 Jin Zhao†
Jin Zhao† Qingzhong Li
Qingzhong Li Shaoli Liu
Shaoli Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 26 September 2022
Sec. Green and Sustainable Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1027465
This article is part of the Research TopicPreparation of Functional Materials and Utilization of Renewable Resources in Green SolventsView all 12 articles
Reactions of PtX+ (X = F, Cl, Br, I) with methane have been investigated at the density functional theory (DFT) level. These reactions take place more easily along the low-spin potential energy surface. For HX (X = F, Cl, Br, I) elimination, the formal oxidation state of the metal ion appears to be conserved, and the importance of this reaction channel decreases in going as the sequence: X = F, Cl, Br, I. A reversed trend is observed in the loss of H2 for X = F, Cl, Br, while it is not favorable for PtI+ in the loss of either HI or H2. For HX eliminations, the transfer form of H is from proton to atom, last to hydride, and the mechanisms are from PCET to HAT, last to HT for the sequence of X = F, Cl, Br, I. One reason is mainly due to the electronegativity of halogens. Otherwise, the mechanisms of HX eliminations also can be explained by the analysis of Frontier Molecular Orbitals. While for the loss of H2, the transfer of H is in the form of hydride for all the X ligands. Noncovalent interactions analysis also can be explained the reaction mechanisms.
Catalysts that can convert methane directly into higher-value-added commodities have long been sought, but breaking the thermodynamically strong, kinetically inert C-H bonds in a controlled way under mild conditions remains a central challenge (Geng et al., 2017). Reactivity studies of transition-metal ions in the gas phase, and, in particular, aspects related to the ongoing challenge of selective activation of inert C-H and C-C bonds, have been studied intensely over the past decades (Howell and Burkinshaw, 1983; Dubois, 1989; Eller and Schwarz, 1991; Balcells et al., 2010; Dobereine and Crabtree, 2010; Roithova and Schröder, 2010; Jana et al., 2011). In recent years, how ligation affects the electronic structure at the transition-metal center has been systematically investigated (Howell and Burkinshaw, 1983; Dubois, 1989; Schlangen et al., 2007; Schlangen et al., 2007; Schlangen and Schwarz, 2008; Dede et al., 2009; Li et al., 2016a; Sun et al., 2016; Zhou et al., 2016; Zhou et al., 2017a; Zhou et al., 2017b; Zhou et al., 2017c; Schwarz et al., 2017; Schwarz et al., 2017; Yue et al., 2017). The ligand can change the electronic structure of the metal center through a shift in the electronic state, or provide a more efficient reaction center, so the addition of a single ligand to a metal center has been widely used to prepare reactants for C-H bonds activation (Chen et al., 1997; Rodgers et al., 2000; Li et al., 2009).
Irikura and Beauchamp (Irikura and Beauchamp, 1989; Irikura and Beauchamp, 1991a; Irikura and Beauchamp, 1991b) discovered that Pt+ as a 5d transition metal dehydrogenates methane to yield the corresponding carbene complexes Pt (CH2)+. Bare Pt+ also has been found to catalyze the reaction of methane with molecular oxygen in the gas phase to produce methanol, formaldehyde and other oxidation products (Wesendrup et al., 1994). Subsequently, a series of activation studies around transition metal Pt+ were carried out (Achatz et al., 2000; Wheeler et al., 2016). Recently, it has been reported that Pt− is able to selectively activate one C-H bond in methane, which represents the first example of methane activation by atomic anions (Liu et al., 2019).
Open-shell ligands X form a covalent bond with the metal cation and thereby increase the formal oxidation state, for example, X = F, Cl, Br, I, OH, NH, O (Schlangen et al., 2007; Dede et al., 2009), which often increases reactivity. For example, bare Cr+ is one of the least reactive transition metal cations, whereas CrCl+ is significantly more reactive (Mandich et al., 1986). Clearly, this example demonstrates that an appropriately chosen ligand can enhance the selectivity of a reagent at the expense of reactivity (Schlangen et al., 2007). Similarly, the naked cations M+ (M = Fe, Co, Ni, Ru, Rh, Pd) do not bring about thermal C-H bond activation of methane (Halle et al., 1982; Tolbert and Beauchamp, 1986; Tolbert et al., 1986; Schultz et al., 1988; Musaev et al., 1993; Musaev and Morokuma, 1994; Westerberg and Blomberg, 1998), but the corresponding MH+ cations (Schilling et al., 1986; Elkind and Armentrout, 1987; Schilling et al., 1987; Schilling et al., 1987; Ohanessian et al., 1990; Zhang and Bowers, 2004; Li et al., 2009; Wang and Andrews, 2009) give rise to efficient H/CH3 ligand switches.
It is not surprising that the nature of the ligand X controls the outcome of a given ion-molecule reaction, as, for example, demonstrated in a systematic investigation of FeX+ cations with acetone (Schröder et al., 1993). The number of ligands also affects the reaction activity. With respect to the activation of methane, CrF+ is not sufficient, and CrF2+ does not react with CH4, whereas CrF3+ and CrF4+ are able to activate the C-H bonds of methane (Mazurek et al., 1998).
Schlangen et al. have reported the studies on ligand and substrate effects in gas-phase reactions of NiX+/RH couples (X = F, Cl, Br, I; R = CH3, C2H5, n-C3H7, n-C4H9) (Schlangen et al., 2007). The results indicate that NiF+ is the only NiⅡ halide complex that brings about thermal activation of methane to eliminate HF, whereas the nickel-halide cations NiCl+, NiBr+, and NiI+ react only with large alkanes. In the elimination of HX (X = F, Cl, Br, I), the formal oxidation state of the metal ion appears to be conserved, and the importance of this reaction channel decreased in going from NiF+ to NiI+. A reversed trend is observed in the losses of H2, which dominate the gas-phase ion chemistry of NiI+/RH couples. Schröder and Schwarz (2005) reported the reactions of methane with PtX+ (X = H, Cl, Br and CHO) using mass spectrometry and found that these species are able to activate methane.
Here, we report our calculated results for the PtX+/CH4 (X = F, Cl, Br, I) systems. The key issues for our study are the mechanistic details of methane catalyzed by ligated transition metal PtX+/CH4 (X = F, Cl, Br, I).
Full optimization of geometries for all stationary points involved in methane dehydrogenation by PtX+ (X = F, Cl, Br, I) has been calculated using the density functional theory (DFT) method based on the hybrid of Becke’s three-parameter exchange functional and the Lee, Yang, and Parr correlation functional (B3LYP) (Becke, 1988; Lee et al., 1988; Becke, 1993), Becke hybrid with correlation functional Perdew (B3P86) (Perdew, 1986a; Perdew, 1986b; Michael et al., 2008) and M06-2X (Zhao and Truhlar, 2008; Zhao and Truhlar, 2008). For carbon and hydrogen, also for F, Cl, and Br, the large 6-311+G** basis set is applied. The Stuttgart/Dresden relativistic effective core potentials (ECP) of SDD were adopted to describe the metal Pt and the halogen I (Andrae et al., 1990). For each optimized stationary point, vibrational analysis was performed at the same level with the geometry optimizations to determine its character (minimum or saddle point). Unscaled harmonic frequencies were employed to obtain entropy corrections and the zero-point vibrational energy (ZPVE) which is included in all relative energies. Furthermore, intrinsic reaction coordinate (IRC) calculations (Gonzalez and Schlegel, 1989) were performed to confirm that the optimized transition states correctly connect the relevant reactants and products. Energies were corrected for (unscaled) zero-point vibrational energy contributions and were given relative to the separated reactant couples PtX+/CH4 in the most stable spin state of PtX+ (Ye and Neese, 2010; Lawson Daku et al., 2012; Vargas et al., 2013). Between two different potential energy surfaces (PES), a configuration that structures are similar with almost the same energy was found, which is called minimum energy crossing point (MECP) (Poli and Harvey, 2003; Harvey, 2007). All computations reported are carried out using the GAUSSIAN 09 program suit (Frisch et al., 2009). The topological parameters of electron density(ρ), its Laplacian (▽2ρ), and energy density at the bond critical point (BCP) were analyzed with the AIM2000 program (Bader, 2000). The molecular electrostatic potentials (MEP) of the various monomers were calculated on the 0.001 a. u. isodensity surfaces using the wave function analysis–surface analysis suite (WFA-SAS) program (Bulat et al., 2010).
