 Gaoxue Wang
Gaoxue Wang Enrique R. Batista
Enrique R. Batista Ping Yang
Ping Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 05 January 2023
Sec. Catalytic Reactions and Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1051496
Ammonia (NH3) is one of the basic chemicals in artificial fertilizers and a promising carbon-free energy storage carrier. Its industrial synthesis is typically realized via the Haber−Bosch process using traditional iron-based catalysts. Developing advanced catalysts that can reduce the N2 activation barrier and make NH3 synthesis more efficient is a long-term goal in the field. Most heterogeneous catalysts for N2-to-NH3 conversion are multicomponent systems with singly dispersed metal clusters on supporting materials to activate N2 and H2 molecules. Herein, we report single-component heterogeneous catalysts based on 5f actinide dioxide surfaces (ThO2 and UO2) with oxygen vacancies for N2-to-NH3 conversion. The reaction cycle we propose is enabled by a dual-site mechanism, where N2 and H2 can be activated at different vacancy sites on the same surface; NH3 is subsequently formed by H− migration on the surface via associative pathways. Oxygen vacancies recover to their initial states after the release of two molecules of NH3, making it possible for the catalytic cycle to continue. Our work demonstrates the catalytic activities of oxygen vacancies on 5f actinide dioxide surfaces for N2 activation, which may inspire the search for highly efficient, single-component catalysts that are easy to synthesize and control for NH3 conversion.
The industrial synthesis of ammonia (NH3) from gaseous N2 and H2 molecules is typically realized via the Haber−Bosch process, one of the most significant catalytic reactions discovered in the early 20th century (Grunze, 1982; Logadottir and Nørskov, 2003; Schlögl, 2015). Over 200 million tons of NH3 is produced annually, 80% of which is used in artificial fertilizers for agriculture to feed Earth’s growing population (Giddey et al., 2017). Since NH3 contains 17.6 wt% hydrogen, it is also considered to be a promising carbon-free energy storage intermediate that can be more easily stored and transported than gaseous H2. The industrial Haber−Bosch process, activated by Fe- and Ru-based catalysts, must be conducted under harsh conditions (high pressure: 150–300 atm and high temperature: 300–600 °C) to overcome kinetic limitations and to achieve a sufficient yield of NH3 (Hammer and Nørskov, 2000; Galloway et al., 2008; Cherkasov et al., 2015). This is because of the inertness of the triply bonded, non-polar N2 molecule, which exhibits a large bond energy of 941 kJ mol−1, negative electron affinity, and high ionization energy (Gambarotta and Scott, 2004; Pool et al., 2004; Walter, 2016). For Fe- and Ru-based catalysts, NH3 is synthesized via a dissociative mechanism, where N2 first dissociates at active sites, and the dissociated *N is subsequently hydrogenated step-by-step to produce NH3 (Bozso et al., 1977; Ertl et al., 1982; Logadottir and Nørskov, 2003; Reuter et al., 2017). The dissociation of N2 into two *N is generally accepted as the rate-determining step for the synthesis of NH3 (Temkin and Pyzhev, 1940; Stoltze and Nørskov, 1985; Ertl, 2008; Kitano et al., 2015). The development of new catalysts to reduce the N2 activation barrier, enabling the synthesis of NH3 with a high yield under mild conditions, is highly desired in the field.
During the search for new catalysts, scientists have been inspired by the biological N2 fixation process using nitrogenase enzymes under ambient temperature and pressure conditions. These enzymes contain metal clusters, such as the Fe−Mo cofactor, providing active sites for the binding and reduction of N2 (Rodriguez et al., 2011; Foster et al., 2018). New approaches to synthesize NH3 using electrocatalysis, photocatalysis, and plasma catalysis have been developed (Patil et al., 2015; Li et al., 2017; Guo et al., 2018; Hong et al., 2018; Yang et al., 2018; Li et al., 2020). Several metal cluster and molecular catalysts have been used in these reactions (Anderson et al., 2013; Kerpal et al., 2013; Shima et al., 2013; Grubel et al., 2014; Hölscher and Leitner, 2015; McWilliams and Holland, 2015; Tyler, 2015; Araake et al., 2017; Ling et al., 2019). Although very promising, NH3 yields obtained using these alternative methods are very low, and the processes are still in their infancy, failing to meet the requirements for practical use (Li et al., 2020). Recently, enlightened by the booming use of single-atom catalysts or single-cluster catalysts (Geng et al., 2018; Ling et al., 2018; Wang et al., 2018; Hannagan et al., 2020; Mitchell and Pérez-Ramírez, 2020), several multicomponent heterogeneous catalysts, composed of atomically dispersed metal atoms or clusters on supporting substrates for N2 reduction, have been proposed (Choi et al., 2018; Niu et al., 2020). Li et al. proposed the use of singly dispersed Fe3 or Rh1Co3 supported by Al2O3(010) or CoO(011) for NH3 synthesis (Liu et al., 2018; Ma et al., 2018; Shi et al., 2022). Different from the dissociative mechanism of the Haber−Bosch process, the synthesis of NH3 using Fe3 or Rh1Co3 occurs via an associative mechanism—mimicking the mechanism in nitrogenase-based synthesis (Liu et al., 2018; Ma et al., 2018). Recent work has demonstrated that the Ni-loaded LaN surface enables stable and highly efficient NH3 synthesis (Ye et al., 2020a). It was found that NH3 was synthesized through a dual-site mechanism, with N2 activation by surface N vacancies and H2 activation by Ni clusters. Since singly dispersed metal clusters are highly unstable and prone to agglomeration due to their high surface energy (Yang et al., 2013), precise control of the morphology, size, and stability of these multicomponent heterogeneous catalysts remains challenging (Speck et al., 2021).
Significant technological advances will be gained by identifying a highly efficient, single-component system for N2-to-NH3 conversion that is easy to synthesize and manufacture. Inspired by our recent observation of a unique phenomenon in early 5f-element materials (Wang et al., 2019), we hypothesize that AnO2 (An = Th and U) surfaces are promising candidates for such catalysts. In that work, we reported that excess electrons due to atomic oxygen vacancies on the surfaces of 5f-element materials can spontaneously induce catalytic water splitting (Wang et al., 2019). Our computational studies found that excess electrons remained at the vacancy site on ThO2 and UO2 instead of moving to the metal centers. These excess electrons at the vacancy site are extremely reactive and can spontaneously induce chemical reactions even with inert molecules. Hence, it is intriguing to investigate if these excess electrons can activate N2 molecules and simplify the N2-to-NH3 conversion by using a single-component system. If the results are positive, what is the unique role of the 5f orbitals in the activation of N2? For example, uranium exhibits a large range of oxidation states, from +II to + VI, which expands the scope of its accessible intermediates and products for chemical reactions.
