 Nazli Mert-Ozupek
Nazli Mert-Ozupek Gizem Calibasi-Kocal
Gizem Calibasi-Kocal Nur Olgun3
Nur Olgun3 Levent Cavas
Levent Cavas
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 06 December 2022
Sec. Medicinal and Pharmaceutical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1046313
This article is part of the Research Topic Computational Drug Discovery of Medicinal Compounds for Cancer Management View all 16 articles
Caulerpa spp. secrete more than thirty different bioactive chemicals which have already been used in cancer treatment research since they play a pivotal role in cancer metabolism. Colorectal cancer is one of the most common cancer types, thus using novel and effective chemicals for colorectal cancer treatment is crucial. In the cheminformatics pipeline of this study, ADME-Tox and drug-likeness tests were performed for filtering the secondary metabolites of Caulerpa spp. The ligands which were selected from the ADME test were used for in silico molecular docking studies against the enzymes of the oxidative branch of the pentose phosphate pathway (glucose-6-phosphate dehydrogenase and 6-phosphoglutarate dehydrogenase), which is of great importance for colorectal cancer, by using AutoDock Vina. Pharmacophore modeling was carried out to align the molecules. Molecular dynamic simulations were performed for each target to validate the molecular docking studies and binding free energies were calculated. According to the ADME test results, 13 different secondary metabolites were selected as potential ligands. Molecular docking studies revealed that vina scores of caulerpin and monomethyl caulerpinate for G6PDH were found as −10.6 kcal mol-1, −10.5 kcal mol-1, respectively. Also, the vina score of caulersin for 6PGD was found as −10.7 kcal mol-1. The highest and the lowest binding free energies were calculated for monomethyl caulerpinate and caulersin, respectively. This in silico study showed that caulerpin, monomethyl caulerpinate, and caulersin could be evaluated as promising marine phytochemicals against pentose phosphate pathway enzymes and further studies are recommended to investigate the detailed activity of these secondary metabolites on these targets.
Caulerpa, a green siphonous macroalgae, belongs to the Caulerpaceae family with 97 species. It was first described by J V Lamourox in 1809 and derived from the Greek words caulos (stalk/stem) and erpos (creep) (Mehra et al., 2019). Especially, C. taxifolia and C. cylindracea (previously known as C. racemosa var. cylindracea) have attracted attention for the last 30 years due to their invasive properties (Montefalcone et al., 2015) and, more importantly, the various properties of bioactive (especially secondary metabolites) chemicals for defense, communication, growth and development regulation, reproduction, competition, and infection (Erb and Kliebenstein, 2020; Ramawat and Goyal, 2020). Caulerpa secrete linear or monocyclic terpenoids that have aldehyde and enol-acetate functional groups (Mehra et al., 2019). The structure of caulerpin (C24H18N2O4), a secondary metabolite and pigment of the C. cylindracea species, was first described by Aguilar-Santos in 1970 (Aguilar-Santos, 1970). Its molecular weight is 398.418 g/mol and its characterized structure is ‘dimethyl-6,13-dihydrodibenzo [b,i] phenazin-5,12-dicarboxylate methyl ester. Caulersin (C21H14N2O3) is another bis-indole alkaloid which is isolated from C. serrulata (Su et al., 1997) and from C. racemosa (Yang et al., 2014). Its isomers are caulersin A, B, and C. The molecular weight of caulersin is 342.1004 g/mol (Su et al., 1997). It is characterized by its “central troponoid bridging” bisindole structure (Su et al., 1997). Caulerpenyne is a sesquiterpenoid-structured secondary metabolite which has some bioactivities such as antiproliferative and apoptotic activities (Cavas et al., 2006) and inhibitors of lipoxygenase (Cengiz et al., 2011) and 5-lipoxygenase (Richter et al., 2014), etc. Secondary metabolites of genus Caulerpa are responsible for complex modulation network induced in AMPK, ER Stress, mitochondrial stress, PTP1B inhibition and cell cycle stop pathways, metabolic reprogramming in cancer cells, apoptosis and cell cycle arrest in cancer metabolism (Mehra et al., 2019).
Colorectal cancer (CRC) is the second and third most common diagnosed cancer type in women and men in the world, respectively (Dekker et al., 2019). According to the World Health Organization data, in 2018, CRC caused 1.80 million new cases and 862,000 deaths all around the world (WHO, 2018). Since CRC is a common and fatal cancer type, using novel and effective chemicals for treatment is essential.
The pentose phosphate pathway (PPP) is the pivotal pathway for ribonucleotide synthesis and is the main source of NADPH (the reduced form of Nicotinamide Adenine Dinucleotide Phosphate) (Stincone et al., 2015), which is of great importance for fatty acid synthesis and reactive oxygen species scavenging. PPP branches from glycolysis, and it plays a key role in cancer cells (Patra and Hay, 2014). The oxidative phase of PPP is initiated with hexokinase, which converts glucose to glucose 6-phosphate (G6P). G6P oxidizes (dehydrogenated) to 6-phosphogluconolactone by the rate-limiting enzyme (glucose 6-phosphate dehydrogenase (G6PDH)) to yield NADPH (by reducing NADP+) (Stincone et al., 2015). The other NADPH source of PPP is the conversion of 6-phosphogluconate into ribose (ribulose) 5-phosphate by 6-phosphogluconate dehydrogenase (6PGD). Eventually, the regeneration of GSH, synthesis of DNA, fatty acids, and sterols is achieved by producing 2 mol of NADPH per mole of G6P entering the oxidative phase of PPP (Patra and Hay, 2014). In many solid tumors, overexpression of PPP (especially the enzyme 6PGD) has already been observed (Patra and Hay, 2014; Jin and Zhou, 2019). Furthermore, targeting oxidative phase of PPP for mutant KRAS colorectal carcinomas prevents the recurrence (Gao et al., 2019). Thus, targeting the PPP is a potentially new target for CRC treatment.
In-silico computer-aided methods are commonly used to predict and elucidate the molecular-level behavior of a compound (Dege et al., 2022). Molecular docking is a convenient in-silico method which can be used to evaluate the binding affinity of the ligand on the receptor and can predict the position of the these molecules (Trott and Olson, 2010; Mert Ozupek and Cavas, 2017). ADME provides information about in-silico ADME behavior which is important for medicinal chemistry (Bocci et al., 2017; Dege et al., 2022; Gokce et al., 2022; Pantaleão et al., 2022). Drug-likeness analysis using in-silico is of great importance for evaluating the pharmacokinetic features of fast and cheap (Gokce et al., 2022; Hasan et al., 2022).
In this study, the anticancer activity of phytochemicals of Caulerpa spp. were tested on potential targets (G6PDH and 6PGD) against CRC by using in-silico pharmacokinetic and pharmacodynamic tools. The aim of this study was to investigate the potential Caulerpa-based phytochemicals against fundamental targets (G6PDH and 6PGD) for colorectal cancer treatment.
