 Kai Wei
Kai Wei Xinhua Zheng
Xinhua Zheng Hongbin Zhang
Hongbin Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 12 December 2022
Sec. Medicinal and Pharmaceutical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1030541
This article is part of the Research Topic Advances in Natural Product Chemistry: Yunnan University 100th Anniversary View all 11 articles
Dioxinone derivatives, a class of acetoacetate derivatives, have attracted widespread attention because of their multiple reactive sites, high reactivity, unique chemical properties, and potential synthetic applications. The dioxinone group is also stable under a wide range of reaction conditions, including strong acids, as well as a variety of transition-metal-catalysed processes, such as olefin metathesis and Pd-mediated cross-coupling. The inherent reactivity and diverse applications of dioxinones make them valuable reactive intermediates in organic synthesis. The conversion of dioxinones to acylketenes and their subsequent nucleophilic capture is also an excellent strategy for synthesising β-keto acid derivatives, which can be applied even in complex molecular synthesis. This review focuses on the recent advances in the application of dioxinones in synthetic method research and the total synthesis of natural products, highlighting the exceptional utility of these synthetic methodologies in the construction of macrocyclic cores and terpenoid skeletons. In particular, successful transformations of dioxinone fragments are discussed.
Chemistry has developed rapidly since its inception as a field of study in the 17th century. Chemists have made outstanding progress in the development of new reagents, reactions, and strategies for selectively and efficiently transforming organic compounds. These advances have been so profound that many highly complex natural products have been obtained through chemical synthesis. The development and utilisation of new reagents remain an area of intense interest in organic chemistry to maximise efficiency and practicability in total synthesis.
Among the numerous reagents used in organic chemistry, dioxinone derivatives have received considerable attention from organic chemists because of their multiple reactive sites, high reactivity, unique chemical properties, and potential synthetic applications. Moreover, dioxinone derivatives are increasingly being applied across a variety of research areas, including agrochemical development, natural product synthesis, and as chemical tools for a wide range of biological investigations.
Several factors have contributed to the popularity of dioxinone derivatives. Compound 1 is practical to use because it is inexpensive to prepare on a large scale using readily available commercial raw materials. Indeed, many chemical suppliers currently market 1 at reasonable prices ($ 290/kg for bulk quantities). More importantly, the steps used to synthesize various organic synthetic building blocks from 1 are typically robust, straightforward, and broad in scope.
Various innovative methods have been developed for the preparation of dioxinone derivatives. Currently, two highly practical procedures (Scheme 1) are used by a large majority of chemical suppliers to produce 1. The first method is a one-pot procedure using tert-butyl acetoacetate derivatives 6 as the starting materials in the presence of concentrated sulfuric acid, acetic anhydride, and acetone to obtain the dioxinone derivatives (route A) (Fuse et al., 2014). The other method uses meldrum’s acid and acid chloride 7 by a practical two-step procedure (route B) (Aoki et al., 2015).
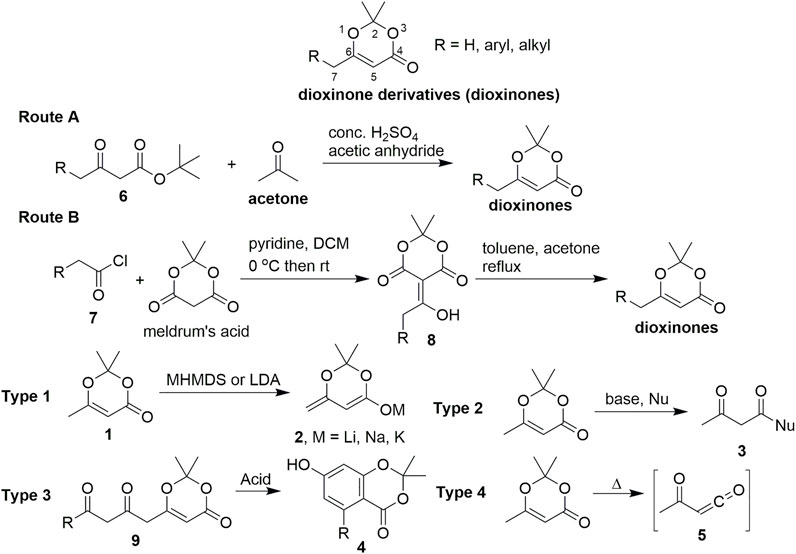
SCHEME 1. General sequence for the synthesis of dioxinone derivatives and their reactivity.
Dioxinone derivatives first entered the purview of chemists as organic synthetic building blocks in the 1950s and have gradually become widely used. In 1989, Winkler reported the asymmetric synthesis of perhydrohistrionicotoxin (Winkler Harshberger, 1989), which was the first application of dioxinone in the total synthesis of a natural product. Dioxinone derivatives demonstrate multiple chemical properties because they can be considered a product of acetoacetate protected by acetone. There are several distinct types of this chemistry (using compound 1 as an example in Scheme 1): 1) The direct reaction of C-7 in 1 with a wide range of bases (e.g., MHMDS or LDA) proceeds to give 2 or enol silyl ethers (Scheme 1, type 1) in high yields. Kalesse reviewed the application of dioxinones in vinylogous aldol reactions in 2005 (Kalesse, 2005). 2) Compound 1 enables clean and high-yield additions of a very wide range of diverse nucleophiles, including organo-magnesium, lithium, and zinc reagents; stabilised carbanions exemplified by enolates; and numerous hydride reagents (Scheme 1, type 2). Compound 1 can also be converted to an acylketene reactive intermediate 5) under high-temperature conditions, exhibiting rich chemistry (Scheme 1, type 4). Reaction types 2 and 4, which provide direct access to β-keto lactones and β-keto lactams, respectively, have been effectively utilised in complex, target-directed synthesis. Intermolecular or intramolecular trapping of reactive acylketenes by nucleophiles gives rise to valuable structures and enables the execution of challenging and delicate bond formations that might be difficult to achieve using alternative synthetic strategies. Sorensen reviewed the application of dioxinones to bond formation by intermolecular and intramolecular trappings of acylketenes in the total synthesis of large-ring natural products in 2009 (Reber et al., 2009). 3) Moreover, a cyclisation–aromatisation cascade process can be accomplished by an acid-catalysed reaction with the dioxinone derivatives 9, which serves as an electron-rich reagent widely used in the synthesis of resorcylate natural products (Scheme 1, type 3).
This review aims to highlight recent strategic applications of dioxinone derivatives in natural product synthesis that were not covered in previous reviews (Kalesse, 2005; Reber et al., 2009), and emphasise the significant role they play in generating molecular skeletons. Only representative examples in which dioxinones are used as a crucial step in the construction of either the core structure or the key structural motif of the target molecule are presented. General applications of the selected examples of total synthesis are grouped based on the natural product types, including macrocyclic natural products, terpenoids, and some applications in research on synthetic methods, with a particular focus on studies from the last decade.
The most common application of acylketenes in organic synthesis is the preparation of β-keto acid derivatives for construction of macrocyclic natural product frameworks, which are widely used in the synthesis of macrolides and macrolactams. Boeckman and Pruitt were the first to use dioxinones as precursors to acylketenes in the synthesis of complex natural products featuring macrolactones and macrolactams, as reviewed by Sorensen (Reber et al., 2009). Here, we briefly introduce other studies. In this section, select recent examples are presented to illustrate contemporary solutions to problems involving dioxinones.
In 2009, Scheidt and coworkers reported the successful synthesis of neopeltolide (Scheme 2, Custar et al., 2008, 2009). Dioxinone 1 was used as an important functional block to construct intermediate 11 via a vinylogous aldol reaction. After condensation, deprotection of TBS, and oxidation, key intermediate 12 was obtained. Macrocyclic 13 was successfully acquired in 25% yield by cyclisation via an intramolecular Prins reaction promoted by Sc(OTf)3.