Actinide-containing materials have been used in catalytic applications (Jackson and Hargreaves, 2008; Ismagilov and Lazareva, 2013; Falcone et al., 2017; Leduc et al., 2019). Uranium-containing compounds are efficient catalysts in organic syntheses (Barbier-Baudry et al., 2000) and in the activation of small molecules such as CO, H2O, CH4, and HCl (Leduc et al., 2019). This is because of the large ionic radii of actinides and the active participation of 5f electrons in the valence space that can lead to unique reactivity patterns and product distributions not accessible to transition metal complexes (Leduc et al., 2019). Haber first applied metallic uranium and uranium nitrides to synthesize NH3 in 1909, which also had to be conducted under high pressure and temperature conditions (Rossignol, 1910; Leduc et al., 2019). Recently, a number of uranium complexes that can bind to the N2 molecule have been reported (Evans et al., 2003; Falcone and Mazzanti, 2018; Lu et al., 2019; Jori et al., 2021; Keener et al., 2021; Xin et al., 2021); however, the full catalytic cycle of NH3 formation has not been realized using these molecular uranium complexes. However, actinide dioxide surfaces containing oxygen defects have never been tested for NH3 synthesis.
In this work, we studied the activation of N2 for NH3 synthesis using singly dispersed atomic vacancies on ThO2 and UO2 surfaces that can be easily generated by Ar ion sputtering (Senanayake et al., 2007a; Senanayake et al., 2007b). We demonstrate, using first-principle calculations, that the atomic oxygen vacancy can serve as the active site for NH3 synthesis from N2 and H2 gases. We show that N2 can be chemically adsorbed at the vacancy site. H2 can dissociate directly at the vacancy site, forming two H− ions. The migration of H− on the surface can lead to the hydrogenation of *NN and the subsequent formation of NH3 through an associative mechanism. Our work demonstrates that early 5f-element materials are promising candidates as high-efficiency single-component catalysts for N2-to-NH3 conversion.
All calculations were performed with the use of density functional theory (DFT) and the projector augmented-wave (PAW) method (Kresse and Joubert, 1999), implemented in the Vienna Ab initio Simulation Package (VASP) (Kresse and Furthmüller, 1996). The generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) (Perdew et al., 1996) functional was used to represent the exchange−correlation interaction. Since the PBE functional does not capture the van der Waals (vdW) dispersion energy (Giovannetti et al., 2007; Sachs et al., 2011), the DFT-D3 method described by Grimme (Grimme, 2006) was included in the calculations. Plane wave basis sets with a cutoff energy of 520 eV were employed (Kresse and Joubert, 1999). The energy convergence was set to 10–6 eV and the residual force on each atom upon geometry optimization was below 0.01 eV/Å. The (111) surfaces of ThO2 and UO2 were represented with slab models, with a vacuum gap in the direction normal to the surface (Wang et al., 2017). The vacuum distance normal to the slab was larger than 30 Å, in order to eliminate interactions between the periodic images due to the periodic boundary conditions. Dipole correction was included to nullify the artificial field imposed on the slab by the periodic boundary conditions (Bengtsson, 1999). For UO2 surfaces, a Hubbard-like on-site Coulomb interaction was included, namely, the DFT + U method (Dudarev et al., 1998), to treat the correlated electrons. The DFT + U (U = 4.0 eV) method overcomes deficiencies of the pure LDA/GGA functionals and has achieved a wide range of success in treating the UO2 system (Dorado et al., 2009; Wen et al., 2013). The reciprocal space was sampled by Monkhorst−Pack k points in the Brillouin zone, with a grid size of 3 × 3 × 1. Spin-orbit coupling was ignored in this work since previous studies have demonstrated that it introduces only minor corrections in reaction energies of both actinide surfaces and molecular systems (Batista et al., 2004; Rak et al., 2013; Goldman and Morales, 2017; Kelley et al., 2017; Deblonde et al., 2018; McSkimming et al., 2018; Han and Kaltsoyannis, 2022). To reduce the computational cost, the 1k collinear antiferromagnetic (AFM) order was used for UO2 surfaces, and the ThO2 surface was non-magnetic. A recent study has shown that non-collinear magnetism is important to completely understand the electronic structure of AnO2 (Pegg et al., 2020). Oxygen vacancies were created by removing a top-surface oxygen atom, corresponding to a vacancy coverage of one-fourth of the monolayer (Θv = 1/4), following our previous work (Wang et al., 2019). The chemical composition at the top surface of this model is U4O7, which has been reported experimentally (UO2−x; x < 0.3) (Senanayake et al., 2007a; Senanayake et al., 2007b). Transition states were searched using the climbing image nudged elastic-band method (CI-NEB) (Henkelman et al., 2000).
The formal oxidation states of the actinide and oxygen atoms in ThO2 and UO2 are +4 and −2, respectively. Therefore, a neutral oxygen vacancy is expected to generate two excess electrons on the surface, which can lead to high chemical activities of the materials. In a previous study, we found that the two excess electrons remained at the vacancy site on ThO2 and moved to the 5f orbitals of Pu on the PuO2 surface. Whereas on the UO2 surface, one of the excess electrons preferred to localize on a nearby U 5f orbital, leaving one excess electron at the vacancy site. This behavior is due to the different reduction potentials of the materials; from +4 to +3 as a result of the contraction and energy lowering of the 5f orbital level going from Th to Pu. These readily available excess electrons are extremely reactive and can spontaneously induce the reduction of water and its splitting (Wang et al., 2019). In this work, we investigate the activation of N2 molecules by these excess electrons at the vacancy site in the first section, followed by a discussion on the activation of H2 molecules in the next section. We conclude with a proposed NH3 formation mechanism in the last section.
Figure 1 shows the adsorption of N2 on ThO2 and UO2 (111) surfaces via oxygen vacancies. From the energy diagram in Figure 1A, it is seen that it is thermodynamically favorable for N2 to chemisorb at the vacancy site, overcoming an energy barrier smaller than 0.1 eV. The adsorption energies are −1.40 eV and −1.10 eV on ThO2 and UO2 surfaces, respectively. Upon N2 adsorption on the ThO2 surface, as shown in Figure 1B, the N−N bond length is stretched from 1.10 Å in the gas phase to 1.23 Å, indicative of a reduction in the bond order from three to two and the activation of the N≡N bond. The *NN distance and the typical N≡N, N=N, and N−N bond lengths are summarized in Supplementary Table S1. From the density of state (DOS) plots in Figure 1C, one can see that the π* orbital of the free N2 molecule is well-aligned with the unoccupied 5f orbitals of Th atoms before N2 adsorption, and π*−5f hybrid orbitals are formed near the Fermi level after N2 adsorption. Before N2 adsorption, there is a small peak in the DOS slightly below the Fermi level, corresponding to the excess electrons at the vacancy sites (Wang et al., 2019). After N2 adsorption, the excess electrons transfer to the π*−5f hybrid orbitals. Hybridization of the N2 σ* and π orbitals with the Th 2p orbitals is also observed from the broadening of these orbitals after N2 adsorption. The transfer of excess electrons and the orbital hybridization between N and Th atoms result in the stretching and weakening of the N−N bond, which is a critical step for the activation of N2.