The secondary metabolites found in Caulerpa spp. Were selected from the literature and organism-specific natural product lists of PubChem; Lotus-the natural products occurrence database (https://pubchem.ncbi.nlm.nih.gov). The three-dimensional (3D) structures of chemicals found in Caulerpa spp. were extracted from the PubChem Database. The Canonical smiles formats of the secondary metabolites were drawn using ACD/ChemSketch software. To optimize the geometry and minimize the energy for the selected ligands (secondary metabolites), Open Babel (O’Boyle et al., 2011) minimization tool was used. As force field, uff (universal force field) was selected. Conjugate gradients were selected as optimization algorithm, and total number of steps was set as 200.
The proteins used in the molecular modeling studies were retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/). The crystal structure of receptors against human colorectal cancer G6PDH (PDBID: 6E08; resolution: 1.90 Å) and 6PGD (PDBID: 4GWK; resolution: 1.534 Å) were extracted. The crystal structures were rebuilt and both water and small molecules were removed. To perform energy minimization and geometry optimization, polar hydrogens were added, and non-polar hydrogens were merged into the molecules by using AutoDock Tools-1.5.6. Before the docking studies, Kollman charges were added, and the related receptors were saved in the PDBQT format.
The drug-likeness of the compounds found in Caulerpa spp. was calculated using SwissADME (http://www.swissadme.ch/)(Daina et al., 2017). The molecular structures of compounds were converted into SMILES format. Only the ligands that could Lipinski’s five rule variations (calculated Log P (CLog P) should be less than five, polar surface area, the number of hydrogen bond donors should be less than five, hydrogen acceptors should be less than ten and the molecular weight should be less than 500) with no more than one violation were used for molecular docking experiments. Toxicity Estimation Software Tool (T.E.S.T.) (Martin et al., 2008) and ProTox-II (http://tox.charite.de/protoc_II; Banerjee et al., 2018) server were used to determine the toxicity estimation of secondary metabolites of Caulerpa which were selected from ADME results.
To examine the selected ligands on related receptors, molecular docking experiments were carried out with AutoDock Vina. After the minimization process, the grid box resolution was set at 29.7030, 17.7197, and 29.5355 along the x, y, and z points, respectively, for G6PDH (PDBID: 6E08). To define the binding site for conducting the docking for 6PGD (PDBID: 4GWK), grid box resolution was set at 21.6405, 23.5892, and −2.9280 along the x, y, and z points, respectively. The grid dimensions of all receptors were adjusted to 25 × 25×25 for all molecular docking studies. DHEA and 6aminonicotinamide (6ANA) were used as control ligands. The results of the graphical representations were prepared via Maestro Schrödinger.
The compounds of Caulerpa spp. that showed binding affinities less than −10 kcal/mol (threshold value), were analyzed for pharmacophoric features using the PharmaGist web server (Schneidman-Duhovny et al., 2008). For further studies, ZINCPharmer server (Koes and Camacho, 2012) was used to visualize the best pairwise alignment of ligands (compounds from Caulerpa spp.) with the pivot molecule (DHEA or 6ANA). Scores were calculated for each pharmacophore feature by PharmaGist server.
The molecular docking simulations of caulerpin, monomethyl caulerpinate and caulersin with glucose-6-phosphate dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PGD) proteins were performed using a web-based MD simulation package WebGRO for Macromolecular Simulations (https://simlab.uams.edu/) Simlab, the University of Arkansas for Medical Sciences (UAMS), Little Rock, United States provided by GROMACS-2019.2 (Abraham et al., 2015). PRODRG server (Schüttelkopf and Van Aalten, 2004) was used for the generation of the ligand topology files. GROMOS96 43a1 force field was used for the approximation of the protein-ligand (G6PDH-caulerpin; G6PDH-monomethylcaulerpinate; 6PGD-caulersin) interactions. The triclinic box was filled with SPC water and 0.15 M NaCl (counter ions) to neutralize the system for each ligand-protein complex. The equilibration type was NVT/NPT and Parrinello-Danadio-Bussi thermostat and Parrinello-Rahmanbarostat were used to control the temperature (300 K) and the pressure (atmospheric pressure-1 bar). 5,000 steepest descent was used to minimize the energy of the system. Each protein-ligand complex (G6PDH-caulerpin; G6PDH-monomethyl caulerpinate; 6PGD-caulersin) was simulated for 100 ns. H bonds, the Radius of gyration (Rg), Root Mean Square Deviation (RMSD) and SASA were tested to estimate the complex stability.
The best docking poses for each ligand (caulerpin, monomethyl caulerpinate, and caulersin) were rescored. In this study, the binding free energy of the ligands was identified to determine the performance of MM/PB(GB)SA by using Amber package (Fast Amber Rescoring for PPI inhibitors-farPPI; http://cadd.zju.edu.cn/farppi; (Wang et al., 2019). The input files were generated using AutoDock Tools. The force field parameter was set as GAFF2 (for ligand) + ff14SB (for a receptor) and the rescoring procedure was set as PB3 (radii = parse, γ = 0.00542, β = 0.9200). AM1-BCC method was used to calculate the partial charge of the ligands by using antechamber module of Amber.
Lipophilicity, water solubility, drug-likeness, medicinal chemistry (leadlikeness) values of 36 metabolites from Caulerpa spp. Were obtained using the SwissADME server. The results reveal that the logP of 31 compounds were in the range of 0–5, on the other hand, five of the secondary metabolites (transphytol, alpha-tocospiroA, alpha-tocospirone, trifarin and caulerpicin) were not in the range of Lipinski’s Rule of five (LRo5): 2≤logP≤5). According to the rule of 5, the molecular weight should be 200 ≤ MW ≤ 500. The molecular weights of the 32 compounds were in the acceptable range. However, the MW of sulfoquinovosyldiacyl glycerol, amBiosome, alpha-tocoxylenoxy and caulerpicin do not satisfying the Lipinski Ro5. The number of H-bond acceptors (≤10) and donors (≤5) for 34 (except sulfoquinovosyldiacyl glicerol and amBiosome) and 35 (except sulfoquinovosyldiacyl glicerol) compounds falling in acceptable range, respectively. All the compounds (except sulfoquinovosyldiacyl glicerol) were found to be the range of topological polar surface area (TPSA; <140). The minimum and the maximum numbers of rotatable bonds were found to be 0 and 39, respectively (Table 1). Only the chemicals that were acceptable for LRo5 with no violation were selected for the cheminformatic pipeline and further pharmacodynamic studies. Considering all the obtained results, 13 Caulerpa-based phytochemicals (caulerpin, caulerpenyne, caulersin, 10,11-epoxycaulerpenyne, flexilin, racemosin C, racemosin B, caulerprenylol B, caulerprenylol A, monomethyl caulerpinate, α-tocospiro A, α-tocospirone and furocaulerpin) were chosen and used in subsequent steps.