The geometries were optimized using density functional theory with B3P86, B3LYP, and M06-2X functional. The comparisons show that the results obtained by the three methods are very similar in terms of geometric optimization, energy, and potential energy surfaces. The data are shown in Table 1 and the Supporting Information. Among them, B3LYP shows a more systematic process in a high-spin state and is also more resource-efficient. Otherwise, we also calculated the single point energy of the reaction at the CCSD(T)/aug-cc-pVTZ (PP) level. The trend of the single point energy is similar to the previous potential energy surface except for the energy of 1Pt (CH3)+ in the last step, which is inconsistent with the experimental results (Schröder and Schwarz, 2005). So, all the data used are obtained based on the B3LYP method.
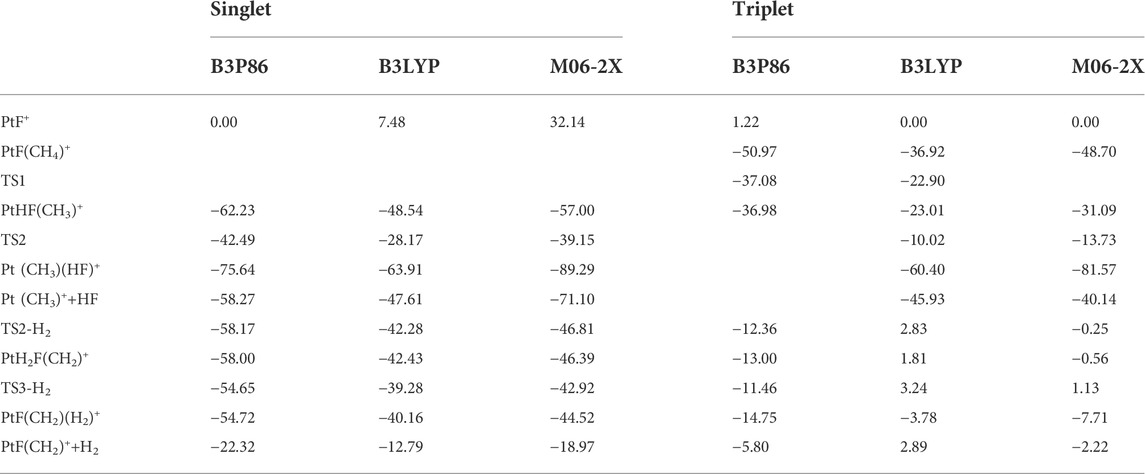
TABLE 1. Calculated relative energies (kcal/mol) of stationary points on the potential energy surfaces of the reaction PtF+ and CH4 in the singlet and triplet states in three methods.
In this section, we discuss the reactivity of the PtX+ (X = F, Cl, Br, I) in the activation process of CH4 and present a brief discussion of the most abundant or interesting processes for the PtX+/CH4 systems. Both low- and high-spin states have been considered. The potential energy surfaces of the reaction PtX+ + CH4 in the low- and high-spin states are summarized in Figure 1 and Figure 4, and the energetics (in kcal/mol) of the intermediates and transition states, relative to the ground state PtX+ plus CH4 have been summarized in Table 2. Geometries of these structures, including bond distances and bond angles, are summarized in the Supporting Information.
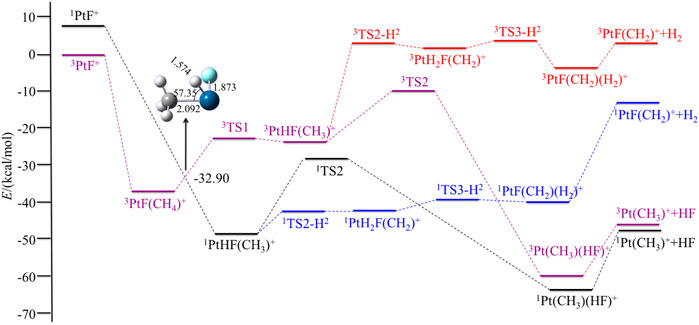
FIGURE 1. Potential energy surfaces of the reaction PtF+ + CH4 in the low- and high-spin states. The structure is the minimum energy crossing point (MECP).
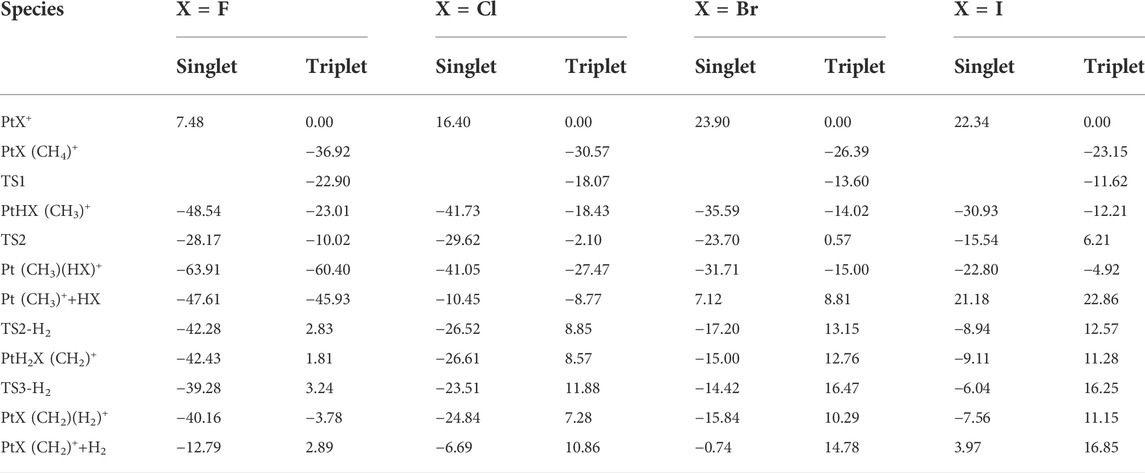
TABLE 2. Calculated relative energies (kcal/mol) of stationary points on the potential energy surfaces of the reaction PtX+ (X = F, Cl, Br, I) and CH4 in the singlet and triplet states.
The reactions observed in Figure 1 can be classified into two different categories: 1) reactions involving Pt-X bond cleavage, namely, the expulsions of HX and 2) bond activation of CH4 without obvious occurrence of Pt-X bond cleavage, that is, the loss of H2 (summarized in Eq. 1 and 2).
For the sake of simplicity, each species is labeled with its spin multiplicity as a superscript preceding the formula.
For PtF+, the ground electronic state has been found to be a triplet, and the singlet electronic excitation state of 1PtF+ has a relative energy of 7.48 kcal/mol. The reaction starts with the formation of a methane complex PtF(CH4)+. Based on Figure 1, the spin-conserving dehydrogenation of CH4 along the ground state route 3PtF+ + CH4 → 3PtF(CH2)+ + H2 is endothermic by 2.89 kcal/mol and cannot occur for the high efficiency of the reaction. So, the reaction would obtain HF through the ground route or would be a two-state reaction scenario (TSR) (Roithova et al., 2010).
