 Miaomiao Zhou1,2†
Miaomiao Zhou1,2† Gui-Juan Cheng
Gui-Juan Cheng- 1Warshel Institute for Computational Biology, School of Medicine, The Chinese University of Hong Kong (Shenzhen), Shenzhen, China
- 2Department of Chemistry, City University of Hong Kong, KowloonTong, Hong Kong SAR, China
- 3School of Life and Health Sciences, School of Medicine, The Chinese University of Hong Kong (Shenzhen), Shenzhen, China
- 4Shenzhen Key Laboratory of Steroid Drug Development, School of Medicine, The Chinese University of Hong Kong (Shenzhen), Shenzhen, China
Selective defunctionalization of synthetic intermediates is a valuable approach in organic synthesis. Here, we present a theoretical study on the recently developed B(C6F5)3/hydrosilane-mediated reductive deamination reaction of primary amines. Our computational results provide important insights into the reaction mechanism, including the active intermediate, the competing reactions of the active intermediate, the role of excess hydrosilane, and the origin of chemoselectivity. Moreover, the study on the substituent effect of hydrosilane indicated a potential way to improve the efficiency of the reductive deamination reaction.
Introduction
In the search for renewable alternatives, biomass feedstock is usually a promising sustainable carbon source to produce fuels, chemicals, and materials. In general, biomass-derived feedstocks, such as sugars, alcohol, phenol, and amines, are over-functionalized. Therefore, defunctionalization has become an important way to produce useful downstream chemicals, which has attracted wide attention. (Huber et al., 2006; Corma et al., 2007; Zhou et al., 2011; Zhang et al., 2017). Over last 2 decades, the deoxygenation of alcohols or derivatives has been well developed to access simple hydrocarbons. (Adlington et al., 1976; Doyle et al., 1976; Orfanopoulos and Smonou, 1988; Yasuda et al., 2001; Nimmagadda and McRae, 2006; McLaughlin et al., 2013; Dai and Li, 2016). Conversely, although amines are also one of the most common feedstock chemicals accessible by biomass conversion, the deaminative transformation of amines is poorly developed, which highlights great challenges, particularly in the development of deaminative strategies for alkyl amines and primary amines to construct C–X (X = C, O, S, B, P, H, etc.) bonds.
Deaminases, such as L-amino acid deaminases (LAAD), are essential biocatalyst in living cells. In the human body, deamination, as a common metabolic process, usually takes place to break down amino acids for the generation of their corresponding α-keto acids and ammonia by deaminases, involving in nucleotide sequence, immunity and cancer. (Massad et al., 1995; Petersen-Mahrt et al., 2009; Vesely et al., 2012; Molla et al., 2017; Nshimiyimana et al., 2019). In contrast, deamination in laboratory is very rare and difficult. Although the challenges are daunting, systematic efforts toward deamination of primary amines have recently begun to emerge, and some progress has been made. Earlier work on C–NH2 bond activation usually requires to pre-activate primary amines into reactive intermediates, such as active pyridinium salts (Katritzky salts) (Hu et al., 2018; Ni et al., 2019; Wang et al., 2019; Correia et al., 2020; Pang et al., 2020), electron-rich diazos (Mitsuhashi et al., 2000; Geoffroy et al., 2001; Barluenga et al., 2009; Peng et al., 2009; Wu et al., 2014), and isonitrile compounds (Barton et al., 1980; Barton et al., 1992), for further deamination, which results in a more complicated and expensive reaction process (Scheme 1A). Based on the principles of green chemistry and sustainable development, the direct activation of C–N bond of primary amines without pre-activation affords a very attractive approach to obtain valuable functionalized building blocks even though it is more challenging.
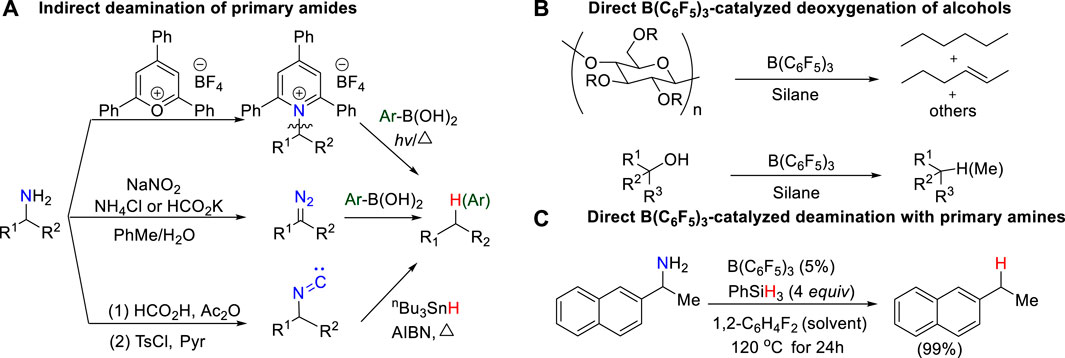
SCHEME 1. (A) Indirect deamination of primary amine; (B) Direct B(C6F5)3-catalyzed deoxygenation of primary alcohols; (C) Direct B(C6F5)3-catalyzed deamination of primary amines.
The combination of B(C6F5)3 and hydrosilanes was recently discovered for selective deoxygenations of 1,2-diols and polyols by the Gagné (Adduci et al., 2014; Adduci et al., 2015; Bender et al., 2016a; Bender et al., 2016b; Seo and Gagné, 2018; Seo et al., 2019), Morandi (Drosos and Morandi, 2015; Nikolaos Drosos and Morandi, 2015; Drosos et al., 2016), Yamamoto (Gevorgyan et al., 1999; Gevorgyan et al., 2000; Gevorgyan et al., 2001), and Oestreich group (Chatterjee et al., 2017; Fang and Oestreich, 2020a; Richter and Oestreich, 2021) (Scheme 1B). However, the reaction of B(C6F5)3 and hydrosilanes with amines generally does not give deamination product; instead, it affords N-silylation product which is formed via B(C6F5)3-catalyzed dehydrogenative coupling of amines and hydrosilanes. (Hermeke et al., 2013; Greb et al., 2014; Zhang et al., 2018; Zhou et al., 2020). Recently, the Oestreich group reported the first metal-free, B(C6F5)3/hydrosilane-mediated deamination reaction of primary amines, which was a breakthrough for the direct C–NH2 bond defunctionalization (Scheme 1C). (Fang and Oestreich, 2020b) Their study showed that the amount of silane reagent is essential to the reaction: 4 equivalents of PhSiH3 were required to obtain high yields, and less PhSiH3 would result in poor yields. Additionally, they found the substitution degree of benzylamines significantly affects the reactivity. But the role of excess hydrosilane and substituent effect on reactivity remain elusive. In addition, the stoichiometric experiments indicated the existence of silylammonium borohydride which was proposed to undergo C–N cleavage to afford deamination product. However, it is unclear which species among mono-, di-, or tri-silylammonium borohydrides (i.e., int2, int6 or int10 shown in Figure 1) is the active intermediate. Moreover, it remains unknown how does deamination compete over the dehydrogenative coupling to successfully afford desired product. A deeper understanding of the reaction mechanism may provide useful information for the optimization and development of deamination reactions.