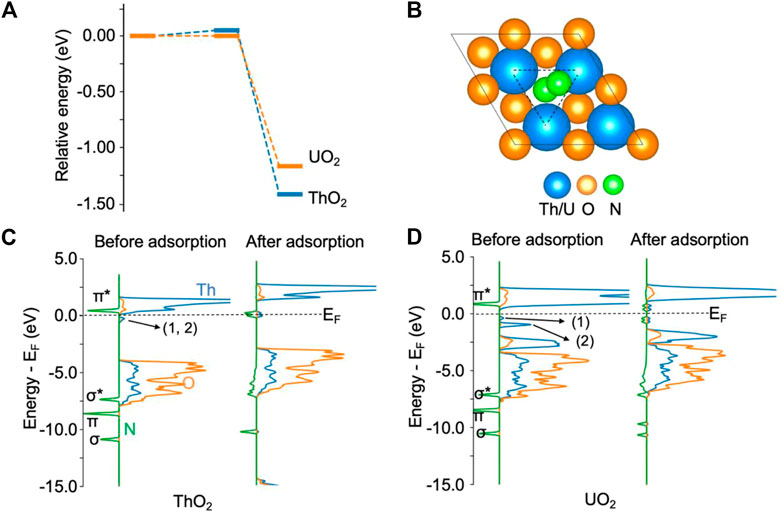
FIGURE 1. N2 adsorption on ThO2 and UO2 (111) surfaces containing oxygen vacancies. (A) Adsorption energy diagram. The total energy of the initial state is set to zero. (B) Illustration of structures after N2 adsorption. The oxygen vacancy site is highlighted by the triangle. (C) Density of states before and after N2 adsorption on the ThO2 surface. (D) Density of states before and after N2 adsorption on the UO2 surface. The two excess electrons on the ThO2 surface are located at the vacancy sites labeled 1 and 2 in the DOS, directly below the Fermi level (EF) in (C). On the UO2 surface (D), one of the excess electrons (1) is located at a vacancy site and the other (2) in uranium 5f orbitals.
On the UO2 surface containing an oxygen vacancy, one of the excess electrons is localized in the 5f orbital of the uranium atom near the vacancy. This is evidenced from the DOS before N2 adsorption, as shown in Figure 1D. The first small peak, labeled 1) in Figure 1D, directly below the Fermi energy, corresponds to the delocalized excess electron at the vacancy site. The second peak, labeled 2) in Figure 1D, below the Fermi energy, corresponds to the other excess electron that is localized in the uranium 5f orbital. The magnetic moment of this uranium atom is 2.8 μB, as expected for U(III). The remaining uranium atoms maintain the U(IV) magnetic moment of 2.0 μB, similar to surfaces without oxygen vacancies. After N2 adsorption, the magnetic moments of all uranium atoms are 2.0 μB, indicating the transfer of the excess electrons to *NN. The N≡N bond is activated, as evidenced by a bond length of 1.24 Å after adsorption. Bader charge analysis shows that *NN receives 1.2 e− transferred from the surface. Similarly, the calculated bond length of *NN is also in agreement with the reported bond length of 1.28 for N2− (Thulstrup and Andersen, 1968-1987), and is close to the reported bond length of 1.25–1.28 Å for the N=N double bond (Smieja et al., 1987; Hill et al., 1991). The adsorbed *NN is spin polarized, with magnetic moments of 0.3 and 0.5 μB on each N atom, respectively.
Likewise, the H2 molecule can be easily activated by excess electrons and can spontaneously dissociate at an oxygen vacancy site on both ThO2 and UO2 (111) surfaces. The activation barrier for this process is less than 0.2 eV, as shown in Figure 2A. On the ThO2 surface, after dissociation, one *H occupies a vacancy site and the other is located between two actinide atoms, as shown in Figure 2B. On most metal surfaces, *H is located at the fcc/top site, as summarized by Greeley et al. (Greeley and Mavrikakis, 2005). For the Rh1Co3/CoO(011) system, H2 dissociates at the Rh site and forms two Rh−H bonds (Ma et al., 2018). For the Ni-loaded LaN surface, H2 dissociates at the Ni cluster and forms a Ni−H bond with one Ni atom (Ye et al., 2020a). We have performed Hessian calculations for this structure and did not find imaginary vibrational frequencies, indicating that the optimization converged to a minimum energy structure. The vibrational frequency for *H at the vacancy site is in the range of 750–1243 cm−1, and that for *H at the bridge site of the two metal atoms is in the range of 630–907 cm−1. Bader charge analysis reveals that each *H receives 0.62 e− from the surface, see Supplementary Table S2. We have considered the structure to contain a lattice O protonated by *H, see Supplementary Figure S1. It is found that this structure is 1.9 eV higher in energy than the structure in Figure 2B. The Bader charge of each *H is in line with the reported Bader charges in metal hydrides (Aboud and Wilcox, 2010), indicating the formation of two H− ions on the surface. The formation of H− has also been recently observed on the CeN surface containing nitrogen vacancies, where a Bader charge of 0.7 e− was determined (Ye et al., 2020b). On the ThO2 surface, the H−H bond length is increased from 0.75 Å in the gas phase to 2.04 Å after dissociation. The DOS in Figure 2C shows that the σ orbital of H2 disappears after H2 adsorption, indicating full dissociation of H2. This dissociation process releases −1.68 eV of energy. A similar process is observed for H2 on the UO2 surface, where each dissociated *H has a charge of −0.56 e−. The formation of the two H− ions is also evidenced by the disappearance of the two peaks directly below the Fermi level, as shown in Figure 2D. In contrast to state-of-the-art heterogeneous catalysts such as Ni-loaded LaN for NH3 synthesis (Ye et al., 2020a), where H2 is activated by the Ni cluster on the surfaces, and N2 by the N vacancy on the LaN surface, both N2 and H2 molecules can be activated by the same type of oxygen vacancy site on ThO2 and UO2. Hence, this provides the first evidence that these single-component materials containing oxygen vacancies may be able to catalyze the N2-to-NH3 conversion.