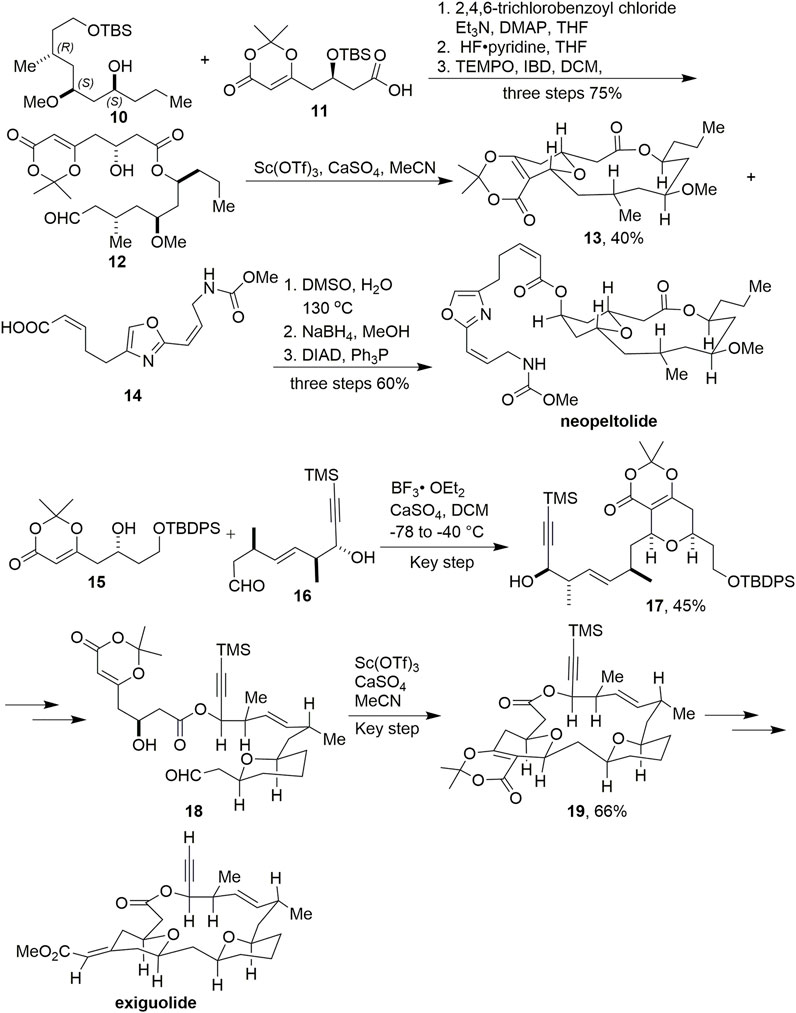
SCHEME 2. Scheidt’s synthesis of neopeltolide and exiguolide.
In 2011, This group constructed the exiguolide skeleton using the same strategy via two Prins cyclisations and ultimately completed the total synthesis of exiguolide (Scheme 2, Crane et al., 2011). The first Prins reaction was successfully mediated by BF3·Et2O with compounds 15 and 16 to produce 17 in 45% yield. After multiple transformations, the second key Prins reaction was promoted by Sc(OTf)3 to construct macrocyclic intermediate 19 in 66% yield.
Dioxinone was also used as an important synthetic block to provide the skeleton in the synthesis of okilactomycin by Scheidts’ group (Scheme 3, Tenenbaum et al., 2011). Using copper-catalyzed vinylogous aldol reaction conditions with dioxinone silyl enol ether and aldehyde 20, β-hydroxy dioxinone 21 was formed in 70% yield and 10:1 diastereomeric ratio favoring the desired product. After multistep transformations to obtain 22, treatment of it with KOEt smoothly provided a β-ketoester, where the protecting group was removed with HFpy to afford 23 without any observed lactonization. The conditions of the stereoselective coupling of 23 and 24 were TMSOTf in DCM, and led to the desired 25 in 60% yield and as a 13:1 mixture of diastereomers favoring the desired 2,6-cis isomer. Ultimately, (–)-okilactomycin has been achieved successfully.
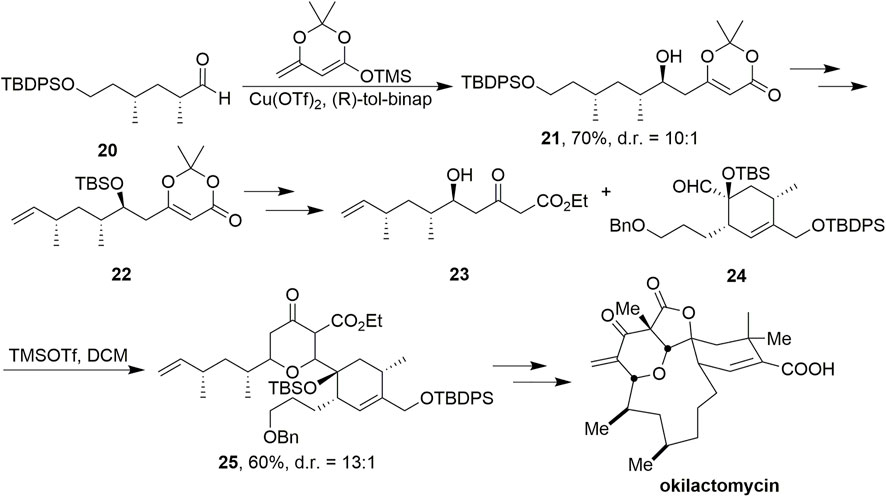
SCHEME 3. Scheidt’s synthesis of okilactomycin.
Callipeltoside is a popular target for total synthesis because of its complex architecture and promising anti-tumour bioactivity (Zampella et al., 1996). Like the related compounds lyngbouilloside and lyngbyaloside, these two natural products feature a 14-membered macrolactone with a transannular hemiketal. In 2010, Hoye et al. (2010) reported the asymmetric total synthesis of the macrolide natural product callipeltoside A (Scheme 4). After a vinylogous aldol reaction with 26, acylketene macrolactonisation took place with a high degree of regioselectivity. Acylketene precursor 27 contains two unprotected hydroxyl groups, yet only the single constitutional isomer 28 was observed after heating this substrate in refluxing benzene.
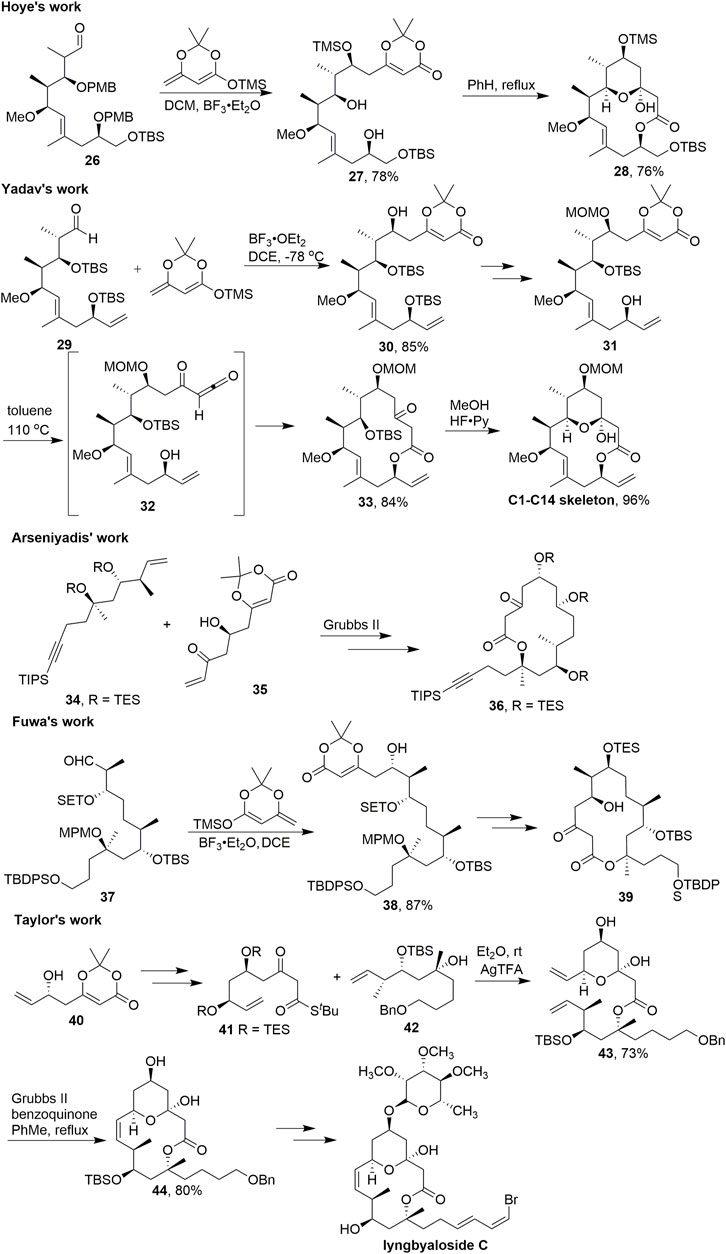
SCHEME 4. Synthesis of a 14-membered macrolactone central macrolide.