In the triplet state, the relative energy of the complex 3PtF(CH4)+ is −36.92 kcal/mol and is found to have a η2 coordination, the θC-Pt-F = 179.89°, which indicates that the carbon atom attacks to Pt along the Pt-F axis. Then, Pt inserts into one of the C-H bonds of methane, resulting in a hydridomethyl complex 3PtHF(CH3)+, which has an energy of −23.01 kcal/mol. TS1 is the transition state of the oxidative addition of the first C-H bond on the reaction path. On the 3TS1, the activated C-H bond is almost broken with the C-H bond length of 1.775 Å and the Pt-H bond is nearly formed with the Pt-H bond length of 1.565 Å, indicating that 3TS1 is a typical three-centered late transition state, which is 14.02 kcal/mol above the encounter complex 3PtF(CH4)+ but only 0.11 kcal/mol above the 3PtHF(CH3)+.
The low-spin 1PtF+ with methane tends to form 1PtHF(CH3)+ intermediate directly. No activation transition state has been found on the singlet surface. The results indicate that the first C-H bond is activated spontaneously on the singlet surface. Energetically, the 1PtHF(CH3)+ is 25.53 kcal/mol lower than that of the triplet 3PtHF(CH3)+. A curve crossing is required from the triplet state to the singlet state via an MECP. As shown in Figure 1, due to the higher energies of the triplet-spin state in the process of the expulsions of HF, and the processes in the triplet-spin state being similar to the singlet-spin paths, later, the triplet surface is not considered in the expulsion of HF.
In the following pages of this section, we will first discuss the process of the expulsions of HX, namely, HF. On the singlet surface, the next step is a reductive elimination step to form an HF molecule complex; that is, the H and F rearrange to form an HF molecule electrostatically bound to Pt to obtain the 1Pt (CH3)(HF)+ with a barrier of 20.37 kcal/mol. In the last step, HF can be eliminated in an exothermic reaction by 17.98 kcal/mol. This last step leaves 1Pt (CH3)+ in its ground singlet state.
For the elimination of H2, a migration of hydrogen from CH3 to Pt, leading to 1PtH2F(CH2)+ with an energy barrier of 6.26 kcal/mol. The transition state 1TS2-H2 (it represents the transition state in the process of the elimination of H2) is a three-centered late transition state. Then, the two hydrogens rearrange easily to form the 1PtF(CH2)(H2)+. Afterward, the molecule H2 is eliminated. The calculated dissociation energy of H2 to 1PtF(CH2)+ is 27.38 kcal/mol.
Generally, the energy barrier controls the reaction rate in a channel. Comparing the above two reaction channels, the energy barrier of H2 elimination is 14.11 kcal/mol lower than that of the HF expulsions. However, in the subsequent steps, the calculated ligand dissociation energy of H2 to 1PtF(CH2)+ is much higher than any energies of the complexes in the path to produce HF. Namely, the favorable path for the reaction of PtF+ + CH4 is the channel of the elimination of HF.
For the overall process, the energetically most favorable route involves a two-state reactivity scenario. The favorable route is the elimination of HF via the route 3PtF+ + CH4 → 3PtF(CH4)+ → MECP → 1PtHF(CH3)+ → 1TS2 → 1Pt (CH3)(HF)+ → 1Pt (CH3)++ HF.
As to the CH4 activation on PtX+ (X = Cl, Br, I), the mechanisms are very similar to those on PtF+, as discussed earlier. Indeed, the critical geometrical parameters in the intermediates and transition state are all very similar to the corresponding structures in the case of PtF+, as can be seen clearly by comparing the figures in the Supporting Information. Therefore, we shall not discuss their geometries in further detail but show some differences and their characteristics.
For PtX+ (X = Cl, Br, I) with CH4, as calculated by the results, the ground low-lying state is all the triplet state. The excitation energies to the excited singlet state are 16.40, 23.90, and 22.34 kcal/mol, respectively, for PtX+ (X = Cl, Br, I). The low-spin 1PtX+ with methane tends to form a 1PtHX (CH3)+ intermediate directly. No activation transition state has been found on the singlet surface. The results are similar to the PtF+-CH4 system and indicate that C-H is activated spontaneously on the singlet surface. Energetically, the 1PtHX (CH3)+ is lower than the triplet 3PtHX (CH3)+. Since the triplet state is the ground state of PtX+, the methane activation starting from the ground state again requires an intersystem crossing as described in the case of PtF+ via a minimum energy crossing point, as shown in Figures 2–4.
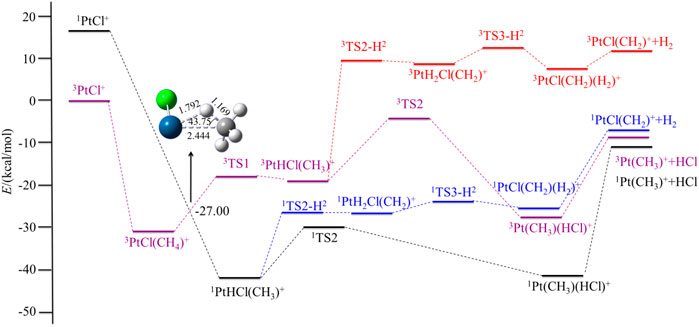
FIGURE 2. Potential energy surfaces of the reaction PtCl+ + CH4 in the low- and high-spin states. The structure is the minimum energy crossing point (MECP).
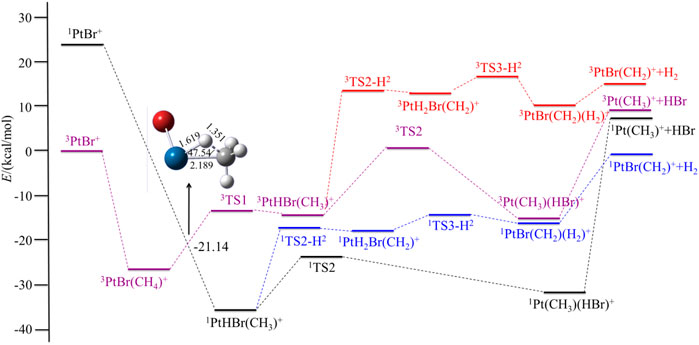
FIGURE 3. Potential energy surfaces of the reaction PtBr+ + CH4 in the low- and high-spin states. The structure is the minimum energy crossing point (MECP).
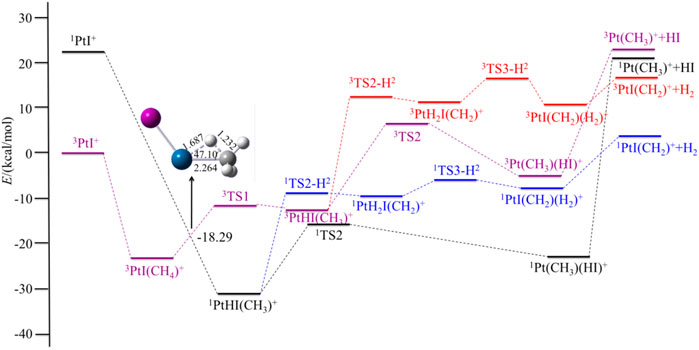
FIGURE 4. Potential energy surfaces of the reaction PtI+ + CH4 in the low- and high-spin states. The structure is the minimum energy crossing point (MECP).
Different from PtF+, as for the other three PtX+ (X = Cl, Br, I), on the singlet surface, the next step is the rearrangement of hydrogen and halogen to obtain HX, which has an activation energy of 12.11, 11.89, and 15.39 kcal/mol, respectively, for PtX+ (X = Cl, Br, I). This activation energy is 3.10, 6.50, and 6.60 kcal/mol lower than that in the path of the elimination of H2.