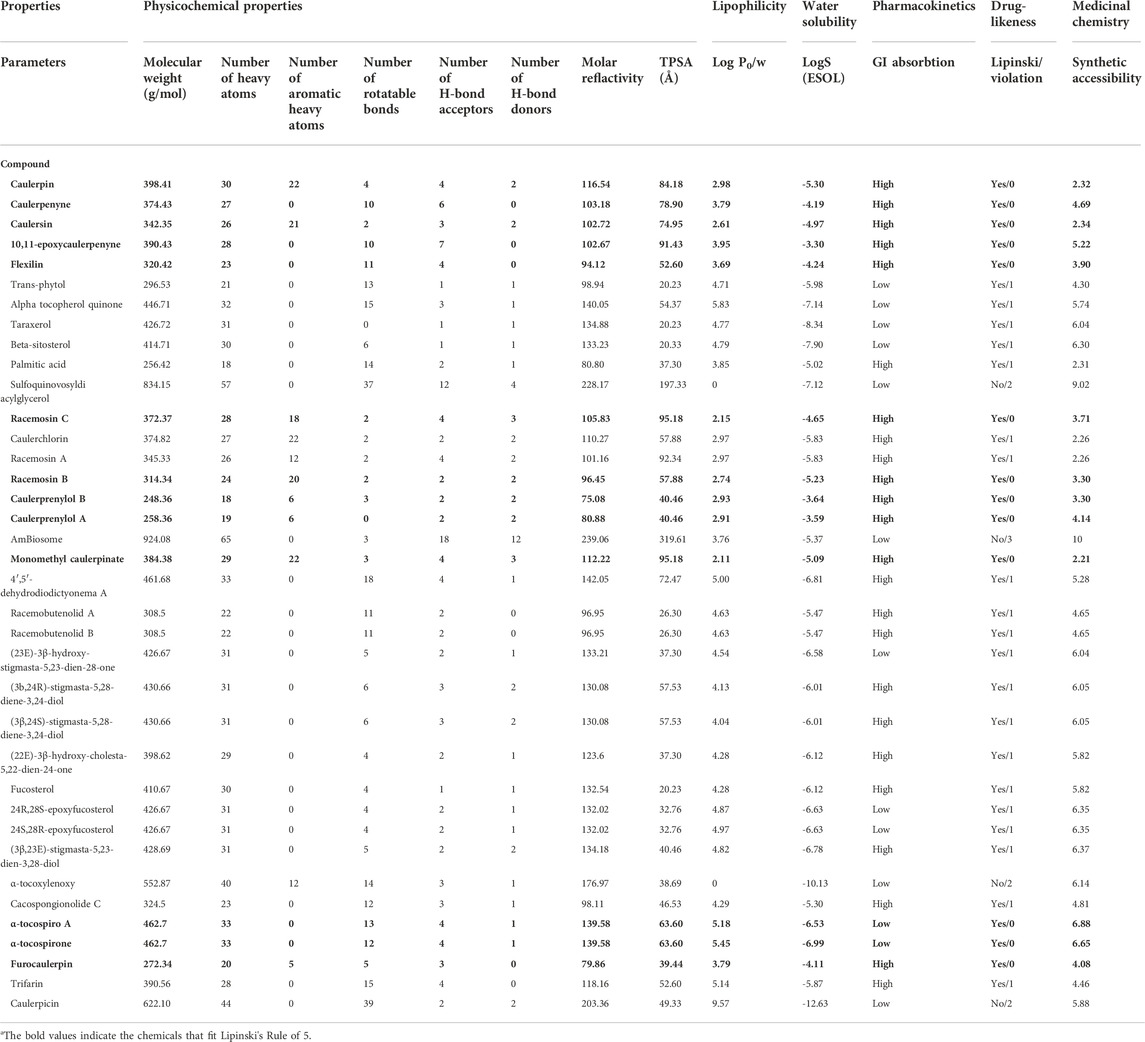
TABLE 1. List of pharmacokinetic properties of 36 compounds from Caulerpa spp.
In the cheminformatic pipeline of the study, computational based in-silico toxicity was also used. T.E.S.T. tool and ProTox-II servers were used to identify the adverse effects and toxicity of the 13 selected compounds to evaluate several toxicological parameters (acute toxicity, carcinogenicity, cytotoxicity, hepatotoxicity, immunotoxicity, predicted median lethal dose; LD50 and mutagenicity). ProTox-II results revealed that caulerpin, caulerpenyne, caulersin, flexilin, racemosin C, racemosin B and monomethyl caulerpinate belonging to the toxicity class 4, LD50 range from 500 to 1760 mg/kg, these would be harmful in case oral delivery. (Table 2).
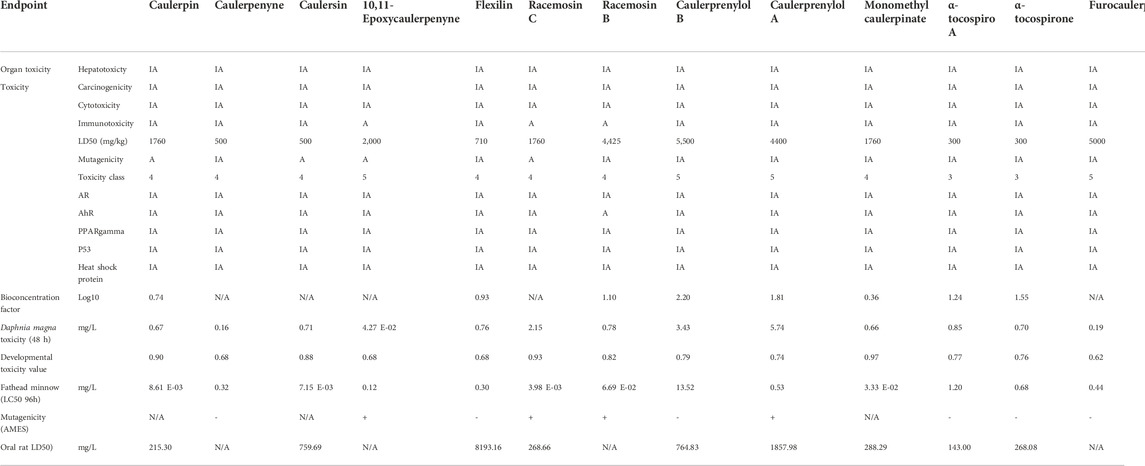
TABLE 2. List of toxicity properties of selected Caulerpa-based phytochemicals.
In this study, molecular docking studies were applied for the investigation of anticancer activity of caulerpin, caulerpenyne, 10,11-epoxycaulerpenyne, caulersin, flexilin, racemosin C, racemosin B, caulerprenylol B, caulerprenylol A, monomethyl caulerpinate, α-tocospiro A, α-tocospirone and furocaulerpin. For anticancer studies, G6PDH and 6PGD, which are crucial for CRC, were selected as receptors.
G6PDH is a cytosolic rate-limiting enzyme that converts G6P into 6-phosphoglucono-δ-lactone in the pentose phosphate pathway. In this study, human G6PDH (PDBID:6E08) was selected as a target. Both caulerpin and monomethyl caulerpinate, which have docking scores less than −10 kcal mol−1 were selected as ligands with the highest activity. According to the results, the lowest and the highest binding energies on G6PDH were found as −10.6 and −5.8 kcal/mol for caulerpin and 6-aminonicotinamide, respectively (Table 3). Inside the binding cavity of G6PDH, the methyl ester group of caulerpin makes H-bond with Lys171. Also, the indole ring of caulerpin docked in G6PDH makes pi-pi staking with Phe253 (Figure 1A). Furthermore, the indole ring of monomethyl caulerpinate makes pi-pi stacking with Phe253 (Figure1B).