In 2012, Yadav et al. (2012) used almost the same strategy to build the skeleton in the total synthesis of callipeltoside A (Scheme 4). By employing a diastereoselective aldol addition from the C5–C14 aldehyde segment 29 and dienyl silyl ether, afforded the adduct 30 as the only product in 85% yield. After several steps, compound 31 was received in good yield. Thus, refluxing of a dilute solution of 31 in toluene induced the loss of acetone through thermal decomposition to evolve the acylketene intermediate 32, which was then trapped intramolecularly by the secondary hydroxy at C13 to generate the 14-membered lactone 33 in 84% yield. The final synthetic operation was carried out by using HF·Py to transform 33 to the C1–C14 skeleton of (–) callipeltoside A by removing the silyl group to form the requisite tetrahydropyran ring.
Similar strategies targeting the central macrolide were reported by Arseniyadis (ElMarrouni et al. 2011), Fuwa (Fuwa et al., 2015), and Taylor (Chang et al., 2015) (Scheme 4). Arseniyadis successfully used C–C bond formation via olefin metathesis with key intermediates 34 and 35; 36 was obtained by heating the dioxinone intermediate. Vinylogous aldol reaction was used to construct the key intermediate 38 to accomplish the subsequent 14-membered macrocyclic 39 in Fuwa’s study. Taylor and coworkers prepared 41 from 40 to obtain key intermediate 43 using AgTFA; Then 43 served as the precursor for olefin metathesis. After the 14-membered macrocyclic 44 was obtained by olefin metathesis cyclization, lyngbyaloside C was synthesized multi-step functional group transformation.
In 2008, Barrett’s group developed a new route (Navarro et al., 2008) to resorcylate natural products which was inspired by biosynthetic considerations and was based on macrocyclisation and transannular aromatisation of the dioxinone fragment (Scheme 5). The lactones 48 containing these units utilizing tandem late stage aromatization was obtained from 2,4,6-triketo-ester precursors 47, which was prepared with the dioxinone derivatives 45 by efficient trapping of the resultant ketene 46 with alcohols.
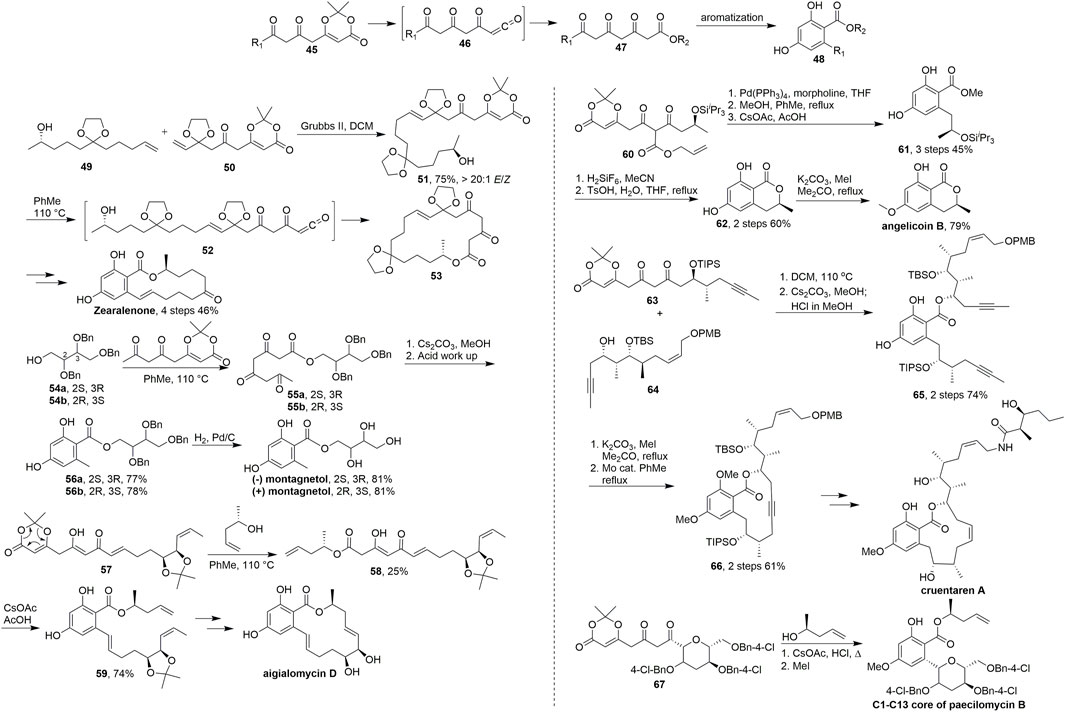
SCHEME 5. Barrett’s synthesis of resorcylate natural products.
In their research (Miyatake-ondozabal and Barrett. 2010), key advanced intermediate 51 was obtained in 75% yield (E/Z > 20/1) via olefin metathesis using a Grubbs II catalyst. Then, 51 was converted into acyl ketene intermediate 52 under refluxing toluene, and intramolecular hydroxyl capture led to the formation of 18-membered macrocyclic lactone 53. The synthesis was completed via multistep transformations and transannular aromatisation. Barrett et al. have used this strategy from 2010 to 2018 in some brilliant studies on the synthesis of resorcylate type natural products.
Thermolysis of dioxinone in the presence of benzyl-protected erithritols 54a and 54b gave the triketo-esters 55a and 55b, respectively. Cyclisation and aromatisation followed by hydrogenolysis of the benzyl groups gave (+)-montagnetol. (+)-Erythrin also could be prepared based on the same route in this work (Scheme 5, Basset et al., 2010). Upon heating in toluene, the dioxinones 57 was trapped with chiral alcohol to generate the ketene 58 and directly aromatized by reaction with cesium acetate followed by acetic acid to give the resorcylates 59 respectively in 74% overall yields. After the key RCM and deprotection of the acetonide moiety, aigialomycin D was achieved successfully (Scheme 5, Calo et al., 2009). In 2011, Barrett and coworkers (Scheme 5, Anderson et al., 2011) used one pot palladium(0)-catalyzed deallyation-decarboxylation-ketene trapping-aromatization to give the desired resorcylate 61 in 45% yield over three steps. Deprotection of the silyl ether and acid catalyzed cyclisation gave lactone 62 in 60% yield over two steps. Finally, regioselective methylation of 62 provided angelicoin B. The core resorcylate unit 65 of cruentaren A was synthesized by thermolysis of diketo-dioxinone 63 afforded the corresponding highly reactive ketene, which was trapped with 64 in 74% yield over two steps. After the ring closing by alkyne metathesis and methylation, the key intermediate 66 was obtained and finally cruentaren A was achieved in several steps from 66 (Scheme 5, Fouché et al., 2012). The C1 to C13 tetrahydropyranyl-resorcylate core of paecilomycin B was also synthesized by this strategy in 2018 (Scheme 5, Cookson et al., 2018).
In addition to its usefulness for macrolide synthesis, dioxinone is also an excellent fragment for the nucleophilic addition of amines to form macrolactams. In 2016, Kalesse reported the synthesis of aetheramide A, which is a highly potent anti-HIV reagent (Scheme 6, Gerstmann and Kalesse, 2016). In their research, to access precursor 72, dioxinone 69 was used as a Horner–Wadsworth–Emmons (HWE) resource in an HWE reaction. The dioxinone moiety was introduced through this olefination, and the final TBS deprotection step completed the synthesis of polyketidic fragment 70 in 67% yield. After condensation with acid 71, mesitylene led to the formation of the acylketene intermediates, which were trapped intramolecularly by the secondary amine. Deprotection was then carried out using HF-pyridine to give aetheramide A. This synthesis is particularly interesting because although there are various well-established procedures for macrolactamisations using unsubstituted dioxinones, examples with dioxinones bearing a methyl group are rare, possibly because of the considerably higher temperatures necessary to initiate the retro Diels–Alder reaction.
