
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Chem. , 10 October 2022
Sec. Medicinal and Pharmaceutical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1002547
This article is part of the Research Topic Coumarins: New Synthetic Approaches and New Pharmacological Applications View all 6 articles
 Leonardo Pisani1†
Leonardo Pisani1† Marco Catto1†
Marco Catto1† Giovanni Muncipinto2
Giovanni Muncipinto2 Orazio Nicolotti1
Orazio Nicolotti1 Antonio Carrieri1
Antonio Carrieri1 Mariagrazia Rullo1
Mariagrazia Rullo1 Angela Stefanachi1*
Angela Stefanachi1* Francesco Leonetti1*Cosimo Altomare1
Francesco Leonetti1*Cosimo Altomare1The coumarin core (i.e., 1-benzopyran-2 (2H)-one) is a structural motif highly recurrent in both natural products and bioactive molecules. Indeed, depending on the substituents and branching positions around the byciclic core, coumarin-containing compounds have shown diverse pharmacological activities, ranging from anticoagulant activities to anti-inflammatory, antimicrobial, anti-HIV and antitumor effects. In this survey, we have reported the main scientific results of the 20-years investigation on the coumarin core, exploited by the research group headed by Prof. Angelo Carotti (Bari, Italy) either as a scaffold or a pharmacophore moiety in designing novel biologically active small molecules.
Due to its occurrence in both natural products and drugs, and the synthetic accessibility of its derivatives as well, the coumarin core (i.e., 1-benzopyran-2 (2H)-one) has been considered a privileged scaffold for drug design. Coumarin was isolated from tonka bean by Vogel in 1820. After its structural elucidation, this chemical scaffold was found in many bioactive natural compounds. Noteworthy, the anticoagulants warfarin, the choleretics armillarisin A and the antibiotic novobiocin must be mentioned as marketed drugs based on the coumarin scaffold. However, coumarin based derivatives are not devoid of drawbacks concerning with solubility and multidrug resistance (Wu et al., 2020; Al-Abass et al., 2021) and this makes the design even more challenging. Indeed, depending upon feature and substitution pattern of the functional groups, the coumarin nucleus has been widely decorated to develop compounds showing diverse pharmacological activities, spanning from anticoagulant activities to anti-inflammatory, antimicrobial, anti-HIV and antitumor effects (Stefanachi et al., 2018).
Herein, we review the scientific results in the last 2 decades of investigation around the coumarin nucleus, developed in the research group of Prof. Angelo Carotti (Bari, Italy).
The interest toward coumarin derivatives by the Carotti’s group began in the early 2000 s from a project aimed at generating ligand-based models of neurotoxicity of MAO substrates, using the classical Hansch quantitative structure-activity relationships (QSARs) and comparative molecular field analysis (CoMFA) (Youdim et al., 2006). In details, the QSAR/CoMFA analyses focused on the MAO-catalyzed oxidation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to 1-methyl-4-phenylpyridinium (MPP+), which produces Parkinson-like symptoms in primates (Altomare et al., 1992). The toxication of MPTP involves a two-step oxidation into the brain by MAO B, and to a lesser extent by MAO A. These CoMFA studies, combined with the Hansch 2D-QSAR analyses, allowed to spot the main steric effects, along with the electrostatic and hydrophobic interactions, most discriminating the binding sites in the two isozymes.
Besides the 3D QSAR studies on MPTP analogs, Carotti and coworkers investigated a number of fused azaheterocycles, like 5H-indeno [1,2-c] pyridazines and other condensed pyridazines and pyrimidines (Altomare et al., 1991), as MAO inhibitors, and derived QSAR and CoMFA models which highlighted key physicochemical interactions responsible for inhibition potency and A/B isoform selectivity (Kneubühler et al., 1993; Kneubühler et al., 1995) and, among them, coumarins showed really interesting results (Altomare et al., 1998; Gnerre et al., 2000)
Indeed, Carotti’s synthetic efforts towards several coumarin derivatives, along with 3D QSAR models, helped to unravel the key molecular features responsible for binding affinity to MAO A/B, inhibition potency and isoform selectivity (Altomare et al., 1998; Gnerre et al., 2000), even before that the co-crystal structures of MAOs with small molecule binders (Binda et al., 2007) opened the way to structure-based drug design of novel inhibitors. Since then, as discussed below, many of the group’s publications focused on potential applications of diverse coumarin-containing compounds in neurodegeneration and oncology.
