 Jian-Wei Han
Jian-Wei Han Wei-Yue Bian1
Wei-Yue Bian1 Yue-Yu Zhang
Yue-Yu Zhang Meng Zhang
Meng Zhang- 1School of Physics, East China University of Science and Technology, Shanghai, China
- 2Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, Zhejiang, China
A novel single-atom catalyst of Fe adsorbed on χ3-borophene has been proposed as a potential catalyst for CO oxidation reaction (COOR). Quantitative pictures have been provided of both the stability of Fe@χ3-borophene and various kinetic reaction pathways using first-principles calculations. Strong adsorption energy of -3.19 eV and large diffusion potential of 3.51 eV indicates that Fe@χ3-borophene is highly stable. By exploring reaction mechanisms for COOR, both Eley-Ridel (E-R) and trimolecule E-R (TER) were identified as possible reaction paths. Low reaction barriers with 0.49 eV of E-R and 0.57 eV of TER suggest that Fe@χ3-borophene is a very promising catalyst for COOR. Charge transfer between the χ3-borophene and CO, O2 and CO2 gas molecules plays a key role in lowering the energy barrier during the reactions. Our results propose that Fe@χ3-borophene can be a good candidate of single-atom catalyst for COOR with both high stability and catalytic activity.
Introduction
The CO oxidation reaction (COOR) is a simple yet fundamental reaction to reveal the intrinsic mechanism of multiphase catalytic conversion reactions and to test the reactivity of new catalysts (Alavi et al., 1998). The conversion of CO to non-toxic gases at room temperature has attracted considerable attention in recent years due to the seriously growing environmental problems and energy shortages (Ernst and Zibrak, 1998; Prockop and Chichkova, 2007). In the past few years, many studies have been focusing on the mechanism of COOR using noble metal nanoparticles and alloys as catalysts such as Pd (Kalita and Deka, 2009), Ag (Kim et al., 2010), Pt (Zhang and Hu, 2001), Au (Lopez and Nørskov, 2002) and others. However, due to the high cost and scarcity of these metals, they have never been commercialized. Therefore, it is crucial to find suitable catalytic methods and catalysts for COOR to improve the catalytic effect and reduce the cost.
A new class of heterogeneous catalysis emerges in the past decade by supporting metal nanoparticles on a surface. Among these systems, single-atom catalysts (SACs) on two-dimensional substrates has been a popular research topic since the single-atom catalyst Pt1/FeOx was first reported to be synthesized in 2009 by Zhang et al. (Qiao et al., 2011). The advantages of SACs include clear active sites, high utilization of metals, high-efficiency in catalytic performance, wide applicability and so on (Yang et al., 2013; Liu et al., 2018; Wang et al., 2018). A fundamental understanding of the reaction pathways on SACs can not only further guide the experimentalist to find more efficient catalysts but also provide valuable theoretical research ideas for COOR.
Due to the outstanding stability and large surface area, graphene has become a promising candidate for anchoring SACs since its discovery in 2004 (Wang et al., 2012). Up to now, the COOR of the noble metal [Au (Lu et al., 2009), Cu (Song et al., 2011), Pt (Tang et al., 2013)], and other transition metal atoms (Pd (Jia et al., 2014), Mo (Tang et al., 2015)) adsorbed or doped on the surface of graphene has been studied successively. In these systems, the reaction path based on Langmuir–Hinshelwood (L-H) mechanism (Lu et al., 2009), Eley-Ridel (E-R) mechanism (Tang et al., 2017) and trimolecule E-R (TER) mechanism (Zhang et al., 2015) has been confirmed. Recently, an increasing amount of efforts has been devoted to finding catalysis for SACs based on other two-dimensional materials, such as graphyne (Wu et al., 2015), MoS2 (Du et al., 2015), and hexagonal boron nitride monolayer (h-BN) (Lin et al., 2013; Liu et al., 2014), or to modify graphene to change some of the carbon atoms to N and other atoms (Zhang et al., 2016; Liu et al., 2017), thereby changing the catalytic environment of single metal to achieve better catalytic performance.
As a neighbor of carbon, borophene has attracted increasing interest since it was synthesized recently on a silver substrate under ultrahigh-vacuum with four phases including 2-Pmmn, β12, χ3, and honeycomb phases (Tang and Ismail-Beigi, 2007; Yang et al., 2008; Mannix et al., 2015). Borophene has complex bonding mechanisms and multiple coordination capabilities (Wang et al., 2019). Particularly, the adsorption of metal atoms with electron-donating properties is expected to occur more likely on borophene than on graphene because of the electron deficient property of boron atoms (Liu et al., 2013). Due to the unique physical and chemical properties, borophene has pronounced potential applications in the service of SACs as the substrate. Based on density functional theory, nitrogen reduction reaction (Liu et al., 2019a; Zhu et al., 2019), hydrogen evolution reaction (Xu et al., 2020a), oxygen evolution/reduction reaction (Xu et al., 2021) and electrocatalytic CO2 reduction (Shen et al., 2018; Zhang et al., 2020) about SACs have been extensively studied on different phases of borophene, and promising catalytic results have been obtained, indicating that borophene has great research potential for applications in the field of SACs.
However, these previous studies on the application of borophene on SACs mainly focus on electrocatalysis. Besides, most of these works are about α-borophene and β-borophene phase which are considered as the thermodynamically most stable phase based on the density functional theory simulation (Wu et al., 2012). As far as we know, there are few studies about COOR of SACs on borophene, especially there is a lack of research on the catalytic performance of the other phases of borophene. Here, the χ3-borophene has been selected to investigate the SACs, which was successfully synthesized by Feng’s team in 2016 under ultra-high vacuum conditions (Feng et al., 2016). Fe cation has been reported with good catalytic activity for many reactions in the gas phase, while other cations such as Ti+, Zr+, V+, Nb+, Cr+ have no such activity (Staley, 1981). In spite of extensive investigations in SACs on two dimensional materials, the transition metal atoms with unfilled d electrons and robust magnetic properties are an interesting and challenging topic in catalysis. In this paper, the COOR of Fe atoms adsorbed on χ3-borophene are systematically investigated using density functional theory to explore the COOR effect. We believe that the results of this paper will provide new ideas for the catalytic properties of borophene and the study of COOR.