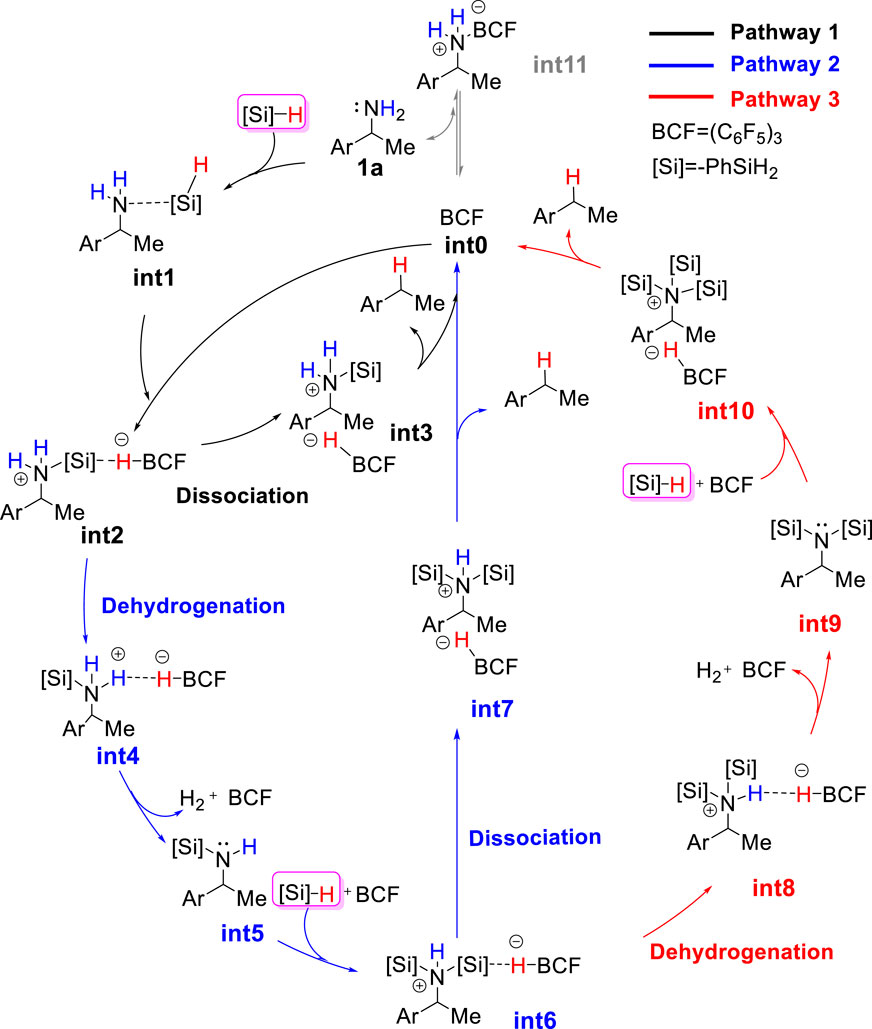
FIGURE 1. The proposed catalytic cycle for B(C6F5)3-catalyzed reductive deamination with hydrosilanes.
Our group is particularly interested in the diverse catalytic capabilities of B(C6F5)3 system. In previous computational study on B(C6F5)3-catalyzed deoxygenation of polyols with silanes, we rationalized the special role of the cyclic siloxane intermediate in promoting reactivity and selectivity, as well as the different product distributions obtained with different silanes. (Drosos et al., 2017; Cheng et al., 2018).
Herein, we disclose the reaction mechanism of B(C6F5)3-catalyzed deamination by theoretical studies, thereby expanding the understanding of B(C6F5)3 catalytic system. The present work provides a detailed mechanistic picture for B(C6F5)3-catalyzed deamination reaction, unveils the role of excess hydrosilane, and explains the substituent effect on reactivity.
Computational details
All the calculations were performed with Gaussian 16 (Frisch et al., 2016) package. All molecular geometries were optimized with B3LYP-D3/def2SVP method in gas phase. (Lee et al., 1988; Schafer et al., 1992; Becke, 1993; Schafer et al., 1994; Grimme et al., 2011). Optimized geometries were verified by frequency computations as minima (zero imaginary frequencies) or transition state (a single imaginary frequency) at the same level of theory. The transition states (TSs) were also confirmed by viewing normal mode vibrational vector and by intrinsic reaction coordinate (IRC) calculation. (Gonzalez and Gonzalez, 1990). All the single point energy calculations in solution phase were carried out by SMD(Marenich et al., 2009) model with 1,2-diflurobenzene as the solvent and B3LYP-D3 method with the def2-TZVP basis set. All of Gibbs energies were corrected at 393.15K. Both relative Gibbs energies and electronic energies were reported in kcal/mol. The 3D structures were generated by CYLview. (Legault, 2009). The conformational space of the system has been extensively explored manually by rotating the torsional angles of the molecule and automatically by using Crest program. (Grimme, 2019).
Results and discussion
Reaction mechanism of B(C6F5)3-catalyzed reductive deamination with PhSiH3 and 1a
As discussed in the introduction, silylammonium borohydride was detected in the stoichiometric experiment and proposed to undergo C–N cleavage to form deamination product. Because it is unknown which one among mono-, bi-, and tri-silylammonium borohydrides (i.e., int2, int6 or int10 shown in Figure 1) is the active intermediate that leads to deamination product, we calculated three possible pathways that involve different silylammonium borohydrides. As shown in Figure 2, the reaction is initiated by the association of substrate 1a with one PhSiH3. This step needs to overcome a Gibbs energy barrier of 14.0 kcal/mol (TS1), generating a thermodynamically unstable amine-silane complex int1. Then, the Lewis acid B(C6F5)3 abstracts a hydride from int1, which is accompanied by the N–Si bond formation to afford the monosilylammonium borohydride species (int2). The activation energy barrier for the silylation process is 27.3 kcal/mol (TS2). The monosilylammonium borohydride intermediate then undergoes C–N bond dissociation (pathway 1), where borohydride (C6F5)3BH− acts as a nucleophile to attack the benzylic carbon of silylammonium moiety via an SN2-type transition state (TS3) to afford the desired deamination product A and monosilazane B. The rate-determining step of pathway one is the silylation step (TS2) and overall reaction barrier is 27.3 kcal/mol.