TABLE 3. Docking scores with G6PDH and 6PGD and the top 13 selected compounds from Caulerpa spp.
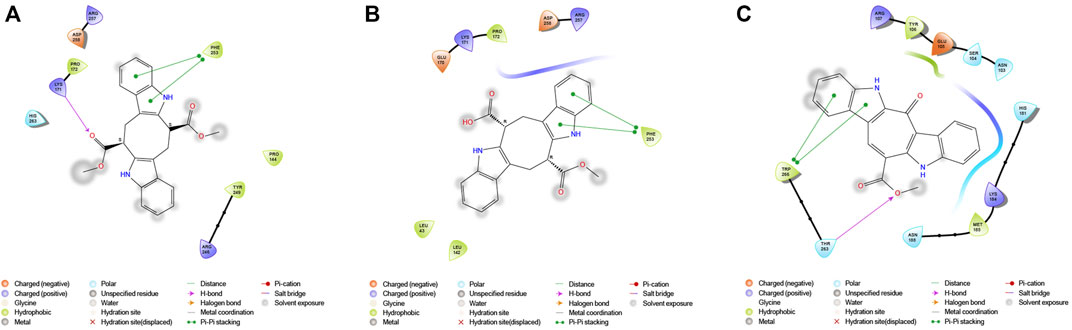
FIGURE 1. 2-D interaction network among (A) CPN and amino acid residues of G6PDH, (B) MMCPNT and amino acid residues of G6PDH, (C) CSN and amino acid residues of 6PGD.
The molecular docking studies on 6PGD reveal that the lowest binding score was found as −10.7 kcal/mol for caulersin as given in Table 3. Also, the highest binding energy was found as −5.8 for 6-aminonicotinamide. Inside the binding cavity of 6PGD, Trp266 forms pi-pi stacking with both the pirole ring of the indole ring of caulersin. Also, Thr263 makes hydrogen bond with double bond oxygen of the methyl ester group of the central traponoid of caulersin. Related information is given in Figure 1C.
In the MD simulations, RMSD (Root Mean Square Deviation) values, the starting position of the backbone of all amino acid residues, were calculated using WebGRO to clarify the stability and overall conformational dynamics of receptor-ligands (G6PDH-caulerpin, G6PDH-monomethyl caulerpinate, 6PGD-caulersin). The results reveal that the average RMSD values for caulerpin-G6PDH, monomethyl caulerpinate-G6PDH and caulersin-6PGD were found as 0.31, 0.46, and 0.43 nm respectively. All the values were comparable and in the physiological environment, indicating the stability of ligand-protein interaction. RMSF (Root Mean Square Fluctuation) values, the standard deviation of atomic positions of each amino acid residues, were also calculated. 0.06, (Figures 2A,E,I). The results from RMSD showed that CPN, MMCPNT and CSN remained positioned at the active sites of the G6PDH and 6PGD with stable interactions.
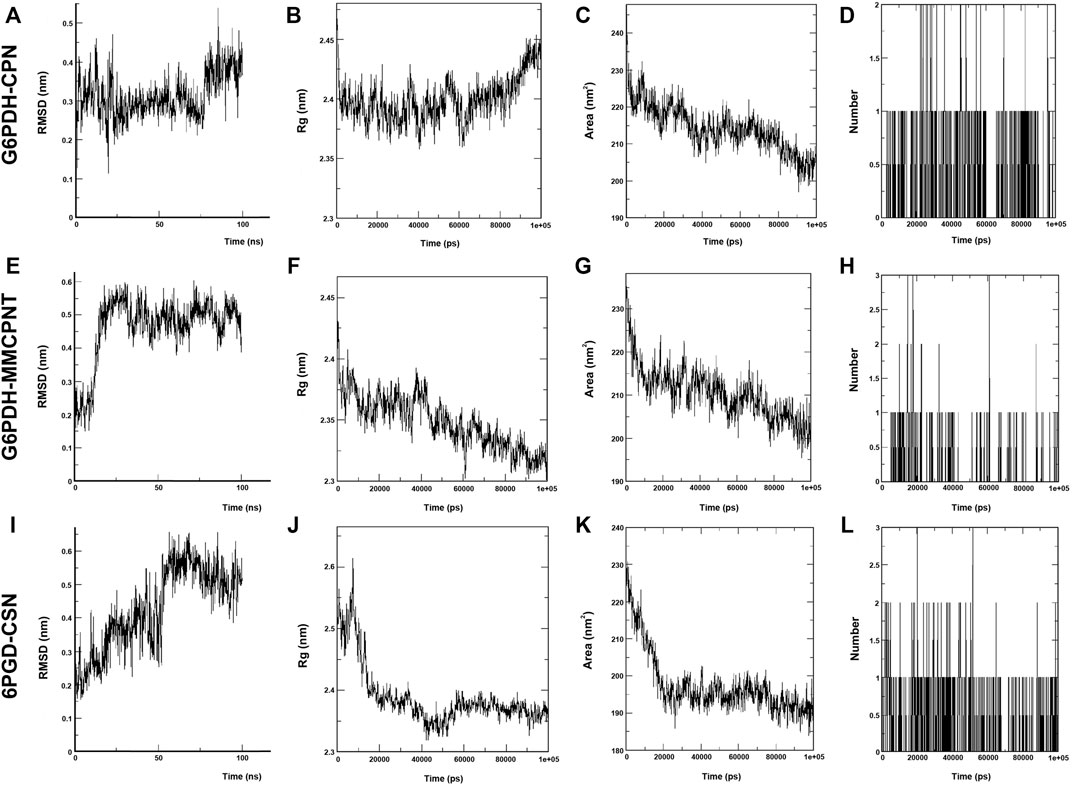
FIGURE 2. MD simulation of CPN, MMCPNT and CSN with G6PDH and 6PGD; (A), (E), and (I) RMSD line plots, (B), (F), and (J) Radius of gyration (Rg) line plots, (C), (G), and (K) SASA line plots, (D), (H), and (L): Line plots of Ligand-protein H bonds for G6PDH-CPN, G6PDH-MMCPNT and 6PGD-CSN, respectively.
Radius of gyration (Rg) computes the structural compactness and dynamic adaptability of the ligand-protein complex about the x-, y- and z-axes, as a function of time. In Figures 2B,F,J, Rg values of CPN, MMCPNT and CSN with G6PDH and 6PGD receptors ranged between −2.35 and 2.45 nm, −2.30–2.43, and −2.32–2.60 nm, respectively. The overall RG results revealed that, G6PDH-CPN receptor-ligand complex had minimum structural compactness variations and this result indicates the stability of the complexes. SASA is an approximate structural stability of the ligand-protein interaction that is accessible to a solvent (water) with respect to simulation time (100 ns). It was observed that the frequencies of SASA of all G6PDH complexes were decreased around 210 nm2 (Figures 2C,G), on the other hand, SASA result of 6PGD-CSN complex was restricted around 190 nm2 (Figure 2K). The maximum numbers of H-bonds of caulerpin-G6PDH, monomethyl caulerpinate-G6PDH, and caulersin-6PGD per time frame were found to be 2, 3 and 4, respectively. Furthermore, H-bond formation dynamics between ligands and proteins reveal that for all complexes, at least one H-bond was found as long-lived all through the simulation (100 ns) (Figures 2D, H, L).