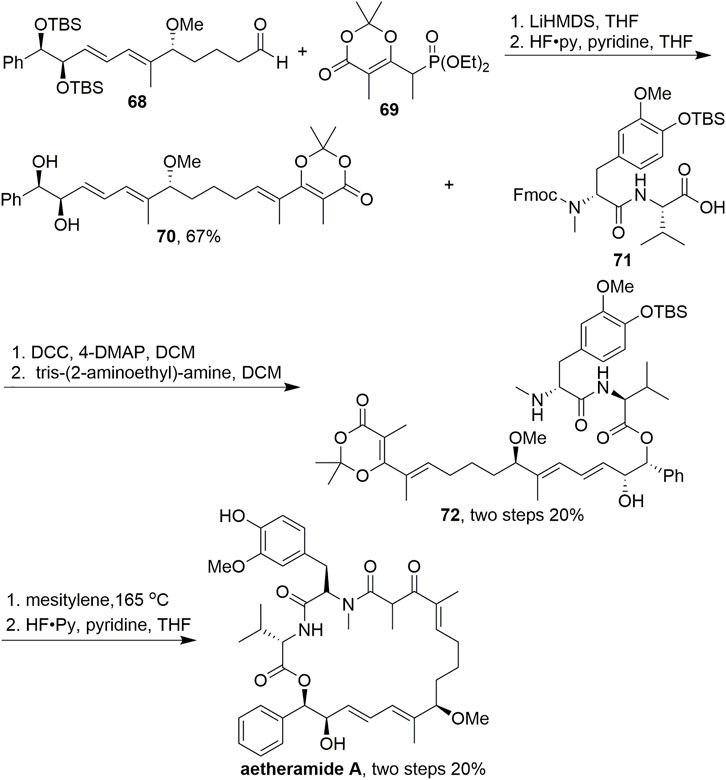
SCHEME 6. Kalesse’s synthesis of aetheramide A.
Captures of acylketenes are particularly useful in a synthetic context because sometimes medium and large rings can be formed, which are often difficult to synthesise using more typical methods. The diverse reactivities and applications of dioxinones make them valuable reactive synthetic blocks for macrocyclic natural products. Many similar synthetic studies have been reported (Rentsch and Kalesse, 2012; Dalby et al., 2013; Ogura et al., 2016), which will not be repeated here, in addition to those described earlier.
Terpenoids are structurally intriguing natural products that have attracted extensive attention owing to their unique and complex structural characteristics and diverse biological activities. Dioxinone derivatives also play an important role in the total synthesis of some complex terpenoids.
In 2006, Narasaka et al. reported the asymmetric synthesis of sordarin and sordaricin (Scheme 7, Chiba et al., 2006). Sordarin has a tetracyclic cage-like structure with a glycosidic moiety which makes it particularly challenging to synthesize. They started with optically active 73, which could prepare 74 in five steps. Dioxinone 75 was used as an alkylation reagent and added dropwise to a mixture of 74 and LDA. After cleavage of N,N-dimethylhydrazone, the resulting ketone 76 was treated with sodium ethoxide in ethanol to give tricyclic keto ester 77 via deprotection of the acetonide group and subsequent condensation. Sordaricin was then obtained through multistep transformations, and glycosylation of sordaricin completed the synthesis of sordarin.
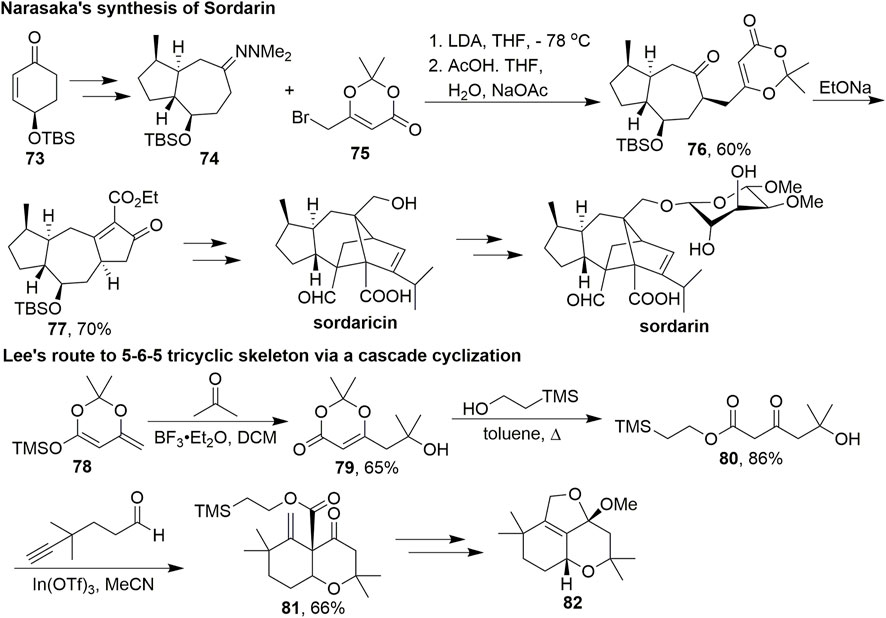
SCHEME 7. Applications of dioxinones in Narasaka and Lee’s studies.
In 2011, Lee’s group developed a novel route to construct the 5-6-5 tricyclic furanochroman skeleton of phomactin A via a Prins/Conia-ene cascade cyclisation (Scheme 7, Huang eta al., 2011). Phomactins represent a new class of platelet-activating factor (PAF) antagonists isolated from the marine fungus Phoma sp. and inhibit PAF-induced platelet aggregation (Sugano et al., 1991, 1996). Dioxinone 79 was obtained via a vinylogous aldol reaction between 78 and acetone. The product was then heated in toluene with the appropriate alcohol to give β-ketoester 80. Subsequently, a variety of factors of this cascade approach, including Lewis acids, solvents, and temperature, were examined. This multifunctional intermediate successfully underwent a Prins/Conia-ene cascade cyclisation with the alkynaldehyde facilitated by In(OTf)3 to give 81 in 66% yield. The desired tricyclic skeleton was obtained after several subsequent steps.
In 2014, Barrett and Barrett (2014) reported the total synthesis of the complex natural product hongoquercin B, which has been isolated from extracts of an unidentified terrestrial fungus and exhibits antibiotic activity against vancomycin-resistant Enterococcus faecium and methicillin-resistant Staphylococcus aureus (Scheme 8, Roll et al., 1998; Ma et al., 2018). The key advanced intermediate 84 was formed via a decarboxylation allylation tandem aromatisation of 83. Treatment of ester 83 with Pd(PPh3)4 at room temperature gave a diketo-dioxinone intermediate, which was readily aromatised over silica gel to give resorcylate 84 in 66% yield over three steps. Notably, only the desired linear E,E-isomer was obtained in this sequence. Subsequent addition of BF3·Et2O in dichloromethane furnished pentacyclic skeleton 85 in 60% yield by cascade cyclisation. The total synthesis of hongoquercin B was completed in two steps through functional group modifications.
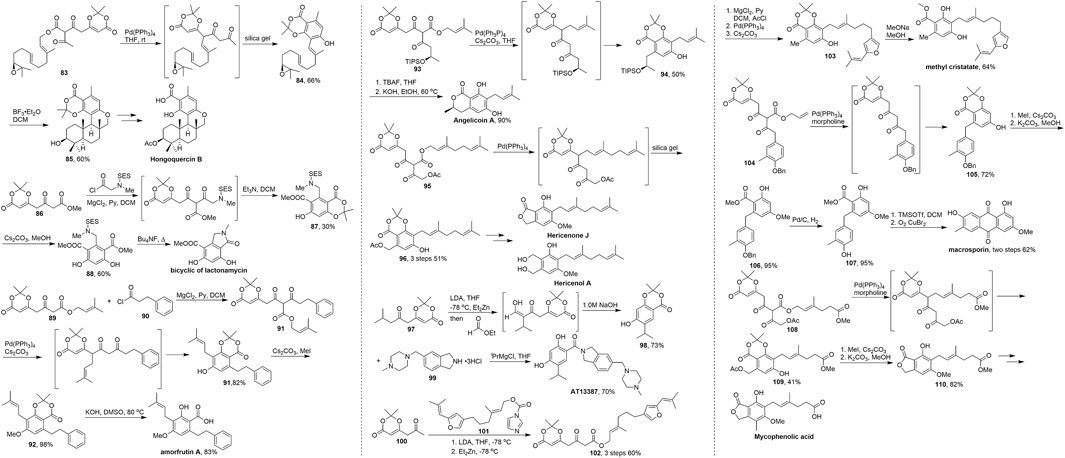
SCHEME 8. Barrett’s synthesis of terpene resorcylates and dihydroxy-isoindolinone derivatives.