Computational details
The first-principles calculations were carried out using the DMol3 package (Delley, 1990) embedded in Material Studio. Here we used χ3-borophene as substrate and a (2 × 4) supercell was built for simulation, which contains total 64 boron atoms. The Perdew–Burke–Ernzerhof (PBE) functional was used as the exchange-correlation functional under the generalized gradient approximation (GGA) (Perdew et al., 1996). The electronic eigenfunctions were expanded in terms of localized atomic orbital DNP (Delley, 1990) basis set, and the core treatment used was DFT semicore pseudopotentials (DSPPs) (Delley, 2002), which replaced the core electrons with individual effective potentials. The real-space cutoff radius was set to 4.7 Å to ensure high-quality results. In the process of geometric optimization, the energy convergence value was 1.0 × 10−5 Ha/atom, the maximum stress convergence value was 0.02 Ha/Å, the maximum displacement convergence value was 0.005 Å, and the threshold of self consistent-field (SCF) density convergence was 1.0 × 10−6 Ha. The K point of the Brillouin zone was selected as 2 × 2 × 1 under the Monkhorst-Pack method (Monkhorst and Pack, 1976). The K point set for PDOS calculation which is 4 × 4 × 1 here in the Fe@χ3-borophene system. The vacuum layer between χ3-borophene layers was set to 20 Å to ensure that there was no interaction between layers. Van der Waals interaction was described by DFT + D2 method throughout all calculations (Grimme, 2006). Mulliken’s net charge analysis has been applied to count charge transfer between the adsorbate atoms and the substrate (Mulliken, 1955). The transition states (TSs) were searched by the linear both synchronous transit (LST) and quadratic synchronous transit (QST) protocol. We first performed LST, and then repeated conjugate gradient minimization and QST maximization until the TS was found (Halgren and Lipscomb, 1977). To check the dynamic stability of the substrate, we ran the ab-initio molecular dynamics simulation using the NVT ensemble.
The adsorption energy Eads of the adsorbent on the substrate is defined as:
where Eadsorbate/substrate is the total energy of the system, Eadsorbate is the energy of the ground state of the adsorbate, and Esubstrate is the energy of the substrate. In order to include the influence of temperature, the change in Gibbs free energy (ΔG) of reaction path is calculated according to
where ΔE and ΔEZPE are the difference between total energy at 0 K and the zero-point vibration energy, respectively. ΔS is the change of entropy, defined as ΔS = ΔStrans + ΔSrot + ΔSvib, where ΔStrans, ΔSrot, and ΔSvib represent the contribution of translation, rotation, and vibration modes, respectively.
According to the transition state theory, the time required for each step of the reaction can be estimated by the Arrhenius law (Arrhenius, 1889):
where Ea is the energy barrier of the reaction, T is the temperature (K),
Results and discussion
Structural stability of Fe@χ3-borophene
The thermodynamic stability of Fe@χ3-borophene is first studied before we further investigate the catalytic activity of this system for COOR. The atomic model of Fe@χ3-borophene include a single Fe atom adsorbed on χ3-borophene. The initial adsorption sites of a single Fe atom in the Fe@χ3-borophene are first placed at top-site, bridge-site or triangle-site and then optimized, as depicted in Figure 1A and Supplementary Figure S1. The result of geometric optimization shows that the heart-site is the most favorable site for Fe atom adsorption on χ3-borophene with an adsorption energy of -3.19 eV. The distance between the Fe atom at the heart-site and the adjacent B atom is 2.067 Å and the protruding height between the Fe atom and the χ3-borophene plane is 1.381 Å.
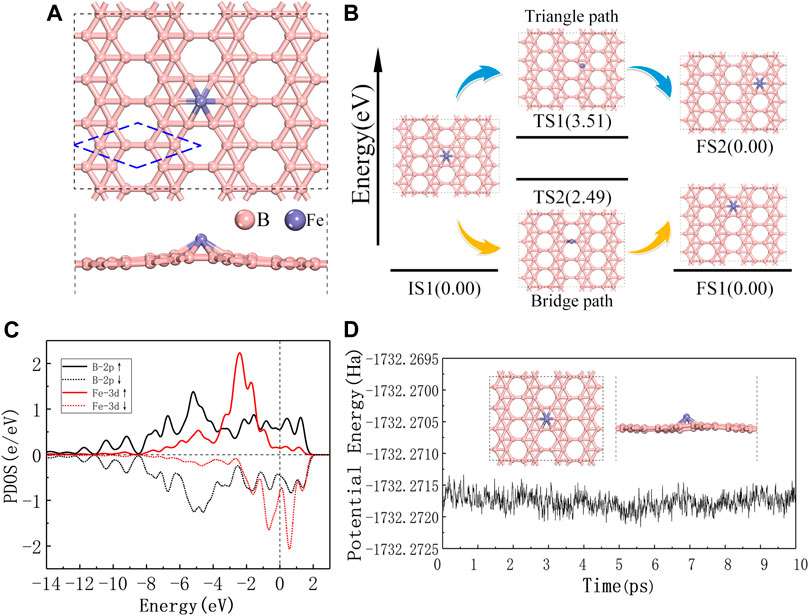
FIGURE 1. (A) Top and side views of the most stable configurations of single Fe atom adsorbed on χ3-borophene, the blue dashed line is the primitive cell of χ3-borophene. (B) The energy profile for Fe atom diffusion on χ3-borophene surface with the configurations involved. (C) The spin polarized partial density of states for Fe atom and the 6 B atoms around it, the vertical dashed line at 0 eV is the Fermi level. (D) Ab-initio molecular dynamics calculation of Fe@χ3-borophene at 300 K, the trajectory length is 10 ps, the inserted figure shows the structure of the system after 10 ps?
The diffusion behavior of Fe atoms on the χ3-borophene surface is studied by evaluating the energy barrier between one heart-site to a neighboring one. Two diffusion paths are identified as the Bridge path and the Triangle path and shown in Figure 1B. The calculated energy barriers of the Bridge path and Triangle path are 2.49 and 3.51 eV, respectively. Therefore, the Bridge path should be preferred as the diffusion path. Higher diffusion energy barrier is shown for Fe@χ3-borophene than those reported on graphene (Li et al., 2010), so that Fe atom is less favorable to migrate on χ3-borophene.
To study the self-aggregation of Fe on χ3-borophene, the structures and energies of nFe (n = 1–4) and Fen (n = 1–4) clusters adsorbed on the surface of both graphene and χ3-borophene (as shown in Supplementary Figures S2 and S3) are calculated for comparison. It can be seen from Supplementary Figures S2 and S3 that the total energy of isolated Fe atoms is significantly lower than that of Fe clusters on χ3-borophene, indicating that the structure of isolated Fe atoms anchored on χ3-borophene’s heart-site is favorited, while Fe tends to form clusters spontaneously on graphene. Thus, we could avoid the problem of self-aggregation of Fe atoms by choosing χ3-borophene as substrate.