For PtCl+, HCl can be eliminated in an exothermic reaction by 10.45 kcal/mol. Due to the path of the expulsions of H2 always being high-lying compared with the process of elimination of HCl, it is an unfavorable path. Namely, the favorable path of PtCl+/CH4 is the process of HCl elimination. This result is in good agreement with the experimental results as reported by Schröder and Schwarz (2005). They reported the branching ratio of HCl is 100%. For PtBr+/CH4, the favorable path is the process of the elimination of HBr, but the calculated ligand dissociation energy of HBr to 1Pt (CH3)+ is 38.83 kcal/mol, different from PtF+, the energy difference between the two processes of the products are only 7.86 kcal/mol, which is lower than others. The activation energy to obtain 1Pt (CH3)(HBr)+ is 6.50 kcal/mol lower than to obtain 1PtH2Br(CH2)+. Due to the lower activation energy, the favorable path of the reaction of PtBr+/CH4 probably is the elimination of HBr, but also has some ratio of H2 in the products, as mentioned in the earlier discussions. As reported by Schröder and Schwarz (2005), the branching ratio of HBr:H2 is 85:15. As for PtI+, although the energies in the potential energy surfaces before the loss of HI are lower than that of H2, the elimination of HI needs much more energy than H2, and it is endothermic at 43.98 kcal/mol, which is not favorable to occur. In the process of the loss of H2, there is a barrier height of 21.99 kcal/mol on the singlet state to form the intermediate 1PtH2I(CH2)+. After overcoming a small barrier of 3.07 kcal/mol, the complex 1PtI(CH2)(H2)+ is formed. The low-spin species 1PtI(CH2)+ has an association energy of 11.53 kcal/mol. This reaction is endothermic of 3.97 kcal/mol, which is also not favorable thermodynamically.
In summary, in the elimination of HX (X = F, Cl, Br, I), the formal oxidation state of the metal ion appears to be conserved, and the importance of this reaction channel decreases in going as the sequence: X = F, Cl, Br, I. A reversed trend is observed in the loss of small closed-shell molecule H2 for X = F, Cl, Br, while it is not favorable for PtI+ in the loss of either HI or H2. The reason for the reactivity along with the abovementioned trends can be explained by the electronegative character of X; on the other hand, by the corresponding reaction enthalpies, which are mostly related to the formation of HX, that is to say, the halogens are heavier the bond-dissociation energies are much lower (Dede et al., 2009). The results can also be seen by the Natural Bond Orbital (NBO) populations and Natural population analysis (NPA) charge, as shown in the Supporting Information, and the results of part of the key structures are shown in Table 3. The electronegativity of the halogens decreases gradually from F to I, and the donor properties increase gradually from F to I, so F forms a strongly polarized covalent bond to Pt, and F increases the formal oxidation of Pt.
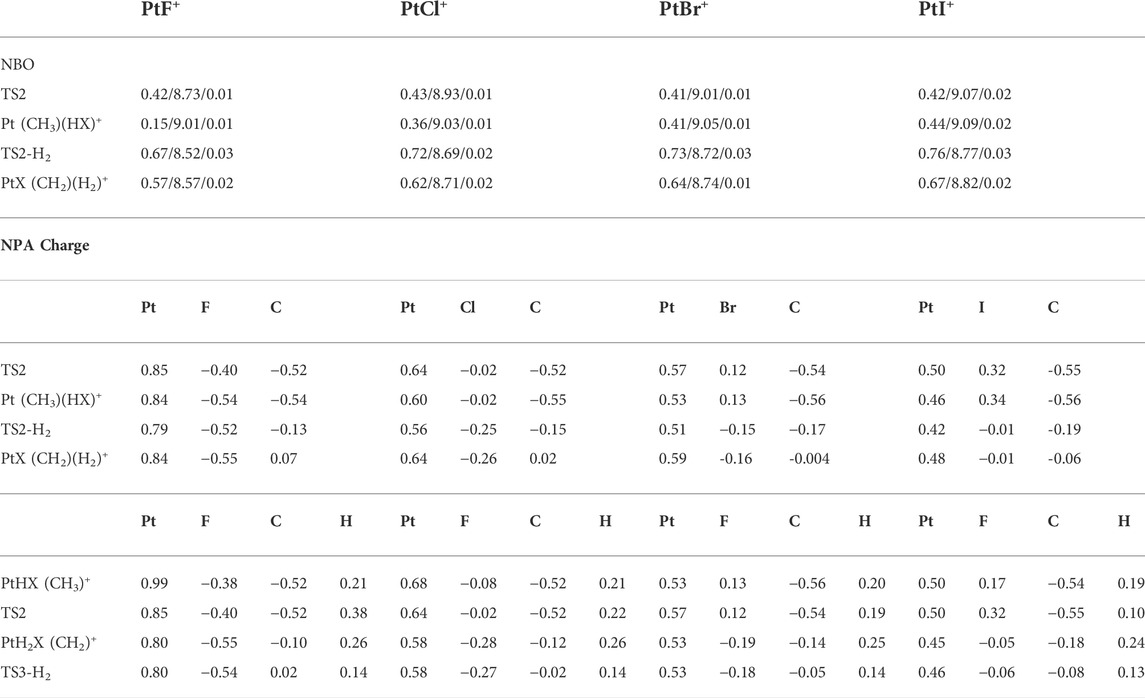
TABLE 3. Valence NBO populations for the 6s/5d/6p orbitals of Pt and the natural population analysis (NPA) charge of the related atoms in part of the key structures in the reaction of PtX+ (X = F, Cl, Br, I) + CH4 in the singlet state.
Reaction mechanisms of these reactions are elucidated by detailed NPA charge and the Frontier Molecular Orbitals (HOMO and LUMO) of the key structures in the rate controlling step.
In the elimination of HF, as shown in Table 3, the F atom carries a significant negative charge, serving as a good proton acceptor, in which the electron is accepted by the metal center, thus, the NPA charge decreases in Pt. This process can be classified as a conventional proton-coupled electron transfer (PCET (Li et al., 2016b)) mechanism. For HCl and HBr eliminations, the charges of all atoms did not change during hydrogen transforms, and the mechanism can be judged as hydrogen atom transfer (HAT (Dietl et al., 2012)), while for I ligand, the NPA charge of I atom increases and that of H atom decreases, and it can be determined that the process is hydride transfer (HT (Li et al., 2016c)) mechanism.
The mechanisms of HX eliminations also can be explained by the analysis of Frontier Molecular Orbitals, which are shown in Figure 5. In the HOMO orbital, the σ(dx2-y2) of Pt and σ(py) of X occupied the main contribution in Pt-X molecular orbital. The coefficient of Pt (σ(dx2-y2)) becomes smaller and X (σ(py)) increases in the sequence of F < Cl < Br < I. From 1PtHX (CH3)+ to 1TS2, the increase of electron density (φ2) on Pt is consistent with the decrease of NPA charge. The decrease of electron density on X also corresponds to the increase of NPA charge. It also can be seen from the LUMO orbital graph of C (σ(py))-Pt (σ(dxy))-X (σ(pX)) that the φ2 of Pt increase and the φ2 of C and X decrease.