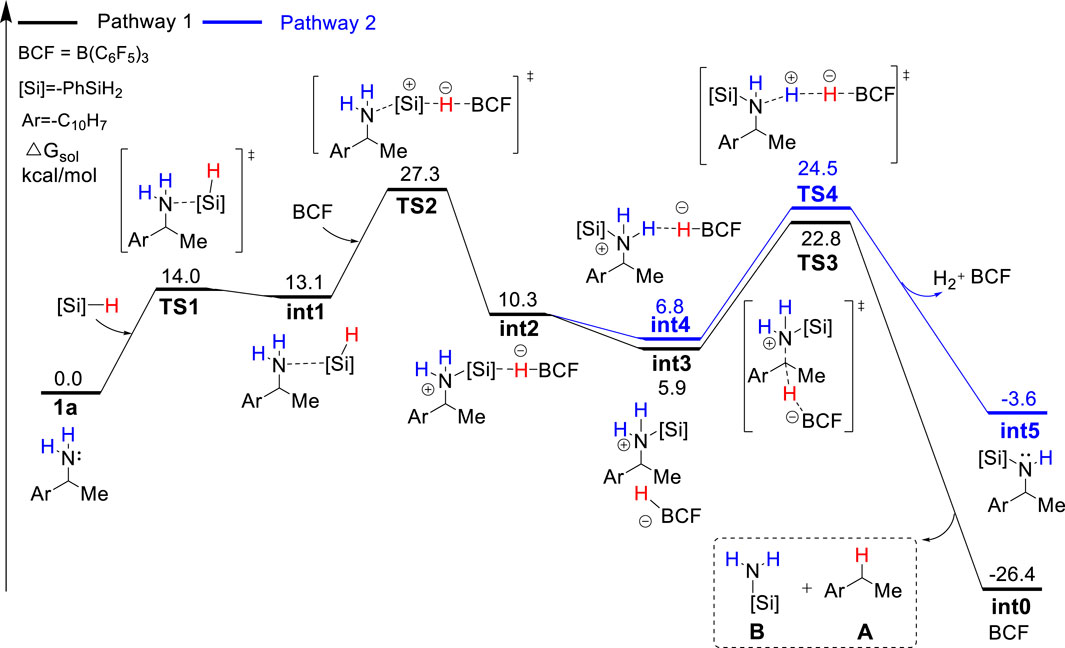
FIGURE 2. Gibbs energy profile for the B(C6F5)3-catalyzed reductive deamination with PhSiH3 and 1a via pathway 1 and 2.
Alternatively, the monosilylammonium borohydride intermediate may occur dehydrogenative reaction (pathway 2, blue color), in which (C6F5)3BH− accepts a proton from the amine group (TS4), which releases a H2 molecule, B(C6F5)3 and mono-silylated amine int5. The Gibbs energy barrier of TS4 is 1.7 kcal/mol higher than that of TS3, indicating that the monosilylammonium borohydride intermediate prefers C–N dissociation than dehydrogenation. Follow the dehydrogenative step, int5 further reacts with another PhSiH3 molecule and B(C6F5)3 catalyst to give the disilylammonium borohydride intermediate, int6 (Figure 3). Like the monosilylammonium borohydride, the disilylammonium borohydride intermediate can undergo an SN2-type C–N cleavage (TS6, ΔG‡ = 26.3 kcal/mol) to yield the deamination species A and disilazane C, completing pathway 2. Pathways one and two bifurcate at the monosilylammonium borohydride intermediate and pathway two is less favorable since the monosilylammonium borohydride intermediate favors C–N dissociation.
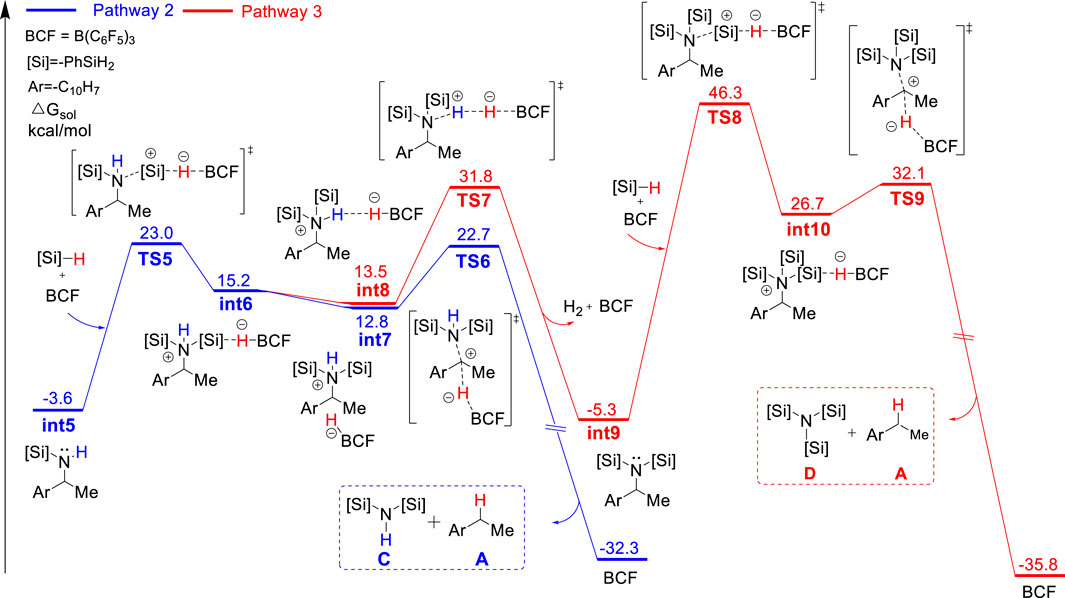
FIGURE 3. Gibbs energy profile for the B(C6F5)3-catalyzed reductive deamination with PhSiH3 and 1a via pathway 2 and 3.