The spin polarized partial density of states (PDOS) for Fe@χ3-borophene has been studied to further understand the interaction between Fe atom and χ3-borophene. PDOS in Figure 1C shows both the 3d-orbital of the guest Fe and the 2p-orbital of the host B atoms cross the Fermi level, showing the metallic characteristics of the Fe atom adsorbed on χ3-borophene system as demonstrated in the pure χ3-borophene (Feng et al., 2016). The superposition of one sharp peak originates from the d orbital of the adsorbed Fe atom and the p orbital of substrate B atoms span from -8.0 to 2.0 eV near the Fermi level. The strong hybridization shown in PDOS verifies the strong interaction between the guest Fe and host borophene and results in the high stability of the structure. We find that the shapes of the PDOS not only in d state of Fe atom but also in p state of borophene are quite different (see Figure 1C). Fe@χ3-borophene carries a large magnetic moment of 3.0
The stability of Fe@χ3-borophene under room temperature is confirmed by ab-initio molecular dynamics calculation (Figure 1D). Within the employed simulation time of 10 ps at 300 K, no structural change is detected, while the instantaneous values of the potential energy fluctuate slightly due to thermal fluctuations. Thus, the lowest energy structure of Fe@χ3-borophene is expected to be stable at room temperature.
Adsorption properties of CO, O2 and CO2 on Fe@χ3-borophene
In order to understand the specific reaction path of COOR on the surface of Fe@χ3-borophene, it is important to further study the adsorption and co-adsorption properties of various gas molecules including CO, O2 and CO2 involved in COOR on Fe@χ3-borophene. The most stable adsorption configurations of various gas molecules absorbed on Fe@χ3-borophene are shown in Figure 2 and Supplementary Figure S4. Their adsorption energies, charge transfers and magnetic moments are listed in Table 1.
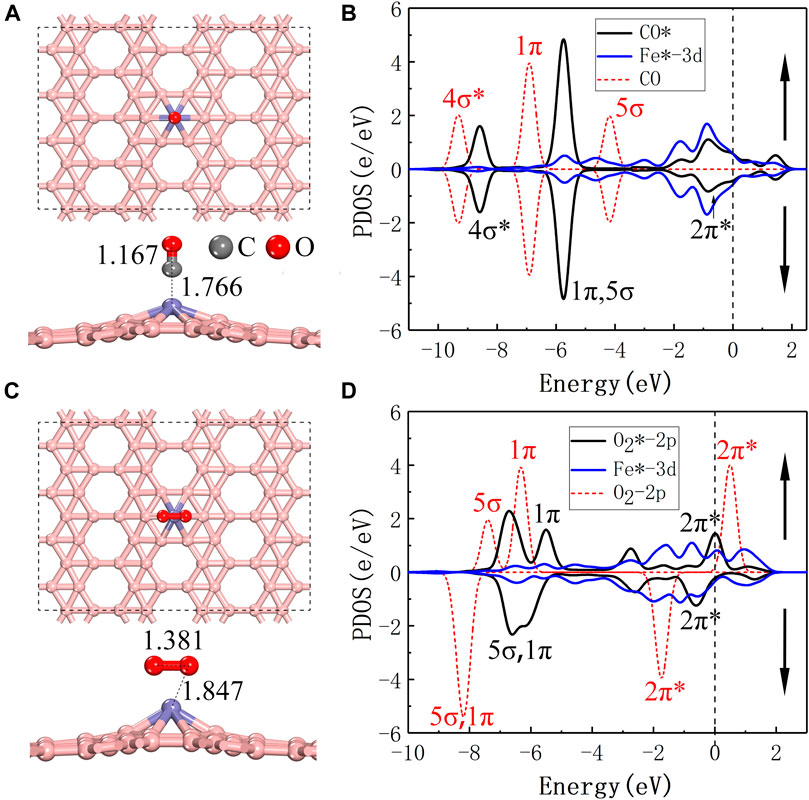
FIGURE 2. (A,C) are top and side views of the most stable configurations of CO/O2 adsorbed on Fe@χ3-borophene, respectively. (B,D) are the spin polarized partial density of states for the CO/O2 and Fe after adsorbed on Fe@χ3-borophene. The vertical dashed line at 0 eV is the Fermi level and the red dashed line is the PDOS for CO and O2 in the gas phase.
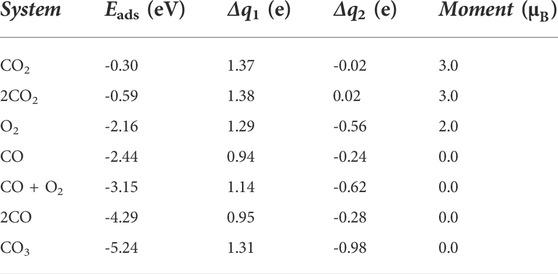
TABLE 1. The related parameters of gas molecules absorbed on Fe@χ3-borophene: adsorption energy (Eads), charge transfer between the Fe atom and the χ3-borophene (Δq1), charge transfer between gas molecules and Fe@χ3-borophene (Δq2), magnetic moment of the system.
The most favorable adsorption sites for CO and O2 are shown in Figures 2A,C, respectively. Both the CO and O2 molecules adsorb on top of the Fe atom as they approach the Fe@χ3-borophene. For CO, the lowest energy adsorption model is that the CO molecule is located vertically above the Fe@χ3-borophene, where Fe, C, O atoms form a line and the C atom is bonded with the Fe atom. The bond length of CO* (1.167 Å) is slightly larger than that of gaseous CO (1.129 Å) and the distance between Fe and C is 1.766 Å (”*” represents the adsorption state). The most stable adsorption position for O2* is the horizontal configuration, where two O atoms are adsorbed on the Fe@χ3-borophene and form an inverted triangle with the Fe atom. The bond length of O2* increases significantly from 1.208 Å to 1.381 Å compared to the gas phase and the bond length of O and Fe is 1.847 Å. The lowest energy adsorption positions for other gas molecules or combinations of gas molecules including 2CO, CO + O2, CO3, CO2 and 2CO2 are drawn in Supplementary Figure S4.
