 Boao Li1
Boao Li1 Xiangyu Zhang
Xiangyu Zhang- 1Department of Colorectal Surgery, Liaoning Cancer Hospital, Shenyang, China
- 2State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing, China
The LSD1 protein is an oxidase that regulates protein methylation, which regulates gene expression and triggers tumors. Previously, inhibiting LSD1 has been found to be an effective treatment strategy for opposing tumors caused by overexpression of LSD1. Our recent study found that compound 17i was a suitable LSD1 inhibitor with potential anti-tumor activity. However, its extremely insoluble nature limits further validation of its anti-tumor activity at the clinical level. In this study, a unique carrier-free supramolecular nanoassemblies of pure compound 17i is expected to enhance therapeutic efficacy. Aqueous-insoluble compound 17i was mixed with a small quantity of DSPE-PEG2000 into an organic solvent and was prepared as nanoassemblies in water via the one-step nanoprecipitation method. The 17i nanoassemblies have a similar effect on its cytotoxicity when compared with 17i solution in vitro. Importantly, the PEGylated 17i nanoassemblies exhibit significant superiorities over 17i solutions in therapeutic efficiency, anti-tumor immune response and systemic toxicity in BALB/c mice bearing CT-26 colorectal tumors. We envision that the fabrication of pure drug nanoassemblies offers an efficient platform for reforming the undesirable characteristics of drug-like compounds to potentiate the anti-tumor therapeutic effect.
Introduction
In histone lysine specific demethylase 1 (LSD1), the methyl groups are removed from the lysine residues (H3K4 and H3K9) by an oxidative enzyme (Shi et al., 2004; Suzuki and Miyata, 2011; Schmitt et al., 2013; Sorna et al., 2013; Tortorici et al., 2013; Zheng et al., 2013). As an epigenetic regulator, gene expression and cancer initiation are influenced by LSD1 (Ma et al., 2015; Zheng et al., 2015; Vianello et al., 2016). Therefore, inhibiting LSD1 is an effective strategy for anti-tumor treatment (Wu et al., 2016; Mould et al., 2017). In our previous study, various LSD1 inhibitors were reported (Wang X. et al., 2020; Zhang et al., 2021), in which the compound 17i (Figure 1) (IC50 = 0.065 μM) exhibited a significant effect on target LSD1. The solubility of compound 17i in organic solvents was good. However, it has been tested that 17i is almost insoluble in water. This impeded further validation of its anti-tumor activity at the clinical level.
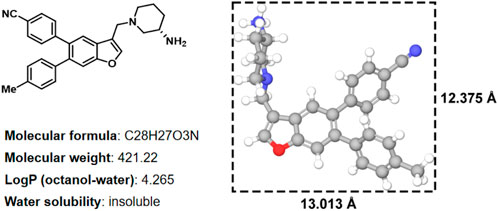
FIGURE 1. Two-dimensional chemical structure, physicochemical properties, three-dimensional structures and sizes of compound 17i (Å, angstrom).
The wide application of nanotechnology in the medical field has significantly enriched the delivery strategies of anti-tumor drugs (Sun M. et al., 2019; Sun et al., 2022). Especially, the construction of a novel nanodrug delivery system can significantly improve the druggability of chemotherapeutics, prolong the systemic circulation time and enhance tumor-specific accumulation by increasing permeability and retention (EPR) effect, thus augmenting the anti-tumor effect and reduce the side effects (Sun B. et al., 2019; Wang Q. et al., 2020; Yang et al., 2020; Zhang et al., 2020; Zhao et al., 2021). Herein, to solve the dilemma of clinical transformation of compound 17i, we put forward a fascinating nanoassembly based on compound 17i for effective anti-tumor therapy. Firstly, we confirm tight interactions between LSD1 and compound 17i through molecular docking and molecular dynamic simulation, elaborating that compound 17i had effective inhibitory activity for LSD1. The supramolecular nanoassemblies of 17i were fabricated by a one-step nanoprecipitation approach. A small amount of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethyleneglycol)-2000] (DSPE-PEG2000) (Supplementary Figure S3) was attached on the surface to improve surface hydrophilicity of nanoassemblies and extend the period of blood circulation. The PEGylated 17i nanoassemblies exhibited comparable cytotoxicity when compared with 17i solution in vitro, but showed particular superiorities in terms of efficient anti-tumor therapy and anti-tumor immune response and less side effect in BALB/c mice bearing CT-26 tumors. As far as we are aware, this is the first time pure LSD1 inhibitor has been fabricated into nanoassemblies without the addition of carrier excipients. Such a potent nanoplatform holds promising clinical application prospects for drug-like compounds.
