 Lewis P. M. Green1
Lewis P. M. Green1 Tasha R. Steel1
Tasha R. Steel1 Mie Riisom1,2
Mie Riisom1,2 Muhammad Hanif1
Muhammad Hanif1 Tilo Söhnel1
Tilo Söhnel1 Stephen M. F. Jamieson2
Stephen M. F. Jamieson2 L. James Wright1
L. James Wright1 James D. Crowley3
James D. Crowley3 Christian G. Hartinger1*
Christian G. Hartinger1*- 1School of Chemical Sciences, University of Auckland, Auckland, New Zealand
- 2Auckland Cancer Society Research Centre, University of Auckland, Auckland, New Zealand
- 3Department of Chemistry, University of Otago, Dunedin, New Zealand
Multimetallic complexes have been shown in several examples to possess greater anticancer activity than their monometallic counterparts. The increased activity has been attributed to altered modes of action. We herein report the synthesis of a series of heterodimetallic compounds based on a ditopic ligand featuring 2-pyridylimine chelating motifs and organometallic half-sandwich moieties. The complexes were characterized by a combination of 1H NMR spectroscopy, electrospray ionization mass spectrometry, elemental analysis and single crystal X-ray diffraction. Investigations into the stability of representative complexes in DMSO-d6 and 10% DMSO-d6/D2O revealed the occurrence of solvent-chlorido ligand exchange. Proliferation assays in four human cancer cell lines showed that the Os-Rh complex possessed minimal activity, while all other complexes were inactive.
1 Introduction
Metals play various roles in biological processes (Buccella et al., 2019), e.g., proteins often use metal centers to adopt certain structures or as catalytically active sites (Haas and Franz, 2009; Wodrich and Hu, 2017; Ghosh et al., 2021). Metal complexes, most often platinum compounds, have been used for a long time to treat cancer (Jakupec et al., 2008; Kenny and Marmion, 2019; Simpson et al., 2019; Boros et al., 2020; Tremlett et al., 2021). In addition to the clinically successful DNA-targeting cis-, carbo- and oxaliplatin (Johnstone et al., 2014), other mononuclear species studied include Ru, Os, Rh and Ir-based compounds (Sava et al., 2003; Arango et al., 2004; Hartinger et al., 2008; Geldmacher et al., 2012; Maillet et al., 2013). The non-Pt complexes have been headlined by various Ru-based examples, including the clinically explored KP1339 (Jakupec et al., 2008; Heffeter et al., 2010).
Synthetic attempts to link one or more metal-containing fragments have been made in order to obtain compounds with higher potency and modes of action different from those of the established anticancer agents. The tris-Pt compound BBR3464 (Figure 1) features Pt centers which contribute to DNA binding through electrostatic or covalent interactions (Manzotti et al., 2000). Indeed, the DNA binding of BBR3464 was found to be considerably different to that of cisplatin and the compound was found to show no cross-resistance in cancer cells (Manzotti et al., 2000; Ruhayel et al., 2012). Inspired by this approach, we reported organometallic compounds containing two metal centers coordinated to a maltol-derived bis(3-hydroxy-2-methyl-4-pyridone) ligand bridged by a spacer of varying length (Figure 1) (Mendoza-Ferri et al., 2008; Parveen et al., 2019b). The IC50 value for the dodecane-bridged bis-Ru complex in SW480 cancer cells was similar to that of platinum reference compounds and was an order of magnitude lower than of the clinically-studied Ru(III) compound KP1019 (Mendoza-Ferri et al., 2008; Mendoza-Ferri et al., 2009; Nováková et al., 2009; Nazarov et al., 2018), while the mononuclear analog gave IC50 values greater than 100 µM, indicating a lack of cytotoxic activity (Mendoza-Ferri et al., 2008). Multimetallic triruthenium-carbonyl clusters (Gonchar et al., 2020) have also been found to be cytotoxic, and the most active of these clusters was shown to be a system that featured a glucose-inspired phosphorus ligand. The IC50 values for the clusters were in the sub-µM range for wildtype and cisplatin-resistant human ovarian cancer cells (Gonchar et al., 2020). Further to this triruthenium cluster, we have investigated the use of easily synthesized ditopic ligands to form homodimetallic complexes (Figure 1, top left). These structures were important as they highlighted the possibility for the rapid formation of a range of homo- and heterodimetallic species. Although the compounds had poor cytotoxicity in comparison to other dimetallic compounds (Steel et al., 2021), synthetic alterations to the ditopic ligand being used could easily yield more potent metal-based chemotherapeutics. Alternatively, scaffold structures can be adopted to form dimetallic species of greater complexity. Relevant examples include a trimeric scaffold consisting of alkylated 1,3,5-triaza-7-phosphaadamantane moieties or other phosphine or nitrogen donor-based ligand systems (Burgoyne et al., 2016; Batchelor et al., 2019; Curado et al., 2019). Moreover, tetra- and octanuclear Ir and Rh complexes can be created using dendritic structures that have Schiff-base ligands based on poly(propyleneimine) scaffolds (Payne et al., 2013). These metallodendrimers highlight an interesting trend whereby increasing multinuclearity resulted in an analogous increase in cytotoxicity as compared to the mono-metallic derivatives (Payne et al., 2013).