The PDOS for CO-Fe@χ3-borophene is shown in Figure 2B. After the CO molecule is absorbed, the 3d spin-up and spin-down lines of Fe atom in the graph are almost identical in shape. A CO molecule adsorbed on the Fe@χ3-borophene is found to exhibit no magnetic moment, which means the magnetic moment of 3.0
The value of the adsorption energy of the individual CO and O2 molecules on Fe@χ3-borophene are -2.44 eV and -2.21 eV, respectively. These large absolute values of the adsorption energy suggest that there are strong interactions between CO/O2 and Fe@χ3-borophene. The strong interaction between the gas molecules and borophene substrate is a prerequisite for the subsequent catalytic reaction of COOR. It should be noted that the difference between the adsorption energies of CO and O2 molecules is only 0.23 eV. As a result, either CO or O2 molecules can be absorbed firstly on the substrate. If a single CO is absorbed at the first time, the adsorption state of 2CO* will be formed when another CO approaches, as shown in Supplementary Figure S4A, where 2 C atoms are simultaneously bonded to Fe with the adsorption energy of -4.29 eV. We also simulate the structure of the classical co-adsorbed state of CO + O2* when a CO molecule is first absorbed and then an O2 molecule approaches (Supplementary Figure S4B). In this case, a Fe atom is bonded with two O atoms and a C atom at the same time with the adsorption energy of -3.15 eV. Since the adsorption energy of the CO + O2* is 1.14 eV smaller than the case of 2CO*, the CO + O2* is more difficult to occur. Another possible adsorption pathway is that a single O2 is first adsorbed on the substrate. In this case, an intermediate state of CO3* will easily form when CO approaches the system with a large adsorption energy of -5.24 eV, which is 2.09 eV more negative than the case of CO + O2*, so that the CO + O2* is unlikely to form in this case either. Furthermore, the adsorption energies of one or two CO2 molecules are -0.33 eV and -0.59 eV respectively, which is quite small compared to that of the other gas molecules. Only -0.02 e and 0.02 e are transferred between CO2 and the substrate, indicating that CO2 is physically adsorbed on the substrate by van der Waals interactions, and can be separated from the substrate very easily.
Reaction mechanism of CO oxidation on Fe@χ3-borophene
According to the adsorption energy calculations of different gas molecules, the possible paths for COOR on the Fe@χ3-borophene are summed up as follows.
E-R:
TER:
where “*” represents the adsorption state.
1) E-R reaction
The E-R reaction path is shown in Figure 3. We place the CO molecule above the O2-Fe@χ3-borophene as the initial state (IS), representing the CO in the gas phase. In the first step of the reaction path, CO molecule combines with O2* to form an intermediate state (MS) CO3*. The energy barrier for this step is only 0.13 eV, and this reaction releases a large amount of heat (3.76 eV). In the conventional E-R reaction, the next step is the decomposition of CO3* into O* and CO2 (see Figure 3A). We simulate this reaction and find that the reaction barrier for CO3* to form CO2 by breaking the C-O bond directly requires 1.02 eV, which indicates that this step is unlikely to occur. However, there is another possible path for CO3* to decompose with lower energy barrier. Chen’s team found that the direct separation of CO3* requires 1.34 eV, while the path to form 2CO2 via another CO only needs to cross an energy barrier of 0.15 eV (Chen et al., 2020) when they studied the COOR on the 3Si-graphene-Ni. According to Chen’s result, here we also investigated the reaction path of forming 2CO2 by adding a second CO molecule in our system and the reaction pathway is shown in Figure 3B, where the reaction barrier is found to be reduced. In this reaction path way, the gas phase CO suspended around the CO3* is used as the IS. When the second CO molecule approaches, one of the C-O bonds in CO3* breaks. As a result, a CO2 is subsequently detached and the remaining single O atom combines with the CO molecule to form the final state (FS) 2CO2*. This reaction requires climbing an energy barrier of 0.49 eV and gives off 0.82 eV of heat as shown in Figure 3B, which is nearly half of the energy barrier of 1.02 eV for direct decomposition, indicating that the presence of gas-phase CO does effectively facilitate the dissociation of CO3*.
2) TER reaction
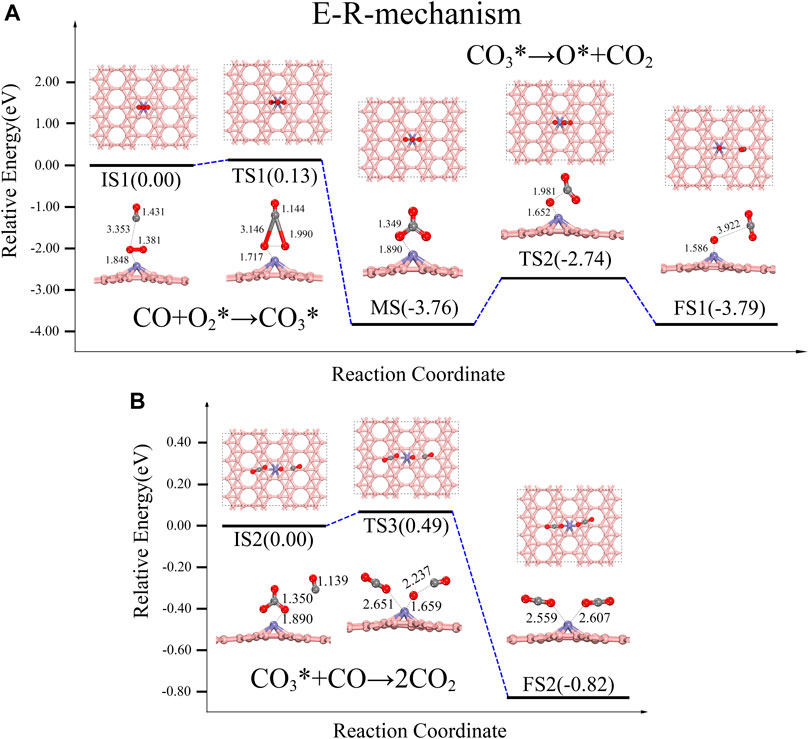
FIGURE 3. Energy distribution diagram under the E-R path and its corresponding structure diagram, the rate-determining step is IS2-FS2, turning over an energy barrier of 0.49 eV.
From the adsorption energies discussed above, it is apparent that two CO molecules are more likely to be adsorbed on Fe@χ3-borophene due to its ∼1.14 eV greater adsorption energy than that of the CO + O2*. Figure 4 shows that the TER reaction path, which starts from the IS of the reaction with the gas-phase O2 physiosorbed on 2CO-Fe@χ3-borophene. The two co-adsorbed CO effectively activate the gas-phase O2 to cross an energy barrier of about 0.57 eV. The product is an OCO-Fe-OCO* intermediate state releasing an energy of 0.49 eV. This OCO-Fe-OCO* intermediate state has a pentagonal structure, which is similar to the OCO-Pt-OCO* intermediate state reported by the Zhang team in the TER path on Pt/NG (Zhang et al., 2015). During the reaction, it is found that two oxygen atoms in O2 will approach the 2 C atoms of the CO molecules progressively and eventually form bonds to the C atom with a bond length of 1.406 Å.