Materials and methods
Materials
Compound 17i was self-prepare. 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) and 3-(4,5-Dimthyl-2-thiazolyl)-2,5-dipphenyl-2H-terazolium bromide (MTT) were obtained from Dalian Meilun Biotech Co., Ltd. (Dalian, China). Cell culture media RPMI 1640, penicilline-streptomycin, and fetal bovine serum were available from GIBCO, (Carlsbad, United States). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethyleneglycol)-2000] (DSPE-PEG2000) was purchased from Shanghai Advanced Vehicle Technology Co., Ltd. All other chemical components and solvents applied in this study are of analytical grade.
Molecular docking
To predict the binding mode of the target molecule with the binding site, we performed molecular docking using Glide 9.7 module (Friesner et al., 2004; Friesner et al., 2006), which the protein structure PDB 5YJB (residues 172–833) used for docking. The inhibitor 17i was docked into the binding pocket of LSD1 using the standard precision module to get initial binding predictions and docking sores.
MD simulation
The 100 ns MD simulations were carried out of LSD1-17i complex by using Desmond v3.8 (Wang et al., 2019). This system is dissolved in a cubic box (8 Å ✕30 Å ✕8 Å) with a simple point charge (SPC) water, adding an appropriate amount of Na+ counter ions to achieve neutralization. Based on the OPLS-2005 force field, the energy of the system was minimized. Finally, the 100 ns MD simulations were performed in NPT ensemble. Other parameters are default.
Preparation of supramolecular 17i nanoassemblies
Nanoassemblies based on pure 17i compound were prepared by the one-step nanoprecipitation method. 8 mg 17i was dissolved in 1 ml methanol to acquire 17i methanol solution. Then, 500 μL mixtures were dropped slowly into the 2 ml aqueous solution under the stirring for 5 min. Following that, methanol was removed from the colloidal solution in a vacuum at 37°C. The PEGylated 17i nanoassemblies were prepared in above protocol using a mixed methanol solution of 17i and DSPE-PEG2000 (15%, w/w). The prepared nanoassemblies would be stored at 4°C. In addtion, the 17i reagents were dissolved in 0.5 ml acetonitrile. Then, the mother liquor was diluted with PBS (1:9), prepared as the 17i solution.
Characterization of supramolecular 17i nanoassemblies
The dynamic light scattering particle size of 17i nanoassemblies was measured through a Zetasizer (Malvern Co., UK). The prepared nanoassemblies were diluted with phosphate buffer solution (PBS) and the particle size was measured three times. The 17i nanoassemblies were diluted 1/20 with deionized water and dropped onto a copper mesh (300 mesh) covered by carbon film. After natural drying, negative staining was performed with 2% phosphotungstate acid. Transmission electron microscopy (TEM) (Hitachi, HT7700, Japan) was used to observe the appearance and morphology of 17i nanoassemblies.
Binding conformation and binding energy of two molecules of 17i together was calculated with molecular docking. Docking was performed using Glide module in Schrödinger. One molecule of 17i was selected as the receptor and receptor grid was set to cover the whole receptor molecule, and then another molecule of 17i was docked onto 17i receptor, and binding energy were calculated.
Molecular dynamic (MD) simulation was performed with Materials Studio Program (Accelrys Inc.). First, amorphous cell module was used to construct molecular aggregation models in which 16 17i molecules and 11170 water molecules were put into cubic box with side length equals 7ns. Then 50000 steps energy minimization were performed followed with 50 ns molecular dynamic with compass force field in temperature of 298K, pressure of 1.01325 bar and NPT ensemble. Root mean square derivation (RMSD) value were calculated using forcite module.
Simulation study of assembly
Computational simulations of intermolecular interactions between 17i molecules were performed. The two-dimensional (2D) structure of 17i was established by Marvin sketch software, and the three-dimensional (3D) structure of 17i was optimized by Sybyl 6.9.1 software package. In the previous work, we have introduced the runtime simulation environment and other method parameters in detail.
Cell viability
CT-26 cells were cultured with RPMI-1640 cells containing 10% FBS and 100 μg/ml 3 × 103 cells were cultured in 96-well plates for 12 h to evaluate the cellular viability of 17i nanoparts. Next, the medium was replaced with different concentrations of 17i solutions and 17i nanocomposites containing 10% alcohol.
24 or 48 h later, MTT solution (1 mg/ml) was placed in an incubator for further incubation for about 4 h. Drained from supernatant, DMSO was then added to each well, and the purple crystals were fully dissolved by slowly shaking for 5 min. The absorbance value of each well was measured at 492 nm by the multifunctional microplate analyzer.