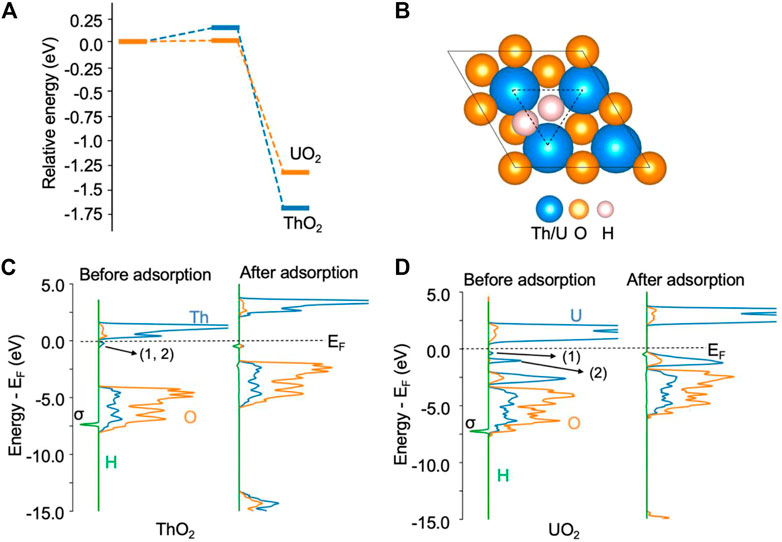
FIGURE 2. H2 dissociation on ThO2 and UO2 (111) surfaces containing oxygen vacancies. (A) Adsorption energy profile. The total energy of the initial state is set to zero. (B) Illustration of structures after H2 adsorption. The oxygen vacancy site is highlighted by the triangle. (C) Density of states before and after H2 adsorption on the ThO2 surface. (D) Density of states before and after H2 adsorption on the UO2 surface. The two excess electrons on the ThO2 surface are located at the vacancy sites labeled 1 and 2 in the DOS, directly below the Fermi level (EF) in (C). On the UO2 surface (D), one of the excess electrons (1) is located at a vacancy site and the other (2) in uranium 5f orbitals.
As shown in the earlier sections, both N2 and H2 can be activated at the vacancy site on ThO2 and UO2 (111) surfaces. The N2 bond length is stretched to 1.24 Å after adsorption on the surface, indicating activation of the triple N≡N bond and formation of the N=N double bond (Smieja et al., 1987; Hill et al., 1991). This is a key step toward the breaking of the N2 molecule for NH3 synthesis. The chemically adsorbed *NN is still bound together, and direct dissociation has large energy barriers of 3.64 eV and 2.91 eV on ThO2 and UO2 surfaces, respectively, see Supplementary Figure S2. Therefore, NH3 synthesis is unlikely to occur via the dissociative mechanism on either surface. However, the H2 molecule can be directly dissociated to form H− at another oxygen vacancy site, and the migration of H− can hydrogenate *NN to form NH3 through associative pathways. To verify this, we calculated the migration barrier of the dissociated H− on the surfaces. It is found that the dissociated H− that is located between two actinide atoms has a migration barrier of 0.54 eV on ThO2 and 0.91 eV on the UO2 surface, see Figure 3. Thus, the migration of H− can be driven by a relatively low temperature and can lead to the hydrogenation of *NN. We have also determined the energy for dissociated H− migration to surface oxygen sites. We found that it is not energetically preferred since over1.9 eV is required for H− to diffuse to the oxygen sites. It should be noted that N2 adsorption on the H2 pre-dissociated vacancy site is possible since H− is reactive. However, H− migration on the surface is still necessary to supply enough hydrogen for the formation of NH3. Based on these observations, we propose a dual-site mechanism for NH3 synthesis, where N2 and H2 are activated at different but neighboring oxygen vacancy sites. We limited ourselves to the thermodynamics of NH3 formation in this work. While the reaction barriers during the reaction cycle are important for the kinetics and NH3 formation rates, a large number of reaction paths need to be considered to sample possible reaction processes, which is computationally expensive at the DFT theory level. More computationally efficient methods, such as Density Functional based Tight Binding (DFTB), are under development to model the dynamics of these reactions (Aguirre et al., 2020; Carlson et al., 2020).
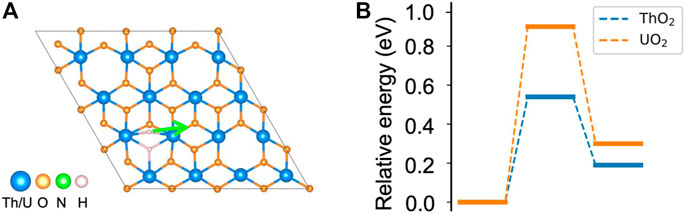
FIGURE 3. Migration of dissociated H− on ThO2 and UO2 surfaces. (A) Migration path of H− to a nearby site. (B) Corresponding energy diagram. The total energy of the initial state is set to zero.
Figure 4 shows the calculated potential energy diagram for the NH3 synthesis cycle on ThO2 and UO2 surfaces containing oxygen vacancies. The most favorable computed pathway is N2 → *NN (II) → *NNH (III) → *NNH2 (IV) → *NHNH2 (V) → *NHNH3 (VI) → *NH + NH3 (VII) → *NH2 + NH3 (VIII) → *NH3 + NH3 (IX) → 2NH3. After the adsorption of N2 at the vacancy site, *NN is hydrogenated step-by-step through the associative pathway to form NH3. From the adsorption of N2 (I and II), to formation of the first *NH3 on the surface (VI), the reactions are exothermic except for the third hydrogenation process. The first (III) and second (IV) hydrogenations occur on the N atom with a higher position on the surface. Hydrogenation of the other N atom requires an additional 0.18 eV, as shown in Supplementary Figure S3A. The third (V) hydrogenation occurs on the other N atom, as seen from the energy comparison of the different structures in Supplementary Figure S3B. After the third (V) hydrogenation, the N−N bond lenght increases from 1.10 Å in the gas phase to 2.55 Å on ThO2 and 2.88 Å on UO2, see Figure 5 and Supplementary Table S1. This is a clear indication that *NN is fully dissociated during this step. The first *NH3 is formed on the surface after the fourth (VI) hydrogenation, as shown in Supplementary Figure S3C. The release of the first NH3 molecule requires ∼0.8 eV on both ThO2 and UO2 surfaces. The subsequent hydrogenations, from VII to XI, require increasing energies. The required energy from VI to IX is lower on the UO2 surface than on the ThO2 surface, indicating that NH3 synthesis is more efficient on the UO2 surface.
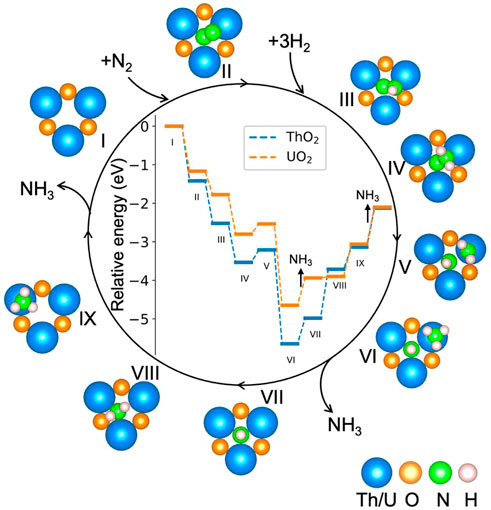
FIGURE 4. Potential energy diagram for the NH3 synthesis cycle on ThO2 and UO2 surfaces containing oxygen vacancies. The relative energy is defined by
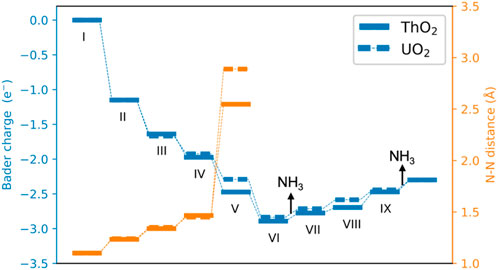
FIGURE 5. Bader charges and N−N distance during the catalytic cycle of NH3 formation. A negative value indicates that *NN obtains electrons from surfaces.