In the alternative pathway 3 (in red color), the dehydrogenation of disilylammonium borohydride intermediate via TS7 (ΔG‡ = 35.4 kcal/mol) releases one H2 molecule and a disilyated amine (int9). int9 then reacts with another molecule of PhSiH3 and B(C6F5)3 for further silylation. The silylation step proceeds via a very high energy barrier transition state, TS8, (ΔG‡ = 51.6 kcal/mol) and yields a highly unstable trisilylammonium borohydride species int10. Finally, the C–N dissociation of the trisilylammonium borohydride via TS9 releases product A and trisilazane D. In summary, the computational results suggest the direct C–N dissociation of the monosilylammonium borohydride to generate monosilazane and deamination product (pathway 1) is the most favorable pathway for this reductive deamination reaction. The first silylation step via TS2 is the rate-limiting step and the overall energy barrier is 27.5 kcal/mol. Moreover, the TSs of dehydrogenation of both mono- and di-silylammonium borohydride are higher in energy than their corresponding TSs of C–N dissociation energy (TS3 vs. TS4 and TS6 vs. TS7), suggesting the deamination is more favorable than dehydrogenation reaction which is consistent with experimental results. The lower activation barrier of deamination can be attributed to the weaker bond strength of C–N bond compared with the N–H bond. The calculated bond dissociation energy of C–N bond is lower than that of N–H bond by 8.5 and 13.2 kcal/mol for mono- and di-silylammonium borohydride, respectively (Supplementary Table S1). Thus, the C–N bond is easier to be cleavage than the N–H bond. In addition, well-ordered π-π stacking interactions between the naphthyl ring of substrate, the phenyl ring of silane and the pentafluorophenyl group of the catalyst were identified in TS3 and TS6 (Figure 4), which help to stabilize the deamination TSs. The NCI analysis (Humphrey et al., 1996; Lu and Chen, 2012) supports the existence of attractive π-π stacking interactions in TS3 and TS6.
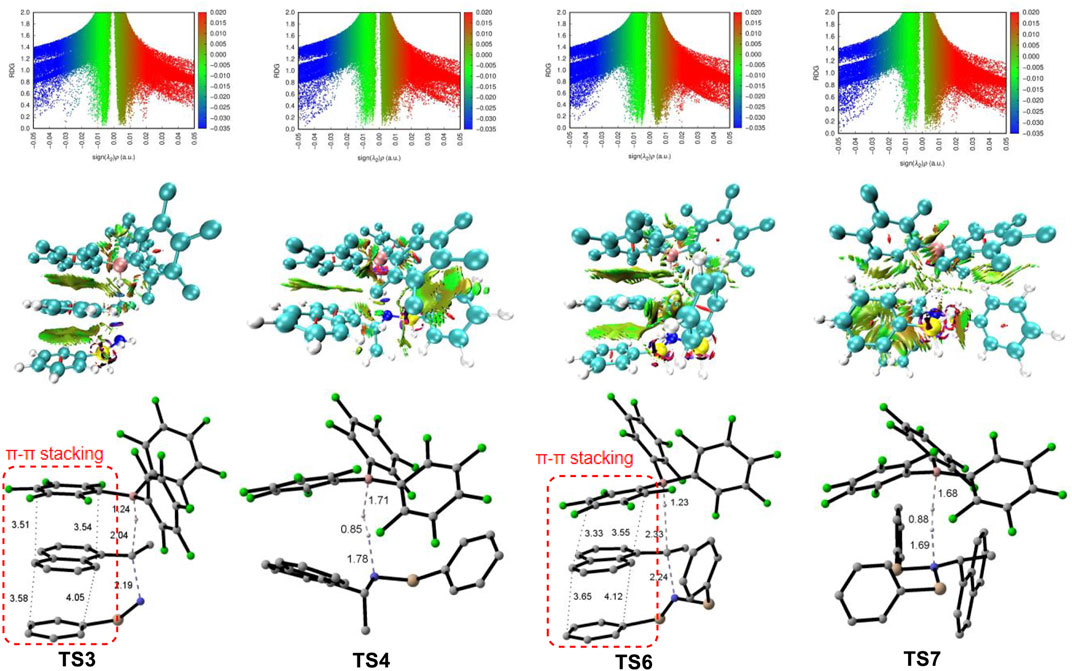
FIGURE 4. Top: Overlapping troughs in s(ρ) plots can be distinguished when sign (λ2)ρ is used as the ordinate. Favorable interactions appear on the left, unfavorable on the right, and van der Waals near zero; Medium: NCI surfaces of TS3, TS4, TS6 and TS7 correspond to s = 0.5 au and a colour scale of −0.05 < ρ < 0.05 au for SCF densities; Bottom: structures of TS3, TS4, TS6 and TS7 (Non-reacting hydrogen atoms are omitted for clarity).
Based on the theoretical calculation, the most favorable pathway (i.e., pathway 1) only consumes one equivalent of PhSiH3 to afford the deamination product A, which contradicts with the experimental observation that four equivalents of PhSiH3 are required to achieve good yields. Because both PhSiH3 and B(C6F5)3 are Lewis acids, we envision that PhSiH3 and B(C6F5)3 may compete to bind with the amine substrate to form amine-silane ([N-Si]) and amine-boron ([N-B]) Lewis adducts, respectively. As shown in Figure 5, the binding of 1a with B(C6F5)3 has a Gibbs energy barrier lower by 2.0 kcal/mol than with PhSiH3 and leads to a very stable [N-B] Lewis adduct int11 (ΔGo = −11.1 kcal/mol). Thus, computational results suggest that the formation of [N–B] Lewis adduct (int11) is kinetically and thermodynamically more favorable than [N–Si] Lewis adduct (int1). The favorable formation of [N–B] Lewis adduct can be attributed to the stronger Lewis acidity of B(C6F5)3. Frontier molecular orbital analysis supports that B(C6F5)3 is a much stronger Lewis acid than PhSiH3 and thus easier to accept a lone pair electron from 1a (Supplementary Figure S2).
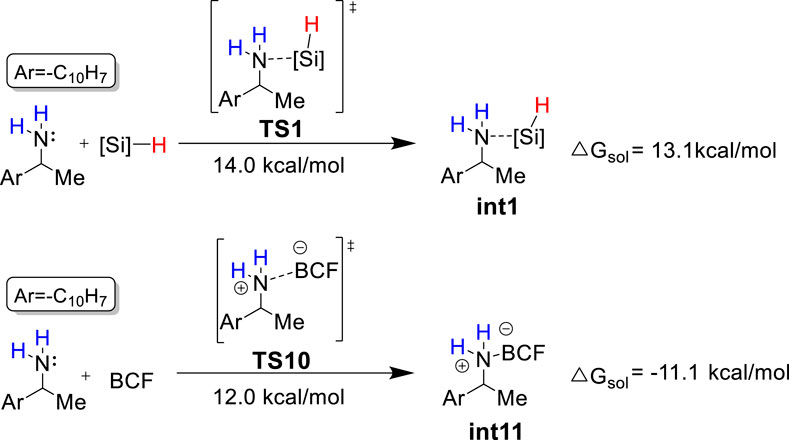
FIGURE 5. Competition between the formation of int1 and int11. BCF = B(C6F5)3; [Si] = −PhSiH2.