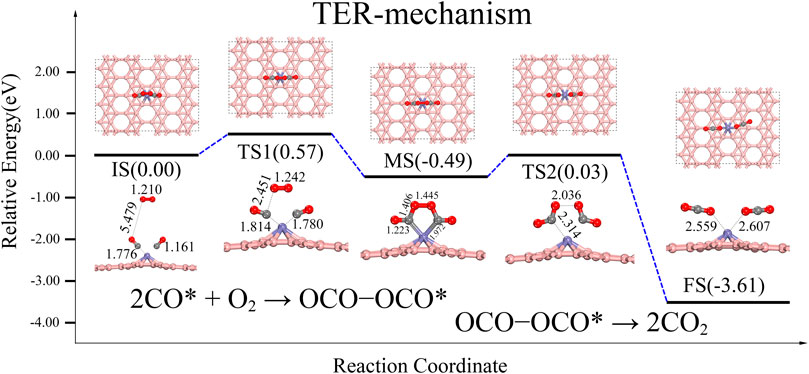
FIGURE 4. Energy distribution diagram under TER path and its corresponding structure diagram, the rate-determining step is IS-MS2 turning over an energy barrier of about 0.57 eV.
The second step of the reaction path is that the intermediate state of OCO-Fe-OCO* proceeds to the final state of 2CO2*. It can be seen from Figure 4 that the MS needs to cross an energy barrier of about 0.52 eV to form two CO2, which is almost the same as the energy barrier of 0.57 eV required from IS to MS. The breaking of the O-O bond and C-Fe bond releases about 3.12 eV of heat. The O-O bond grows from 1.352 Å to 2.036 Å, and finally breaks completely, while the C-Fe bond grows from 1.973 Å to 2.314 Å, and finally breaks completely. The final product 2CO2 is physically adsorbed on the system and can be easily dissociated to allow the cycle to continue.
3) Discussion of catalytic results and electronic properties
Here we summarize the rate-determining steps (RDS) of both reactions described above. The important parameters of each reaction are listed in Table 2. In the E-R reaction, the RDS is2-FS2, climbing an energy barrier of about 0.49 eV, which is less than the energy barrier needed for the E-R reaction on Fe-SV-graphene (Li et al., 2010; Xu et al., 2020b). The RDS in the TER reaction is IS-MS, requiring an energy barrier of about 0.57 eV.
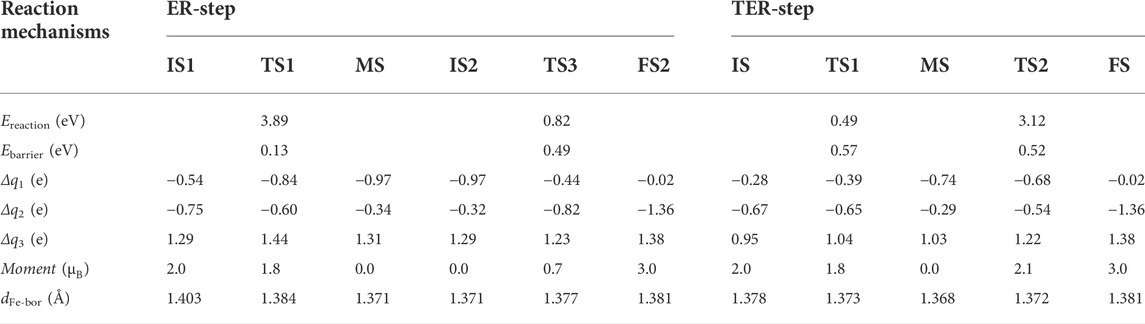
TABLE 2. The relevant parameters of each basic reaction path: the energy released by the reaction (Ereaction), the energy barrier required for the reaction (Ebarrier), the charge transfer between reactants and substrates (Δq1), the charge obtained by χ3-borophene (Δq2), the charge loss of Fe atom (Δq3), the magnetic moment of the system (Moment), the distance between Fe atom and the χ3-borophene surface (dFe-bor).
We analyze the electronic structural property of the system to provide deeper understanding for the reaction. As can be seen from the charge transfer between reactants and substrates (Δq1) in Table 2, the adsorbate will gain the largest number of electrons from the fe@χ3-borophene when O2 needs to be activated in both E-R and TER processes, with 0.97 e and 0.74 e obtained for the MS state in the E-R and TER reactions, respectively. Most of the obtained electrons will occupy the 2π* anti-bonding orbital of O2, leading to a longer bond length of O2, which makes subsequent catalytic reactions easier. In addition, it can be found from Δq2 which denotes the charge obtained by the χ3-borophene that it always gets electrons during the whole reaction process, and the number of electrons gained decreases significantly when the reaction requires a large number of electrons to activate O2, with 0.34 e and 0.29 e obtained for the MS state in the E-R and TER reactions, respectively. It is not difficult to find that χ3-borophene plays an important role in both donating and obtaining electrons in the whole reaction. Finally, it can be observed by the charge loss of Fe atom (Δq3) that the charge transfer of Fe atom is not very large in the E-R reaction. One possible reason is that Fe plays the role of a transport medium during the charge transfer process. Due to the strong electron deficiency of χ3-borophene, when a single Fe atom is adsorbed on the surface of χ3-borophene, the Fe atom will lose 1.34 e, and these electrons will be temporarily stored in the χ3-borophene. When O2 requires electrons to be activated in the subsequent catalytic process, charge transfer occurs from χ3-borophene to O2, so as to provide electrons to active the catalytic reaction. Therefore, not only does the Fe atom that directly interacts with the gas molecules play an important role in catalysis, but also the χ3-borophene as the substrate is the key to the catalytic reaction.
It is widely known that the analysis of the electronic state of the reaction path is of great significance for understanding the COOR process. It can be seen from Table 2 that no matter whether it is the E-R reaction or the TER reaction, the magnetic moments of the system undergo a process from presence to disappearance, and finally regain. O2 in the gas phase has a triplet state, resulting in a total magnetic moment of 2.0
During the COOR catalytic reaction, the distance between the Fe atom and the χ3-borophene surface is slightly changed. The initial distance is 1.381 Å after the geometry optimization. In the catalytic reaction, the distance of the Fe atom from the χ3-borophene surface varies as shown in Table 2. The distances show a pattern of first getting smaller and then larger.