In this study, combined structure- and ligand-based pharmacophore modeling was performed to evaluate Caulerpa-based phytochemicals with potential activity against G6PDH and 6PGD. PharmaGist server was used for pharmacophore modeling to enlighten the three-dimensional pharmacophoric features of top hit ligands for each receptor. Pharmacophore modeling is of great importance for specific receptors to elucidate if the interaction blocks or triggers a biological response.
In this study, top hits (caulerpin and monomethyl caulerpinate for G6PDH and caulersin for 6PGD) were used for each compound in the same orientation at the same binding pocket. The pairwise structural alignment details are given in Table 4. DHEA and 6-aminonicotinamide (6ANA) were used as pivot molecules for G6PDH and 6PGD, respectively. The hit compounds, “caulerpin and monomethyl caulerpinate” and “caulersin” were modeled for G6PDH and 6PGD, respectively. The results revealed that both caulerpin and monomethyl caulerpinate shared the maximum feature number with DHEA. The pairwise structural alignment of DHEA and 6ANA with the top hits is shown in Figure 3.
TABLE 4. Pairwise structural alignment showing common pharmacophoric features of secondary metabolites (pivot molecule) and top hit compounds against G6PDH and 6PGD
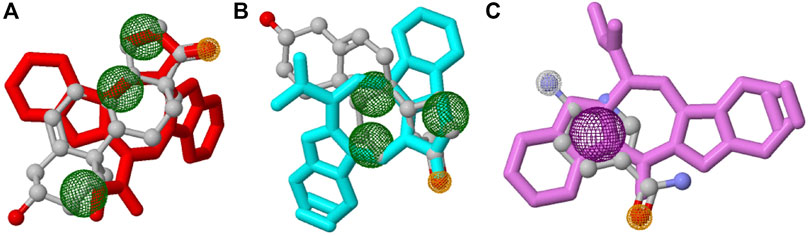
FIGURE 3. Structural alignment of pivot molecule, (A) DHEA (gray) with CPN (red), (B) DHEA (gray) with MMCPNT (blue), and (C) 6ANA (gray) with CSN (violet). DHEA and 6ANA were displayed in ball and stick style, CPN, MMCPNT and CSN were shown in sticks style. (A,B) yellow spheres represent hydrogen bond acceptors, green spheres represent hydrophobic features, (C) white spheres represent hydrogen bond donors, yellow spheres represent hydrogen bond acceptors, and purple spheres specify aromatic features.
The binding free energies of caulerpin and monomethyl caulerpinate with G6PDH using MM-PB(GB)SA were calculated as -38.43 and -40.94 kcal/mol, respectively (Figures 4A,B). Also, the binding free energy of caulersin with 6PGD using MM-PB (GB) SA was calculated as −20.20 kcal/mol (Figure 4C).
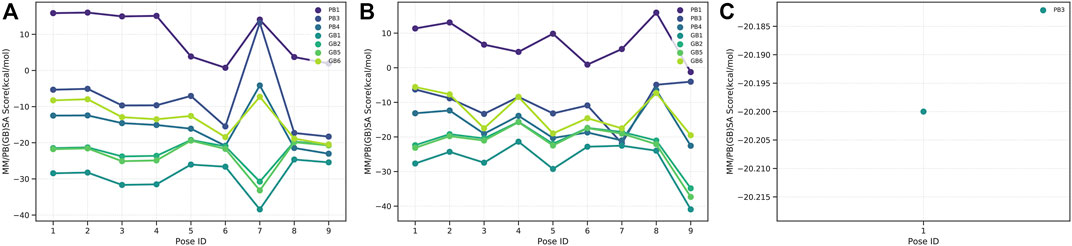
FIGURE 4. MM-PB(BB)SA results of (A) CPN, (B) MMCPNT, and (C) CSN.
In this study, 13 of 36 different secondary metabolites of Caulerpa (caulerpin, caulerpenyne, 10,11-epoxycaulerpenyne, caulersin, flexilin, racemosin C, racemosin B, caulerprenylol B, caulerprenylol A, monomethyl caulerpinate, α-tocospiro A, α-tocospirone and furocaulerpin) against crucial targets (glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase) for colorectal cancer were carried out by using in-silico pharmacokinetic and pharmacodynamic methods. Caulerpin and monomethyl caulerpinate were found to be the most effective metabolites against G6PDH. Caulersin had the lowest affinity score against 6PGD. The pentose phosphate pathway is fundamental for colorectal cancer, thus caulerpin, monomethyl caulerpinate and caulersin play important roles in colorectal cancer treatment through the pentose phosphate pathway.
ADME is an important medicinal chemistry tool that provides information about in-silico ADME behavior (Bocci et al., 2017; Pantaleão et al., 2022). The detailed pharmacokinetic ADME-Tox and drug-likeness results are given in Table 1 and Table 2. The physicochemical properties (molecular weight (g/mol), the number of heavy atoms, the number of aromatic heavy atoms, the number of rotatable bonds, number of H-bond acceptors, the number of H-bond donors, molar refractivity and TPSA (Å)) were calculated for 36 Caulerpa-based phytochemicals. ADME results reveal that the compounds with low molecular weight (≤500 g/mol) tend to absorb well (Daina et al., 2017). In our study, 4 of 36 phytochemicals (sulfoquinovosyldiacyl glycerol, amBiosome, alpha-tocoxylenoxy and caulerpicin) from Caulerpa spp. have high molecular weight (low absorption capacity). The flexibility of bioactive molecules is determined using the number of rotatable bonds (Daina et al., 2017) which should be between 0 and 9. In this study, all the chemicals except sulfoquinovosyldiacyl glycerol and caulerpicin are in the range of this value. Topological polar surface area (TPSA) is based on the fragmental system of phosphorous atoms and polar sulfur. TPSA value should be between 20 and 130 Å for polarity (Daina et al., 2017). In this study, all the 13 phytochemicals were found in this range. Lipophilicity (logP) is crucial for clarifying the effect of chemicals’ absorption, distribution, transportation on physiological systems. In this study, all the samples (36) out of 5 phytochemicals were in the range of logP (-2≤logP≤5). For water solubility, logS (ESOL) was tested. Daina et al. defined the scale of water solubility as insoluble <−10 < poorly <−6 < moderately <−4 < soluble <−2 < very <0 < highly (Daina et al., 2017). The results reveal that caulerpin, caulerpenyne, caulersin, flexilin, racemosin C, racemosin B, monomethyl caulerpinate, and furocaulerpin were found as moderately soluble; 10,11-epoxycaulerpenyne, caulerprenylol B and caulerprenylol A were found as soluble and α-tocospiro A and α-tocospirone were found as poorly soluble.
Before the clinical trials of drug candidates, in-silico toxicity measurement procedure is quite important for better selecting the lead compound (Han et al., 2019). These computational-based toxicity measurement procedures are accurate, accessible, rapid, and common. Both ProTox-II and T.E.S.T servers (freely accessible) were used to identify the adverse effects and toxicity (acute toxicity, carcinogenicity, cytotoxicity, hepatotoxicity, immunotoxicity and mutagenicity) of selected phytochemicals from ADME results. Our results reveal that the toxicity classes of the selected phytochemicals were found to be more than three. For all selected phytochemicals, hepatotoxicity, carcinogenicity, cytotoxicity androgen receptor results were found as inactive.