The computational results demonstrate that when B(C6F5)3 and PhSiH3 were added with a ratio of 1:1, the amine substrate prefers to bind with B(C6F5)3 and form a very stable Lewis adduct (int11) which is the resting state of catalyst and substrate. Taking the resting state int11 as the start point for the deamination reaction, the overall reaction barrier is as high as 38.6 kcal/mol (int11 to TS2), which is difficult to overcome. This result is in line with the experimental observation that int11 rather than the deamination product was obtained as main product in the stoichiometric experiments with stepwise addition of B(C6F5)3 and PhSiH3.
In the catalytic reaction with 20% mol of B(C6F5)3, the use of four equivalents of PhSiH3 afforded deamination product as main product. We speculate this is related to the concentration effect which influences the competition between the formation of int1 and int11. Based on the reaction rate equation, the reaction rate (r1 and r2) for the formation of int1 and int11 can be calculated by (Eqs 1, 2), respectively. Thus, their ratio (r1/r2) is determined by Eq. 3 which is affected by the ratio of rate constants (k1/k2) and the concentration ([PhSiH3]/[BCF]). k1/k2 is calculated based on the Erying equation (Eq. 4)
As the activation energy difference between TS1 and TS10 is 2.0 kcal/mol (Figure 5), k1/k2 is calculated to be 0.077 at 120°C. Therefore, r1/r2 for the reaction with equivalent amount of PhSiH3 and B(C6F5)3 is as small as 0.077, suggesting the formation of int11 is dominant. However, under the catalytic reaction condition with 20% mol of B(C6F5)3 and 4 equivalents of PhSiH3, the concentration ratio [PhSiH3]/[ BCF] increases to 20. As a result, r1/r2 is increased by 20 folds compared to that of the stoichiometric reaction with equivalent of PhSiH3 and B(C6F5)3, and thus the formation of int1 is 1.54 times faster than int11. This result is in good agreement with the experimental observation that increasing the equivalent of PhSiH3 gradually increases the yields of silylammonium borohydride intermediate and deamination product in the stoichiometric reaction with 1 equivalent of B(C6F5)3. Therefore, the excess PhSiH3 plays a crucial role to maintain a high [PhSiH3]/[BCF] ratio so that PhSiH3 can compete with B(C6F5)3 for binding with the amine substrate, avoiding the deactivation of catalyst and substrate.
Reactivity of amines with different α-substitutions
In the original experimental work, control experiments were performed to assess the relative reactivity of a variety of benzylamines. As shown in Scheme 2, the competition reaction with one equivalent of an equimolar benzylamines mixture (1b, 1c, 1d) demonstrates that the relative reactivity of benzylamines follows the order: 1b < 1c < 1d. To understand the relative reactivity of benzyl amines with different degrees of substitution at the α-carbon atom, we calculated the reaction pathway one for all substrates. Pathway one involves three main steps, i.e., the amine-silane binding, amine silylation, and deamination (Figure 6). It is worth noting that, for substrate 1d, the silylation step (TS2d, ΔG‡ = 26.2 kcal/mol) remains as the rate-limiting step, like the reaction with 1a. However, for substrate 1b and 1c, the C–N bond cleavage of monosilylammonium borohydride (i.e., deamination) via TS3b (ΔG‡ = 28.7 kcal/mol) and TS3c (ΔG‡ = 27.2 kcal/mol) becomes the rate-determining step. Thus, the overall reaction energy barriers for the deamination of 1b/1c/1d are calculated to be 28.7, 27.2 and 26.2 kcal/mol, respectively, which is consistent with the experimental observed reactivity order. Figure 6 clearly shows that the C–N bond cleavage (TS3) of 1d is more favorable than 1b and 1c, which is because the reacting benzylic carbon in TS3d is stabilized by more methyl substituents, leading to stronger stabilization effect on the transition state.
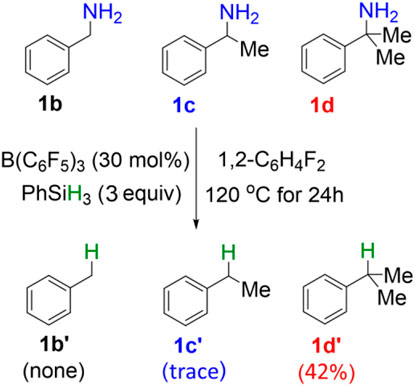
SCHEME 2. Reactivity study on B(C6F5)3-catalyzed reductive deamination with equimolar mixture of benzylamines (1b, 1c, 1d) and PhSiH3.
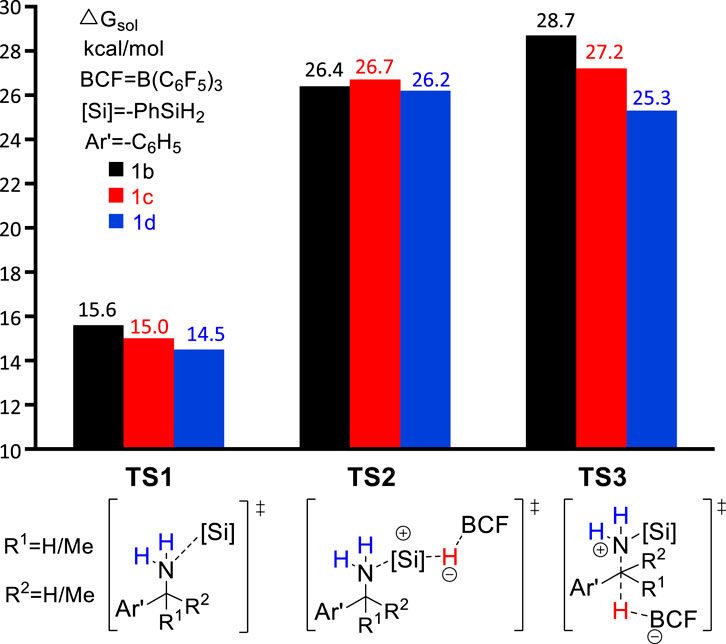
FIGURE 6. Gibbs energy of transition states TS1, TS2, and TS3 for different substrates.