Due to higher synthetic feasibility, seminal explorations focusing on MAO inhibitors mainly addressed the coumarin substituent at the 7-position, while limiting the structural variations at position 3 and 4 to methyl groups (Figure 1, general structures 1–2). The modulation of MAO B activity was linearly correlated to the substituent’s lipophilicity at C7 (Carotti et al., 2006). Different linkers were installed at C7, probing both size and electronic features, with the OCH2Ph group being the most effective in promoting MAO B inhibitory activity. Among 7-benzyloxy derivatives (Figure 1, general structure 3), activity was highly dependent on the phenyl meta-substituent, whose lipophilicity governed MAO B inhibitory potencies rather than electronic properties, as outlined by a reliable QSAR model showing a parabolic correlation of potency with the hydrophobic Hansch constant π (Gnerre et al., 2000). These findings were later confirmed by disclosing a linear correlation between MAO B potency and calculated partition coefficient (ClogP) from a data set of coumarin derivatives exploring even more diverse substituents (De Colibus et al., 2005). A more extensive CoMFA-GOLPE study encompassing larger substitution patterns at the 7-position of the coumarin allowed to derive easily interpretable isocontour maps for both MAO A and MAO B affinity (Catto et al., 2006) (Figure 1). All steric, lipophilic, and electrostatic potential maps were found to slightly differ in the two isozymes, with some common features (e.g., a detrimental effect of ortho substitution). Noteworthy, CoMFA maps were successfully applied to shed light on MAO B/A selectivity, which was mainly modulated by the electronic and steric properties of the bridge connecting the coumarin core to the aryl substituent. Interestingly, the insertion of 7-phenylsulfonate esters reversed the selectivity profile yielding potent MAO A inhibitors (Catto et al., 2006). Complex stabilization was strongly promoted by sulfonate-group hydrogen bonding to the sidechain of Gln215 as supported by both molecular dynamics and molecular mechanics generalized Born surface area (MM-GBSA) calculations (Mangiatordi et al., 2017). With the aim of improving the drug-like properties of neutral coumarin hits, differently sized and substituted polar and/or protonatable moieties were introduced at the 4-position of 7-benzyloxy derivatives (Figure 1, general structure 5–6) leading to the discovery of NW-1772 (Pisani et al., 2009), an in vivo potent, bioavailable, reversible and CNS-acting MAO B inhibitor devoid of safety issues. X-ray crystallography pointed out its binding within the MAO enzymatic cleft, fully buried from the entrance cavity to FAD in a safinamide-superimposable pose (Binda et al., 2007). While maintaining a meta-halobenzyloxy group at C7 (Figure 1, general structure 4), fine-tuning the substituent at the 4-position returned nanomolar inhibitors that laid the groundwork for developing a robust and highly predictive Gaussian field-based 3D-QSAR model (Pisani et al., 2015), indicating that target recognition was dominated by steric hindrance whereas hydrogen bonding and electrostatic interactions tempered affinity to a lesser extent. During our investigation, serendipitously, while reacting 4-chloromethyl-7-substituted coumarins with excess primary amines, we faced an unexpected lactone opening reaction followed by intramolecular nucleophilic substitution (Pisani et al., 2013). This mechanism gave straightforward and efficient access to a novel scaffold for MAO inhibition, namely 6′-substituted (E)-2-(benzofuran-3 (2H)-ylidene)-N-alkylacetamides. Depending on the linker, sulfonate and benzyl derivatives confirmed A/B selectivity profiles obtained for the coumarin isomers.
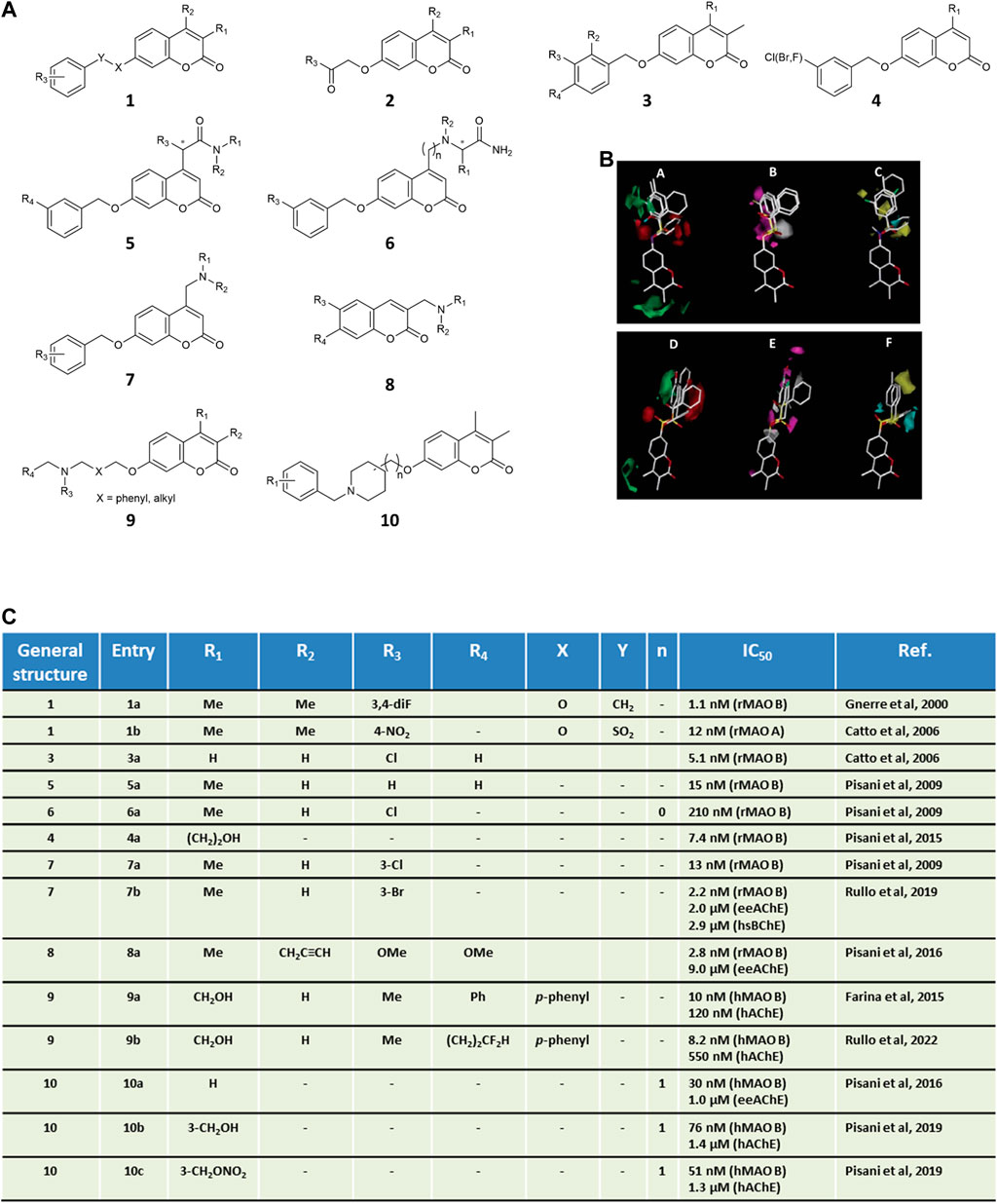
FIGURE 1. Panel (A) general structures of coumarin-based MAO inhibitors and MTDLs. Panel (B) CoMFA isocontour maps for MAO B (A–C) and MAO A (D–F) affinity of coumarin derivatives of general structure 1. In steric maps A and D, green and red regions show allowed and forbidden regions, respectively. Electronic density areas (B,E) are depicted as favorable (magenta) and unfavorable (grey), respectively. Lipophilic interactions (C,F) are reported as favorable (yellow) and unfavorable (cyan), respectively. CoMFA images were adapted with permission from Ref. (Catto et al., 2006). Copyright 2006, American Chemical Society. Panel (C) in vitro inhibitory data for the selected coumarins.