The Bader charges of nitrogen atoms during the catalytic reaction cycle are summarized in Figure 5. It is seen that after N2 adsorption, *NN possesses a Bader charge of 1.2 e− due to charge transfer from the excess surface electrons to the hybrid π*−5f orbitals. *NN is further reduced step-by-step during hydrogenation, to give a Bader charge of 2.83 e− in configuration VI. After the first NH3 release from the surface, the Bader charge of the remaining nitrogen atom on the surface becomes less negative, from 2.83 e− to 2.30 e− because it is gradually driven away from the vacancy site during hydrogenation from steps VII to IX, and the excess electrons return to the vacancy site in this part of the synthesis cycle. The surfaces containing oxygen vacancies recover to their initial states and are ready for the next catalytic cycle of NH3 synthesis.
The participation of excess electrons in the chemical reactions is critical to enable the proposed catalytic NH3 formation on ThO2 and UO2 surfaces. An illustration of the participation of excess electrons in the reactions is shown in Supplementary Figure S4. The excess electrons immediately transfer to N2 after adsorption at the vacancy site, weakening the N≡N bond. The hydrogenation of *NN further breaks the N−N bond and forms NH3. This associative *NN bond breaking process has been reported for NH3 synthesis using singly dispersed Fe3 or Rh1Co3 supported by Al2O3(010) or CoO(011) (Liu et al., 2018; Ma et al., 2018). After the release of NH3, the excess electrons return to the vacancy to enable the next catalytic cycle for NH3 synthesis. It should be noted that an oxygen vacancy has a formation energy of 6.95−2.43 eV on ThO2 and UO2 surfaces (Wang et al., 2019); however, oxygen vacancies can be artificially created by high energy collisions with the surfaces, for example, by artificial bombardment with Ar ions, as demonstrated in previous experiments (Senanayake et al., 2007a; Senanayake et al., 2007b). Compared with multicomponent heterogeneous catalysts composed of singly dispersed metal atoms or clusters on supporting substrates, the single-component ThO2 and UO2 surfaces containing oxygen vacancies are easier to experimentally synthesize and control. Since early actinides, in particular thorium and uranium, are relatively abundant in nature, ThO2 and UO2 surfaces are promising candidates for industrialization toward catalytic applications Table 1.
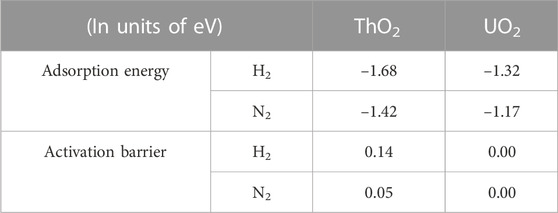
TABLE 1. Adsorption energies and activation barriers of H2 and N2 molecules on ThO2 and UO2 surfaces containing oxygen vacancies.
In summary, we explored the prospect of actinide dioxide surfaces containing oxygen vacancies as catalysts for N2 molecule activation and NH3 synthesis using first-principle calculations. Our calculations reveal that N2 and H2 can be chemically adsorbed at surface oxygen vacancy sites on ThO2 and UO2, with a very smaller energy barrier. H2 can be directly dissociated at the vacancy site to form two H−. N2 can be activated by excess electron transfer to the molecule. Subsequently, NH3 can be formed by H− migration on the surfaces through associative mechanisms. This is the first demonstration of the synthesis of NH3 by utilizing surface oxygen vacancy sites on actinide dioxide surfaces. Compared to single-atom catalysts, where precise control of the structure and the stability of the catalysts are extremely challenging (Speck et al., 2021), the proposed oxygen vacancies on ThO2 and UO2 surfaces can be fabricated by Ar ion sputtering, which are relatively easier to realize in experiments and, in this application, would serve as activators of the surface (Senanayake et al., 2007a; Senanayake et al., 2007b). These results demonstrate that 5f-element materials can serve as promising single-component catalysts for challenging chemical reactions that generally require multiple-component catalysts, thereby simplifying the synthetic process of catalysts.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
PY, EB, and GW initiated the project. GW performed the calculations and analyzed the results along with PY and EB. GW wrote the manuscript. All authors revised the manuscript.
The authors gratefully acknowledge funding from the U.S. DOE Office of Basic Energy Science under the Heavy Element Chemistry program at LANL. LANL is operated by the Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy (Contract No. 89233218CNA000001). The calculations were performed using the high-performance computing facilities at LANL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, are not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1051496/full#supplementary-material
Aboud, S., and Wilcox, J. (2010). A density functional theory study of the charge state of hydrogen in metal hydrides. J. Phys. Chem. C 114 (24), 10978–10985. doi:10.1021/jp911811r
Aguirre, N. F., Morgenstern, A., Cawkwell, M. J., Batista, E. R., and Yang, P. (2020). Development of density functional tight-binding parameters using relative energy fitting and particle swarm optimization. J. Chem. Theory Comput. 16 (3), 1469–1481. doi:10.1021/acs.jctc.9b00880
Anderson, J. S., Rittle, J., and Peters, J. C. (2013). Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 501 (7465), 84–87. doi:10.1038/nature12435
Araake, R., Sakadani, K., Tada, M., Sakai, Y., and Ohki, Y. (2017). [Fe4] and [Fe6] hydride clusters supported by phosphines: Synthesis, characterization, and application in N2 reduction. J. Am. Chem. Soc. 139 (15), 5596–5606. doi:10.1021/jacs.7b01965
Barbier-Baudry, D., Bouazza, A., Desmurs, J., Dormond, A., and Richard, S. (2000). UraniumIV and uranyle salts, efficient and reusable catalysts for acylation of aromatic compounds. J. Mol. Catal. A Chem. 164 (1-2), 195–204. doi:10.1016/s1381-1169(00)00410-6
Batista, E. R., Martin, R. L., Hay, P. J., Peralta, J. E., and Scuseria, G. E. (2004). Density functional investigations of the properties and thermochemistry of UF 6 and UF 5 using valence-electron and all-electron approaches. J. Chem. Phys. 121 (5), 2144–2150. doi:10.1063/1.1768518
Bengtsson, L. (1999). Dipole correction for surface supercell calculations. Phys. Rev. B 59 (19), 12301–12304. doi:10.1103/physrevb.59.12301
Bozso, F., Ertl, G., Grunze, M., and Weiss, M. (1977). Interaction of nitrogen with iron surfaces: I. Fe (100) and Fe (111). J. Catal. 49 (1), 18–41. doi:10.1016/0021-9517(77)90237-8