Substituent effect of hydrosilanes
In the end, we turn our attention to the reactivity of hydrosilanes, another important reactant for the deamination reaction. To study the substituent effect of PhSiH3, we calculated the reaction of a series of PhSiH3 derivatives that carry electron-withdrawing groups (EWGs) or electron-donating groups (EDGs) at the phenyl ring. Figure 7 summarizes the Gibbs energies of all TSs of deamination reaction with different silanes (PhSiH3, C6F5SiH3, 1,3,5-C6H3F2SiH3, 1,3,5-C6H3Cl2SiH3, 1,3,5-C6H3Br2SiH3, and 1,3,5-C6H3Me2SiH3). In all reactions, the silylation step (TS2) is the rate-determining step. Compared to unsubstituted PhSiH3, hydrosilanes with EWGs, such as F, Cl, and Br substituents, lower the barrier of TS2 by 0.4–2.0 kcal/mol. Moreover, the formation of amine-silane complex (TS1) becomes more favorable than the generation of amine-boron Lewis product as TS1 for the EWG-substituted hydrosilanes are lower in energy than TS10 by 0.7–1.2 kcal/mol. This indicates that the reaction with these hydrosilanes may not need excess amount of silane reagent. The increased reactivity and binding affinity of hydrosilanes caused by the EWGs may because the EWGs increases the acidity of hydrosilanes which makes them more reactive toward amine substrates. On the contrary, EDGs will decrease the acidity of hydrosilane and thus lower the reactivity. Indeed, the potential energy surface of 1,3,5-C6H3Me2SiH3 lies above the energy surface of PhSiH3.
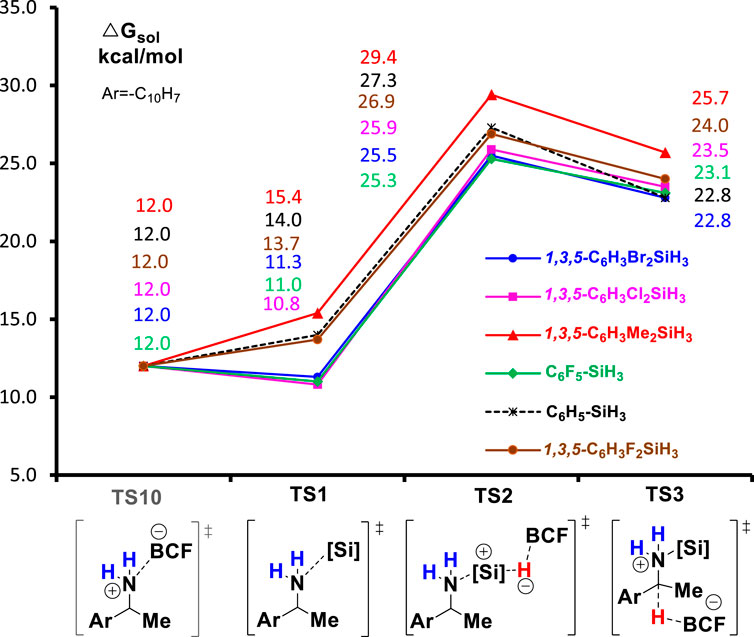
FIGURE 7. The B(C6F5)3-catalyzed reductive deamination with different hydrosilanes.
Conclusion
In the present study, we perform DFT calculations on the reaction of B(C6F5)3-catalyzed reductive deamination of benzylic amines with hydrosilanes. Three possible reaction pathways (shown in Figure 1) involving mono-, bi- or tri-silylammonium borohydride as active intermediate were explored. The computational results reveal that the pathway one which includes the deamination of monosilylammonium borohydride is most favorable. Pathway one consists of three steps: first, amine and silane associate to form an amine-silane Lewis adduct; then, the amine-silane complex is catalyzed by B(C6F5)3 to undergo silylation reaction, affording monosilylammonium borohydride intermediate; finally, the C–N dissociation of monosilylammonium borohydride intermediate generates the desired deamination product. The second step (silylation process) is the rate-determining step. The monosilylammonium borohydride prefers to undergo SN2 C–N bond dissociation rather than the dehydrogenation, probably because the C–N is weaker than the N–H bond and the π-π stacking interaction stabilizes the transition state for C–N bond dissociation.
Our computational results suggest that B(C6F5)3 acts as stronger Lewis acid than hydrosilane to bind with amine substrate, which will deactivate the catalyst and substrate. The excess silanes used in the experiment play an essential role to maintain a high concentration of silane which enables PhSiH3 to compete with B(C6F5)3 for binding with amine substrate, avoiding the deactivation of catalyst and substrate. Furthermore, the calculated relative reactivity of benzylamines with different degrees of substitution agrees well with the experimental observed reactivity order. In addition, our DFT studies on the substituent effect of silanes indicate that the introduction of electron-withdrawing groups on the phenyl ring of PhSiH3 could lower the reaction energy barrier of the reductive deamination reaction, which may improve the reaction efficiency. Overall, this work promotes the understanding of mechanism of deamination reaction and lays the theoretical foundation for the development of new deamination methodology.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
MZ and TW performed calculations and data analysis. G-JC designed the project and wrote the paper. All authors reviewed the manuscript.
Funding
This work is supported by the National Natural Science Foundation of China (no. 21803047), the Shenzhen Science and Technology Program (Grant No. RCYX20200714114736199), and the research grant from Shenzhen Key Laboratory Project (no. ZDSYS20190902093417963).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1025135/full#supplementary-material
Supplementary Figure S1 | Gibbs energy profile for the B(C6F5)3-catalyzed reductive deamination with PhSiH3 and 1b/1c/1d by pathway 2.
Supplementary Figure S2 | Frontier molecular orbital analysis of 1a, B(C6F5) and PhSiH3 (top) and the NCI surfaces of int1 and int11 (bottom).
Supplementary Figure S3 | The formation of int12 and int13.
Supplementary Figure S4 | Gibbs energy profile for the structural rearrangement between int2 and int4.
Supplementary Figure S5 | Gibbs energy profile for the structural rearrangement between int6 and int8.
Supplementary Table S1 | Corrections to zero point energies, enthalpies, free energies and electronic potential energies (in Hartree) and imaginary frequencies (IF) (cm-1) of optimized structures which were calculated at B3LYP-D3/def2-SVP//B3LYP-D3/def2-TZVP level in solvent (1,2- diflurobenzene) at 298.15 K and 393.15K and 1 atm.
References
Adduci, L. L., Bender, T. A., Dabrowski, J. A., and Gagné, M. R. (2015). Chemoselective conversion of biologically sourced polyols into chiral synthons. Nat. Chem. 7, 576–581. doi:10.1038/nchem.2277
Adduci, L. L., McLaughlin, M. P., Bender, T. A., Becker, J. J., and Gagné, M. R. (2014). Metal-free deoxygenation of carbohydrates. Angew. Chem. Int. Ed. 53, 1646–1649. doi:10.1002/anie.201306864
Adlington, M. G., Orfanopoulos, M., and Fry, J. L. (1976). A convenient one-step synthesis of hydrocarbons from alcohols through use of the organosilane-boron trifluoride reducing system. Tetrahedron Lett. 17, 2955–2958. doi:10.1016/s0040-4039(01)85498-8
Barluenga, J., Tomás-Gamasa, M., Aznar, F., and Valdés, C. (2009). Metal-free carbon–carbon bond-forming reductive coupling between boronic acids and tosylhydrazones. Nat. Chem. 1, 494–499. doi:10.1038/nchem.328
Reductive deaminative conversion involving isonitrilation-reduction: Barton, D. H. R., Bringmann, G., Lamotte, G., Motherwell, W. B., Hay Motherwell, R. S., and Porter, A. E. A. (1980). J. Chem. Soc. Perkin Trans. 1, 2657–2664.
