 3-hydroxy-3-methylglutaryl coenzyme a reductase activity in Arabidopsis. Plant Cell 25, 728–743. doi:10.1105/tpc.112.108696
Edel-Hermann, V., and Lecomte, C. (2019). Current status of Fusarium oxysporum formae speciales and races. Phytopathology 109, 512–530. doi:10.1094/PHYTO-08-18-0320-RVW
Fujiwara, A., Nishi, M., Yoshida, S., Hasegawa, M., Yasuma, C., Ryo, A., et al. (2016). Eucommicin A, a beta-truxinate lignan from Eucommia ulmoides, is a selective inhibitor of cancer stem cells. Phytochemistry 122, 139–145. doi:10.1016/j.phytochem.2015.11.017
Gordon, T. R., and Martyn, R. D. (1997). The evolutionary biology of Fusarium oxysporum. Annu. Rev. Phytopathol. 35, 111–128. doi:10.1146/annurev.phyto.35.1.111
Guo, P., Tang, h., Liu, C., Wei, S., and Huang, J. (2021). Differential Expression Analysis of Genes Related to Flesh Color in <i>Hylocereu polyrhizus</i> and <i>Hylocereu undatus</i>. Mol. Plant Breed. 19, 4311–4326. doi:10.5376/mpb.2021.12.0006
Harding, H. P., Calfon, M., Urano, F., Novoa, I., and Ron, D. (2002). Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 18, 575–599. doi:10.1146/annurev.cellbio.18.011402.160624
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397 (6716), 271–274. doi:10.1038/16729
Helenius, A., and Aebi, M. (2004). Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049. doi:10.1146/annurev.biochem.73.011303.073752
Howell, S. H. (2013). Endoplasmic reticulum stress responses in plants. Annu. Rev. Plant Biol. 64, 477–499. doi:10.1146/annurev-arplant-050312-120053
Hussain, T., Tan, B., Rahu, N., Hussain Kalhoro, D., Dad, R., and Yin, Y. (2017). Protective mechanism of Eucommia ulmoids flavone (EUF) on enterocyte damage induced by LPS. Free Radic. Biol. Med. 108, S40. doi:10.1016/j.freeradbiomed.2017.04.152
Jahanshir, A., and Dzhalilov, S. (2010). The effects of fungicides on Fusarium oxysporum F. SP. Lycopersici associated with Fusarium wilt of tomato. J. Plant Prot. Res. 50, 172–178. doi:10.2478/v10045-010-0029-x
Jing, M. F., and Wang, Y. C. (2020). Plant pathogens utilize effectors to hijack the host endoplasmic reticulum as part of their infection strategy. Engineering 6 (5), 500–504. doi:10.1016/j.eng.2020.03.003
Kamauchi, S., Nakatani, H., Nakano, C., and Urade, R. (2005). Gene expression in response to endoplasmic reticulum stress in Arabidopsis thaliana. FEBS J. 272, 3461–3476. doi:10.1111/j.1742-4658.2005.04770.x
Kim, D., Langmead, B., and Salzberg, S. L. (2015). Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi:10.1038/nmeth.3317
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi:10.1038/nmeth.1923
Lee, G. H., Lee, H. Y., Choi, M. K., Choi, A. H., Shin, T. S., and Chae, H. J. (2018). Eucommia ulmoides leaf (EUL) extract enhances NO production in ox-LDL-treated human endothelial cells. Biomed. Pharmacother. 97, 1164–1172. doi:10.1016/j.biopha.2017.11.035
Li, B., and Dewey, C. N. (2011). Rsem: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinforma. 12, 323. doi:10.1186/1471-2105-12-323
Li, J., Zhao-Hui, C., Batoux, M., Nekrasov, V., Roux, M., Chinchilla, D., et al. (2009). Specific ER quality control components required for biogenesis of the plant innate immune receptor EFR. Proc. Natl. Acad. Sci. U. S. A. 106, 15973–15978. doi:10.1073/pnas.0905532106
Liu, S. H., Zhao, D. G., and Song, B. A. (2007). Effect of Eucommia bark protein against plant pathogenic fungi. Agrochemicals 46, 848–850. doi:10.16820/j.cnki.1006-0413.2007.12.020
Liu, S. H., Zhao, D. G., and Song, B. A. J. A. (2008). Study on inhibition activity of Eucommia ulmoides antifungal protein to Botrytis cinerea. Agrochemicals 11, 836–838. doi:10.16820/j.cnki.1006-0413.2008.11.021
Luber, C. A., Cox, J., Lauterbach, H., Fancke, B., Selbach, M., Tschopp, J., et al. (2010). Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 32, 279–289. doi:10.1016/j.immuni.2010.01.013
Ma, S. (2019). Study on mannose regulating ER stress and alleviating islet inflammation in early stage of type I diabetes. Third Military Medical University. doi:10.27001/d.cnki.gtjyu.2019.000451