Acetylcholinesterase (AChE) is the enzyme responsible for the hydrolysis of the neurotransmitter acetylcholine (ACh) at the synaptic cleft. Because of many physiological effects mediated both in central and peripheral tissues, AChE is by far a validated pharmacological target. AChE inhibitors (AChEIs) are currently the first-line drugs for the symptomatic treatment of Alzheimer’s disease (AD), where AChEIs may sustain the cholinergic transmission in early to moderate AD stages (Mufson et al., 2008). The early crystal structures of AChE-inhibitor complexes revealed the presence of two binding pockets, a catalytic active site (CAS), surrounded by aromatic amino acids and neighbouring the so-called catalytic triad, and a peripheral binding site (PAS), separated by a deep gorge (Sussman et al., 1991). The presence of ammonium cationic warheads warrants the tight binding with both CAS and PAS, by means of cation-π interactions, as suggested by decamethonium and other neuromuscular blockers. To limit the peripheral effects of AChEIs and obtain CNS-active drugs, a protonatable nitrogen usually replaces the net positive charge of the ammonium ions for binding at the CAS, while a lipophilic moiety brought as the distal substituent acts as a second pharmacophore motif for π-π interactions with aromatic amino acids at the PAS. Such archetypal dual binding site (DBS) feature is represented by donepezil (Figure 2), the reference drug for the treatment of AD. Since the PAS is involved in the aggregation of beta-amyloid (Aβ) protein (Inestrosa et al., 1996), DBS inhibitors may exert antiamyloidogenic activity by disrupting protein-protein interactions triggered at the PAS.
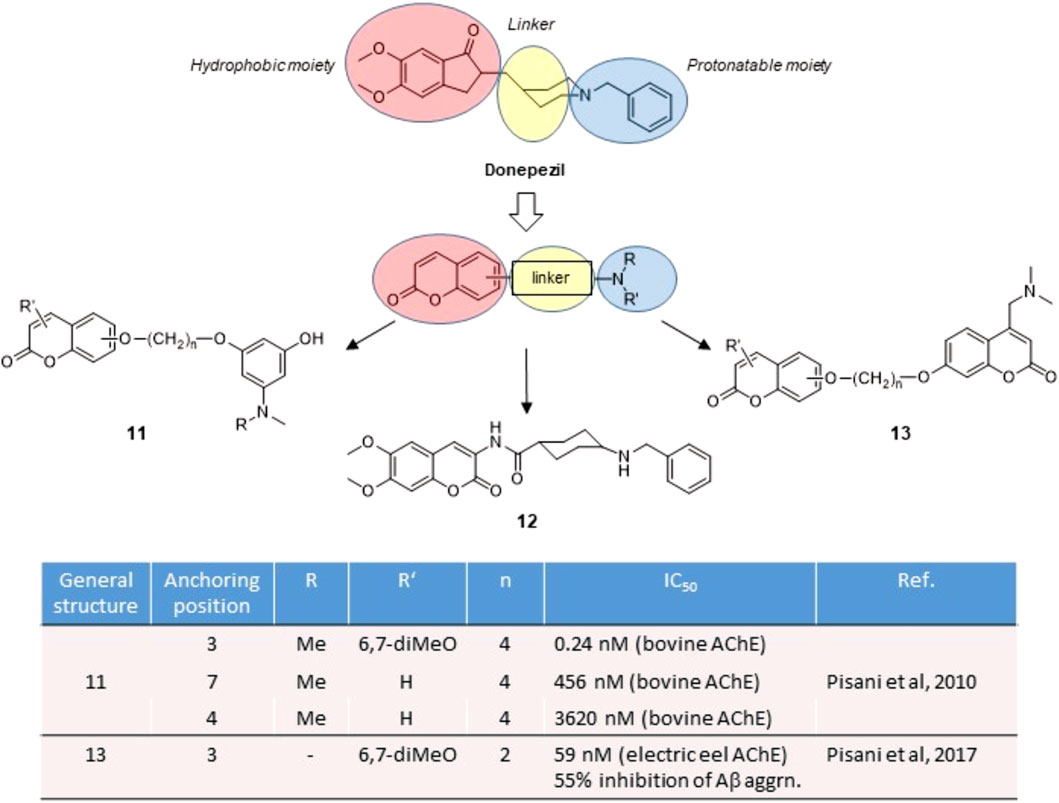
FIGURE 2. Rational design of AChE inhibitors by conjugative approach.