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Bender, T. A., Dabrowski, J. A., and Gagné, M. R. (2016). Delineating the multiple roles of B(C6F5)3 in the chemoselective deoxygenation of unsaturated polyols. ACS Catal. 6, 8399–8403. doi:10.1021/acscatal.6b02551
Bender, T. A., Dabrowski, J. A., Zhong, H., and Gagné, M. R. (2016). Diastereoselective B(C6F5)3-catalyzed reductive carbocyclization of unsaturated carbohydrates. Org. Lett. 18, 4120–4123. doi:10.1021/acs.orglett.6b02050
Chatterjee, I., Porwal, D., and Oestreich, M. (2017). B(C6F5)3-Catalyzed chemoselective defunctionalization of ether-containing primary alkyl tosylates with hydrosilanes. Angew. Chem. Int. Ed. 56, 3389–3391. doi:10.1002/anie.201611813
Cheng, G., Drosos, N., Morandi, B., and Thiel, W. (2018). Computational study of B(C6F5)3-catalyzed selective deoxygenation of 1, 2-diols: Cyclic and noncyclic pathways. ACS Catal. 8, 1697–1702. doi:10.1021/acscatal.7b04209
Corma, A., Iborra, S., and Velty, A. (2007). Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 107, 2411–2502. doi:10.1021/cr050989d
Correia, J. T. M., Fernandes, V. A., Matsuo, B. T., Delgado, J. A. C., de Souza, W. C., and Paixão, M. W. (2020). Chem. Commun. 56 (4), 503–514.
Dai, X., and Li, C. (2016). En route to a practical primary alcohol deoxygenation. J. Am. Chem. Soc. 138, 5433–5440. doi:10.1021/jacs.6b02344
Doyle, M. P., West, T. C., Donnelly, J. S., and McOsker, C. C. (1976). Silane reductions in acidic media. J. Organomet. Chem. 117, 129–140. doi:10.1016/s0022-328x(00)87264-2
Drosos, N., Cheng, G., Ozkal, E., Cacherat, B., Thiel, W., and Morandi, B. (2017). Catalytic reductive pinacol-type rearrangement of unactivated 1, 2-diols through a concerted, stereoinvertive mechanism. Angew. Chem. Int. Ed. 56, 13377–13381. doi:10.1002/anie.201704936
Drosos, N., and Morandi, B. (2015). Boron-catalyzed regioselective deoxygenation of terminal 1, 2-diols to 2-alkanols enabled by the strategic formation of a cyclic siloxane intermediate. Angew. Chem. Int. Ed. 54, 8814–8818. doi:10.1002/anie.201503172
Fang, H., and Oestreich, M. (2020). Defunctionalisation catalysed by boron Lewis acids. Chem. Sci. 11, 12604–12615. doi:10.1039/d0sc03712e
Fang, H., and Oestreich, M. (2020). Reductive deamination with hydrosilanes catalyzed by B(C6F5)3. Angew. Chem. Int. Ed. 59, 11394–11398. doi:10.1002/anie.202004651
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, revision A.03. Wallingford CT: Gaussian, Inc.
Geoffroy, O. J., Morinellib, T. A., and Meier, G. P. (2001). Chemoselective one-pot reductive deamination of aryl amines. Tetrahedron Lett. 42, 5367–5369. doi:10.1016/s0040-4039(01)01027-9
Gevorgyan, V., Liu, J., Rubin, M., Benson, S., and Yamamoto, Y. (1999). A novel reduction of alcohols and ethers with a HSiEt3catalytic B(C6F5)3 system. Tetrahedron Lett. 40, 8919–8922. doi:10.1016/s0040-4039(99)01757-8
Gevorgyan, V., Rubin, M., Benson, S., Liu, J., and Yamamoto, Y. (2000). A novel B(C6F5)3-catalyzed reduction of alcohols and cleavage of aryl and alkyl ethers with hydrosilanes. J. Org. Chem. 65, 6179–6186. doi:10.1021/jo000726d
Gevorgyan, V., Rubin, M., Liu, J., and Yamamoto, Y. (2001). A direct reduction of aliphatic aldehyde, acyl chloride, ester, and carboxylic functions into a methyl group. J. Org. Chem. 66, 1672–1675. doi:10.1021/jo001258a
Gonzalez, C., and Gonzalez, H. B. (1990). Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 94, 5523–5527. doi:10.1021/j100377a021
Greb, L., Tamke, S., and Paradies, J. (2014). Catalytic metal-free Si–N cross-dehydrocoupling. Chem. Commun. 50, 2318–2320. doi:10.1039/c3cc49558b
Grimme, S., Ehrlich, S., and Goerigk, L. J. (2011). Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465. doi:10.1002/jcc.21759
Grimme, S. (2019). Exploration of chemical compound, conformer, and reaction space with meta-dynamics simulations based on tight-binding quantum chemical calculations. J. Chem. Theory Comput. 15, 2847–2862. doi:10.1021/acs.jctc.9b00143
Hermeke, J., Mewald, M., and Oestreich, M. (2013). Experimental analysis of the catalytic cycle of the borane-promoted imine reduction with hydrosilanes: Spectroscopic detection of unexpected intermediates and a refined mechanism. J. Am. Chem. Soc. 135, 17537–17546. doi:10.1021/ja409344w
Hu, J., Wang, G., Li, Sh., and Shi, Zh. (2018). Selective C−N borylation of alkyl amines promoted by Lewis base. Angew. Chem. Int. Ed. 57, 15227–15231. doi:10.1002/anie.201809608
Huber, G. W., Iborra, S., and Corma, A. (2006). Synthesis of transportation fuels from Biomass: chemistry, catalysts, and engineering. Chem. Rev. 106, 4044–4098. doi:10.1021/cr068360d
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi:10.1016/0263-7855(96)00018-5
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi:10.1103/physrevb.37.785
Legault, C. Y. (2009). CYLview, 1.0b. Sherbrooke, Canada: Université de Sherbrooke. Available at: http://www.cylview.org.