Malhotra, J. D., and Kaufman, R. J. (2007). The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18, 716–31. doi:10.1016/j.semcdb.2007.09.003
Mann, M., Hendrickson, R. C., and Pandey, A. (2001). Analysis of proteins and proteomes by mass spectrometry. Annu. Rev. Biochem. 70, 437–473. doi:10.1146/annurev.biochem.70.1.437
Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. doi:10.1101/gad.1250704
Martinez, I. M., and Chrispeels, M. J. (2003). Genomic analysis of the unfolded protein response in arabidopsis shows its connection to important cellular processes[W]. Plant Cell 15 (2), 561–576. doi:10.1105/tpc.007609
Michielse, C. B., and Rep, M. (2010). Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol. 10, 311–324. doi:10.1111/j.1364-3703.2009.00538.x
Mishiba, K., Nagashima, Y., Suzuki, E., Hayashi, N., Ogata, Y., Shimada, Y., et al. (2013). Defects in IRE1 enhance cell death and fail to degrade mRNAs encoding secretory pathway proteins in the Arabidopsis unfolded protein response. Proc. Natl. Acad. Sci. U. S. A. 110, 5713–5718. doi:10.1073/pnas.1219047110
Monti, M., Orru, S., Pagnozzi, D., and Pucci, P. (2005). Functional proteomics. Clin. Chim. Acta 357 (2), 140–150. doi:10.1016/j.cccn.2005.03.019
Mukhtar, I. (2007). Comparison of phytochemical and chemical control of Fusarium oxysporium f. sp. ciceri. Mycopath 5, 107–110.
Nekrasov, V., Li, J., Batoux, M., Roux, M., Chu, Z. H., Lacombe, S., et al. (2009). Control of the pattern-recognition receptor EFR by an ER protein complex in plant immunity. EMBO J. 28, 3428–3438. doi:10.1038/emboj.2009.262
Ni, M., Zhou, H., Wey, S., Baumeister, P., and Lee, A. S. (2009). Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS One 4 (8), e6868. doi:10.1371/journal.pone.0006868
Park, C. J., and Seo, Y. S. (2015). Heat shock proteins: A review of the molecular chaperones for plant immunity. Plant Pathol. J. 31, 323–333. doi:10.5423/PPJ.RW.08.2015.0150
Pietro, A. D., Madrid, M. P., Caracuel, Z., Delgado-Jarana, J., and Roncero, M. I. G. (2010). Fusarium oxysporum: Exploring the molecular arsenal of a vascular wilt fungus. Mol. Plant Pathol. 4, 315–325. doi:10.1046/j.1364-3703.2003.00180.x
Pobre, K. F. R., Poet, G. J., and Hendershot, L. M. (2019). The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 294, 2098–2108. doi:10.1074/jbc.REV118.002804
Pollier, J., Moses, T., Gonzalez-Guzman, M., De Geyter, N., Lippens, S., Vanden Bossche, R., et al. (2013). The protein quality control system manages plant defence compound synthesis. Nature 504 (7478), 148–152. doi:10.1038/nature12685
Qian, Z. Z., Yang, D., Liu, Y. Z., and Peng, Y. (2010). Pharmacopoeia of the people's Republic of China (2010 edition):a milestone in development of China's healthcare. Chin. Herb. Med. 2, 157–160.
Qiang, X., Liu, X., Wang, X., Zheng, Q., Kang, L., Gao, X., et al. (2021). Susceptibility factor RTP1 negatively regulates Phytophthora parasitica resistance via modulating UPR regulators bZIP60 and bZIP28. Plant Physiol. 186, 1269–1287. doi:10.1093/plphys/kiab126
Qiang, X., Zechmann, B., Reitz, M. U., Kogel, K. H., and Schafer, P. (2012). The mutualistic fungus Piriformospora indica colonizes Arabidopsis roots by inducing an endoplasmic reticulum stress-triggered caspase-dependent cell death. Plant Cell 24, 794–809. doi:10.1105/tpc.111.093260
Saijo, Y., Tintor, N., Lu, X. L., Rauf, P., Pajerowska-Mukhtar, K., Haweker, H., et al. (2009). Receptor quality control in the endoplasmic reticulum for plant innate immunity. EMBO J. 28, 3439–3449. doi:10.1038/emboj.2009.263
Saleh, S., Staes, A., Deborggraeve, S., and Gevaert, K. (2019). Targeted proteomics for studying pathogenic bacteria. Proteomics 19, e1800435. doi:10.1002/pmic.201800435
Sarwar, N., Zahid, C. M., Haq, I., and Jamil, F. F. (2005). Induction of systemic resistance in chickpea against Fusarium wilt by seed treatment with Salicylic acid and Bion. Pak J. Bot. 37, 989–995. doi:10.1378/chest.10732