The influence of temperature
In order to get a more realistic picture of the catalytic reaction, it is important to consider the effect of temperature. According to Peng team’s research, χ3-borophene remains thermodynamically stable at 1000 K (Peng et al., 2017), thus we plot the change of adsorption energy, reaction energy, and energy barrier for the whole system at 0–1000 K as shown in Figure 5. As can be seen from Figure 5A, when the temperature ranges from 0 to 300 K, the value of adsorption energy is almost the same compared to the adsorption energy at 0 K. When the temperature is greater than 300 K, the absolute value of the adsorption energy increases, but the order of the adsorption energy values does not change. The absolute value of the adsorption energy of O2+CO* is still about 1 eV lower than 2CO*, so the L-H reaction will not occur, indicating that the reaction path we obtained at 0 K is unchanged. The changes in the Gibbs free energy of the E-R and TER reactions are all negative, so all steps are exothermic as depicted in Figure 5B. Finally in Figure 5C we find that as the temperature reaches 200 K, the reaction energy barrier of IS1→TS1 in the E-R reaction becomes negative. It indicates that CO3* is more likely to form spontaneously under the influence of temperature. The energy barrier of MS→TS2 is always greater than IS2→TS3 in the E-R reaction, so the E-R reaction still requires a second external CO to participate in the presence of temperature. The energy barrier of IS→TS1 in the TER reaction increases slightly with temperature, but the value of the energy barrier is not large, and the reaction can still proceed. Overall, the RDS of each reaction step does not change with the presence of temperature and most of the reaction energy barriers decrease significantly with the increasing temperature.
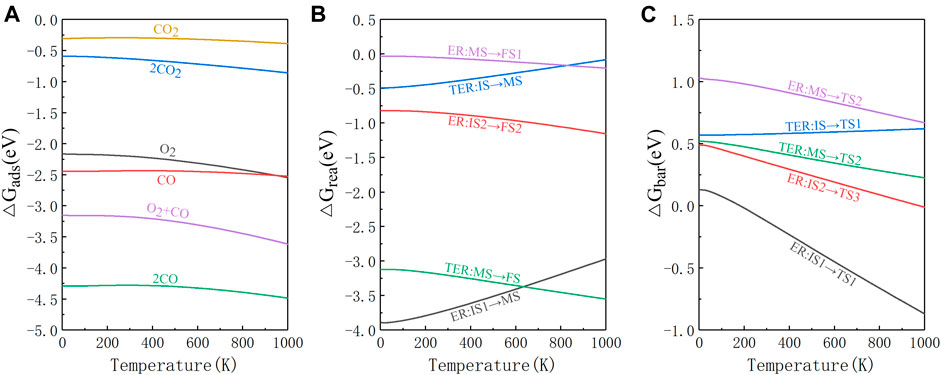
FIGURE 5. The change in Gibbs free energy of each reaction path at 0–1000 K. (A) The gas molecules adsorption energy. (B) The energy released by the reaction. (C) The energy barrier required for the reaction.
Reaction rate
Table 3 shows the specific time required for each step of the reaction process obtained by the Arrhenius law. In the E-R reaction, it can be seen that the time required for IS1-MS is very short at any temperature, which is consistent with the low energy barrier. In addition, the time required for IS2-FS2 is much less than that for MS-FS1, which means that the direct dissociation of CO3* is very difficult and the second CO is always required in the actual reaction. In the TER reaction, when the temperature is 200 K, the time taken by IS-MS is about 3.11 × 102 s, thus the reaction is not easy to occur. However, the entire TER reaction can proceed normally at higher temperature. In summary, the reaction rate of the E-R reaction is fast at any temperature, while the TER reaction is more suitable when temperature is above 200 K.
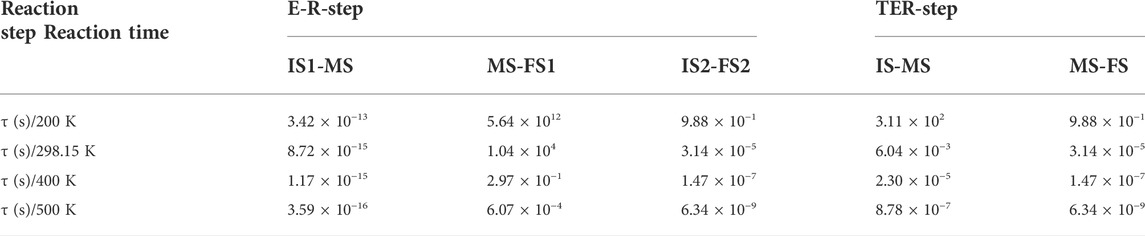
TABLE 3. Time required for each step of the reaction path in E-R and TER reaction at 200 K, 298.15 K, 400 K, 500 K.
Conclusion
The DFT calculations have been performed to investigate the mechanism of CO oxidation on Fe@χ3-borophene. In this work we have ensured the stability of the system with formation energy calculation, diffusion path calculation and Ab-Initio molecular dynamics simulation. Two possible reaction paths have been identified by absorption energy comparisons and their energy barriers have been calculated. The results show that the COOR on the Fe@χ3-borophene substrate has good catalytic performance. Further electronic structure analysis indicates that charge transfers between χ3-borophene and CO, O2 and CO2 molecules are the major cause of both the low energy barrier and magnetic moment difference. First-principles study of Fe@χ3-borophene as a catalyst for COOR provides new possibilities and ideas for the COOR, and proposes new opportunities for the application of borophene in the field of heterogeneous catalysis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
MZ, Y-YZ, and J-WH designed the research and wrote the paper. J-WH and W-YB carried out the simulation. All authors entered the discussion and commented on the manuscript.