Animal studies
All animal protocols were evaluated and approved by the Animal Laboratory Ethics Committee of the Liaoning Cancer Hospital. The BALB/c mice bearing CT-26 tumors were established. PBS, 17i solution and PEGylated 17i nanoassemblies (20 mg/kg 17i) were intraperitoneal-injected into the mice separately. The drug was administered every other day for five times, and the tumor volume was measured and weighed daily. On the last day of the efficacy trial, animals were killed, and tumor tissues were isolated, weighed, and photographed to compare the effects of different preparations on tumor growth.
Flow cytometry analysis
Tumor tissues after different treatments were extracted and collected from CT-26-bearing BALB/c mice. Tumor tissue were conducted to prepare a single-cell suspension. Then, the cells were stained with fluorescence-labeled antibodies CD3, CD4 and CD8 in compliance with the instructions of manufacturers. The proportion of stained cells from tumor tissues were measured using flow cytometry and data were analyzed using FlowJo software.
Statistical analysis
Statistical analysis was carried out using Graphpad Prism software. All data results were showed as mean ± standard deviation (SD). Student’s T-test was used to analyze differences between groups. The p < 0.05 was deemed statistically significant (Sun B. et al., 2019; Sun M. et al., 2019; Wang Q. et al., 2020; Sun et al., 2022).
Results
Computational simulation studies
The binding affinity of inhibitor 17i in LSD1 (PDB code 5YJB) was first evaluated using a combined docking and molecular dynamics protocol. Figure 2 illustrates the possible binding schema between 17i and LSD1 was predicted by glide 9.7 software with a high binding affinity (SP score = -9.109 kcal/mol), consisting of its bioactivity (IC50 = 65 nM).
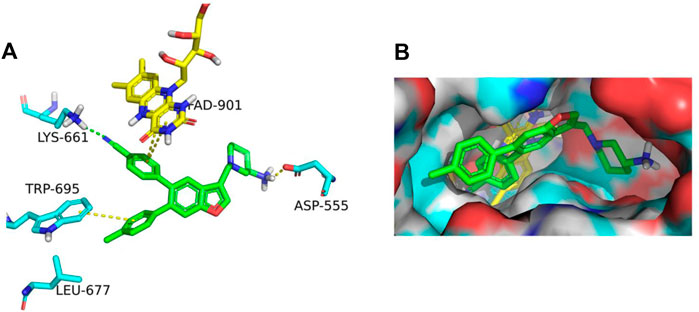
FIGURE 2. (A) In LSD1, 17i (green) binds to its active site. (B) 17i represents the binding surface of the LSD1 pocket (PDB: 5YJB).
Next, 100 ns MD simulations of the protein-ligand complex (LSD1 protein and compound 17i) were performed to predefined binding modes using Desmond v3.8. As depicted in Figure 3A, the LSD1-17i complex reached equilibrium at about 80 ns and the fluctuation of RMSD values (Å) was found to be 2–4 Å, indicating that compound 17i was stabilized favorably with the active site during the binding process. Meanwhile, the contributions of amino acid interactions were also analyzed during the MD simulation in Figures 3B,C. Hydrogen bonds were formed between the piperidine-3-amine moiety of 17i and Asp555 and Pro808, accounting for 100 and 51% respectively. The nitrogen atom on the piperidine ring also interacted with Ala539 through water molecules to form the hydrogen bond, counting for 16%. Surprisingly, the Lys661-mediated hydrogen bond interaction was weak (less than 10%). There was little that Lys661 contributed to the activity of the compound 17i during the binding process. In addition, some new residues not determined by molecular docking were observed, such as His564. It was positioned in proximity of compound 17i and participated in π-π stacking interactions with the ring of benzofuran ring.
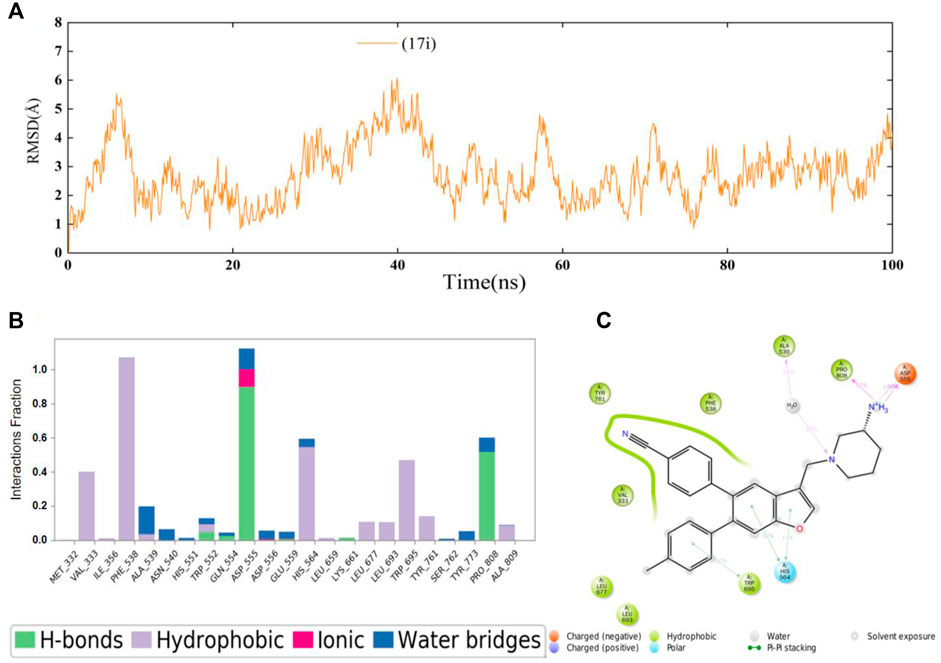
FIGURE 3. (A) Time evolution of the RMSD value of compound 17i analyzed by 100 ns molecular dynamic (MD) simulations. (B) Statistical protein-ligand contacts of compound 17i and (C) Two-dimensional interactions of compound 17i during the whole MD simulations.