Carlson, R. K., Cawkwell, M. J., Batista, E. R., and Yang, P. (2020). Tight-binding modeling of uranium in an aqueous environment. J. Chem. Theory Comput. 16 (5), 3073–3083. doi:10.1021/acs.jctc.0c00089
Cherkasov, N., Ibhadon, A., and Fitzpatrick, P. (2015). A review of the existing and alternative methods for greener nitrogen fixation. Chem. Eng. Process. Process Intensif. 90, 24–33. doi:10.1016/j.cep.2015.02.004
Choi, C., Back, S., Kim, N. Y., Lim, J., Kim, Y. H., and Jung, Y. (2018). Suppression of hydrogen evolution reaction in electrochemical N2 reduction using single-atom catalysts: A computational guideline. Acs Catal. 8 (8), 7517–7525. doi:10.1021/acscatal.8b00905
Deblonde, G. J. P., Kelley, M. P., Su, J., Batista, E. R., Yang, P., Booth, C. H., et al. (2018). Spectroscopic and computational characterization of diethylenetriaminepentaacetic acid/transplutonium chelates: Evidencing heterogeneity in the heavy actinide (III) series. Angew. Chem. Int. Ed. 57 (17), 4611–4616. doi:10.1002/ange.201709183
Dorado, B., Amadon, B., Freyss, M., and Bertolus, M. (2009). DFT+Ucalculations of the ground state and metastable states of uranium dioxide. Phys. Rev. B 79 (23), 235125. doi:10.1103/physrevb.79.235125
Dudarev, S., Botton, G. A., Savrasov, S. Y., Humphreys, C. J., and Sutton, A. P. (1998). Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+ U study. Phys. Rev. B 57 (3), 1505–1509. doi:10.1103/physrevb.57.1505
Ertl, G., Lee, S., and Weiss, M. (1982). Kinetics of nitrogen adsorption on Fe (111). Surf. Sci. 114 (2-3), 515–526. doi:10.1016/0039-6028(82)90702-6
Ertl, G. (2008). Reactions at surfaces: From atoms to complexity (Nobel lecture). Angew. Chem. Int. Ed. 47 (19), 3524–3535. doi:10.1002/anie.200800480
Evans, W. J., Kozimor, S. A., and Ziller, J. W. (2003). A monometallic f element complex of dinitrogen:(C5Me5) 3U (η-N2). J. Am. Chem. Soc. 125 (47), 14264–14265. doi:10.1021/ja037647e
Falcone, M., Chatelain, L., Scopelliti, R., Zivkovic, I., and Mazzanti, M. (2017). Nitrogen reduction and functionalization by a multimetallic uranium nitride complex. Nature 547 (7663), 332–335. doi:10.1038/nature23279
Falcone, M., and Mazzanti, M. (2018). Four-electron reduction and functionalization of N2 by a uranium (iii) bridging nitride. CHIMIA Int. J. Chem. 72 (4), 199–202. doi:10.2533/chimia.2018.199
Foster, S. L., Sergio, I., Perez, B., Royce, D. D., Sharad, M., Ross, D., et al. (2018). Catalysts for nitrogen reduction to ammonia. Nat. Catal. 1 (7), 490–500. doi:10.1038/s41929-018-0092-7
Galloway, J. N., Townsend, A. R., Erisman, J. W., Bekunda, M., Cai, Z., Freney, J. R., et al. (2008). Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 320 (5878), 889–892. doi:10.1126/science.1136674
Gambarotta, S., and Scott, J. (2004). Multimetallic cooperative activation of N2. Angew. Chem. Int. Ed. 43 (40), 5298–5308. doi:10.1002/anie.200301669
Geng, Z., Liu, Y., Kong, X., Li, P., Li, K., Liu, Z., et al. (2018). N2 electrochemical reduction: Achieving a record-high yield rate of 120.9 μgNH3 mgcat.−1 h−1 for N2 electrochemical reduction over Ru single-atom catalysts (adv. Mater. 40/2018). Adv. Mater. 30 (40), 1870301. doi:10.1002/adma.201870301
Giddey, S., Badwal, S. P. S., Munnings, C., and Dolan, M. (2017). Ammonia as a renewable energy transportation media. ACS Sustain. Chem. Eng. 5 (11), 10231–10239. doi:10.1021/acssuschemeng.7b02219
Giovannetti, G., Khomyakov, P. A., Brocks, G., Kelly, P. J., and van den Brink, J. (2007). Substrate-induced band gap in graphene on hexagonal boron nitride: Ab initio density functional calculations. Phys. Rev. B 76 (7), 073103. doi:10.1103/physrevb.76.073103
Goldman, N., and Morales, M. A. (2017). A First-principles study of hydrogen diffusivity and dissociation on δ-Pu (100) and (111) surfaces. J. Phys. Chem. C 121 (33), 17950–17957. doi:10.1021/acs.jpcc.7b04992
Greeley, J., and Mavrikakis, M. (2005). Surface and subsurface hydrogen: Adsorption properties on transition metals and near-surface alloys. J. Phys. Chem. B 109 (8), 3460–3471. doi:10.1021/jp046540q
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27 (15), 1787–1799. doi:10.1002/jcc.20495
Grubel, K., Brennessel, W. W., Mercado, B. Q., and Holland, P. L. (2014). Alkali metal control over N–N cleavage in iron complexes. J. Am. Chem. Soc. 136 (48), 16807–16816. doi:10.1021/ja507442b
Grunze, M. (1982). “Synthesis and decomposition of ammonia,” in The chemical physics of solid surfaces and heterogeneous catalysis (Elsevier scientific publishing company), 143.
Guo, C., Ran, J., Vasileff, A., and Qiao, S. Z. (2018). Rational design of electrocatalysts and photo (electro) catalysts for nitrogen reduction to ammonia (NH 3) under ambient conditions. Energy & Environ. Sci. 11 (1), 45–56. doi:10.1039/c7ee02220d
Hammer, B., and Nørskov, J. K. (2000). Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 45, 71–129. doi:10.1016/s0360-0564(02)45013-4
Han, X., and Kaltsoyannis, N. (2022). Computational study of the energy landscape of water on the ThO2 {111} surface. J. Nucl. Mater. 559, 153476. doi:10.1016/j.jnucmat.2021.153476
Hannagan, R. T., Giannakakis, G., Flytzani-Stephanopoulos, M., and Sykes, E. C. H. (2020). Single-atom alloy catalysis. Chem. Rev. 120 (21), 12044–12088. doi:10.1021/acs.chemrev.0c00078
Henkelman, G., Uberuaga, B. P., and Jónsson, H. (2000). A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113 (22), 9901–9904. doi:10.1063/1.1329672
Hill, J. E., Fanwick, P. E., and Rothwell, I. P. (1991). Formation of a terminal aryl-imido compound of titanium by cleavage of the nitrogen nitrogen double bond in benzo [c] cinnoline. Inorg. Chem. 30 (5), 1143–1144. doi:10.1021/ic00005a048
Hölscher, M., and Leitner, W. (2015). Reduction of metal coordinated N2 to NH3 with H2 by heterolytic hydrogen cleavage induced by external lewis bases–a DFT study. Z. für Anorg. Allg. Chem. 641 (1), 72–77. doi:10.1002/zaac.201400337
Hong, J., Prawer, S., and Murphy, A. B. (2018). Plasma catalysis as an alternative route for ammonia production: Status, mechanisms, and prospects for progress. ACS Sustain. Chem. Eng. 6 (1), 15–31. doi:10.1021/acssuschemeng.7b02381
Ismagilov, Z., and Lazareva, S. (2013). Synthesis and characterization of uranium-containing catalysts. Catal. Rev. 55 (2), 135–209. doi:10.1080/01614940.2013.776858
Jackson, S. D., and Hargreaves, J. S., Metal oxide catalysis, 2 volume set. Vol. 1. 2008: John Wiley & Sons.