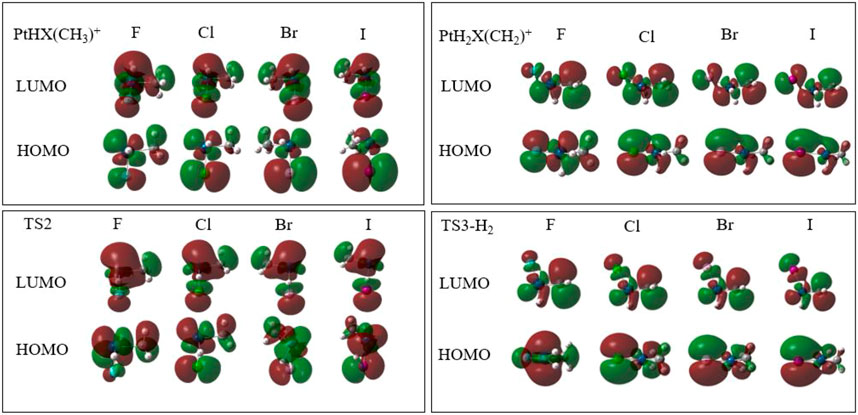
FIGURE 5. HOMO/LUMO orbital graphs in part of the structures of rate controlling step in the reaction of PtX+ (X = F, Cl, Br, I) + CH4 in the singlet state.
In the course of the formation of H2, the positive charge of H diminishes, indicating that it may be an HT mechanism. Corresponding to this, electron density has been transferred from methane to hydrogen, as shown in the HOMO orbital graph in Figure 5. As the transferred electron density takes the same route as that of the concurrently transferred hydrogen atom, it can be described as a hydride transfer mechanism. For different halogens, the change of charges is the same, that is to say, in terms of rate controlling step, different halogen ligands have no significant effect on the process of elimination of H2.
In sum, in the eliminations of HX, the mechanisms are different. The transfer form of H is from proton to atom, last to hydride. The reason is mainly due to the electronegativity of halogens, while for the loss of H2, the transfer of H is in the form of hydride for all the X ligands.
To further investigate the mechanisms, the interactions between the complexes in the reactions were also discussed. The MEP diagrams of singlet and triplet PtX+ on the 0.001 a. u. isodensity surface are displayed in Figure 6. Since the complexes have positive charge, the overall electrostatic potential is in the red region. There is a deep red region (σ-hole) along the Pt-F axis around the Pt+, which correspond to the site where carbon atoms attack PtX+. It is consistent with the previously optimized structure. In addition, the σ-hole strength decreases in the order PtF+> PtCl+> PtBr+> PtI+ owing to the different electron-withdrawing ability among the halogen.
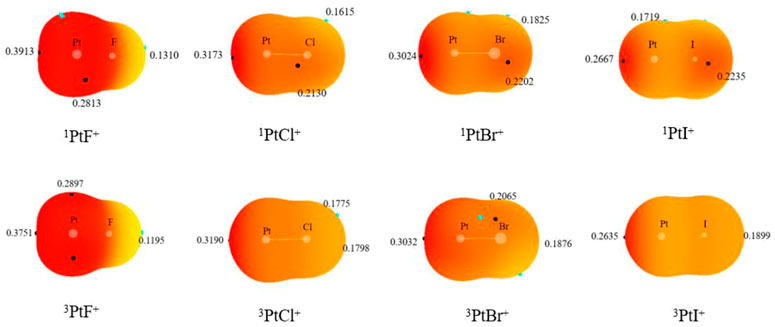
FIGURE 6. Molecular electrostatic potential (MEP) maps on the 0.001 a. u. isodensity surface of the monomers.
In the AIM theory, electron density at the bond critical point (BCP) is one of the indicators of interaction strength. The electron density, Laplacian, and total energy density at the XPt-CH4 (X = F, Cl, Br, I) complexes are listed in Table 4. The values of the density at the BCP lie in the range around 0.07 a. u., Laplacian is positive and energy density is negative in the methane complexes, which indicates that there exhibits a property of a partially covalent interaction. The charge transfer from CH4 to PtX+ decreases in the order PtF+> PtCl+> PtBr+> PtI+, which is consistent with the σ-hole strength of PtX+. The charge transfer and the interaction energy of methane complexes in the potential energy surface have similar changes.
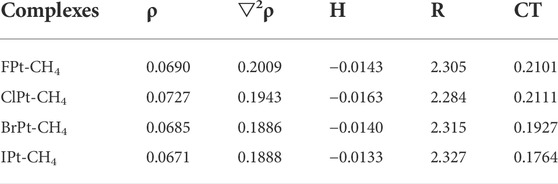
TABLE 4. Electron density (ρ, a.u.), Laplacian (▽2ρ, a.u.), energy density (H, a.u.), intermolecular distance (R, Å), and charge transfer (CT, e) at the XPt-CH4 (X = F, Cl, Br, I) complexes.
Table 5 shows the electron density, Laplacian, and total energy density at the complexes before the expulsions of HX or H2. It is generally believed that the greater the electron density between two atoms in a composite, the more concentrated the charge between the two atoms, which also means that the bond between these two atoms has a stronger tendency (Kraka and Cremer, 1990; Alkorta et al., 1998). As shown in Table 5, for all the complexes before dissociating the H2 or HX molecule, Laplacian is positive and energy density is negative, indicating that there exists a partially covalent interaction between H2 or HX molecule and Pt atom. For the same complex, the values of ρ, ▽2ρ, and energy density are obviously stronger in the singlet state than those in the triplet state, indicating a stronger interaction between the Pt and the H2 or HX molecule in the singlet state, and more energy is required to expel H2 or HX, which is consistent with the energy barrier of the reaction in the potential energy surfaces. Through the analysis of the interaction between atoms in the transition metal–ligand complex, the reaction path can be better explained.
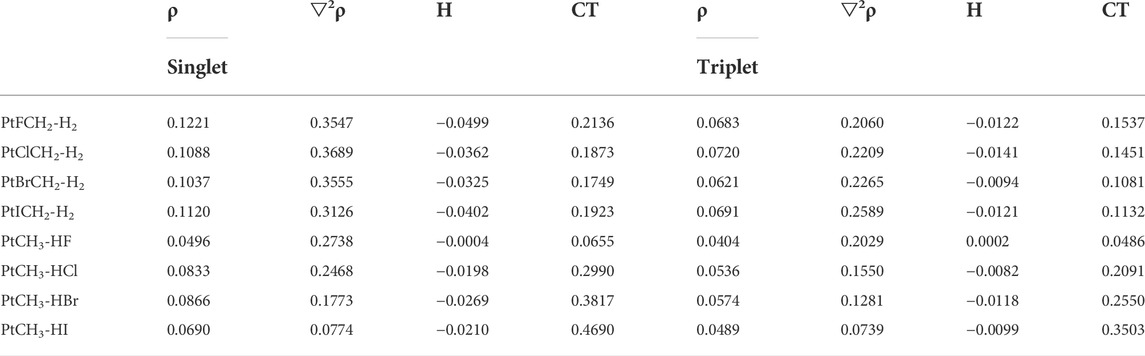
TABLE 5. Electron density (ρ, a.u.), Laplacian (▽2ρ, a.u.), energy density (H, a.u.), and charge transfer (CT, e) in the PtXCH2-H2 and PtCH3-HX (X = F, Cl, Br, I) complexes.
From Table 5, one also can see that for the complexes before H2 was removed, the values of charge transfer (CT) are between 0.17 e and 0.21 e, and the differences between PtXCH2-H2 (X = F, Cl, Br, I) are small. However, for the complexes before HX was expelled, the values of charge transfer increased from PtCH3-HF to PtCH3-HI, which was generally consistent with the interaction energy. Namely, the complexes before the expulsions of HI had larger interaction energy than the others.