Starting from the disclosure of a fair AChE inhibitory activity for the 7-benzyloxy coumarin derivatives (Brühlmann et al., 2001), our group focused on the exploitation of the synthetic versatility of the coumarin scaffold to obtain DBS reversible inhibitors. By conjugating in a fragment-based design an edrophonium-like moiety with hydroxycoumarin through a polymethylene bridge, we obtained chimeric derivatives 11 (Figure 2) (Leonetti et al., 2008; Pisani et al., 2010), with good IC50 values in the submicromolar range, and in some cases hitting subnanomolar values. Docking studies evidenced the interaction of the coumarin residue at the PAS through hydrophobic and π-stacking interactions and of the cationic head at the CAS as the key interactions for binding. A systematic exploration of the substitution pattern around the coumarin core outlined the best solutions for its decoration. An “extended” substitution in 3-, 6- and 7-position of the coumarin led to higher potencies than in case of “folded” substitution in 4-, 5- and 8-position.
The same activity trend was later confirmed by coumarins 12 and 13 (Figure 2), where the putative toxicophore fragment represented by the hydroxyaniline moiety was replaced with a simple benzylamine substituent or a protonatable coumarin building block. Such substitution, still matching the donepezil-like pharmacophore, retained an effective binding at both the CAS and the PAS (Catto et al., 2013), and was later confirmed by X-ray analysis of AChE in complex with inhibitor 12 (Catto et al., 2020). By following a conjugation approach, we disclosed a series of homo- and heterodimers 13 (Figure 2) (Pisani et al., 2017) where at least one coumarin residue presented a protonatable nitrogen as the key substituent. Moreover, the DBS interaction of these coumarin hybrids was accompanied with a direct antiaggregating activity against Aβ self-aggregation, thus representing an appealing example of multitarget activity directly related to AChE inhibition.
Our contribution to the field of multi-targeting ligands has long been devoted to the development of dual coumarin-based AChE-MAO B inhibitors as potential polypharmacological weapons to combat AD. Apart from the well-established role of cholinergic transmission in AD, the rationale lies in the synergistic or additive effect coming from the oxidative stress restriction as a consequence of MAO blockade. In a seminal paper by some of us, the in vitro screening enrolling a small subset of 7-benzyloxycoumarins (Figure 1, general structure 3), previously characterized as nanomolar MAO inhibitors, led us identifying AChE inhibitors in the low micromolar IC50 range and with a non-competitive mechanism (Brühlmann et al., 2001). These encouraging preliminary results draw attention to the likelihood of decorating the coumarin skeleton to obtain dual-acting compounds with a particular attention to the substitution pattern branching position 3, 4, and 7 in line with steric prerequisites and synthetic handling.
The introduction of diverse basic functionalities at the 3-position of the 6,7-dimethoxycoumarin furnished promising molecules (Figure 1, general structure 8), although their activity was biased against MAO B inhibition at the nanomolar level while showing only moderate anti-AChE activity in the low micromolar range (Pisani et al., 2016). Similarly, unbalanced profiles were observed after the design and the screening of a small collection bearing head-or-tail modifications on 4-aminomethyl-7-benzyloxy-2H-chromen-2-one motif (Figure 1, general structure 7) (Rullo et al., 2019). The SAR indicated a preference for halogens as the meta-substituents of the benzyl ring to increase multitargeting activity. Interestingly, during these efforts triple-acting AChE/BChE/MAO B inhibitors were discovered, which could benefit from the additional inhibition of BChE whose increased expression and activity has been documented in chronic AD (Arendt et al., 1992). Upon applying “design-in” approaches (Farina et al., 2015) the molecular framework of coumarin-based multitarget ligands was built by exploiting the ability of the MAO B active site to accommodate 4-unhindered 7-benzyloxy-2H-chromen-2-one rings, which were tethered to N-benzylaminomethyl basic heads in order to improve AChE inhibition (Figure 1, general structure 9). By hybridizing the coumarin core with a protonatable group and a nitrate ester, we obtained triple-acting chemotypes able to inhibit AChE and MAO B, and to release low neuroprotective doses of nitric oxide (Pisani et al., 2019). Furthermore, structural variations led to improve the drug-likeness of the hybrid hit compounds, e.g., water solubility (Pisani et al., 2016). To this scope, the entropic penalties introduced by more flexible spacers were mitigated by the xylyl/polymethylene chain to piperidine ring exchange that allowed more favorable pKa values thanks to the N-benzylpiperidine moiety (Figure 1, general structure 10), inspired by donepezil. Cell-based assays showed that some of the hits presented low cytotoxicity and increased cell viability in neuroblastoma cell lines insulted by pro-oxidative toxins such as hydrogen peroxide, along with in vitro dual target’s inhibition from sub-micromolar to low nanomolar range and outstanding MAO B/A selectivity. Moreover hits 9a (Farina et al., 2015) (IC50 = 10 and 120 nM toward MAO B and AChE, respectively; MAO B/A selectivity >1,000) and 10a (Pisani et al., 2016) (IC50 = 30 nM and 1.0 μM toward MAO B and AChE, respectively; MAO B/A selectivity >90) behaved as CNS-permeant compounds in a BBB-mimicking model without efflux pump liabilities arising from P-glycoprotein interactions. For dual-targeting compound 9a the earliest X-ray crystallographic structures upon binding mutually to MAO B and AChE have been recently reported (Ekström et al., 2022). As challenging alternative, H/F and OH/CF2H bioisosteric mimicry led to a potent, reversible, and drug-like derivative as proved in early-ADME studies (compound 9b; IC50 = 8.2 and 550 nM toward MAO B and AChE, respectively; MAO B/A selectivity >1,200 (Rullo et al., 2022).