Preparation and characterization of 17i nanoassemblies
For the fabrication of 17i nanoassemblies, one-step nanoprecipitation was used. The hydrophobic 17i molecules spontaneously assembled into uniform 17i nanoassemblies formed without the aid of any carrier excipients. We constructed PEGylated 17i nanoassemblies with a small quantity DSPE-PEG2000 (15 wt%). Obviously, nanoassemblies composed of 17i molecules were obviously the main components, and PEGylated 17i nanoassemblies loaded drugs more than 80 wt% efficiently.
17i molecules themselves were the main component of nanoassemblies, and the drug loading efficiency of the PEGylated 17i nanoassemblies was more than 80 wt%. The hydrated particle size of the nanoassemblies by dynamic light scattering (DLS) was 188.4 ± 0.332 nm. The image showed the irregular spheres and a particle size of <200 nm in dehydrated diameter, as determined by transmission electron microscopy (TEM) (Figures 4A,B). The critical aggregation concentration (CAC) of 17i-based nanoassemblies was equal to 1 μg/ml compound 17i. Subsequently, the assembly mechanisms of pure compound 17i were then evaluated using molecular docking. Multiple intermolecular interactions led to the formation of hydrogen bonds, hydrophobic interactions, and stacking interactions in this nanosystem (Figure 4C). It was assumed that the hydrophobic interactions among compound 17i molecules could drive to gather, and the hydrophilic amino groups in 17i molecules were exposed and coexisted stably with water by hydrogen bonds.
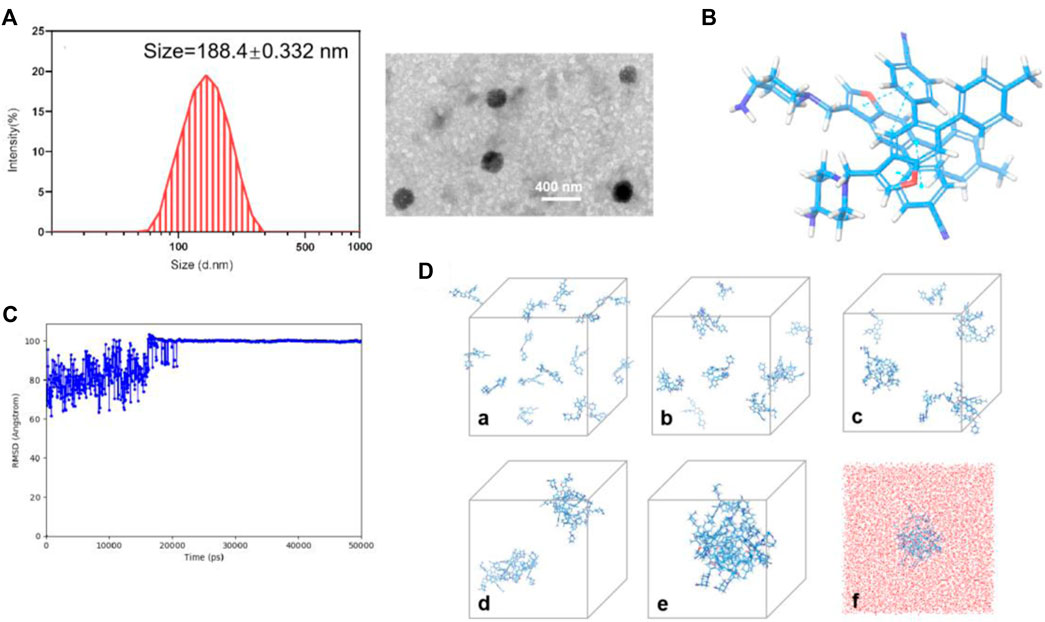
FIGURE 4. (A) Particle size distribution and TEM images of PEGylated 17i nanoassemblies. (B) Docking diagram of the assembly mechanism of compound 17i. (C) Root Mean Square Deviation (RMSD) values of the nanoassembly system. (D) The aggregation process of 17i in water at (a) 0 ns, (b) 2.5 ns, (c) 6.1 ns, (d) 14 ns, (e) 50 ns and (f) 50 ns contained water beads (red beads).