Since the first example reported by Barrett in 2011, more than ten molecules have been synthesized using this approach via the cascade cyclisation-aromatization, which is an efficient and concise method for constructing dioxinone-resorcylate especially the terpene resorcylates and dihydroxy-isoindolinone derivatives. Barrett’s works (Scheme 8) were carried out using dioxinone derivatives as an important synthetic building block to achieve the diketo-dioxinone ester, palladium catalyzed migratory, decarboxylative prenylation–aromatization sequence as the key cascade process to establish the core frameworks of natural products with the mild conditions, easy work-up, wide scopes and high yield.
In 2011, they developed a concise five-step synthesis to the E, F-ring system of lactonamycin (Jacques et al., 2011). Dioxinone ketoester 89 and chloride 90 were used to provide the key in intermediate. Then subsequent reaction of diketoester–dioxinone 91 with Pd(PPh3)4 and cesium carbonate resulted in decarboxylative prenyl migration and formation of the resorcylate 92. Amorfrutin A was obtained after the phenol methylation and saponification (Laclef et al., 2012). In 2012, Barrett and coworkers used different dioxinone-diketoesters 93 and 95 to provide the corresponding products 94 and 96 of the tandem process in good yields over several steps. Finally, they accomplished three terpene resorcylates angelicoin A, hericenone J and hericenol A in just five steps (Cordes et al., 2012). In 2012, Patel and Barrett (2012) completed the total synthesis of a Hsp90 nnhibitor AT13387. They started with ketoester 97 and gave the dioxinone-resorcylate 98 in 73% yield and obtained AT13387 after the saponification and condensation in total 3 steps from 97. Barrett’s group finished the total synthesis of macrosporin (Cordes and Barrett, 2013), mycophenolic acid (Brookes et al., 2013) and methyl cristatate (George et al., 2013) utilizing this tandem strategy in 2013. In general, this conversion strategy has great advantages in the synthesis of natural products containing aryl-phenol structures and provides a new convenient route for this kind of natural product.
In 2015, Zhao and Maimone (2015) used dioxinone as a key building block to construct key intermediate 114 in the asymmetric total synthesis of chatancin (Scheme 9). A vinylogous adol reaction was employed to convert aldehyde 111 and an enol ether into a secondary alcohol, followed by treatment with DMP to furnish dioxinone 112. After heating in toluene and the intramolecular capture of acylketenes, treatment of the product with Tf2O successfully gave 113. Pd-catalysed carbonylation of 113 dissolved in a mixture of acetonitrile and methanol successfully provided desired product 114. It was discovered that heating a toluene solution of the ester for 4 days smoothly elicited a [4 + 2] cycloaddition in high yield. This process forged four stereocentres in a single operation. Equimolar amounts of diastereomers 114 and 114′ were formed during this process.
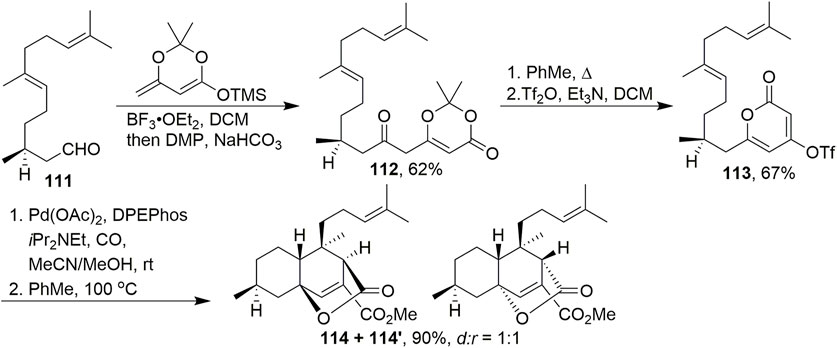
SCHEME 9. Maimone’s protocol to synthesise the tricyclic skeleton of chatancin.
Since early efforts on the synthesis of perhydrohistrionicotoxin in 1989, using dioxinone to build the β-keto acid derivatives used to access macrocyclic and terpenoid natural products has been investigated in many groups around the world. In this section, select recent examples illustrate contemporary methodologies for applications involving dioxinone derivatives.
In 2013, a new cycloaddition methodology to synthesise novel multisubstituted γ-butyrolactones was developed by Leleu et al. (Scheme 10, Peixoto et al., 2013). In this study, multisubstituted γ-butyrolactones 116 were prepared in a one-step procedure by capturing the thermal fragmentation of dioxinones in the presence of hydroxy ketones 115 under basic conditions. Various parameters were considered for the one-step synthesis of γ-butyrolactones, including the amount of base or dioxinone and the nature of the base. Ultimately, the use of 0.5 equiv. triethylamine and 1.5 equiv. dioxinone resulted in the highest yields. Under these conditions, acylfuranone 120 was prepared in one step from 117, 118, and 119 in 77% yield. This method was extended to the one-pot multicomponent synthesis of densely functionalized γ-butyrolactones. This diversity-oriented approach provided expeditious access to various small-ring compounds with potentially high antimicrobial activities under mild conditions with easy handling procedures and a wide scope of substrates. This will most certainly find a broad range of applications in medicinal chemistry.
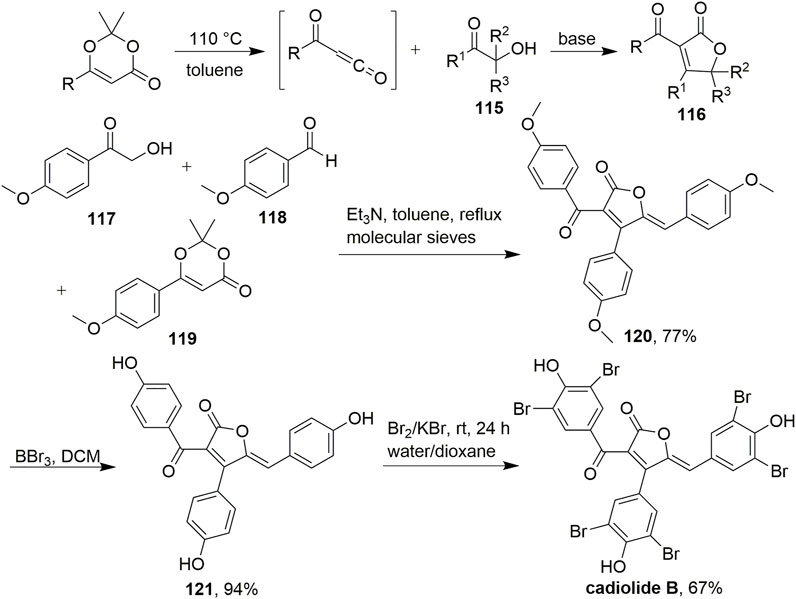
SCHEME 10. A new cycloaddition to prepare novel multisubstituted γ-butyrolactones.