Coumarin itself or many of its derivatives showed antitumor activity by inhibiting the proliferation of the tumor cells at very low concentration without showing toxicity towards healthy cells. Their antitumor activity is due to (Wu et al., 2020) the inhibition of many signalling pathways such as PI3K/AKT/mTOR (Ma et al., 2020), microtubule polymerization (Madari et al., 2003; Liu et al., 2015) and angiogenesis. They also act on apoptotic proteins (Chuang et al., 2007) and on ROS regulation (Kontogiorgis et al., 2007), and through the inhibition of enzymes such as aromatase (Chen et al., 2004) and estrogen sulfatase (Stanway et al., 2006) both involved in breast cancer progression.
Aromatase (AR) is a multienzymatic complex, composed by cytochrome P450 (CYP19) and a NADPH-cytochrome P450 flavoprotein reductase, and it is localized in the endoplasmic reticulum of the cell.
AR controls the conversion of androgens to estrogens, catalyzing the aromatization of the steroidal enone, present in the A ring, into the corresponding phenolic ring trough the oxidation and the consequent elimination of the C19 methyl group. Many AR inhibitors contain an azole ring able to coordinate the iron ion of the AR heme group by means of their lone pair.
The design of our inhibitors started with the evaluation of the introduction of the appropriate azole substituent on our coumarin core. The best results were obtained by 4-imidazolyl coumarin derivatives (Leonetti et al., 2004; Stefanachi et al., 2011) bearing in 7-position a phenoxy or benzyloxy substituent as illustrated in Figure 3 (14).
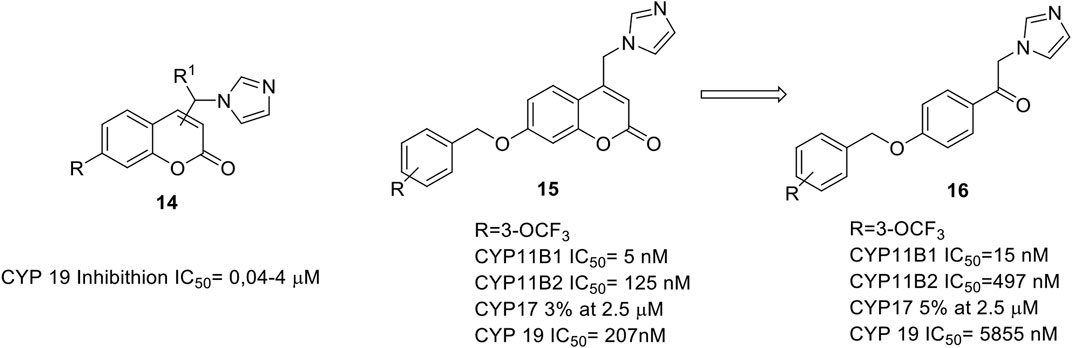
FIGURE 3. General structure of aromatase inhibitors. (Stefanachi et al., 2011; Stefanachi et al., 2015).
A three sites interaction model was proposed: 1) the binding driving interaction is the heme iron coordination by the lone pair of the electron rich imidazole, 2) the coumarin ring is positioned perpendicularly to the imidazole ring, and its lactone group establish a key hydrogen bond with S478, 3) the phenoxy and benzyloxy groups fill a hydrophobic pocket. We were able to obtain inhibitory activities in the range of 40–400 nM, comparable with that of fadrozole, in particular for the phenoxy derivatives.
All the synthesized compounds showed selectivity for AR with no activity towards other cytochromes P such as CYP17 (C17,20-lyase), CYP11B1 (steroid 11β-hydroxylase) and CYP11B2 (aldosterone synthase).
We also designed and synthesized acyclic analogs of coumarin derivatives (16, Figure 3), and the data showed that the absence of the coumarin ring does decrease the aromatase inhibitory activity, while increasing the potency at CYP11B1 and selectivity over CYP11B2, CYP19, and CYP17 (Stefanachi et al., 2015).
Herein, we review the main results of the pioneering scientific journey started about 20 years ago under the wise guide of Prof. Angelo Carotti (Bari, Italy) to explore the bioactivity potential of the coumarin core. Several programs on coumarin derivatives as single-targeting MAO, followed by AChE inhibitors as well as MTDLs, have been carried out focusing on the potential pharmacological treatment of neurodegenerative syndromes. Moreover, some coumarin derivatives also showed aromatase inhibitory activity and, therefore, have been developed as potential therapeutic agents for the treatment of breast cancer.Cao et al., 2016, Pisani et al., 2013.
Dedicated to prof. Angelo Carotti in the occasion of his 76th birthday.
AS and FL projected and organized the review; LP, MC, CA, AS, ON, and MR wrote the manuscript; GM and AC revised the manuscript. All authors gave final approval to the manuscript.