Drug design and discovery is a step-by-step process, which is costly for companies (Opo et al., 2021). The bioavailability and drug-likeness analysis using in-silico is of great importance for evaluating the pharmacokinetic features of fast and cheap (Hasan et al., 2022).
In this study, the selected metabolites (ligands) have the potential anticancer activity against the selected receptor targets for CRC. According to the results, among the commercial drugs (DHEA and 6ANA), for G6PDH, the best docking energy was exhibited by caulerpin with a vina score of −10.6 kcal/mol, while for G6PDH, the other best docking energy was exhibited by monomethyl caulerpinate with a vina score of −10.5 kcal/mol. For 6PGD, the best docking score among the secondary metabolites of Caulerpa was exhibited by caulersin (−10.7 kcal/mol). (Table 3). In the literature, limited studies are related to the activities of caulerpin on different targets in-silico. In the study by Lorenzo et al. (2015), in-silico molecular docking study of caulerpin and its nine analogs against monoamine oxidase B was carried out. Their results reveal that moldock energy, predicted probability (%) and drug-like score of caulerpin were found as −152%, 58%, and 0.77, respectively. However, the analogs of caulerpin which have non-polar and polar groups showed different moldock energy, predicted probability (%) and drug-like scores. The methods that they used (Volsurf descriptors, structure-based methodology and Random Forest algorithm) are crucial for finding the good drug candidates (caulerpin and its analogs) against monoamine oxidase B (Lorenzo et al., 2015). In the literature, Vitale et al. (2018) carried out an in silico molecular docking evaluation of caulerpin against PPARalpha and PPAR gamma. Their study reveals that the main interaction between ligand (caulerpin) and protein is a hydrophobic interaction. Also, according to their molecular dynamics results, caulerpin makes intermolecular H-bonds with S289 (VI) and S342/R288 (V) (Vitale et al., 2018). Furthermore, antiviral activity of caulerpin against SARS-CoV-2 was tested using in-silico tools (Abdelrheem et al., 2020; Ahmed et al., 2020; Çavaş et al., 2020; El-Mageed et al., 2021).
MM-PBSA analysis is an important and popular method in drug candidate filtration since it is an easy method, and the speed-accuracy balance of the information is high. Estimation of binding free energies with MM-PB(GB) SA for the ligands is automated with the farPPI web server. In our study, the highest and the lowest binding free energies were calculated for monomethyl caulerpinate and caulersin, respectively.
Caulerpin also has some biological activities such as anticancer activity on melanoma cells (Rocha et al., 2007), HIF-1 activation and inhibition of mitochondrial respiration (Liu et al., 2009), antiviral activities against bovine viral diarrhea virus in cattle and herpes simplex virus (Macedo et al., 2012; Pinto et al., 2012; Zhang et al., 2015), pain-sensitizing and spasmolytic effect (Cavalcante-Silva et al., 2014), antituberculosis activity (Canché Chay et al., 2014), antiproliferative activity (Movahhedin et al., 2014), monoamine oxidase inhibitory activity against Alzheimer’s and Parkinson’s disease (Lorenzo et al., 2015), activity on cisplatin-resistant overian cancer and inhibition respiratory complex II activity (Ferramosca et al., 2016), AMPKα1 pathway activation in colorectal cancer cells (Yu et al., 2017) and PPARα and PPARγ agonist activity on hepatocellular cell line (Vitale et al., 2018). Caulersin is a known human protein tyrosine phosphatase-1B inhibitor, which regulates insulin signaling negatively (Yang et al., 2014). The anticancer activity of caulerpin, caulersin, caulerpenyne and 10,11-epoxycaulerpenyne for colorectal cancer data reveal that caulerpin and caulersin are promising anticancer agents against CRC targets and G6PDH and 6PGD could be important targets for CRC.
Inhibition of PPP-enzymes is related with AMPK-activation, HIF-1α degradation, impaired folate metabolism and PP2A-activation (Meskers et al., 2022). G6PDH is the main NADPH production and redox homeostasis contributor (Ghergurovich et al., 2020). The expression level of G6PDH is upregulated and negatively correlated with patients with cancer (Ghergurovich et al., 2020). In different CRC cell lines, the expression levels of G6PDH and 6PGD are quite different. In the study of Polat et al. (2021), the highest and the lowest G6PDH levels were found in HT29 and Caco-2 cell lines, respectively (Polat et al., 2021). Thus, the effectiveness of the selected secondary metabolites could be changed depending on the colorectal cancer cell type.
For the prospects, the not only the in-silico analysis but also the in-vitro experiments of these targets for caulerpin, monomethyl caulerpinate and caulersin should be performed.
Global warming and human activities change the ecosystem structures. Alien members of Genus Caulerpa are widely studied marine algae due to their invasive properties. The present paper proposes an alternative utilization method in medicinal chemistry. The secondary metabolites of Caulerpa spp. attract attention due to their bioactivities. Using the ADME-tox and drug-likeness tests, 13 of 36 secondary metabolites were selected and molecular docking, and molecular dynamics analysis were performed. Caulerpin, monomethyl caulerpinate, and caulersin were found the hit compounds of Caulerpa spp. Against G6PDH and 6PGD, which may play pivotal roles in CRC. Thus, instead of eradication of these algae, the secondary metabolites proposed in this paper (caulerpin, monomethyl caulerpinate) might further be evaluated as promising agents that can be obtained from pharmacy of nature.
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
YB, LC, and HE made significant contributions to the concept or design of the work, NM-O made contributions to the acquisition and analysis data of the study, HE, NO, and GC-K made contributions to the interpretation of data for the work. All authors participated in the drafted and revised the manuscript critically. All authors read and approved the final manuscript. All authors agreed to be responsible for all aspects of the work are appropriately investigated and resolved.
This study was funded by Dokuz Eylül University Scientific Research Projects (Grant number: 2018.KB.SAG.079). NMO is supported by the Council of Higher Education (YOK) 100/2000 Doctoral Project and Scientific and Technological Research Council of Turkey (TUBITAK, 2211-A).