The reactions of NiX+ (X = F, Cl, Br, I) and methane have been investigated at the B3LYP level of theory by Schlangen et al. (Schlangen et al., 2007; Schlangen and Schwarz, 2008). They reported that NiF+ is the only nickel-halide complex capable of activating methane for NiX+ (X = F, Cl, Br, I). The driving force of the reaction NiF+ with methane is provided by the exceptionally high stability of HF (Schlangen et al., 2007). In the present study, we found that the PtX+ (X = F, Cl, Br) can bring about thermal activation of methane to loss HX decreasing in going as the sequence X = F, Cl, Br, and to loss H2 increasing in the reverse sequence.
The reactions of NiF+ and their third-row congeners PtX+ with methane have many features in common; whereas, fundamental differences exist with regard to the details of the potential energy surfaces and, thus, to actual reaction mechanisms. As reported by Schlangen and Schwarz (2008), for the NiF+ and methane systems studied, the energetically most favored variant corresponds to an σ-complex-assisted metathesis (σ-CAM). First, the reactions start with the formation of the encounter complex NiF(CH4)+, and then, the molecule HF is directly eliminated via a multicenter transition state to obtain the product complex, Ni(CH3)(HF)+. However, in the PtX+/CH4 systems, oxidative addition/reductive elimination (OA/RE) is operative. After the formation of the complex PtX (CH4)+, the next step is the cleaving of the C-H bond (oxidative addition), resulting in the insertion product PtHX (CH3)+, and then a reductive elimination step to form an HX molecule complex is obtained, that is, the H and X rearrange to form an HX molecule electrostatically bound to Pt to obtained the 1Pt (CH3)(HX)+.
Another difference in the reaction mechanisms is the potential energy surfaces. In the NiF+/CH4 system, the exothermic ligand exchange proceeds adiabatically only on the one potential energy surface, whereas the reaction of PtX+/CH4 needs a curve crossing, that is to say, it is a two-state reactivity. Otherwise, the NiF+/CH4 system proceeds on the high-spin ground triplet state, while the reaction of PtX+ with methane takes place more easily along the low-spin potential energy surface. As for the reasons for the differences, Schlangen and Schwarz (2008) have reported that the strongly electron-withdrawing F substituent reduces the electron density at the Ni-center and, thus, decreases the repulsive interaction; therefore, the reaction can proceed on the high-spin ground state. Based on this point, we calculated the Mullikan charges of the M-atom (M = Ni, Pt) in the systems MX+/CH4. The results indicate that the Mullikan charges of the Ni-atom in the NiF+/CH4 system increase 0.198, while the Pt-atom in the reaction PtF+/CH4 increases only 0.082.
The gas-phase ion-molecule reactions of PtX+ cations (X = F, Cl, Br, I) with methane have been investigated theoretically at the DFT (B3LYP) level, considering both the low- and high-spin potential energy surfaces. All reactions fall into two major categories: 1) reactions involving Pt-X bond cleavage to expulse HX and 2) bond activation of CH4 without obvious occurrence of Pt-X bond cleavage to loss H2. In the elimination of HX (X = F, Cl, Br, I), this reaction channel decreases in going as the sequence: X = F, Cl, Br, I. A reversed trend is observed in the losses of small closed-shell molecule H2 for X = F, Cl, Br, while it is not favorable for PtI+ in the loss of either HI or H2. The reason for the reactivity along with the abovementioned trends can be explained by the electronegative character of X.
In the eliminations of HX, the transfer form of H is from proton to atom, last to hydride, and the mechanisms are from PECT to HAT, last to HT for the sequence of X = F, Cl, Br, I. One reason is mainly due to the electronegativity of halogens. Otherwise, the mechanisms of HX eliminations also can be explained by the analysis of Frontier Molecular Orbitals, while for the loss of H2, the transfer of H is in the form of hydride for all the X ligands.
The charge transfer from CH4 to PtX+ decreases in the order PtF+> PtCl+> PtBr+> PtI+, which is consistent with the σ-hole strength of PtX+. For the same complex, the values of ρ, ▽2ρ, and energy density are obviously stronger in the singlet state than those in the triplet state, indicating a stronger interaction between the Pt and the H2 or HX molecule in the singlet state, and more energy is required to expel H2 or HX, which is consistent with the energy barrier of the reaction in the potential energy surfaces. The differences in charge transfer between PtXCH2-H2 (X = F, Cl, Br, I) for the complexes before H2 is removed are small. However, for the complexes before HX is expelled, the values of charge transfer increase from PtCH3-HF to PtCH3-HI, namely, the complexes before the expulsions of HI have larger interaction energy than the others. Through the analysis of the interaction between atoms in the transition metal ligand complex, the reaction path can be better explained.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
JZ and LQ: structure optimization and data collection; JC and QL: data analysis; WL and SL: data analysis and manuscript writing and revision. All authors read and approved the submitted version.
The authors thank the National Natural Science Foundation of China (Grant No. 21803052) and the Natural science foundation of Shandong province, China (Grant No. ZR2018LB017). The grants are gratefully acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1027465/full#supplementary-material
Achatz, U., Berg, C., Joos, S., Fox, B. S., Beyer, M. K., Niedner-Schatteburg, G., et al. (2000). Methane activation by platinum cluster ions in the gas phase: Effects of cluster charge on the Pt4 tetramer. Chem. Phys. Lett. 320, 53–58. doi:10.1016/S0009-2614(00)00179-2
Alkorta, I., Rozas, I., and Elguero, J. (1998). Charge-transfer complexes between dihalogen compounds and electron donors. J. Phys. Chem. A 102, 9278–9285. doi:10.1021/jp982251o
Andrae, D., Haeussermann, U., Dolg, M., Stoll, H., and Preuss, H. (1990). Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 77, 123–141. doi:10.1007/BF01114537
Balcells, D., Clot, E., and Eisenstein, O. (2010). C-H bond activation in transition metal species from a computational perspective. Chem. Rev. 110, 749–823. doi:10.1021/cr900315k
Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A . Coll. Park. 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Becke, A. D. (1993). Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Bulat, F. A., Toro-Labbé, A., Brinck, T., Murray, J. S., and Politzer, P. (2010). Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 16, 1679–1691. doi:10.1007/s00894-010-0692-x
Chen, Q., Chen, H. P., Kais, S., and Freiser, B. S. (1997). Gas-phase reactions of Fe(CH2O)+ and Fe(CH2S)+ with small alkanes: An experimental and theoretical study. J. Am. Chem. Soc. 119, 12879–12888. doi:10.1021/ja964234n
Dede, Y., Zhang, X. H., Schlangen, M., Schwarz, H., and Baik, M. H. (2009). A redox non-innocent ligand controls the life time of a reactive quartet excited state-an MCSCF study of [Ni(H)(OH)]+. J. Am. Chem. Soc. 131, 12634–12642. doi:10.1021/ja902093f
Dietl, N., Schlangen, M., and Schwarz, H. (2012). Thermal hydrogen‐atom transfer from methane: The role of radicals and spin states in oxo‐cluster chemistry. Angew. Chem. Int. Ed. 51, 5544–5555. doi:10.1002/anie.201108363
Dobereine, G. E., and Crabtree, R. H. (2010). Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem. Rev. 110, 681–703. doi:10.1021/cr900202j
Dubois, M. R. (1989). Catalytic applications of transition-metal complexes with sulfide ligands. Chem. Rev. 89, 1–9. doi:10.1021/cr00091a001
Elkind, J. L., and Armentrout, P. B. (1987). State-specific reactions of atomic transition-metal ions with H2, HD, and D2: Effects of d orbitals on chemistry. J. Phys. Chem. 91, 2037–2045. doi:10.1021/j100292a012
Eller, K., and Schwarz, H. (1991). Organometallic chemistry in the gas phase. Chem. Rev. 91, 1121–1177. doi:10.1021/cr00006a002
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. revision A. 02. Wallingford CT: Gaussian, Inc.