The authors would like to thank all the professor Carotti’s colleagues and alumni who collaborated for the studies reported in this mini-review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al-Abbas, N. S., and Shaer, N. A. (2021). Combination of coumarin and doxorubicin induces drug-resistant acute myeloid leukemia cell death. Heliyon 7 (3), e06255. doi:10.1016/j.heliyon.2021.e06255
Altomare, C., Carrupt, P.-A., El Tayar, N., Testa, B., and Nagatsu, T. (1991). Electronic and conformational effects on the lipophilicity of isomers and analogs of the neurotoxin 1-methyl-4-phenylpyridinium (MPP+). Helv. Chim. Acta 74, 290–296. doi:10.1002/hlca.19910740205
Altomare, C., Carrupt, P. A., Gaillard, P., El Tayar, N., Testa, B., and Carotti, A. (1992). Quantitative structure-metabolism relationship analyses of MAO-mediated toxication of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine and analogs. Chem. Res. Toxicol. 5 (3), 366–375. doi:10.1021/tx00027a008
Altomare, C., Cellamare, S., Summo, L., Catto, M., Carotti, A., Thull, U., et al. (1998). Inhibition of monoamine oxidase-B by condensed pyridazines and pyrimidines: Effects of lipophilicity and structure-activity relationships. J. Med. Chem. 41 (20), 3812–3820. doi:10.1021/jm981005y
Arendt, T., Brückner, M. K., Lange, M., and Bigl, V. (1992). Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer's disease resemble embryonic development--a study of molecular forms. Neurochem. Int. 21 (3), 381–396. doi:10.1016/0197-0186(92)90189-x
Binda, C., Wang, J., Pisani, L., Caccia, C., Carotti, A., Salvati, P., et al. (2007). Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. Struct. Biol. 509 (23), 584822. doi:10.1038/nsb732
Brühlmann, C., Ooms, F., Carrupt, P. A., Testa, B., Catto, M., Leonetti, F., et al. (2001). Coumarins derivatives as dual inhibitors of acetylcholinesterase and monoamine oxidase. J. Med. Chem. 44 (19), 3195–3198. doi:10.1021/jm010894d
Cao, D., Liu, Y., Yan, W., Wang, C., Bai, P., Wang, T., et al. (2016). Design, synthesis, and evaluation of in vitro and in vivo anticancer activity of 4-substituted coumarins: A novel class of potent tubulin polymerization inhibitors. J. Med. Chem. 59 (12), 5721–5739. doi:10.1021/acs.jmedchem.6b00158
Carotti, A., Altomare, C., Catto, M., Gnerre, C., Summo, L., De Marco, A., et al. (2006). Lipophilicity plays a major role in modulating the inhibition of monoamine oxidase B by 7-substituted coumarins. Chem. Biodivers. 3 (2), 134–149. doi:10.1002/cbdv.200690017
Catto, M., Nicolotti, O., Leonetti, F., Carotti, A., Favia, A. D., Soto-Otero, R., et al. (2006). Structural insights into monoamine oxidase inhibitory potency and selectivity of 7-substituted coumarins from ligand- and target-based approaches. J. Med. Chem. 49 (16), 4912–4925. doi:10.1021/jm060183l
Catto, M., Pisani, L., de la Mora, E., Belviso, B. D., Mangiatordi, G. F., Pinto, A., et al. (2020). Chiral separation, X-ray structure, and biological evaluation of a potent and reversible dual binding site AChE inhibitor. ACS Med. Chem. Lett. 11 (5), 869–876. doi:10.1021/acsmedchemlett.9b00656
Catto, M., Pisani, L., Leonetti, F., Nicolotti, O., Pesce, P., Stefanachi, A., et al. (2013). Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg. Med. Chem. 21 (1), 146–152. doi:10.1016/j.bmc.2012.10.045
Chen, S., Cho, M., Karlsberg, K., Zhou, D., and Yuan, Y. C. (2004). Biochemical and biological characterization of a novel anti-aromatase coumarin derivative. J. Biol. Chem. 279 (46), 48071. doi:10.1074/jbc.M406847200
Chuang, J. Y., Huang, Y. F., Lu, H. F., Ho, H. C., Yang, J. S., Li, T. M., et al. (2007). Coumarin induces cell cycle arrest and apoptosis in human cervical cancer HeLa cells through a mitochondria- and caspase-3 dependent mechanism and NF-kappaB down-regulation. Vivo 21 (6), 1003–1009.