The palladium-catalysed arylation or vinylation of enolisable carbonyl and related compounds represents a viable and useful C–C bond-forming reaction, that is, widely applied in synthetic organic chemistry. In 2015, Lindhardt et al. developed a Pd-catalysed carbonylative coupling of aryl and vinyl halides with vinylogous enolates in which the C–C bond is formed exclusively at the γ-position (Scheme 11, Makarov et al., 2015). In this reaction, the conditions for the carbonylative coupling, including ligand, base, and solvent, were screened under Pd(dba)2 catalysis. Ultimately, using Xantphos as the ligand and LiHMDS as the base, this reaction gave an 82% yield under 3 mol% Pd(dba)2 catalysis in THF. The reaction was performed under mild conditions with various dioxinones and substituted aryl and alkenyl iodides to give aryl and alkenyl ketones 125 and 126, respectively. Dioxinone was coupled to a range of aryl and vinyl iodides to provide 3,5-dicarbonyl acids with complete γ-selectivity. Furthermore, the carbonylation reactions were performed at room temperature with stoichiometric amounts of carbon monoxide. To apply this reaction to natural product synthesis, first, substituted quinoline 127 was readily prepared from 2-nitrobenzoic acid in six steps. Subsequently, 13C-SilaCOgen produced 13C-labeled dioxinone 128 according to the standard conditions for carbonylative coupling in an excellent isolated yield of 85%. Dioxinone opening was then accomplished using ethanol in toluene, which was followed by syn-diastereoselective reduction of the two ketones, ultimately affording the 13C-labeled ethyl ester of (±)-pitavastatin in 31% isolated yield over two steps. The synthesis of this corresponding 13C-labeled product was to indicated that this carbonylative coupling reaction could be used to synthesize drug molecules and also demonstrate the origin of carbonyl groups.
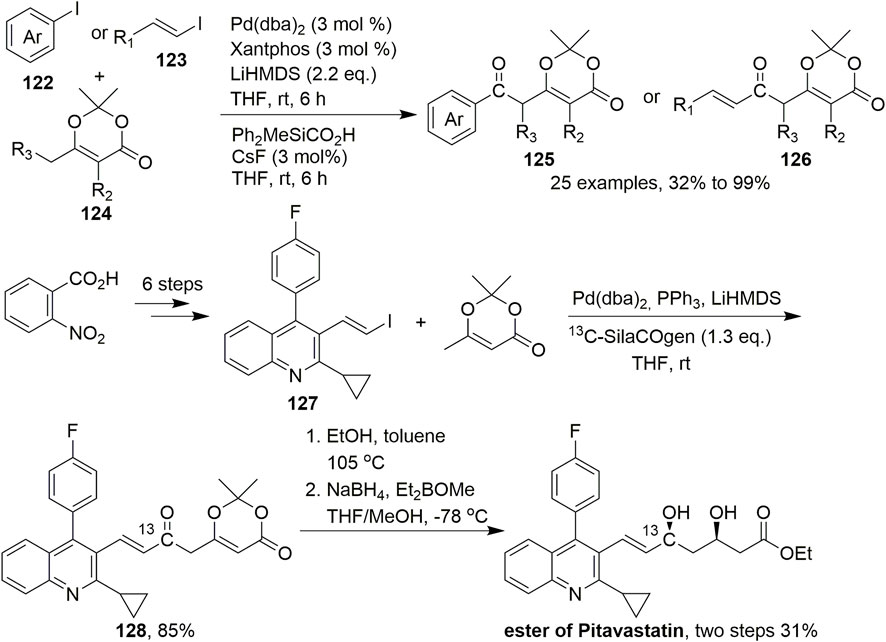
SCHEME 11. Pd-catalysed carbonylative couplings.
In 2017, Zhang et al. reported the full details of a general and practical diastereoselective approach towards the synthesis of δ-amino acid derivatives by vinylogous Mannich reactions between N-tert-butanesulfinyl imines and dioxinone lithium dienolate (Scheme 12, Li et al., 2017). In this study, systematic screening of the reaction conditions, especially the base and Lewis acid, was conducted to optimise the yield and diastereoselectivity of this reaction. It was clear that the base and Lewis acid had a major influence on the reaction. Notably, the corresponding product (130, 130′ and 132, 132’) was isolated in up to 87% yield and 40:1 d.r. in the presence of 2 equiv. BF3·Et2O. With the scope of the diastereoselective vinylogous Mannich reactions having been investigated, a variety of aryl, alkyl, and cyclic substituted N-tert-butanesulfinyl aldimines and ketimines (129 and 131) were obtained in mild to excellent diastereoselectivities (d.r. 1.2:1 to >40:1) and yields (20%–96%) under optimised conditions. Moreover, heterocyclic- and fused-ring–substituted imines gave moderate to excellent yields and diastereoselectivities. In the proposed transition state model, the author inferred that the imine was activated by coordination with BF3, and Si-face addition of the dioxinone-derived lithium dienolate led to major products with S-configuration for the newly formed stereocentre. This reaction provides a novel method for the synthesis of amino acids and chiral amines. Most importantly, the corresponding products can undergo many valuable transformations. Additionally, this method provides a foundation for the synthesis of natural products.
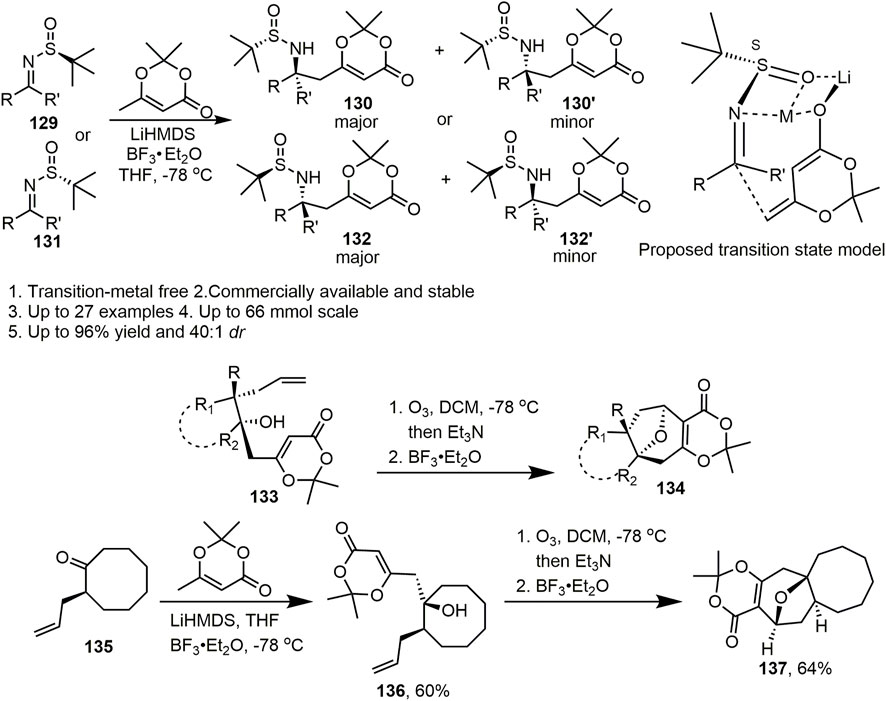
SCHEME 12. The application of dioxinone in Zhang’s work.
Two years later, Zhang’s group developed a practical method for the construction of an oxa-bridged bicyclic ring system via an oxidative-cleavage/Prins-cyclisation approach towards the synthesis of highly functional oxa-bridged seven-, eight-, and nine-membered rings (Scheme 12, Wang et al., 2019). Zhang et al. used various substituted aldehydes, ketones, and Weinreb amides as starting materials. Alcohol 133 was successfully obtained in two steps and set the stage for the proposed oxidative cleavage and Prins cyclisation. In the initial studies, oxidation by ozone provided a semiketal that formed an oxonium ion upon treatment with BF3·Et2O, and cyclisation gave the desired oxa-bridged compound. According to the established route, Zhang et al. extended the general utility of this process to synthesise ring systems of other sizes, including 7/8/9-membered oxa-bridged rings. Substrates bearing cyclopentane, cyclohexane cycloheptane, and cyclooctane also reacted well and provided tetracyclic products. In addition, seven−eight fused, eight−eight fused, and nine-membered ring systems could be constructed using this methodology. Notably, 137 can also be obtained from ketone 135, which is the core skeleton of neoabyssomicin D. This straightforward method for the synthesis of oxa-bridged bicyclic ring systems in many natural products is flexible and enables the entry of virous of highly functionalized fused carbocycles. Reactions are easy to handle, highly diastereoselective, and can be performed on the Gram scale. This process is applicable for the synthesis of natural products containing an oxa-bridged bicyclic core skeleton.