Geng, C. Y., Li, J. L., Weiske, T., Schlangen, M., Shaik, S., and Schwarz, H. (2017). Electrostatic and charge-induced methane activation by a concerted double C-H bond insertion. J. Am. Chem. Soc. 139, 1684–1689. doi:10.1021/jacs.6b12514
Gonzalez, C., and Schlegel, H. B. (1989). An improved algorithm for reaction path following. J. Chem. Phys. 90, 2154–2161. doi:10.1063/1.456010
Halle, L. F., Armentrout, P. B., and Beauchamp, J. L. (1982). Ion beam studies of the reactions of group VIII metal ions with alkanes: Correlation of thermochemical properties and reactivity. Organometallics 1, 963–968. doi:10.1021/om00067a012
Harvey, J. N. (2007). Understanding the kinetics of spin-forbidden chemical reactions. Phys. Chem. Chem. Phys. 9, 331–343. doi:10.1039/b614390c
Howell, J. A. S., and Burkinshaw, P. M. (1983). Ligand substitution reactions at low-valent four-five-and six-coordinate transition metal centers. Chem. Rev. 83, 557–599. doi:10.1021/cr00057a005
Irikura, K. K., and Beauchamp, J. L. (1991b). Electronic structure considerations for methane activation by third-row transition-metal ions. J. Phys. Chem. 95, 8344–8351. doi:10.1021/j100174a057
Irikura, K. K., and Beauchamp, J. L. (1991a). Methane oligomerization in the gas phase by third-row transition-metal ions. J. Am. Chem. Soc. 113, 2769–2770. doi:10.1021/ja00007a070
Irikura, K. K., and Beauchamp, J. L. (1989). Osmium tetroxide and its fragment ions in the gas phase: Reactivity with hydrocarbons and small molecules. J. Am. Chem. Soc. 111, 75–85. doi:10.1021/ja00183a014
Jana, R., Pathak, T. P., and Sigman, M. S. (2011). Advances in transition metal (Pd, Ni, Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev. 111, 1417–1492. doi:10.1021/cr100327p
Kraka, E., and Cremer, D. (1990). Chemical implication of local features of the electron density distribution. Concept Chem. Bond, 116, 453–542. Maksic, ZB, Ed. doi:10.1007/978-3-642-61277-0_12
Lawson Daku, L. M., Aquilante, F., Robinson, T. W., and Hauser, A. (2012). Accurate spin-state energetics of transition metal complexes. 1. CCSD(T), CASPT2, and DFT study of [M(NCH)6]2+ (M = Fe, Co). J. Chem. Theory Comput. 8, 4216–4231. doi:10.1021/ct300592w
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi:10.1103/PhysRevB.37.785
Li, J. L., Zhou, S. D., Schlangen, M., Weiske, T., and Schwarz, H. (2016a). Hidden hydride transfer as a decisive mechanistic step in the reactions of the unligated gold carbide [AuC]+ with methane under ambient conditions. Angew. Chem. Int. Ed. 55, 13072–13075. doi:10.1002/anie.201606707
Li, J. L., Zhou, S. D., Zhang, J., Schlangen, M., Usharani, D., Shaik, S., et al. (2016b). Mechanistic variants in gas-phase metal-oxide mediated activation of methane at ambient conditions. J. Am. Chem. Soc. 138, 11368–11377. doi:10.1021/jacs.6b07246
Li, J. L., Zhou, S. D., Zhang, J., Schlangen, M., Weiske, T., Usharani, D., et al. (2016c). Electronic origins of the variable efficiency of room-temperature methane activation by homo-and heteronuclear cluster oxide cations [XYO2]+ (X, Y= Al, Si, Mg): Competition between proton-coupled electron transfer and hydrogen-atom transfer. J. Am. Chem. Soc. 138, 7973–7981. doi:10.1021/jacs.6b03798
Li, W. Q., Geng, Z. Y., Wang, Y. C., Yan, P. J., Zhang, X., Wang, Z., et al. (2009). Density functional theory studies of thermal activation of methane by MH+ (M= Ru, Rh, and Pd). J. Phys. Chem. A 113, 1807–1812. doi:10.1021/jp808830c
Liu, G. X., Zhu, Z. G., Ciborowski, S. M., Ariyarathna, I. R., Miliordos, E., and Bowen, K. H. (2019). Selective activation of the C-H bond in methane by single platinum atomic anions. Angew. Chem. Int. Ed. 58, 7773–7777. doi:10.1002/anie.201903252
Mandich, M. L., Steigerwald, M. L., and Reents, W. D. (1986). The effects of chloro substitution on the electronic structure of ClCr+, ClMn+, and ClFe+ and their reactivity with small alkanes. J. Am. Chem. Soc. 108, 6197–6202. doi:10.1021/ja00280a015
Mazurek, U., Schröder, D., and Schwarz, H. (1998). Generation and reactivity of chromium fluoride cations (CrFn+, n= 0-4) in the gas phase. Collect. Czech. Chem. Commun. 63, 1498–1512. doi:10.1135/cccc19981498
Michael, B., Christoph, R., Dimitrios, A. P., Thomas, B., and Frank, N. (2008). Geometries of third-row transition-metal complexes from density-functional theory. J. Chem. Theory Comput. 4, 1449–1459. doi:10.1021/ct800172j
Musaev, D. G., Koga, N., and Morokuma, K. (1993). Ab initio molecular orbital study of the electronic and geometric structure of RhCH2+ and the reaction mechanism: RhCH2+ + H2. → Rh+ + CH4. J. Phys. Chem. 97, 4064–4075. doi:10.1021/j100118a022
Musaev, D. G., and Morokuma, K. (1994). Ab initio molecular orbital study of the molecular and electronic structure of FeCH2+ and of the reaction mechanism of FeCH2+ + H2. J. Chem. Phys. 101, 10697–10707. doi:10.1063/1.467883
Ohanessian, G., Brusich, M. J., and Goddard, W. A. (1990). Theoretical study of transition-metal hydrides. 5. Hafnium to mercury (HfH+ through HgH+), barium and lanthanum (BaH+ and LaH+). J. Am. Chem. Soc. 112, 7179–7189. doi:10.1021/ja00176a016
Perdew, J. P. (1986a). Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 33, 8822–8824. doi:10.1103/PhysRevB.33.8822
Perdew, J. P. (1986b). Erratum: Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 34, 7406. doi:10.1103/PhysRevB.34.7406
Poli, R., and Harvey, J. N. (2003). Spin forbidden chemical reactions of transition metal compounds. New ideas and new computational challenges. Chem. Soc. Rev. 32, 1–8. doi:10.1039/b200675h
Rodgers, M. T., Stanley, J. R., and Amunugama, R. (2000). Periodic trends in the binding of metal ions to pyridine studied by threshold collision-induced dissociation and density functional theory. J. Am. Chem. Soc. 122, 10969–10978. doi:10.1021/ja0027923
Roithova, J., and Schröder, D. (2010). Selective activation of alkanes by gas-phase metal ions. Chem. Rev. 110, 1170–1211. doi:10.1021/cr900183p
Schilling, J. B., Goddard, W. A., and Beauchamp, J. L. (1986). Theoretical studies of transition-metal hydrides. 1. Bond energies for MH+ with M= Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, and Zn. J. Am. Chem. Soc. 108, 582–584. doi:10.1021/ja00264a004