Funding
This project is supported by the Startup Grant of Wenzhou Institute and Oujiang Laboratory (WIUCASQD2021014 and WIUCASQD2022025). Calculations are performed at the super computer center in School of Physics, East China University of Science and Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1008332/full#supplementary-material
References
Alavi, A., Hu, P., Deutsch, T., Silvestrelli, P. L., and Hutter, J. (1998). CO oxidation on Pt(111): AnAb InitioDensity functional theory study. Phys. Rev. Lett. 80 (16), 3650–3653. doi:10.1103/physrevlett.80.3650
Alvarez-Quiceno, J. C., Schleder, G. R., Marinho, E., and Fazzio, A. (2017). Adsorption of 3d, 4d, and 5dtransition metal atoms onβ12—Borophene. J. Phys. Condens. Matter 29 (30), 305302. doi:10.1088/1361-648x/aa75f0
Arrhenius, S. (1889). Über die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren. Z. fur Phys. Chem. 4, 226–248. doi:10.1515/zpch-1889-0416
Chen, W., Zhao, G., Wu, B., Tang, Y., Teng, D., and Dai, X. (2020). Theoretical study on the catalytic properties of single-atom catalyst stabilised on silicon-doped graphene sheets. Mol. Phys. 118 (7), e1652368. doi:10.1080/00268976.2019.1652368
Delley, B. (1990). An all‐electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92 (1), 508–517. doi:10.1063/1.458452
Delley, B. (2002). Hardness conserving semilocal pseudopotentials. Phys. Rev. B 66 (15), 155125. doi:10.1103/physrevb.66.155125
Du, C., Lin, H., Lin, B., Ma, Z., Hou, T., Tang, J., et al. (2015). MoS2 supported single platinum atoms and their superior catalytic activity for CO oxidation: A density functional theory study[J]. J. Mat. Chem. A Mat. 3 (46), 23113–23119. doi:10.1039/c5ta05084g
Ernst, A., and Zibrak, J. D. (1998). Carbon monoxide poisoning. N. Engl. J. Med. Overseas. Ed. 339 (22), 1603–1608. doi:10.1056/nejm199811263392206
Feng, B., Zhang, J., Zhong, Q., Li, W., Li, S., Li, H., et al. (2016). Experimental realization of two-dimensional boron sheets. Nat. Chem. 8 (6), 563–568. doi:10.1038/nchem.2491
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27 (15), 1787–1799. doi:10.1002/jcc.20495
Halgren, T. A., and Lipscomb, W. N. (1977). The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 49 (2), 225–232. doi:10.1016/0009-2614(77)80574-5
Jia, T. T., Lu, C. H., Zhang, Y. F., and Chen, W. K. (2014). A comparative study of CO catalytic oxidation on Pd-anchored graphene oxide and Pd-embedded vacancy graphene[J]. J. nanoparticle Res. 16 (2), 1–11. doi:10.1007/s11051-013-2206-0
Jiang, Q., Huang, M., Qian, Y., Miao, Y., and Ao, Z. (2021). Excellent sulfur and water resistance for CO oxidation on Pt single-atom-catalyst supported by defective graphene: The effect of vacancy type. Appl. Surf. Sci. 566, 150624. doi:10.1016/j.apsusc.2021.150624
Jiang, Q., Zhang, J., Ao, Z., Huang, H., He, H., and Wu, Y. (2018). First principles study on the CO oxidation on Mn-embedded divacancy graphene. Front. Chem. 6, 187. doi:10.3389/fchem.2018.00187
Jiang, Q., Zhang, J., Huang, H., Wu, Y., and Ao, Z. (2020). A novel single-atom catalyst for CO oxidation in humid environmental conditions: Ni-Embedded divacancy graphene. J. Mat. Chem. A Mat. 8 (1), 287–295. doi:10.1039/c9ta08525d
Kalita, B., and Deka, R. C. (2009). Reaction intermediates of CO oxidation on gas phase Pd4 clusters: A density functional study. J. Am. Chem. Soc. 131 (37), 13252–13254. doi:10.1021/ja904119b
Kim, H. Y., Han, S. S., Ryu, J. H., and Lee, H. M. (2010). Balance in adsorption energy of reactants steers CO oxidation mechanism of Ag13 and Ag12Pd1 nanoparticles: Association mechanism versus carbonate-mediated mechanism. J. Phys. Chem. C 114 (7), 3156–3160. doi:10.1021/jp9111553
Li, Y., Zhou, Z., Yu, G., Chen, W., and Chen, Z. (2010). CO catalytic oxidation on iron-embedded graphene: Computational quest for low-cost nanocatalysts. J. Phys. Chem. C 114 (14), 6250–6254. doi:10.1021/jp911535v
Lin, S., Ye, X., Johnson, R. S., and Guo, H. (2013). First-principles investigations of metal (Cu, Ag, Au, Pt, Rh, Pd, Fe, Co, and Ir) doped hexagonal boron nitride nanosheets: Stability and catalysis of CO oxidation. J. Phys. Chem. C 117 (33), 17319–17326. doi:10.1021/jp4055445
Liu, C., Li, Q., Zhang, J., Jin, Y., MacFarlane, D. R., and Sun, C. (2019). Conversion of dinitrogen to ammonia on Ru atoms supported on boron sheets: A DFT study. J. Mat. Chem. A Mat. 7 (9), 4771–4776. doi:10.1039/c8ta08219g
Liu, H., Gao, J., and Zhao, J. (2013). From boron cluster to two-dimensional boron sheet on Cu (111) surface: Growth mechanism and hole formation[J]. Sci. Rep. 3 (1), 1–9. doi:10.1038/srep03238
Liu, J. C., Tang, Y., Wang, Y. G., Zhang, T., and Li, J. (2018). Theoretical understanding of the stability of single-atom catalysts. Natl. Sci. Rev. 5 (5), 638–641. doi:10.1093/nsr/nwy094
Liu, J. H., Yang, L. M., and Ganz, E. (2019). Efficient electrocatalytic reduction of carbon dioxide by metal-doped β12-borophene monolayers. RSC Adv. 9 (47), 27710–27719. doi:10.1039/c9ra04135d
Liu, X., Duan, T., Sui, Y., Meng, C., and Han, Y. (2014). Copper atoms embedded in hexagonal boron nitride as potential catalysts for CO oxidation: A first-principles investigation. RSC Adv. 4 (73), 38750–38760. doi:10.1039/c4ra06436d
Liu, Z., He, T., Liu, K., Chen, W., and Tang, Y. (2017). Structural, electronic and catalytic performances of single-atom Fe stabilized by divacancy-nitrogen-doped graphene. RSC Adv. 7 (13), 7920–7928. doi:10.1039/c6ra28387j
Lopez, N., and Nørskov, J. K. (2002). Catalytic CO oxidation by a gold Nanoparticle: A density functional study. J. Am. Chem. Soc. 124 (38), 11262–11263. doi:10.1021/ja026998a
Lu, Y. H., Zhou, M., Zhang, C., and Feng, Y. P. (2009). Metal-embedded graphene: A possible catalyst with high activity. J. Phys. Chem. C 113 (47), 20156–20160. doi:10.1021/jp908829m
Mannix, A. J., Zhou, X. F., Kiraly, B., Wood, J. D., Alducin, D., Myers, B. D., et al. (2015). Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 350 (6267), 1513–1516. doi:10.1126/science.aad1080
Monkhorst, H. J., and Pack, J. D. (1976). Special points for Brillouin-zone integrations. Phys. Rev. B 13 (12), 5188–5192. doi:10.1103/physrevb.13.5188
Mulliken, R. S. (1955). Atomic Dipole moment corrected Hirshfeld population method[J]. J. Chem. Phys. 1833, 23. doi:10.1142/S0219633612500113
Peng, B., Zhang, H., Shao, H., Ning, Z., Xu, Y., Ni, G., et al. (2017). Stability and strength of atomically thin borophene from first principles calculations. Mater. Res. Lett. 5 (6), 399–407. doi:10.1080/21663831.2017.1298539
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77 (18), 3865–3868. doi:10.1103/physrevlett.77.3865
Prockop, L. D., and Chichkova, R. I. (2007). Carbon monoxide intoxication: An updated review. J. neurological Sci. 262 (1-2), 122–130. doi:10.1016/j.jns.2007.06.037
Qiao, B., Wang, A., Yang, X., Allard, L. F., Jiang, Z., Cui, Y., et al. (2011). Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3 (8), 634–641. doi:10.1038/nchem.1095
Shen, H., Li, Y., and Sun, Q. (2018). Cu atomic chains supported on β-borophene sheets for effective CO2 electroreduction[J]. Nanoscale 10 (23), 11064–11071. doi:10.1039/c8nr01855c
Song, E. H., Wen, Z., and Jiang, Q. (2011). CO catalytic oxidation on copper-embedded graphene. J. Phys. Chem. C 115 (9), 3678–3683. doi:10.1021/jp108978c
Staley, R. H. (1981). Gas-phase oxidation catalysis by transition-metal cations[J]. J. Am. Chem. Soc. 103 (5), 1286–1287.