This review illustrates recent advances in the application of dioxinone derivatives to macrocyclic natural products, terpenoid synthesis, and new synthetic methods. Dioxinone derivatives have quickly become powerful, fascinating, and highly efficient tools in organic synthesis. Many researchers have contributed innovative and often practical methods that have established dioxinone derivatives as extremely versatile reagents for the robust and general synthesis of diverse classes of functional compounds, especially β-keto acid derivatives, even in complex natural products. The commercial availability in large quantities at low cost, the robustness and generality of its methods, and the prominence of macrocyclic and terpenoid natural products ensures that dioxinone-based strategies will continue to be some of the most extensively used methods in synthesis. Despite these achievements, new strategies and novel methodologies are both required and expected to facilitate the synthesis of complex molecules. However, due to the sensitivity of the dioxinone derivatives to strong base and high temperature, the application in synthesis is restricted to a certain extent. It is necessary to develop more novel dioxinone derivatives with diverse structure to make up for its defects and adapt to more extensive reaction conditions. We hope that organic chemists will continue to utilise dioxinone derivatives in their endeavours and make use of these multifunctional intermediates in the synthesis of even more complex natural products.
KW conceived and wrote the manuscript. All other authors (XZ and HZ) provided comments and discussion on the manuscript to aid its preparation.
This work was supported by grants from the PhD Research Program of Pingdingshan University (Grant No. PXY-BSDQ-2022038).
We would like to thank Dr Jiuling Li and Dr Jinxu Qi from Pingdingshan University for useful discussion on this topic.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Anderson, K., Calo, F., Pfaffeneder, T., White, A. J. P., and Barrett, A. G. M. (2011). Biomimetic total synthesis of angelicoin A and B via a palladium-catalyzed decarboxylative prenylation aromatization sequence. Org. Lett. 13, 5748–5750. doi:10.1021/ol202320m
Aoki, Y., Ohmori, K., and Suzuki, K. (2015). Dioxinone-fused dienes enable highly endo – selective intramolecular diels–alder reactions. Org. Lett. 17, 2756–2759. doi:10.1021/acs.orglett.5b01172
Barrett, T. N., and Barrett, A. G. M. (2014). Cascade polyketide and polyene cyclizations: Biomimetic total synthesis of hongoquercin B. J. Am. Chem. Soc. 136, 17013–17015. doi:10.1021/ja511534x
Basset, J. F., Leslie, C., Hamprecht, D., White, A. J. P., and Barrett, A. G. M. (2010). Studies on the resorcylates: Biomimetic total syntheses of (+)-montagnetol and (+)-erythrin. Tetrahedron Lett. 51, 783–785. doi:10.1016/j.tetlet.2009.11.134
Brookes, P. A., Cordes, J., White, A. J. P., and Barrett, A. G. M. (2013). Total synthesis of mycophenolic acid by a palladium-catalyzed decarboxylative allylation and biomimetic aromatization sequence. Eur. J. Org. Chem. 7313, 7313–7319. doi:10.1002/ejoc.201300974
Calo, F., Richardson, J., and Barrett, A. G. M. (2009). Total synthesis of aigialomycin D using a one-pot ketene generation-trapping-aromatization sequence. Org. Lett. 11, 4910–4913. doi:10.1021/ol901979x
Chang, C. F., Stefan, E., and Taylor, R. E. (2015). Total synthesis and structural reassignment of lyngbyaloside C highlighted by intermolecular ketene esterification. Chem. Eur. J. 21, 10681–10686. doi:10.1002/chem.201502132
Chiba, S., Kitamura, M., and Narasaka, K. (2006). Synthesis of (-)-Sordarin. J. Am. Chem. Soc. 128, 6931–6937. doi:10.1021/ja060408h
Cookson, R., White, A. J. P., and Barrett, A. G. M. (2018). Synthesis of the C1 to C13 tetrahydropyranyl-resorcylate core of paecilomycin B. Tetrahedron 74, 5040–5048. doi:10.1016/j.tet.2018.05.083
Cordes, J., and Barrett, A. G. M. (2013). Synthesis of macrosporin and related 9, 10-anthraquinones by biomimetic polyketide aromatization and cyclization of 6-benzylresorcylates. Eur. J. Org. Chem. 1318, 1318–1326. doi:10.1002/ejoc.201201480
Cordes, J., Calo, F., Anderson, K., Pfaffeneder, T., Laclef, S., White, A. J. P., et al. (2012). Total syntheses of angelicoin A, hericenone J, and hericenol A via migratory prenyl- and geranylation-aromatization sequences. J. Org. Chem. 77, 652–657. doi:10.1021/jo202354j
Crane, E. A., Zabawa, T. P., Farmer, R. L., and Scheidt, K. A. (2011). Enantioselective synthesis of (-)-Exiguolide by iterative stereoselective dioxinone-directed Prins cyclizations. Angew. Chem. Int. Ed. Engl. 50, 9278–9281. doi:10.1002/ange.201102790
Custar, D. W., Zabawa, T. P., Hines, J., Crews, C. M., and Scheidt, K. A. (2009). Total synthesis and structure-activity investigation of the marine natural product neopeltolide. J. Am. Chem. Soc. 131, 12406–12414. doi:10.1021/ja904604x
Custar, D. W., Zabawa, T. P., and Scheidt, K. A. (2008). Total synthesis and structural revision of the marine macrolide neopeltolide. J. Am. Chem. Soc. 130, 804–805. doi:10.1021/ja710080q
Dalby, S. M., Tindall, J. G., and Paterson, I. (2013). Total synthesis of (-)-Rhizopodin. Angew. Chem. Int. Ed. Engl. 52, 6517–6521. doi:10.1002/anie.201301978
ElMarrouni, A., Lebeuf, R., Gebauer, J., Heras, M., Arseniyadis, S., and Cossy, J. (2011). Total synthesis of nominal lyngbouilloside aglycon. Org. Lett. 14, 314–317. doi:10.1021/ol203064r
Fouché, M., Rooney, L., and Barrett, A. G. M. (2012). Biomimetic total synthesis of cruentaren A via aromatization of diketodioxinones. J. Org. Chem. 77, 3060–3070. doi:10.1021/jo300225z
Fuse, S., Yoshida, H., Oosumi, K., and Takahashi, T. (2014). Rapid and structurally diverse synthesis of multi-substituted β-keto amide derivatives based on a dioxinone scaffold. Eur. J. Org. Chem. 4854, 4854–4860. doi:10.1002/ejoc.201402478
Fuwa, H., Okuaki, Y., Yamagata, N., and Sasaki, M. (2015). Total synthesis, stereochemical reassignment, and biological evaluation of (-)-Lyngbyaloside B. Angew. Chem. Int. Ed. Engl. 54, 882–887. doi:10.1002/ange.201409629
George, N. S., Anderson, K. E., and Barrett, A. G. M. (2013). Total synthesis of cristatic acid based on late-stage decarboxylative allylic migration and biomimetic aromatization of a diketo dioxinone. Eur. J. Org. Chem. 7604, 7604–7610. doi:10.1002/ejoc.201301102
Gerstmann, L., and Kalesse, M. (2016). Total synthesis of aetheramide A. Chem. Eur. J. 22, 11210–11212. doi:10.1002/chem.201602682
Hoye, T. R., Danielson, M. E., May, A. E., and Zhao, H. Y. (2010). Total synthesis of (-)-Callipeltoside A. J. Org. Chem. 75, 7052–7060. doi:10.1021/jo101598y
Huang, S. P., Du, G. Y., and Lee, C. S. (2011). Construction of the tricyclic furanochroman skeleton of phomactin A via the prins/conia-ene cascade cyclization approach. J. Org. Chem. 76, 6534–6541. doi:10.1021/jo200644t