Jori, N., Barluzzi, L., Douair, I., Maron, L., Fadaei-Tirani, F., Z̆ivkovic, I., et al. (2021). Stepwise reduction of dinitrogen by a uranium–potassium complex yielding a U (VI)/U (IV) tetranitride cluster. J. Am. Chem. Soc. 143 (29), 11225–11234. doi:10.1021/jacs.1c05389
Keener, M., Scopelliti, R., and Mazzanti, M. (2021). Nitride protonation and NH 3 binding versus N–H bond cleavage in uranium nitrides. Chem. Sci. 12, 12610–12618. doi:10.1039/d1sc03957a
Kelley, M. P., Su, J., Urban, M., Luckey, M., Batista, E. R., Yang, P., et al. (2017). On the origin of covalent bonding in heavy actinides. J. Am. Chem. Soc. 139 (29), 9901–9908. doi:10.1021/jacs.7b03251
Kerpal, C., Harding, D. J., Lyon, J. T., Meijer, G., and Fielicke, A. (2013). N2 activation by neutral ruthenium clusters. J. Phys. Chem. C 117 (23), 12153–12158. doi:10.1021/jp401876b
Kitano, M., Kanbara, S., Inoue, Y., Kuganathan, N., Sushko, P. V., Yokoyama, T., et al. (2015). Electride support boosts nitrogen dissociation over ruthenium catalyst and shifts the bottleneck in ammonia synthesis. Nat. Commun. 6 (1), 6731–6739. doi:10.1038/ncomms7731
Kresse, G., and Furthmüller, J. (1996). Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6 (1), 15–50. doi:10.1016/0927-0256(96)00008-0
Kresse, G., and Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59 (3), 1758–1775. doi:10.1103/physrevb.59.1758
Leduc, J., Frank, M., Jurgensen, L., Graf, D., Raauf, A., and Mathur, S. (2019). Chemistry of actinide centers in heterogeneous catalytic transformations of small molecules. ACS Catal. 9 (6), 4719–4741. doi:10.1021/acscatal.8b04924
Li, C., Wang, T., and Gong, J. (2020). Alternative strategies toward sustainable ammonia synthesis. Trans. Tianjin Univ. 26 (2), 67–91. doi:10.1007/s12209-020-00243-x
Li, J., Zhan, G., and Zhang, L. (2017). Solar water splitting and nitrogen fixation with layered bismuth oxyhalides. Accounts Chem. Res. 50 (1), 112–121. doi:10.1021/acs.accounts.6b00523
Ling, C., Niu, X., Li, Q., Du, A., and Wang, J. (2018). Metal-free single atom catalyst for N2 fixation driven by visible light. J. Am. Chem. Soc. 140 (43), 14161–14168. doi:10.1021/jacs.8b07472
Ling, C., Ouyang, Y., Li, Q., Bai, X., Mao, X., Du, A., et al. (2019). A General two-step strategy–based high-throughput screening of single atom catalysts for nitrogen fixation. Small Methods 3 (9), 1800376. doi:10.1002/smtd.201800376
Liu, J.-C., Ma, X. L., Li, Y., Wang, Y. G., Xiao, H., and Li, J. (2018). Heterogeneous Fe 3 single-cluster catalyst for ammonia synthesis via an associative mechanism. Nat. Commun. 9 (1), 1610–1619. doi:10.1038/s41467-018-03795-8
Logadottir, A., and Nørskov, J. K. (2003). Ammonia synthesis over a Ru (0001) surface studied by density functional calculations. J. Catal. 220 (2), 273–279. doi:10.1016/s0021-9517(03)00156-8
Lu, E., Atkinson, B. E., Wooles, A. J., Boronski, J. T., Doyle, L. R., Tuna, F., et al. (2019). Back-bonding between an electron-poor, high-oxidation-state metal and poor π-acceptor ligand in a uranium (v)–dinitrogen complex. Nat. Chem. 11 (9), 806–811. doi:10.1038/s41557-019-0306-x
Ma, X.-L., Liu, J. C., Xiao, H., and Li, J. (2018). Surface single-cluster catalyst for N2-to-NH3 thermal conversion. J. Am. Chem. Soc. 140 (1), 46–49. doi:10.1021/jacs.7b10354
McSkimming, A., Su, J., Cheisson, T., Gau, M. R., Carroll, P. J., Batista, E. R., et al. (2018). Coordination chemistry of a strongly-donating hydroxylamine with early actinides: An investigation of redox properties and electronic structure. Inorg. Chem. 57 (8), 4387–4394. doi:10.1021/acs.inorgchem.7b03238
McWilliams, S. F., and Holland, P. L. (2015). Dinitrogen binding and cleavage by multinuclear iron complexes. Accounts Chem. Res. 48 (7), 2059–2065. doi:10.1021/acs.accounts.5b00213
Mitchell, S., and Pérez-Ramírez, J. (2020). Single atom catalysis: A decade of stunning progress and the promise for a bright future. Nat. Commun. 11 (1), 4302–4303. doi:10.1038/s41467-020-18182-5
Niu, H., Wang, X., Shao, C., Zhang, Z., and Guo, Y. (2020). Computational screening single-atom catalysts supported on g-CN for N2 reduction: High activity and selectivity. ACS Sustain. Chem. Eng. 8 (36), 13749–13758. doi:10.1021/acssuschemeng.0c04401
Patil, B., Wang, Q., Hessel, V., and Lang, J. (2015). Plasma N2-fixation: 1900–2014. Catal. today 256, 49–66. doi:10.1016/j.cattod.2015.05.005
Pegg, J. T., Shields, A. E., Storr, M. T., Scanlon, D. O., and de Leeuw, N. H. (2020). Interaction of hydrogen with actinide dioxide (011) surfaces. J. Chem. Phys. 153 (1), 014705. doi:10.1063/5.0010200
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77 (18), 3865–3868. doi:10.1103/physrevlett.77.3865
Pool, J. A., Lobkovsky, E., and Chirik, P. J. (2004). Hydrogenation and cleavage of dinitrogen to ammonia with a zirconium complex. Nature 427 (6974), 527–530. doi:10.1038/nature02274
Rak, Z., Ewing, R. C., and Becker, U. (2013). Hydroxylation-induced surface stability of AnO2 (An= U, Np, Pu) from first-principles. Surf. Sci. 608, 180–187. doi:10.1016/j.susc.2012.10.002
Reuter, K., Plaisance, C. P., Oberhofer, H., and Andersen, M. (2017). Perspective: On the active site model in computational catalyst screening. J. Chem. Phys. 146 (4), 040901. doi:10.1063/1.4974931