Tang, H., and Ismail-Beigi, S. (2007). Novel precursors for boron nanotubes: The competition of two-center and three-center bonding in boron sheets. Phys. Rev. Lett. 99 (11), 115501. doi:10.1103/physrevlett.99.115501
Tang, Y., Chen, W., Shen, Z., Chang, S., Zhao, M., and Dai, X. (2017). Nitrogen coordinated silicon-doped graphene as a potential alternative metal-free catalyst for CO oxidation. Carbon 111, 448–458. doi:10.1016/j.carbon.2016.10.028
Tang, Y., Pan, L., Chen, W., Li, C., Shen, Z., and Dai, X. (2015). Reaction mechanisms for CO catalytic oxidation on monodisperse Mo atom-embedded graphene. Appl. Phys. A 119 (2), 475–485. doi:10.1007/s00339-015-9093-4
Tang, Y., Yang, Z., Dai, X., Ma, D., and Fu, Z. (2013). Formation, stabilities, and electronic and catalytic performance of platinum catalyst supported on non-metal-doped graphene. J. Phys. Chem. C 117 (10), 5258–5268. doi:10.1021/jp400202e
Wang, A., Li, J., and Zhang, T. (2018). Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2 (6), 65–81. doi:10.1038/s41570-018-0010-1
Wang, H., Wang, Q., Cheng, Y., Li, K., Yao, Y., Zhang, Q., et al. (2012). Doping monolayer graphene with single atom substitutions. Nano Lett. 12 (1), 141–144. doi:10.1021/nl2031629
Wang, Z. Q., Lü, T. Y., Wang, H. Q., and Zheng, J. C. (2019). Review of borophene and its potential applications[J]. Front. Phys. 14 (3), 1–20. doi:10.1007/s11467-019-0884-5
Wu, P., Du, P., Zhang, H., and Cai, C. (2015). Graphyne-supported single Fe atom catalysts for CO oxidation. Phys. Chem. Chem. Phys. 17 (2), 1441–1449. doi:10.1039/c4cp04181j
Wu, X., Dai, J., Zhao, Y., Zhuo, Z., Yang, J., and Zeng, X. C. (2012). Two-dimensional boron monolayer sheets. ACS Nano 6 (8), 7443–7453. doi:10.1021/nn302696v
Xu, L., Yang, L. M., and Ganz, E. (2018). Mn–graphene single-atom catalyst evaluated for CO oxidation by computational screening[J]. Theor. Chem. Accounts 137 (7), 1–13. doi:10.1007/s00214-018-2270-8
Xu, X., Hou, X., Lu, J., Zhang, P., Xiao, B., and Mi, J. (2020). Metal-doped two-dimensional borophene nanosheets for the carbon dioxide electrochemical reduction reaction. J. Phys. Chem. C 124 (44), 24156–24163. doi:10.1021/acs.jpcc.0c05998
Xu, X., Si, R., Dong, Y., Fu, K., Guo, Y., He, Y., et al. (2021). Borophene− supported single transition metal atoms as potential oxygen evolution/reduction electrocatalysts: A density functional theory study[J]. J. Mol. Model. 27 (3), 1–10. doi:10.1007/s00894-021-04693-5
Xu, X. Y., Li, J., Xu, H., and Zhao, C. (2016). DFT investigation of Ni-doped graphene: Catalytic ability to CO oxidation. New J. Chem. 40 (11), 9361–9369. doi:10.1039/c6nj00924g
Xu, X. Y., Xu, H., Guo, H., and Zhao, C. (2020). Mechanism investigations on CO oxidation catalyzed by Fe-doped graphene: A theoretical study. Appl. Surf. Sci. 523, 146496. doi:10.1016/j.apsusc.2020.146496
Yang, X., Ding, Y., and Ni, J. (2008). Ab initioprediction of stable boron sheets and boron nanotubes: Structure, stability, and electronic properties. Phys. Rev. B 77 (4), 041402. doi:10.1103/physrevb.77.041402
Yang, X. F., Wang, A., Qiao, B., Li, J., Liu, J., and Zhang, T. (2013). Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 46 (8), 1740–1748. doi:10.1021/ar300361m
Zhang, C. J., and Hu, P. (2001). CO oxidation on Pd(100) and Pd(111): A comparative study of reaction pathways and reactivity at low and medium coverages. J. Am. Chem. Soc. 123 (6), 1166–1172. doi:10.1021/ja002432f
Zhang, P., Xu, X., Song, E., Hou, X., Yang, X., Mi, J., et al. (2020). Transition metal-doped α-borophene as potential oxygen and hydrogen evolution electrocatalyst: A density functional theory study. Catal. Commun. 144, 106090. doi:10.1016/j.catcom.2020.106090
Zhang, X., Lu, Z., Xu, G., Wang, T., Ma, D., Yang, Z., et al. (2015). Single Pt atom stabilized on nitrogen doped graphene: CO oxidation readily occurs via the tri-molecular eley–rideal mechanism. Phys. Chem. Chem. Phys. 17 (30), 20006–20013. doi:10.1039/c5cp01922b
Zhang, X., Lu, Z., and Yang, Z. (2016). Single non-noble-metal cobalt atom stabilized by pyridinic vacancy graphene: An efficient catalyst for CO oxidation. J. Mol. Catal. A Chem. 417, 28–35. doi:10.1016/j.molcata.2016.03.008
Keywords: CO oxidation reaction, borophene, first-principles study, catalytic activity, reaction mechanism
Citation: Han J-W, Bian W-Y, Zhang Y-Y and Zhang M (2022) Fe@χ3-borophene as a promising catalyst for CO oxidation reaction: A first-principles study. Front. Chem. 10:1008332. doi: 10.3389/fchem.2022.1008332
Received: 31 July 2022; Accepted: 24 August 2022;
Published: 13 September 2022.
Edited by:
Jamie Platts, Cardiff University, United KingdomCopyright © 2022 Han, Bian, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue-Yu Zhang, eXVleXUuemhhbmdAZm94bWFpbC5jb20=; Meng Zhang, bXpoYW5nQGVjdXN0LmVkdS5jbg==