Shi, W., Zhao, S. L., Liu, K., Sun, Y. B., Ni, Z. B., Zhang, G. Y., et al. (2020). Comparison of leaf transcriptome in response to Rhizoctonia solani infection between resistant and susceptible rice cultivars. BMC Genomics 21, 245. doi:10.1186/s12864-020-6645-6
Swarupa, V., Ravishankar, K. V., and Rekha, A. (2014). Plant defense response against Fusarium oxysporum and strategies to develop tolerant genotypes in banana. Planta 239, 735–751. doi:10.1007/s00425-013-2024-8
Tokumoto, Y., Kajiura, H., Takeno, S., Harada, Y., Suzuki, N., Hosaka, T., et al. (2016). Induction of tetraploid hardy rubber tree, <i>Eucommia ulmoides</i>, and phenotypic differences from diploid. Plant Biotechnol. (Tokyo). 33, 51–57. doi:10.5511/plantbiotechnology.15.1219a
Ul Haq, S., Khan, A., Ali, M., Khattak, A. M., Gai, W. X., Zhang, H. X., et al. (2019). Heat shock proteins: Dynamic biomolecules to counter plant biotic and abiotic stresses. Int. J. Mol. Sci. 20, 5321. doi:10.3390/ijms20215321
Usman, M. G., Rafii, M. Y., Martini, M. Y., Yusuff, O. A., Ismail, M. R., and Miah, G. (2017). Molecular analysis of Hsp70 mechanisms in plants and their function in response to stress. Biotechnol. Genet. Eng. Rev. 33, 26–39. doi:10.1080/02648725.2017.1340546
Wan, S., and Jiang, L. (2016). Endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) in plants. Protoplasma 253, 753–764. doi:10.1007/s00709-015-0842-1
Wasinger, V. C., Cordwell, S. J., Cerpa-Poljak, A., Yan, J. X., Gooley, A. A., Wilkins, M. R., et al. (1995). Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis 16, 1090–1094. doi:10.1002/elps.11501601185
Wen, Q., and Wei, S. (2015). Research progress on the role of endoplasmic reticulum stress in glucose and lipid metabolism. Editors Zhong Guo, Xu Mu, Shou Yi, and Wen Zhai 31, 52–53. (in chinese). Available at: https://kns.cnki.net/kcms/detail/detail.aspx?FileName=ZXWA201510048&DbName=CJFQ2015.
Wilhelm, B. T., and Landry, J. R. (2009). RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods 48, 249–257. doi:10.1016/j.ymeth.2009.03.016
Williams, B., Verchot, J., and Dickman, M. B. (2014). When supply does not meet demand-ER stress and plant programmed cell death. Front. Plant Sci. 5, 211. doi:10.3389/fpls.2014.00211
Xu, M., Wang, T., Yu, L., Wang, W., Zeng, S., Chen, Z., et al. (2014). Endoplasmic reticulum stress response and related molecular chaperonesProg. Anatomical Sci. Biochem. Physiol, 20147, 38120–38426. doi:10.16695/j.cnki.1006-2947.2014.04.00210.1016/j.pestbp.2017.06.005
Yu, L., Wang, W., Zeng, S., Chen, Z., Yang, A., Shi, J., et al. (2018). Label-free quantitative proteomics analysis of Cytosinpeptidemycin responses in southern rice black-streaked dwarf virus-infected rice. Pestic. Biochem. Physiol. 147, 20–26. doi:10.1016/j.pestbp.2017.06.005
Zhang, H., Wang, D., and Zhang, Z. (2016). Progress and prospects in the research on wheat drought response and resistance mechanisms using transcriptomic and proteomic approaches. J. Triticeae Crops 36, 878–887. doi:10.7606/j.issn.1009-1041.2016.07.08
Zhang, R., Sun, Y., Su, J., Wang, S., Liu, Y., Tong, H., et al. (2021). Advances in endoplasmic reticulum stress in plants. Curr. Biotechnol. 11, 289–296. doi:10.19586/j.2095-2341.2020.0147
Zhang, X., and Wei, H. (1997). Advances in antigen processing and presentation. Foreign Medicine (Immunology) 1, 1–5. (in chinese). Available at: https://kns.cnki.net/kcms/detail/detail.aspx?FileName=GYMF199701000&DbName=CJFQ1997.
Keywords: transcriptome, proteome, Eucommia ulmoides, Fusarium oxysporum, endoplasmic reticulum
Citation: Lu Y, Dong X, Huang X, Zhao D-g, Zhao Y and Peng L (2022) Combined analysis of the transcriptome and proteome of Eucommia ulmoides Oliv. (Duzhong) in response to Fusarium oxysporum. Front. Chem. 10:1053227. doi: 10.3389/fchem.2022.1053227
Received: 25 September 2022; Accepted: 03 October 2022;
Published: 13 October 2022.
Edited by:
Pei Li, Kaili University, ChinaCopyright © 2022 Lu, Dong, Huang, Zhao, Zhao and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuan Dong, eGRvbmdAZ3p1LmVkdS5jbg==; Yichen Zhao, eWN6aGFvQGd6dS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.