Next, we investigated the cellular uptake of PEGylated 17i nanoassemblies by measuring the amount of 17i after incubation with CT-26 murine colorectal tumor cells. Figure 5A shows that, with the passage of time, 17i nanoassemblies treated with PEGylated nanoparticles produced stronger red intracellular fluorescence. In addition, a MTT assay was conducted to determine the cytotoxicity of 17i solution and PEGylated 17i nanoassemblies in vitro (Figure 5B). For CT-26 cells, the cytotoxicity of PEGylated 17i nanoassemblies was comparable to that of 17i compound solutions, demonstrating that 17i nanoassemblies had a negligible effect on its in vitro cytotoxicity. The wound healing assay and apoptosis induction was also used to investigate the anti-cancer activity in vitro (Supplementary Figure S4). Similar to results of the cytotoxicity experiment, the anti-cancer ability of 17i nanoassemblies make it a potent candidate for further in vivo evaluation.
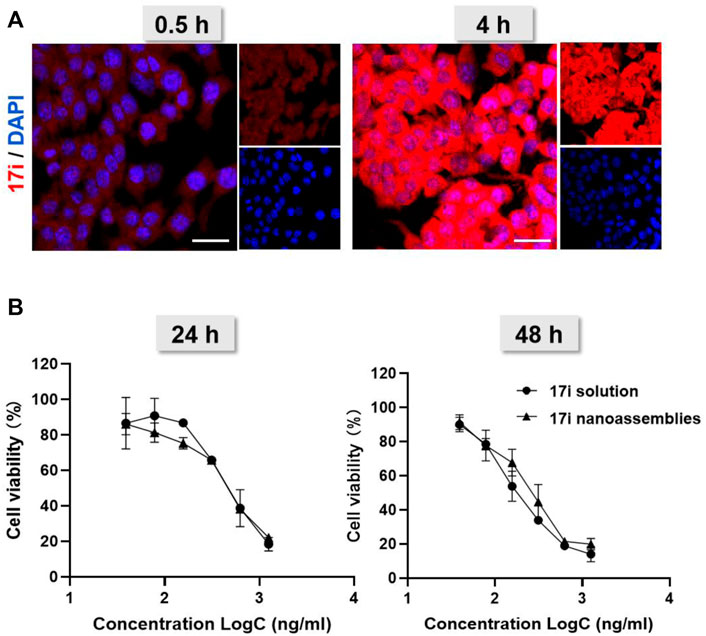
FIGURE 5. (A) Cellular uptake: confocal laser scanning microscopy (CLSM) images of CT-26 cells after being treated for 0.5 and 4 h with 17i nanoassemblies. Scale bar = 20 μm. (B) Viability of CT-26 cells after treated with various concentrations of 17i solution and 17i nanoassemblies for 24 and 48 h, respectively (n = 3).
In vivo antitumor studies
This part evaluated the anti-tumor activity of CT-26 in vivo. PBS, 17i solution and PEGylated 17i nanoassemblies (20 mg kg−1 of 17i) were treated by intraperitoneal administration for a total of five times. As shown in Figure 6, the PBS group could not inhibit the rapid growth of the tumor. In contrast, both 17i solution and PEGylated 17i nanoassemblies had an anti-tumor effect, and the tumor growth rate was significantly slowed down. The PEGylated 17i nanoassemblies had a much stronger anti-tumor effect than the 17i solution. Like the intraperitoneal administrated model, the anti-tumor activity of 17i nanoassemblies in the intravenous administrated model was better than the other groups (Supplementary Figure S5). In addition, as illustrated in Figure 7, In immunofluorescence staining and flow cytometry, C57 mice bearing CT-26 were significantly infiltrated by CD8+ T cells in tumor regions after receiving PEGylated 17i nanoassemblies. It is speculated that the appropriate particle size of PEGylated 17i nanoassemblies was more conducive to drugs enrichment in the tumor site via the EPR effect. An enhanced anti-tumor efficacy was demonstrated by the presence of enriched 17i at tumor sites, resulting in an increased number of CD8+ T cells infiltrating the tumor site. The PEGylated nanoassemblies showed stronger anti-tumor activity than the solution, which is determined by the pharmacologic advantages of nanoassemblies: 1) extremely high drug loading (more than 80%); 2) long circulation time in the body; 3) strong ability of tumor-targeted accumulation. These factors lead to the large area of apoptosis and necrosis in tumor tissue, showing a great anti-tumor effect. In our previous study, the body weight of the nude mice after the treatment of compound 17i at a high dose (20 mg/kg/d) was loss to some extent during the treatment (Zhang et al., 2021). In this study, H&E stained images are provided, and blood tests are conducted to demonstrate the safety of PEGylated 17i nanoassemblies in Supplementary Figure S6, S7. This is mainly due to their biophysical targeting properties and little distribution to other tissues.