De Colibus, L., Li, M., Binda, C., Lustig, A., Edmondson, D. E., and Mattevi, A. (2005). Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. U. S. A. 102 (36), 12684. doi:10.1073/pnas.0505975102
Ekström, F., Gottinger, A., Forsgren, N., Catto, M., Iacovino, L. G., Pisani, L., et al. (2022). Dual reversible coumarin inhibitors mutually bound to monoamine oxidase B and acetylcholinesterase crystal structures. ACS Med. Chem. Lett. 13 (3), 499–506. doi:10.1021/acsmedchemlett.2c00001
Farina, R., Pisani, L., Catto, M., Nicolotti, O., Gadaleta, D., Denora, N., et al. (2015). Structure-based design and optimization of multitarget-directed 2H-Chromen-2-one derivatives as potent inhibitors of monoamine oxidase B and cholinesterases. J. Med. Chem. 58 (14), 5561–5578. doi:10.1021/acs.jmedchem.5b00599
Gnerre, C., Catto, M., Leonetti, F., Weber, P., Carrupt, P. A., Altomare, C., et al. (2000). Inhibition of monoamine oxidases by functionalized coumarin derivatives: Biological activities, QSARs, and 3D-QSARs. J. Med. Chem. 43 (25), 4747–4758. doi:10.1021/jm001028o
Inestrosa, N. C., Alvarez, A., Pérez, C. A., Moreno, R. D., Vicente, M., Linker, C., et al. (1996). Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into alzheimer's fibrils: Possible role of the peripheral site of the enzyme. Neuron 16 (4), 881–891. doi:10.1016/s0896-6273(00)80108-7
Kneubühler, S., Carta, V., Altomare, C., Carotti, A., and Testa, B. (1993). Synthesis and monoamine oxidase inhibitory activity of 3-substituted 5H-indeno[1, 2-c]pyridazines. Helv. Chim. Acta 76, 1956–1963. doi:10.1002/hlca.19930760514
Kneubühler, S., Thull, U., Altomare, C., Carta, V., Gaillard, P., Carrupt, P. A., et al. (1995). Inhibition of monoamine oxidase-B by 5H-indeno[1, 2-c]pyridazines: Biological activities, quantitative structure-activity relationships (QSARs) and 3D-QSARs. J. Med. Chem. 38 (19), 3874–3883. doi:10.1021/jm00019a018
Kontogiorgis, C. A., Xu, Y., Hadjipavlou-Litina, D., and Luo, Y. (2007). Coumarin derivatives protection against ROS production in cellular models of Aβtoxicities. Free Radic. Res. 41 (10), 1168–1180. doi:10.1080/10715760701447884
Leonetti, F., Catto, M., Nicolotti, O., Pisani, L., Cappa, A., Stefanachi, A., et al. (2008). Homo- and hetero-bivalent edrophonium-like ammonium salts as highly potent, dual binding site AChE inhibitors. Bioorg. Med. Chem. 16 (15), 7450–7456. doi:10.1016/j.bmc.2008.06.022
Leonetti, F., Favia, A., Rao, A., Aliano, R., Paluszcak, A., Hartmann, R. W., et al. (2004). Design, synthesis, and 3D QSAR of novel potent and selective aromatase inhibitors. J. Med. Chem. 47 (27), 6792–6803. doi:10.1021/jm049535j
Liu, H., Wang, Y., Sharma, A., Mao, R., Jiang, N., She, J. X., et al. (2015). Dun, BDerivatives containing both coumarin and benzimidazole potently induce caspase-dependent apoptosis of cancer cells through inhibition of PI3K-AKT-mTOR signaling. Anticancer. Drugs 26 (6), 667–677. doi:10.1097/CAD.0000000000000232
Ma, C. C., and Liu, Z. P. (2017). Design and synthesis of coumarin derivatives as novel PI3K inhibitors. Anticancer. Agents Med. Chem. 17 (3), 395–403. doi:10.2174/1871520616666160223120207
Madari, H., Panda, D., Wilson, L., and Jacobs, R. S. (2003). Dicoumarol: A unique microtubule stabilizing natural product that is synergistic with taxol. Cancer Res. 63 (6), 1214.
Mangiatordi, G. F., Alberga, D., Pisani, L., Gadaleta, D., Trisciuzzi, D., Farina, R., et al. (2017). A rational approach to elucidate human monoamine oxidase molecular selectivity. Eur. J. Pharm. Sci. 101, 90–99. doi:10.1016/j.ejps.2017.02.008
Mufson, E. J., Counts, S. E., Perez, S. E., and Ginsberg, S. D. (2008). Cholinergic system during the progression of alzheimer's disease: Therapeutic implications. Expert Rev. Neurother. 8 (11), 1703–1718. doi:10.1586/14737175.8.11.1703
Pisani, L., Barletta, M., Soto-Otero, R., Nicolotti, O., Mendez-Alvarez, E., Catto, M., et al. (2013). Discovery, biological evaluation, and structure-activity and -selectivity relationships of 6'-substituted (E)-2-(benzofuran-3(2H)-ylidene)-N-methylacetamides, a novel class of potent and selective monoamine oxidase inhibitors. J. Med. Chem. 56 (6), 2651–2664. doi:10.1021/jm4000769
Pisani, L., Catto, M., De Palma, A., Farina, R., Cellamare, S., and Altomare, C. D. (2017). Discovery of potent dual binding site acetylcholinesterase inhibitors via homo- and heterodimerization of coumarin-based moieties. ChemMedChem 12 (16), 1349–1358. doi:10.1002/cmdc.201700282
Pisani, L., Catto, M., Giangreco, I., Leonetti, F., Nicolotti, O., Stefanachi, A., et al. (2010). Design, synthesis, and biological evaluation of coumarin derivatives tethered to an edrophonium-like fragment as highly potent and selective dual binding site acetylcholinesterase inhibitors. ChemMedChem 5 (9), 1616–1630. doi:10.1002/cmdc.201000210
Pisani, L., Catto, M., Nicolotti, O., Grossi, G., Di Braccio, M., Soto-Otero, R., et al. (2013). Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur. J. Med. Chem. 70, 723–739. doi:10.1016/j.ejmech.2013.09.034