Schilling, J. B., Goddard, W. A., and Beauchamp, J. L. (1987). Theoretical studies of transition-metal hydrides. 2. CaH+ through ZnH+. J. Phys. Chem. 91, 5616–5623. doi:10.1021/j100306a024
Schilling, J. B., Goddard, W. A., and Beauchamp, J. L. (1987). Theoretical studies of transition-metal hydrides. 3. SrH+ through CdH+. J. Am. Chem. Soc. 109, 5565–5573. doi:10.1021/ja00253a001
Schlangen, M., Schröder, D., and Schwarz, H. (2007). Ligand and substrate effects in gas‐phase reactions of NiX+/RH couples (X= F, Cl, Br, I; R= CH3, C2H5, nC3H7, nC4H9). Chem. Eur. J. 13, 6810–6816. doi:10.1002/chem.200700506
Schlangen, M., Schröder, D., and Schwarz, H. (2007). Pronounced ligand effects and the role of formal oxidation states in the nickel-mediated thermal activation of methane. Angew. Chem. Int. Ed. 46, 1641–1644. doi:10.1002/anie.200603266
Schlangen, M., and Schwarz, H. (2008). Ligand effects on the mechanisms of thermal bond activation in the gas‐phase reactions NiX+/CH4 → Ni(CH3)+/HX (X= H, CH3, OH, F). Short Communication. Helv. Chim. Acta 91, 2203–2210. doi:10.1002/hlca.200890238
Schröder, D., Hrusak, J., and Schwarz, H. (1993). Ligand effects on the reactivity of iron (II) cations FeX+ in the gas phase. Berichte Bunsenges. fur Phys. Chem. 97, 1085–1090. doi:10.1002/bbpc.19930970904
Schröder, D., and Schwarz, H. (2005). Activation of methane by gaseous platinum (II) ions PtX+ (X= H, Cl, Br, CHO). Can. J. Chem. 83, 1936–1940. doi:10.1139/v05-217
Schultz, R. H., Elkind, J. L., and Armentrout, P. B. (1988). Electronic effects in C-H and C-C bond activation. State-specific reactions of Fe+ (6D, 4F) with methane, ethane, and propane. J. Am. Chem. Soc. 110, 411–423. doi:10.1021/ja00210a017
Schwarz, H., Navarrete, P. G., Li, J. L., Schlangen, M., Sun, X. Y., Weiske, T., et al. (2017). Unexpected mechanistic variants in the thermal gas-phase activation of methane. Organometallics 36, 8–17. doi:10.1021/acs.organomet.6b00372
Schwarz, H., Shaik, S., and Li, J. L. (2017). Electronic effects on room-temperature, gas-phase C-H Bond activations by cluster oxides and metal carbides: The methane challenge. J. Am. Chem. Soc. 139, 17201–17212. doi:10.1021/jacs.7b10139
Sun, X. Y., Zhou, S. D., Schlangen, M., and Schwarz, H. (2016). Efficient room‐temperature methane activation by the closed‐shell, metal‐free cluster [OSiOH]+: A novel mechanistic variant. Chem. Eur. J. 22, 14257–14263. doi:10.1002/chem.201601981
Tolbert, M. A., and Beauchamp, J. L. (1986). Homolytic and heterolytic bond dissociation energies of the second row group 8, 9, and 10 diatomic transition-metal hydrides: Correlation with electronic structure. J. Phys. Chem. 90, 5015–5022. doi:10.1021/j100412a029
Tolbert, M. A., Mandich, M. L., Halle, L. F., and Beauchamp, J. L. (1986). Activation of alkanes by ruthenium, rhodium, and palladium ions in the gas phase: Striking differences in reactivity of first-and second-row metal ions. J. Am. Chem. Soc. 108, 5675–5683. doi:10.1021/ja00279a003
Vargas, A., Krivokapic, I., Hauser, A., and Lawson Daku, L. M. (2013). Towards accurate estimates of the spin-state energetics of spin-crossover complexes within density functional theory: A comparative case study of cobalt(II) complexes. Phys. Chem. Chem. Phys. 15, 3752–3763. doi:10.1039/c3cp44336a
Wang, X. F., and Andrews, L. (2009). Infrared spectra and theoretical calculations for Fe, Ru, and Os metal hydrides and dihydrogen complexes. J. Phys. Chem. A 113, 551–563. doi:10.1021/jp806845h
Wesendrup, R., Schroder, D., and Schwarz, H. (1994). Catalytic Pt+‐mediated oxidation of methane by molecular oxygen in the gas phase. Angew. Chem. Int. Ed. Engl. 33, 1174–1176. doi:10.1002/anie.199411741
Westerberg, J., and Blomberg, M. R. A. (1998). Methane activation by naked Rh+ atoms. A theoretical study. J. Phys. Chem. A 102, 7303–7307. doi:10.1021/jp981291p
Wheeler, O. W., Michelle, S., Gao, A., Bakker, J. M., and Armentrout, P. B. (2016). Activation of C-H bonds in Pt+ + x CH4 reactions, where x= 1-4: Identification of the platinum dimethyl cation. J. Phys. Chem. A 120, 6216–6227. doi:10.1021/acs.jpca.6b05361
Ye, S., and Neese, F. (2010). Accurate modeling of spin-state energetics in spin-crossover systems with modern density functional theory. Inorg. Chem. 49, 772–774. doi:10.1021/ic902365a
Yue, L., Li, J. L., Zhou, S. D., Sun, X. Y., Schlangen, M., Shaik, S., et al. (2017). Control of product distribution and mechanism by ligation and electric field in the thermal activation of methane. Angew. Chem. Int. Ed. 56, 10219–10223. doi:10.1002/anie.201703485
Zhang, Q., and Bowers, M. T. (2004). Activation of methane by MH+ (M= Fe, Co, and Ni): A combined mass spectrometric and DFT study. J. Phys. Chem. A 108, 9755–9761. doi:10.1021/jp047943t
Zhao, Y., and Truhlar, D. G. (2008). Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167. doi:10.1021/ar700111a
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi:10.1007/s00214-007-0310-x
Zhou, S. D., Firouzbakht, M., Schlangen, M., Kaupp, M., and Schwarz, H. (2017a). On the electronic origin of remarkable ligand effects on the reactivities of [NiL]+ complexes (L= C6H5, C5H4N, CN) towards methane. Chem. Eur. J. 23, 14430–14433. doi:10.1002/chem.201703767
Zhou, S. D., Li, J. L., Firouzbakht, M., Schlangen, M., and Schwarz, H. (2017b). Sequential gas-phase activation of carbon dioxide and methane by [Re(CO)2]+: The sequence of events matters. J. Am. Chem. Soc. 139, 6169–6176. doi:10.1021/jacs.7b01255
Zhou, S. D., Li, J. L., Schlangen, M., and Schwarz, H. (2016). Thermal dehydrogenation of methane by [ReN]+. Angew. Chem. Int. Ed. 55, 14863–14866. doi:10.1002/anie.201607960
Keywords: PtX +, activation of methane, reaction mechanism, ligand effect, noncovalent interactions
Citation: Zhao J, Qi L, Li W, Cheng J, Li Q and Liu S (2022) CH4 activation by PtX+ (X = F, Cl, Br, I). Front. Chem. 10:1027465. doi: 10.3389/fchem.2022.1027465
Received: 25 August 2022; Accepted: 06 September 2022;
Published: 26 September 2022.
Edited by:
Honglei Fan, Institute of Chemistry (CAS), ChinaCopyright © 2022 Zhao, Qi, Li, Cheng, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenzuo Li, bGl3ZW56dW8yMDA0QDEyNi5jb20=; Qingzhong Li, bGlxaW5nemhvbmcxOTkwQHNpbmEuY29t; Shaoli Liu, bGl1c2hhb2xpNjk4NEBzaW5hLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.