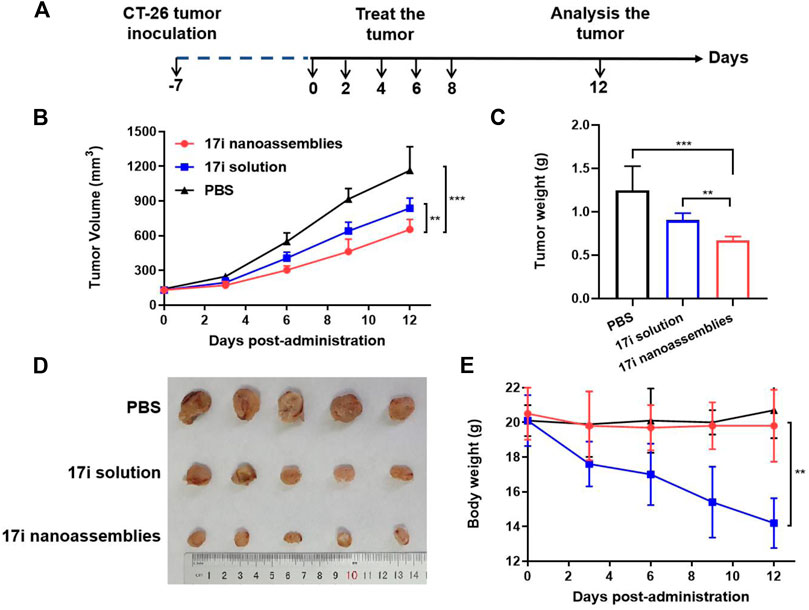
FIGURE 6. In vivo anti-tumor efficacy of 17i solution and PEGylated 17i nanoassemblies against CT-26 tumors. (A) Therapeutic protocol on mouse CT26 subcutaneous tumor xenograft. (B) Tumor volume growth curve after different treatments. (C) Tumor weight (G) of mice after 12 days in different treatments (n = 5). (D) After repeated administration, excised tumors of different groups are shown. (E) Changes in body weight of mice during different treatments (n = 5).
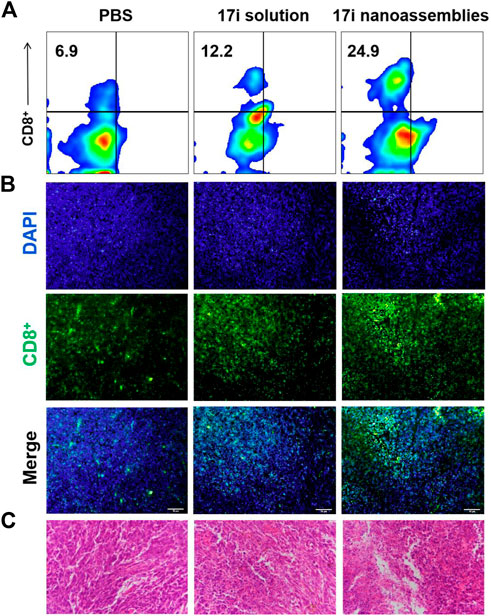
FIGURE 7. (A) Flow cytometry, (B) immunofluorescence and (C) H&E stained images of tumor slices. The nuclei were stained with DAPI (blue) and CD8 antibody (green). Scale bar = 50 μm.
Discussion
In the clinical setting, poorly soluble drug molecules often have low bioavailability issues and absorption problems. Thus, almost 70% of potential drugs were discarded because of their poorly soluble. As the number of poorly soluble drugs increases from discovery, developing technology to enhance their solubility as well as control their release is one of the many challenges facing the pharmaceutical industry. Reducing the size of insoluble molecules is an effective solubilization method. However, there is no simple and reliable technology to manufacture and stabilize nanoparticles in aqueous solution. Polymer carriers are used to load insoluble drug molecules, but they still face problems such as low drug loading capacity. In addition, their pharmacokinetic characteristics are closer to the properties of carrier which also affect key parameters such as distribution and drug release. Further evaluation and modification of carriers are always needed.