The authors would like to thank Caner Karaca from Dokuz Eylül University for improvement of qualities of figures in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelrheem, D. A., Ahmed, S. A., Abd El-Mageed, H. R., Mohamed, H. S., Rahman, A. A., Elsayed, K. N. M., et al. (2020). The inhibitory effect of some natural bioactive compounds against SARS-CoV-2 main protease: insights from molecular docking analysis and molecular dynamic simulation. Journal of Environmental Science and Health Part A 55, 1373–1386. doi:10.1080/10934529.2020.1826192
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1 (2), 19–25. doi:10.1016/j.softx.2015.06.001
Aguilar-Santos, G. (1970). Caulerpin, a New Red Pigment from Green Algae of the Genus Caulerpa. J. Chem. Soc. Perkin 1, 6, 842-843. doi:10.1039/J39700000842
Ahmed, S. A., Abdelrheem, D. A., El-Mageed, H. R. A., Mohamed, H. S., Rahman, A. A., Elsayed, K. N. M., et al. (2020). Destabilizing the structural integrity of COVID-19 by caulerpin and its derivatives along with some antiviral drugs: An in silico approaches for a combination therapy. Struct. Chem. 31, 2391–2412. doi:10.1007/s11224-020-01586-w
Banerjee, P., Eckert, A. O., Schrey, A. K., and Preissner, R. (2018). ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46, W257–W263. doi:10.1093/nar/gky318
Bocci, G., Carosati, E., Vayer, P., Arrault, A., Lozano, S., and Cruciani, G. (2017). ADME-Space: A new tool for medicinal chemists to explore ADME properties. Sci. Rep. 7, 6359–27. doi:10.1038/s41598-017-06692-0
Canché Chay, C. I., Cansino, R. G., Espitia Pinzón, C. I., Torres-Ochoa, R. O., and Martínez, R. (2014). Synthesis and anti-tuberculosis activity of the marine natural product caulerpin and its analogues. Mar. Drugs 12, 1757–1772. doi:10.3390/md12041757
Cavalcante-Silva, L. H. A., Falcão, M. A. P., Vieira, A. C. S., Viana, M. D. M., De Araújo-Júnior, J. X., Sousa, J. C. F., et al. (2014). Assessment of mechanisms involved in antinociception produced by the alkaloid caulerpine. Molecules 19, 14699–14709. doi:10.3390/molecules190914699
Cavas, L., Baskin, Y., Yurdakoc, K., and Olgun, N. (2006). Antiproliferative and newly attributed apoptotic activities from an invasive marine alga: Caulerpa racemosa var. cylindracea. J. Exp. Mar. Biol. Ecol. 339, 111–119. doi:10.1016/j.jembe.2006.07.019
Çavaş, L., Dag, C., Carmena-barreño, M., Martínez-cortés, C., Pedro, J., and Pérez-sánchez, H. (2020). Secondary Metabolites from Caulerpa cylindracea ( Sonder ) Could Be Alternative Natural Antiviral Compounds for COVID-19: A Further in Silico Proof. doi:10.26434/chemrxiv.13353473.v1
Cengiz, S., Cavas, L., Yurdakoc, K., and Pohnert, G. (2011). The Sesquiterpene Caulerpenyne from Caulerpa spp. is a Lipoxygenase Inhibitor. Mar. Biotechnol. 13, 321–326. doi:10.1007/s10126-010-9303-1
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717–13. doi:10.1038/srep42717
Dege, N., Gökce, H., Doğan, O. E., Alpaslan, G., Ağar, T., Muthu, S., et al. (2022). Quantum computational, spectroscopic investigations on N-(2-((2-chloro-4, 5-dicyanophenyl)amino)ethyl)-4-methylbenzenesulfonamide by DFT/TD-DFT with different solvents, molecular docking and drug-likeness researches. Colloids and Surfaces A: Physicochemical and Engineering Aspects 638, 128311. doi:10.1016/j.colsurfa.2022.128311
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M., and Wallace, M. B. (2019). Colorectal cancer. The Lancet 394, 1467–1480. doi:10.1016/S0140-6736(19)32319-0
El-Mageed, H. R. A., Abdelrheem, D. A., Ahmed, S. A., Rahman, A. A., Elsayed, K. N. M., Ahmed, S. A., et al. (2021). Combination and tricombination therapy to destabilize the structural integrity of COVID-19 by some bioactive compounds with antiviral drugs: insights from molecular docking study. Struct. Chem. 32, 1415–1430. doi:10.1007/s11224-020-01723-5
Erb, M., and Kliebenstein, D. J. (2020). Plant Secondary Metabolites as Defenses, Regulators, and Primary Metabolites: The Blurred Functional Trichotomy. Plant Physiol. 184, 39–52. doi:10.1104/PP.20.00433
Ferramosca, A., Conte, A., Guerra, F., Felline, S., Rimoli, M. G., Mollo, E., et al. (2016). Metabolites from invasive pests inhibit mitochondrial complex II: A potential strategy for the treatment of human ovarian carcinoma? Biochem. Biophys. Res. Commun. 473, 1133–1138. doi:10.1016/j.bbrc.2016.04.028
Gao, W., Xu, Y., Chen, T., Du, Z., Liu, X., Hu, Z., et al. (2019). Targeting oxidative pentose phosphate pathway prevents recurrence in mutant Kras colorectal carcinomas. PLos Biol. 17, e3000425. doi:10.1371/journal.pbio.3000425
Ghergurovich, J. M., Esposito, M., Chen, Z., Wang, J. Z., Bhatt, V., Lan, T., et al. (2020). Glucose-6-phosphate dehydrogenase is not essential for K-Ras-driven tumor growth or metastasis. Cancer Res. 80, 3820–3829. doi:10.1158/0008-5472.can-19-2486
Gokce, H., Sen, F., Sert, Y., Abdel-Wahab, B. F., Kariuki, B. M., and El-Hiti, G. A. (2022). Quantum Computational Investigation of (E)-1-(4-methoxyphenyl)-5-methyl-N′-(3-phenoxybenzylidene)-1H-1, 2, 3-triazole-4-carbohydrazide. Molecules 27, 2193. doi:10.3390/molecules27072193
Han, Y., Zhang, J., Hu, C. Q., Zhang, X., Ma, B., and Zhang, P. (2019). In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 10, 434–12. doi:10.3389/fphar.2019.00434
Hasan, R., Alsaiari, A. A., Fakhurji, B. Z., Habibur, M., Molla, R., Asseri, A. H., et al. (2022). Application of Mathematical Modeling and Computational Tools in the Modern Drug Design and Development Process. Molecules 27, 4169. doi:10.3390/molecules27134169
Jin, L., and Zhou, Y. (2019). Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 17, 4213–4221. doi:10.3892/ol.2019.10112
Koes, D. R., and Camacho, C. J. (2012). ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 40, W409–W414. doi:10.1093/nar/gks378
Liu, Y., Morgan, J. B., Coothankandaswamy, V., Liu, R., Jekabsons, M. B., Mahdi, F., et al. (2009). The Caulerpa pigment caulerpin inhibits HIF-1 activation and mitochondrial respiration. J. Nat. Prod. 72, 2104–2109. doi:10.1021/np9005794
Lorenzo, V. P., Filho, J. M. B., Scotti, L., and Scotti, M. T. (2015). Combined structure- and ligand-based virtual screening to evaluate caulerpin analogs with potential inhibitory activity against monoamine oxidase B. Rev. Bras. Farmacogn. 25, 690–697. doi:10.1016/j.bjp.2015.08.005
Macedo, N. R. P. V., Ribeiro, M. S., Villaça, R. C., Ferreira, W., Pinto, A. M., Teixeira, V. L., et al. (2012). Caulerpin as a potential antiviral drug against herpes simplex virus type 1. Rev. bras. farmacogn. 22, 861–867. doi:10.1590/S0102-695X2012005000072
Martin, T. M., Harten, P., Venkatapathy, R., Das, S., and Young, D. M. (2008). A hierarchical clustering methodology for the estimation of toxicity. Toxicol. Mech. Methods 18, 251–266. doi:10.1080/15376510701857353
Mehra, R., Bhushan, S., Bast, F., and Singh, S. (2019). Marine macroalga Caulerpa: role of its metabolites in modulating cancer signaling. Mol. Biol. Rep. 46, 3545–3555. doi:10.1007/s11033-019-04743-5
Mert Ozupek, N., and Cavas, L. (2017). Triterpene glycosides associated antifouling activity from Holothuria tubulosa and H. polii. Reg. Stud. Mar. Sci. 13, 32–41. doi:10.1016/j.rsma.2017.04.003
Meskers, C. J. W., Franczak, M., Smolenski, R. T., Giovannetti, E., and Peters, G. J. (2022). Are we still on the right path(way)?: the altered expression of the pentose phosphate pathway in solid tumors and the potential of its inhibition in combination therapy. Expert Opin. Drug Metab. Toxicol. 18, 61–83. doi:10.1080/17425255.2022.2049234
Montefalcone, M., Morri, C., Parravicini, V., and Bianchi, C. N. (2015). A tale of two invaders: divergent spreading kinetics of the alien green algae Caulerpa taxifolia and Caulerpa cylindracea. Biol. Invasions 17, 2717–2728. doi:10.1007/s10530-015-0908-1
Movahhedin, N., Barar, J., Azad, F. F., Barzegari, A., and Nazemiyeh, H. (2014). Phytochemistry and biologic activities of caulerpa peltata native to Oman sea. Iran. J. Pharm. Res. 13, 515–521.