Rodriguez, M. M., Bill, E., Brennessel, W. W., and Holland, P. L. (2011). N2 reduction and hydrogenation to ammonia by a molecular iron-potassium complex. Science 334 (6057), 780–783. doi:10.1126/science.1211906
Sachs, B., Wehling, T. O., Katsnelson, M. I., and Lichtenstein, A. I. (2011). Adhesion and electronic structure of graphene on hexagonal boron nitride substrates. Phys. Rev. B 84 (19), 195414. doi:10.1103/physrevb.84.195414
Schlögl, R. (2015). Heterogeneous catalysis. Angew. Chem. Int. Ed. 54 (11), 3465–3520. doi:10.1002/anie.201410738
Senanayake, S. D., Waterhouse, G., Chan, A., Madey, T., Mullins, D., and Idriss, H. (2007). The reactions of water vapour on the surfaces of stoichiometric and reduced uranium dioxide: A high resolution xps study. Catal. today 120 (2), 151–157. doi:10.1016/j.cattod.2006.07.040
Senanayake, S. D., Waterhouse, G. I. N., Chan, A. S. Y., Madey, T. E., Mullins, D. R., and Idriss, H. (2007). Probing surface oxidation of reduced uranium dioxide thin film using synchrotron radiation. J. Phys. Chem. C 111 (22), 7963–7970. doi:10.1021/jp068828g
Shi, L., Bi, S., Qi, Y., Ren, K., Zheng, L., Wang, J., et al. (2022). Anchoring Mo single-atom sites on B/N codoped porous carbon nanotubes for electrochemical reduction of N2 to NH3. ACS Catal. 12 (13), 7655–7663. doi:10.1021/acscatal.2c01293
Shima, T., Hu, S., Luo, G., Kang, X., Luo, Y., and Hou, Z. (2013). Dinitrogen cleavage and hydrogenation by a trinuclear titanium polyhydride complex. Science 340 (6140), 1549–1552. doi:10.1126/science.1238663
Smieja, J. A., Gozum, J. E., and Gladfelter, W. L. (1987). Nitrogen-nitrogen double bond cleavage and the ortho metalation of azoarenes using Ru3 (NPh)(CO) 10. Organometallics 6 (6), 1311–1317. doi:10.1021/om00149a029
Speck, F. D., Jae, H K., Geunsu, B, Joo, S H., Mayrhofer, K J. J., Choi, C H., et al. (2021). Single-atom catalysts: A perspective toward application in electrochemical energy conversion. JACS Au 8, 1086. doi:10.1021/jacsau.1c00121
Stoltze, P., and Nørskov, J. (1985). Bridging the" pressure gap" between ultrahigh-vacuum surface physics and high-pressure catalysis. Phys. Rev. Lett. 55 (22), 2502–2505. doi:10.1103/physrevlett.55.2502
Temkin, M., and Pyzhev, V. (1940). Kinetics of ammonia synthesis on promoted iron catalysts. Acta physiochim. URSS 12, 327–356.
Thulstrup, E. W., and Andersen, A. (1968-19871975). Configuration interaction studies of bound, low-lying states of N2-N2, N2+ and N22+. J. Phys. B Atomic Mol. Phys. 8 (6), 965–976. doi:10.1088/0022-3700/8/6/023
Tyler, D. R. (2015). Mechanisms for the Formation of NH3, N2H4, and N2H2 in the Protonation Reaction of Fe (DMeOPrPE) 2N2 {DMeOPrPE= 1, 2-bis [bis (methoxypropyl) phosphino] ethane}. Z. für Anorg. Allg. Chem. 641 (1), 31–39. doi:10.1002/zaac.201400126
Walter, M. (2016). Recent advances in transition metal-catalyzed dinitrogen activation. Adv. Organomet. Chem. 65, 261–377. doi:10.1016/bs.adomc.2016.03.001
Wang, A., Li, J., and Zhang, T. (2018). Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2 (6), 65–81. doi:10.1038/s41570-018-0010-1
Wang, G., Batista, E. R., and Yang, P. (2019). Excess electrons on reduced AnO2 (111) surfaces (An= Th, U, Pu) and their impacts on catalytic water splitting. J. Phys. Chem. C 123 (50), 30245–30251. doi:10.1021/acs.jpcc.9b06543
Wang, G., Pandey, R., Moody, N. A., and Batista, E. R. (2017). Degradation of alkali-based photocathodes from exposure to residual gases: A first-principles study. J. Phys. Chem. C 121, 8399–8408. doi:10.1021/acs.jpcc.6b12796
Wen, X.-D., Martin, R. L., Henderson, T. M., and Scuseria, G. E. (2013). Density functional theory studies of the electronic structure of solid state actinide oxides. Chem. Rev. 113 (2), 1063–1096. doi:10.1021/cr300374y
Xin, X., Douair, I., Zhao, Y., Wang, S., Maron, L., Zhu, C., et al. (2021). Dinitrogen cleavage and hydrogenation to ammonia with a uranium complex. Natl. Sci. Rev. 144. doi:10.1093/nsr/nwac144
Yang, J., Guo, Y., Lu, W., Jiang, R., and Wang, J. (2018). Emerging applications of plasmons in driving CO2 reduction and N2 fixation. Adv. Mater. 30 (48), 1802227. doi:10.1002/adma.201802227
Yang, X.-F., Wang, A., Qiao, B., Li, J., Liu, J., and Zhang, T. (2013). Single-atom catalysts: A new frontier in heterogeneous catalysis. Accounts Chem. Res. 46 (8), 1740–1748. doi:10.1021/ar300361m
Ye, T.-N., Park, S. W., Lu, Y., Li, J., Sasase, M., Kitano, M., et al. (2020). Contribution of nitrogen vacancies to ammonia synthesis over metal nitride catalysts. J. Am. Chem. Soc. 142 (33), 14374–14383. doi:10.1021/jacs.0c06624
Keywords: N2-to-NH3 conversion, excess electrons, point vacancies, actinide dioxide surfaces, DFT density functional theory
Citation: Wang G, Batista ER and Yang P (2023) N2-to-NH3 conversion by excess electrons trapped in point vacancies on 5f-element dioxide surfaces. Front. Chem. 10:1051496. doi: 10.3389/fchem.2022.1051496
Received: 22 September 2022; Accepted: 09 December 2022;
Published: 05 January 2023.
Edited by:
Simon Freakley, University of Bath, United KingdomReviewed by:
Vivek Sinha, Delft University of Technology, NetherlandsCopyright © 2023 Wang, Batista and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaoxue Wang, Z2FveHVld0BsYW5sLmdvdg==; Enrique R. Batista, ZXJiQGxhbmwuZ292; Ping Yang, cHlhbmdAbGFubC5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.