Lu, T., and Chen, F. (2012). Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592. doi:10.1002/jcc.22885
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396. doi:10.1021/jp810292n
Massad, G., Zhao, H., and Mobley, H. L. (1995). Proteus mirabilis amino acid deaminase: Cloning, nucleotide sequence, and characterization of aad. J. Bacteriol. 177, 5878–5883. doi:10.1128/jb.177.20.5878-5883.1995
McLaughlin, M. P., Adduci, L. L., Becker, J. J., and Gagne, M. R. (2013). Iridium-catalyzed hydrosilylative reduction of glucose to hexane(s). J. Am. Chem. Soc. 135, 1225–1227. doi:10.1021/ja3110494
Reductive deaminative conversion involving or diazotization-dediazotization: Mitsuhashi, H., Kawakami, T., and Suzuki, H. (2000). Tetrahedron Lett. 41, 5567–5569.
Molla, G., Melis, R., and Pollegioni, L. (2017). Breaking the mirror: l-Amino acid deaminase, a novel stereoselective biocatalyst. Biotechnol. Adv. 35, 657–668. doi:10.1016/j.biotechadv.2017.07.011
Katritzky salts as intermediate to construct for C-C, C-B and C-S bond: Ni, S., Li, C., Mao, Y., Han, J., Wang, Y., Yan, H., et al. (2019). Ni-catalyzed deaminative cross-electrophile coupling of Katritzky salts with halides via C─N bond activation. Sci. Adv. 5, 1–5. doi:10.1126/sciadv.aaw9516
Nikolaos Drosos, M. Sc., and Morandi, B. (2015). Boron-catalyzed regioselective deoxygenation of terminal 1, 2-diols to 2-alkanols enabled by the strategic formation of a cyclic siloxane intermediate. Angew. Chem. Int. Ed. 54, 8814–8818. doi:10.1002/anie.201503172
Nimmagadda, R. D., and McRae, C. (2006). A novel reduction reaction for the conversion of aldehydes, ketones and primary, secondary and tertiary alcohols into their corresponding alkanes. Tetrahedron Lett. 47, 5755–5758. doi:10.1016/j.tetlet.2006.06.007
Nshimiyimana, P., Liu, L., and Du, G. (2019). Engineering of L-amino acid deaminases for the production of α-keto acids from L-amino acids. Bioengineered 10, 43–51. doi:10.1080/21655979.2019.1595990
Orfanopoulos, M., and Smonou, I. (1988). Selective reduction of diaryl or aryl alkyl alcohols in the presence of primary hydroxyl or ester groups by etherated boron trifluoride-triethylsilane system. Synth. Commun. 18, 833–839. doi:10.1080/00397918808057852
Pang, Y., Moser, D., and Cornella, J. (2020). Pyrylium salts: Selective reagents for the activation of primary amino groups in organic synthesis. Synthesis 52, 489–503. doi:10.1055/s-0039-1690703
Peng, C., Zhang, W., Yan, G., and Wang, J. (2009). Arylation and vinylation of α-diazocarbonyl compounds with boroxines. Org. Lett. 11, 1667–1670. doi:10.1021/ol900362d
Petersen-Mahrt, S. K., Coker, H. A., and Pauklin, S. (2009). DNA deaminases: AIDing hormones in immunity and cancer. J. Mol. Med. 87, 893–897. doi:10.1007/s00109-009-0496-6
Richter, S. C., and Oestreich, M. (2021). Chemoselective deoxygenation of 2° benzylic alcohols through a sequence of formylation and B(C 6 F 5 ) 3 -catalyzed reduction. Eur. J. Org. Chem. 2021, 2103–2106. doi:10.1002/ejoc.202100148
Schafer, A., Horn, H., and Ahlrichs, R. (1992). Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577. doi:10.1063/1.463096
Schafer, A., Huber, C., and Ahlrichs, R. (1994). Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835. doi:10.1063/1.467146
Seo, Y., and Gagné, M. R. (2018). Positional selectivity in the hydrosilylative partial deoxygenation of disaccharides by boron catalysts. ACS Catal. 8, 81–85. doi:10.1021/acscatal.7b02992
Seo, Y., Gudz, A., Lowe, J. M., and Gagné, M. R. (2019). Selective deoxygenation of gibberellic acid with fluoroarylborane catalysts. Tetrahedron 75, 130712–130720. doi:10.1016/j.tet.2019.130712
Vesely, C., Tauber, S., Sedlazeck, F. J., von Haeseler, A., and Jantsch, M. F. (2012). Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res. 22, 1468–1476. doi:10.1101/gr.133025.111
Wang, X., Kuang, Y., Ye, S., and Wu, J. (2019). Photoredox-catalyzed synthesis of sulfones through deaminative insertion of sulfur dioxide. Chem. Commun. 55, 14962–14964. doi:10.1039/c9cc08333b
Wu, G., Deng, Y., Wu, C., Wang, X., Zhang, Y., and Wang, J. (2014). Switchable 2, 2, 2-trifluoroethylation andgem-difluorovinylation of organoboronic acids with 2, 2, 2-trifluorodiazoethane. Eur. J. Org. Chem. 21, 4477–4481. doi:10.1002/ejoc.201402597
Yasuda, M., Onishi, Y., Ueba, M., Miyai, T., and Baba, A. (2001). Direct reduction of Alcohols: highly chemoselective reducing system for secondary or tertiary alcohols using chlorodiphenylsilane with a catalytic amount of indium trichloride. J. Org. Chem. 66, 7741–7744. doi:10.1021/jo0158534
Zhang, J., Park, S., and Chang, S. (2018). Catalytic access to bridged sila-N-heterocycles from piperidines via cascade sp3 and sp2 C–Si bond formation. J. Am. Chem. Soc. 140, 13209–13213. doi:10.1021/jacs.8b08733
Zhang, Z., Song, J., and Han, B. (2017). Catalytic transformation of lignocellulose into chemicals and fuel products in ionic liquids. Chem. Rev. 117, 6834–6880. doi:10.1021/acs.chemrev.6b00457
Zhou, C. H., Xia, X., Lin, C. X., Tong, D. S., and Beltramini, J. (2011). Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 40, 5588–5617. doi:10.1039/c1cs15124j
Keywords: B(C6F5)3, reductive deamination, reaction mechanism, substituent effect, DFT
Citation: Zhou M, Wang T and Cheng G-J (2022) Mechanistic insights into reductive deamination with hydrosilanes catalyzed by B(C6F5)3: A DFT study. Front. Chem. 10:1025135. doi: 10.3389/fchem.2022.1025135
Received: 22 August 2022; Accepted: 20 September 2022;
Published: 16 November 2022.
Edited by:
Israel Fernandez, Complutense University of Madrid, SpainReviewed by:
Miquel Solà, University of Girona, SpainCina Foroutan-Nejad, Institute of Organic Chemistry (PAN), Poland
Copyright © 2022 Zhou, Wang and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gui-Juan Cheng, Z3VpanVhbmNoZW5nQGZveG1haWwuY29t
†These authors have contributed equally to this work