Carrier-free supramolecular nanoassemblies of pure compound 17i is expected to enhance its solubility without causing problems such as low drug loading capacity as mentioned above. The drug content is greater than 80%, and the preparation process is controllable and simple. Importantly, the preparation process does not involve the use of organic solvents for dissolving hydrophobic precursors, thereby solving safety issues (such as possible toxicity) from carriers or organic solvents. On the one hand, 17i nanoassemblies showed similar in vitro antitumor activity to free drugs. On the other hand, PEGylated 17i nanoassemblies showed longer circulation times than free drugs. We speculate that its nanostructure are not substrates for some enzymes, thus the adverse metabolism of 17i nanoassemblies caused by enzymes are reduced, which prolong the blood circulation time. In addition, we found that 17i nanoassemblies exhibit satisfactory targeting ability, possibly due to the EPR effect. We also observed that tumors regions in C57 mice were significantly infiltrated by CD8+ T cells after treatment of PEGylated 17i nanoassemblies in immunofluorescence staining and flow cytometry. Obviously, 17i nanoassemblies elevated tumor immune response when killing tumor cells. The principle of this pure nano-drugs may open up the way to provide inspiration for maximizing the therapeutic potential of drug-like compounds.
Conclusion
In this paper, insoluble LSD1 inhibitor, pure compound 17i, was prepared into nanoassemblies by one-step nano-precipitation method without the addition of carrier material, and DSPE-PEG2000 was modified on the surface to improve stability and prolong blood circulation time. The characterization of the DLS and TEM proved that the ∼200 nm PEGylated 17i nanoassemblies were successfully constructed. Multiple intermolecular forces in assemblies were observed using computational simulation. The PEGylated 17i nanoassemblies had a negligible impact on the cytotoxicity of 17i solution. As expected, the PEGylated 17i nanoassemblies exhibited distinct advantages over 17i solution in terms of therapeutic efficiency, anti-tumor immune response and side effects in vivo. This is the first time that the one-step nanoprecipitation method overcomes the problem of low water solubility of anti-tumor drug-likeness LSD1 inhibitor. Such a simple and practical nanoplatform of pure LSD1 inhibitor holds a promising application prospect for clinical cancer therapy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
All animal protocols were evaluated and approved by the Animal Laboratory Ethics Committee of the Liaoning Cancer Hospital.
Author contributions
BL: Data curation, writing and original draft. XZ: Conceptualization and investigation. JL: Writing, conceptualization, investigation, review, editing and funding acquisition.
Funding
This work was supported by the Guiding plan of Liaoning Natural Science Foundation (No. 20180550277).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1012882/full#supplementary-material
References
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi:10.1021/jm0306430
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for Protein−Ligand complexes. J. Med. Chem. 49, 6177–6196. doi:10.1021/jm051256o
Ma, L.-Y., Zheng, Y.-C., Wang, S.-Q., Wang, B., Wang, Z.-R., Pang, L.-P., et al. (2015). Design, synthesis, and structure-activity relationship of novel LSD1 inhibitors based on pyrimidine-thiourea hybrids as potent, orally active antitumor agents. J. Med. Chem. 58, 1705–1716. doi:10.1021/acs.jmedchem.5b00037
Mould, D. P., Alli, C., Bremberg, U., Cartic, S., Jordan, A. M., Geitmann, M., et al. (2017). Development of (4-cyanophenyl)glycine derivatives as reversible inhibitors of lysine specific demethylase 1. J. Med. Chem. 60, 7984–7999. doi:10.1021/acs.jmedchem.7b00462
Schmitt, M. L., Hauser, A.-T., Carlino, L., Pippel, M., Schulz-Fincke, J., Metzger, E., et al. (2013). Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity. J. Med. Chem. 56, 7334–7342. doi:10.1021/jm400792m
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. doi:10.1016/j.cell.2004.12.012
Sorna, V., Theisen, E. R., Stephens, B., Warner, S. L., Bearss, D. J., Vankayalapati, H., et al. (2013). High-throughput virtual screening identifies novel N′-(1-Phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 56, 9496–9508. doi:10.1021/jm400870h