Jacques, S. A., Patel, B. H., and Barrett, A. G. M. (2011). Biomimetic synthetic studies on lactonamycin: An expedient synthesis of dihydroxy-isoindolinone-carboxylates. Tetrahedron Lett. 52, 6072–6075. doi:10.1016/j.tetlet.2011.08.173
Kalesse, M. (2005). Recent advances in vinylogous aldol reactions and their applications in the syntheses of natural products. Top. Curr. Chem. (Cham). 244, 43–76. doi:10.1007/b96887
Laclef, S., Anderson, K., White, A. J. P., and Barrett, A. G. M. (2012). Total synthesis of amorfrutin A via a palladium-catalyzed migratory prenylation–aromatization sequence. Tetrahedron Lett. 53, 225–227. doi:10.1016/j.tetlet.2011.11.019
Li, G. J., Xu, X. L., Tian, H. C., Liu, X. T., Chen, W., Yang, X. D., et al. (2017). Asymmetric synthesis of δ-amino acid derivatives via diastereoselective vinylogous Mannich reactions between N-tert-butanesulfinyl imines and dioxinone-derived lithium dienolate. RSC Adv. 7, 50822–50828. doi:10.1039/c7ra10529k
Ma, T. K., Elliott, D. C., Reid, S., White, A. J. P., Parsons, P. J., and Barrett, A. G. M. (2018). Meroterpenoid synthesis via sequential polyketide aromatization and cationic polyene cyclization: Total syntheses of (+)-Hongoquercin A and B and related meroterpenoids. J. Org. Chem. 83, 13276–13286. doi:10.1021/acs.joc.8b02095
Makarov, I. S., Kuwahara, T., Jusseau, X., Ryu, I., Lindhardt, A. T., and Skrydstrup, T. (2015). Palladium-catalyzed carbonylative couplings of vinylogous enolates: Application to statin structures. J. Am. Chem. Soc. 137, 14043–14046. doi:10.1021/jacs.5b09342
Miyatake-Ondozabal, H., and Barrett, A. G. M. (2010). A novel biomimetic synthesis of (S) (-)-zearalenone: Via macrocyclization and transannular aromatization. Tetrahedron 66, 6331–6334. doi:10.1016/j.tet.2010.05.084
Navarro, I., Basset, J. F., Hebbe, S., Major, S. M., Werner, T., Howsham, C., et al. (2008). Biomimetic synthesis of resorcylate natural products utilizing late stage aromatization: Concise total syntheses of the marine antifungal agents 15G256ι and 15G256β. J. Am. Chem. Soc. 130, 10293–10298. doi:10.1021/ja803445u
Ogura, Y., Sato, H., and Kuwahara, S. (2016). Total synthesis of amphirionin-4. Org. Lett. 18, 2399–2402. doi:10.1021/acs.orglett.6b00883
Patel, B. H., and Barrett, A. G. M. (2012). Total synthesis of resorcinol amide Hsp90 inhibitor AT13387. J. Org. Chem. 77, 11296–11301. doi:10.1021/jo302406w
Peixoto, P. A., Boulangé, A., Leleu, S., and FranckVersatile, X. (2013). Versatile synthesis of acylfuranones by reaction of acylketenes with α-hydroxy ketones: Application to the one-step multicomponent synthesis of cadiolide B and its analogues. Eur. J. Org. Chem. 3316, 3316–3327. doi:10.1002/ejoc.201300166
Reber, K. P., Tilley, S. D., and Sorensen, E. J. (2009). Bond formations by intermolecular and intramolecular trappings of acylketenes and their applications in natural product synthesis. Chem. Soc. Rev. 38, 3022–3034. doi:10.1039/b912599j
Rentsch, A., and Kalesse, M. (2012). The total synthesis of corallopyronin A and myxopyronin B. Angew. Chem. Int. Ed., 51, 11381–11384. doi:10.1002/anie.201206560
Roll, D. M., Manning, J. K., and Carter, G. T. (1998). Hongoquercins A and B, new sesquiterpenoid antibiotics: Isolation, structure elucidation, and antibacterial activity. J. Antibiot. 51, 635–639. doi:10.7164/antibiotics.51.635
Sugano, M., Sato, A., Iijima, Y., Oshima, T., Furuya, K., Kuwano, H., et al. (1991). Phomactin A; a novel PAF antagonist from a marine fungus Phoma sp. J. Am. Chem. Soc. 113, 5463–5464. doi:10.1021/ja00014a053
Sugano, M., Sato, A., Saito, K., Takaishi, S., Matsushita, Y., and Iijima, Y. (1996). Structure−Activity relationships of phomactin derivatives as platelet activating factor antagonists. J. Med. Chem. 39, 5281–5284. doi:10.1021/jm950640q
Tenenbaum, J. M., Morris, W. J., Custar, D. W., and Scheidt, K. A. (2011). Synthesis of (-)-Okilactomycin by a prins-type fragment-assembly strategy. Angew. Chem. Int. Ed. Engl. 50, 5892–5895. doi:10.1002/anie.201102037
Wang, M. S., Wang, Z., Chen, W., Yang, X. D., and Zhang, H. B. (2019). Synthesis of oxa-bridged medium-sized carbocyclic rings via Prins cyclization. Org. Lett. 21, 1881–1884. doi:10.1021/acs.orglett.9b00491
Winkler, J. D., and Hershberger, P. M. (1989). A stereoselective synthesis of (-)-Perhydrohistrionicotoxin. J. Am. Chem. Soc. 111, 4852–4856. doi:10.1021/ja00195a042
Yadav, J. S., Haldar, A., and Maity, T. (2012). Towards the synthesis of (-)-Callipeltoside A: Stereoselective synthesis of the C1-C14 macrolactone core. Eur. J. Org. Chem. 2012, 2062–2071. doi:10.1002/ejoc.201101635
Zampella, A., D’Auria, M. V., Minale, L., Debitus, C., and Roussakis, C. (1996). Callipeltoside A: A cytotoxic aminodeoxy sugar-containing macrolide of a new type from the marine lithistida sponge callipelta sp. J. Am. Chem. Soc. 118, 11085–11088. doi:10.1021/ja9621004
Zhao, Y. M., and Maimone, T. J. (2015). Short, enantioselective total synthesis of chatancin. Angew. Chem. Int. Ed. 54, 1223–1226. doi:10.1002/anie.201410443
Ac Acetyl
Ar Aryl
Bn Benzyl
Boc tert-Butyloxycarbonyl
Bu Butyl
Bz Benzoyl
Cbz Carbobenzoxyl
DBU 1,8-Diazabicyclo[5,4,0]undec-7-ene
DCC N,N′-Dicyclohexylcarbodiimide
DCE Dichloroethane
DCM Dichloromethane
DDQ 2,3-Dicyano-5,6-dichlorobenzoquinone
DIBAL-H Diisobutyl aluminium hydride
DIPEA N,N-Diisopropylethylamine
DMAP 4-Dimethylaminopyridine
DMF N,N-Dimethylformamide
DMP Dess-Martin Periodinane
DMSO Dimethyl sulfoxide
dr Diasetereomer ratio
EA Ethyl acetate
ee Enantiomeric excess
eq. Equivalent
Et Ethyl
IBX 2-Iodoxybenzoic Acid
im Imidazole
i-Pr isopropyl
KHMDS Potassium Hexamethyldisilazide
LDA Lithium diisopropylamide
LHMDS Lithium Hexamethyldisilazide
m-CPBA meta-Chloroperbenzoic Acid
Me Methyl
MOM methoxymethyl acetal
PCC Pyridinium chlorochromate
PE Petroleum ether
Ph phenyl
PMB p-methoxybenzyl
PMBCl para-Methoxybenzyl
PPTS Pyridinium toluene-4-sulphonate
p-TsOH p-Toluenesulfonic acid
Py pyridine
rt Room temperature
TBAF Tetrabutylammonium fluoride
TBS tert-Butyldimethylsilyl
TBDPS tert-Butyldiphenylchlorosilyl
TEA Triethylamine
Tf trifluoromethanesulfonate
TFA Trifluoroacetic acid
TFAA trifluoroacetic anhydride
THF Tetrahydrofuran
TLC Thin layer chromatography
TIPS Triisopropylsilyl
TMS Trimethylsilyl
Ts p-toluenesulfonyl
Keywords: dioxianone, macrocyclic, macrolactam, terpenoid, macrolide
Citation: Wei K, Zheng X and Zhang H (2022) Recent applications of dioxinone derivatives for macrocyclic natural product and terpenoid synthesis. Front. Chem. 10:1030541. doi: 10.3389/fchem.2022.1030541
Received: 29 August 2022; Accepted: 01 December 2022;
Published: 12 December 2022.
Edited by:
Siva S. Panda, Augusta University, United StatesReviewed by:
Pankaj Khanna, University of Delhi, IndiaCopyright © 2022 Wei, Zheng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Wei, d2Vpa2FpMTk4N0AxMjYuY29t; Hongbin Zhang, emhhbmdoYkB5bnUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.