Pisani, L., Farina, R., Catto, M., Iacobazzi, R. M., Nicolotti, O., Cellamare, S., et al. (2016). Exploring basic tail modifications of coumarin-based dual acetylcholinesterase-monoamine oxidase B inhibitors: Identification of water-soluble, brain-permeant neuroprotective multitarget agents. J. Med. Chem. 59 (14), 6791–6806. doi:10.1021/acs.jmedchem.6b00562
Pisani, L., Farina, R., Nicolotti, O., Gadaleta, D., Soto-Otero, R., Catto, M., et al. (2015). In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur. J. Med. Chem. 89, 98–105. doi:10.1016/j.ejmech.2014.10.029
Pisani, L., Farina, R., Soto-Otero, R., Denora, N., Mangiatordi, G. F., Nicolotti, O., et al. (2016). Searching for multi-targeting neurotherapeutics against alzheimer's: Discovery of potent AChE-MAO B inhibitors through the decoration of the 2H-Chromen-2-one structural motif. Mol. (Basel, Switz. 21 (3), 362. doi:10.3390/molecules21030362
Pisani, L., Iacobazzi, R. M., Catto, M., Rullo, M., Farina, R., Denora, N., et al. (2019). Investigating alkyl nitrates as nitric oxide releasing precursors of multitarget acetylcholinesterase-monoamine oxidase B inhibitors. Eur. J. Med. Chem. 161, 292–309. doi:10.1016/j.ejmech.2018.10.016
Pisani, L., Muncipinto, G., Miscioscia, T. F., Nicolotti, O., Leonetti, F., Catto, M., et al. (2009). Discovery of a novel class of potent coumarin monoamine oxidase B inhibitors: Development and biopharmacological profiling of 7-[(3-chlorobenzyl)oxy]-4-[(methylamino)methyl]-2H-chromen-2-one methanesulfonate (NW-1772) as a highly potent, selective, reversible, and orally active monoamine oxidase B inhibitor. J. Med. Chem. 52 (21), 6685–6706. doi:10.1021/jm9010127
Rullo, M., Catto, M., Carrieri, A., de Candia, M., Altomare, C. D., and Pisani, L. (2019). Chasing ChEs-MAO B multi-targeting 4-aminomethyl-7-benzyloxy-2H-Chromen-2-ones. Molecules 24 (24), 4507. doi:10.3390/molecules24244507
Rullo, M., Cipolloni, M., Catto, M., Colliva, C., Miniero, D. V., Latronico, T., et al. (2022). Probing fluorinated motifs onto dual AChE-MAO B inhibitors: Rational design, synthesis, biological evaluation, and early-ADME studies. J. Med. Chem. 65 (5), 3962–3977. doi:10.1021/acs.jmedchem.1c01784
Stanway, S. J., Purohit, A., Woo, L. W., Sufi, S., Vigushin, D., Ward, R., et al. (2006). Phase I study of STX 64 (667 coumate) in breast cancer patients: The first study of a steroid sulfatase inhibitor. Clin. Cancer Res. 12 (5), 1585–1592. doi:10.1158/1078-0432.CCR-05-1996
Stefanachi, A., Favia, A. D., Nicolotti, O., Leonetti, F., Pisani, L., Catto, M., et al. (2011). Design, synthesis, and biological evaluation of imidazolyl derivatives of 4, 7-disubstituted coumarins as aromatase inhibitors selective over 17-α-hydroxylase/C17-20 lyase. J. Med. Chem. 54 (6), 1613–1625. doi:10.1021/jm101120u
Stefanachi, A., Hanke, N., Pisani, L., Leonetti, F., Nicolotti, O., Catto, M., et al. (2015). Discovery of new 7-substituted-4-imidazolylmethyl coumarins and 4'-substituted-2-imidazolyl acetophenones open analogues as potent and selective inhibitors of steroid-11β-hydroxylase. Eur. J. Med. Chem. 89, 106–114. doi:10.1016/j.ejmech.2014.10.021
Stefanachi, A., Leonetti, F., Pisani, L., Catto, M., and Carotti, A. (2018). Coumarin: A natural, privileged and versatile scaffold for bioactive compounds. Mol. (Basel, Switz. 23 (2), 250. doi:10.3390/molecules23020250
Sussman, J. L., Harel, M., Frolow, F., Oefner, C., Goldman, A., Toker, L., et al. (1991). Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Sci. (New York, N.Y.) 253 (5022), 872–879. doi:10.1126/science.1678899
Vogel, A. (1820). Darstellung von Benzoesaure aus der Tonka-Boline und aus den MeliIoten-oder Steinklee-Blumen. Ann. Phys. Phys. Chem. 64, 161–166. doi:10.1002/andp.18200640205
Wu, Y., Xu, J., Liu, Y., Zeng, Y., and Wu, G. (2020). A review on anti-tumor mechanisms of coumarins. Front. Oncol. 10, 592853. doi:10.3389/fonc.2020.592853
Keywords: cholinesterases, monoamine oxidases, coumarin, neurodegeneration, cancer
Citation: Pisani L, Catto M, Muncipinto G, Nicolotti O, Carrieri A, Rullo M, Stefanachi A, Leonetti F and Altomare C (2022) A twenty-year journey exploring coumarin-based derivatives as bioactive molecules. Front. Chem. 10:1002547. doi: 10.3389/fchem.2022.1002547
Received: 25 July 2022; Accepted: 14 September 2022;
Published: 10 October 2022.
Edited by:
Nenad Filipović, University of Belgrade, SerbiaReviewed by:
Sonia Malik, Université d’Orléans, FranceCopyright © 2022 Pisani, Catto, Muncipinto, Nicolotti, Carrieri, Rullo, Stefanachi, Leonetti and Altomare. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Stefanachi, YW5nZWxhLnN0ZWZhbmFjaGlAdW5pYmEuaXQ= Francesco Leonetti, ZnJhbmNlc2NvLmxlb25ldHRpQHVuaWJhLml0
†These authors have contributed equally to this work and share first authorship.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.