O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open Babel: An open chemical toolbox. J. Cheminform. 3, 33–14. Available at: https://jcheminf.biomedcentral.com/track/pdf/10.1186/1758-2946-3-33. doi:10.1186/1758-2946-3-33
Opo, F. A. D., Rahman, M. M., Ahammad, F., Ahmed, I., Bhuiyan, M. A., and Asiri, A. M. (2021). Structure based pharmacophore modeling , virtual screening , molecular docking and ADMET approaches for identification of natural anti - cancer agents targeting XIAP protein. Sci. Rep. 11, 4049–18. doi:10.1038/s41598-021-83626-x
Pantaleão, S. Q., Fernandes, P. O., Gonçalves, J. E., Maltarollo, V. G., and Honorio, K. M. (2022). Recent Advances in the Prediction of Pharmacokinetics Properties in Drug Design Studies: A Review. ChemMedChem 17, e202100542. doi:10.1002/cmdc.202100542
Patra, K. C., and Hay, N. (2014). The pentose phosphate pathway and cancer. Trends Biochem. Sci. 39, 347–354. doi:10.1016/j.tibs.2014.06.005
Pinto, A. M. V., Leite, J. P. G., Ferreira, W. J., Cavalcanti, D. N., Villaça, R. C., Giongo, V., et al. (2012). Marine natural seaweed products as potential antiviral drugs against bovine viral diarrhea virus. Rev. bras. farmacogn. 22, 813–817. doi:10.1590/S0102-695X2012005000060
Polat, I. H., Tarrado-Castellarnau, M., Benito, A., Hernandez-Carro, C., Centelles, J., Marin, S., et al. (2021). Glutamine modulates expression and function of glucose 6-phosphate dehydrogenase via nrf2 in colon cancer cells. Antioxidants 10, 1349. doi:10.3390/antiox10091349
Ramawat, K. G., and Goyal, S. (2020). Co-evolution of Secondary Metabolites During Biological Competition for Survival and Advantage: An Overview. Ref. Ser. Phytochem., 3–17. doi:10.1007/978-3-319-96397-6_45
Richter, P., Schubert, G., Schaible, A. M., Cavas, L., Werz, O., and Pohnert, G. (2014). Caulerpenyne and related bis-enol esters are novel-type inhibitors of human 5-lipoxygenase. ChemMedChem 9, 1655–1659. doi:10.1002/cmdc.201402065
Rocha, F. D., Soares, A. R., Houghton, P. J., Pereira, R. C., Kaplan, M. A. C., and Teixeira, V. L. (2007). Potential Cytotoxic Activity of Some Brazilian Seaweeds on Human Melanoma Cells. Phytother. Res. 21, 170–175. doi:10.1002/ptr.2038
Schneidman-Duhovny, D., Dror, O., Inbar, Y., Nussinov, R., and Wolfson, H. J. (2008). PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 36, 223–228. doi:10.1093/nar/gkn187
Schüttelkopf, A. W., and Van Aalten, D. M. F. (2004). PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363. doi:10.1107/S0907444904011679
Stincone, A., Prigione, A., Cramer, T., Wamelink, M. M. C., Campbell, K., Cheung, E., et al. (2015). The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 90, 927–963. doi:10.1111/brv.12140
Su, J. Y., Zhu, Y., Zeng, L. M., and Xu, X. H. (1997). A new bisindole from alga Caulerpa serrulata. J. Nat. Prod. 60, 1043–1044. doi:10.1021/np970149x
Trott, O., and Olson, A. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. doi:10.1002/jcc.21334
Vitale, R. M., D’aniello, E., Gorbi, S., Martella, A., Silvestri, C., Giuliani, M. E., et al. (2018). Fishing for targets of alien metabolites: A novel peroxisome proliferator-activated receptor (PPAR) agonist from a marine pest. Mar. Drugs 16, 431–446. doi:10.3390/md16110431
Wang, Z., Wang, X., Li, Y., Lei, T., Wang, E., Li, D., et al. (2019). FarPPI: A webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics 35, 1777–1779. doi:10.1093/bioinformatics/bty879
Yang, H., Liu, D. Q., Liang, T. J., Li, J., Liu, A. H., Yang, P., et al. (2014). Racemosin C, a novel minor bisindole alkaloid with protein tyrosine phosphatase-1B inhibitory activity from the green alga Caulerpa racemosa. J. Asian Nat. Prod. Res. 16, 1158–1165. doi:10.1080/10286020.2014.965162
Yu, H., Zhang, H., Dong, M., Wu, Z., Shen, Z., Xie, Y., et al. (2017). Metabolic reprogramming and AMPKα1 pathway activation by caulerpin in colorectal cancer cells. Int. J. Oncol. 50, 161–172. doi:10.3892/ijo.2016.3794
Keywords: Caulerpa, colorectal cancer, in silico, pentose phosphate pathway, oncoinformatics
Citation: Mert-Ozupek N, Calibasi-Kocal G, Olgun N, Basbinar Y, Cavas L and Ellidokuz H (2022) In-silico molecular interactions among the secondary metabolites of Caulerpa spp. and colorectal cancer targets. Front. Chem. 10:1046313. doi: 10.3389/fchem.2022.1046313
Received: 16 September 2022; Accepted: 22 November 2022;
Published: 06 December 2022.
Edited by:
Sibhghatulla Shaikh, Yeungnam University, South KoreaCopyright © 2022 Mert-Ozupek, Calibasi-Kocal, Olgun, Basbinar, Cavas and Ellidokuz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hulya Ellidokuz, aHVseWEuZWxsaWRva3V6QGRldS5lZHUudHI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.