Sun, B., Luo, C., Zhang, X., Guo, M., Sun, M., Yu, H., et al. (2019a). Probing the impact of sulfur/selenium/carbon linkages on prodrug nanoassemblies for cancer therapy. Nat. Commun. 10, 3211. doi:10.1038/s41467-019-11193-x
Sun, M., Jiang, H., Liu, T., Tan, X., Jiang, Q., Sun, B., et al. (2022). Structurally defined tandem-responsive nanoassemblies composed of dipeptide-based photosensitive derivatives and hypoxia-activated camptothecin prodrugs against primary and metastatic breast tumors. Acta Pharm. Sin. B 12, 952–966. doi:10.1016/j.apsb.2021.08.008
Sun, M., Zhang, X., Gao, Z., Liu, T., Luo, C., Zhao, Y., et al. (2019b). Probing a dipeptide-based supramolecular assembly as an efficient camptothecin delivering carrier for cancer therapy: Computational simulations and experimental validations. Nanoscale 11, 3864–3876. doi:10.1039/C8NR07014H
Suzuki, T., and Miyata, N. (2011). Lysine demethylases inhibitors. J. Med. Chem. 54, 8236–8250. doi:10.1021/jm201048w
Tortorici, M., Borrello, M. T., Tardugno, M., Chiarelli, L. R., Pilotto, S., Ciossani, G., et al. (2013). Protein recognition by short peptide reversible inhibitors of the chromatin-modifying LSD1/CoREST lysine demethylase. ACS Chem. Biol. 8, 1677–1682. doi:10.1021/cb4001926
Vianello, P., Botrugno, O. A., Cappa, A., Dal Zuffo, R., Dessanti, P., Mai, A., et al. (2016). Discovery of a novel inhibitor of histone lysine-specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J. Med. Chem. 59, 1501–1517. doi:10.1021/acs.jmedchem.5b01209
Wang, Q., Sun, M., Li, D., Li, C., Luo, C., Wang, Z., et al. (2020a). Cytochrome P450 enzyme-mediated auto-enhanced photodynamic cancer therapy of co-nanoassembly between clopidogrel and photosensitizer. Theranostics 10, 5550–5564. doi:10.7150/thno.42633
Wang, X., Zhang, C., Zhang, X., Yan, J., Wang, J., Jiang, Q., et al. (2020b). Design, synthesis and biological evaluation of tetrahydroquinoline-based reversible LSD1 inhibitors. Eur. J. Med. Chem. 194, 112243. doi:10.1016/j.ejmech.2020.112243
Wang, Y., Hu, B., Peng, Y., Xiong, X., Jing, W., Wang, J., et al. (2019). In silico exploration of the molecular mechanism of cassane diterpenoids on anti-inflammatory and immunomodulatory activity. J. Chem. Inf. Model. 59, 2309–2323. doi:10.1021/acs.jcim.8b00862
Wu, F., Zhou, C., Yao, Y., Wei, L., Feng, Z., Deng, L., et al. (2016). 3-(Piperidin-4-ylmethoxy)pyridine containing compounds are potent inhibitors of lysine specific demethylase 1. J. Med. Chem. 59, 253–263. doi:10.1021/acs.jmedchem.5b01361
Yang, Y., Sun, B., Zuo, S., Li, X., Zhou, S., Li, L., et al. (2020). Trisulfide bond-mediated doxorubicin dimeric prodrug nanoassemblies with high drug loading, high self-assembly stability, and high tumor selectivity. Sci. Adv. 6, 1725. doi:10.1126/sciadv.abc1725
Zhang, X., Huang, H., Zhang, Z., Yan, J., Wu, T., Yin, W., et al. (2021). Design, synthesis and biological evaluation of novel benzofuran derivatives as potent LSD1 inhibitors. Eur. J. Med. Chem. 220, 113501. doi:10.1016/j.ejmech.2021.113501
Zhang, X., Li, N., Zhang, S., Sun, B., Chen, Q., He, Z., et al. (2020). Emerging carrier‐free nanosystems based on molecular self‐assembly of pure drugs for cancer therapy. Med. Res. Rev. 40, 1754–1775. doi:10.1002/med.21669
Zhao, D., Tao, W., Li, S., Chen, Y., Sun, Y., He, Z., et al. (2021). Apoptotic body-mediated intercellular delivery for enhanced drug penetration and whole tumor destruction. Sci. Adv. 7, 880. doi:10.1126/sciadv.abg0880
Zheng, Y.-C., Duan, Y.-C., Ma, J.-L., Xu, R.-M., Zi, X., Lv, W.-L., et al. (2013). Triazole-dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J. Med. Chem. 56, 8543–8560. doi:10.1021/jm401002r
Keywords: drug delivery, supramolecular nanoassemblies, therapeutic efficiency, systemic toxicity, drug-like compound
Citation: Li B, Zhang X and Li J (2022) Carrier-free supramolecular nanoassemblies of pure LSD1 inhibitor for effective anti-tumor therapy. Front. Chem. 10:1012882. doi: 10.3389/fchem.2022.1012882
Received: 06 August 2022; Accepted: 14 September 2022;
Published: 30 September 2022.
Edited by:
Khurshid Ahmad, Yeungnam University, South KoreaReviewed by:
Piotr G Rychahou, University of Kentucky, United StatesQiwen Wang, Zhejiang University, China
Barnali Maiti, VIT University, India
Praveen Nekkar Rao, University of Waterloo, Canada
Copyright © 2022 Li, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangyu Zhang, eGlhbmd5dXpoYW5nc3lwaHVAMTYzLmNvbQ==; Jibin Li, bGVlamliaW5AMTI2LmNvbQ==