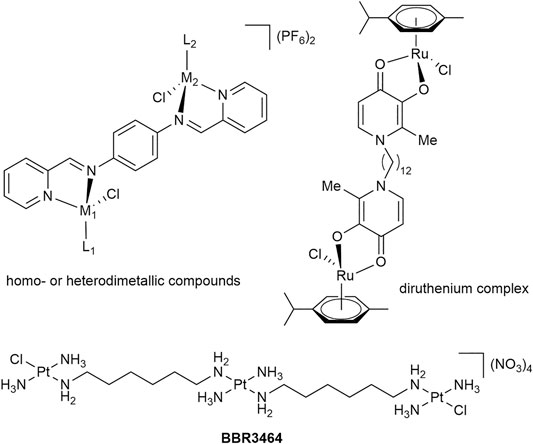
FIGURE 1. Homo- and heterometallic complexes with anticancer activity.
The nature of the anticancer activity observed for multimetallic compounds is likely to be different from those found for the corresponding monometallic analogs due to altered molecular modes of action, as discussed above for BBR3464 and closely related compounds (Ruhayel et al., 2012). Besides DNA, bis- and tris-Pt complexes can also bind to proteoglycans through sulfate anchoring (Gorle et al., 2018; Gorle et al., 2021). The resulting metalloshielding stops enzyme degradation and has recently been shown to be biologically relevant in cells (Gorle et al., 2018; Gorle et al., 2021). This method of template protection, a strategy analogous to complex–DNA binding, heralds a new avenue in the mechanisms of action for multimetallic compounds (Gorle et al., 2021).
Recent investigations have also demonstrated that cytotoxic efficacy can be altered when heterodimetallic systems are employed or could be used to release cytotoxic payload (Herry et al., 2019; Jain, 2019; Lisboa et al., 2020; Lisboa et al., 2021). The distinct properties of each metal center in terms of inertness, redox properties and affinity to bioligands will determine the lipophilic character and biological target binding ability of heterometallic compounds (Benjamin Garbutcheon-Singh et al., 2011; Parveen et al., 2019a; Batchelor et al., 2019). Mixing and matching these characteristics within one compound carves out new chemical space in the development of chemotherapeutic agents. For example, a Fe-Ru phosphane-bridged complex in ovarian carcinoma cell lines showed increased activity over the Ru-Ru analogue (Herry et al., 2019). The difference in activity was hypothesized to be the result of the Fe center facilitating the cellular uptake of the compound (Herry et al., 2019). Heteronuclear transition metal systems have also been investigated as theranostics (Redrado et al., 2021), in which the therapeutic can be traced via the diagnostic component of the molecule. Most often in theranostics, a fluorophore is conjugated to the metal-based therapeutic, for example, as in the recently reported gold- and ruthenium-based complexes with therapeutic and imaging capability (Bertrand et al., 2016). Polyaryl derivates like anthracene or pyrene are common fluorophores in theranostics, as is BODIPY (Bertrand et al., 2018). The versatility of BODIPY has allowed it to be incorporated into Pt-, Au-, Ru-, Ti- and Ir-based therapeutics (Bertrand et al., 2018). The formation of dimetallic theranostic compounds is also possible, yet slightly more complex (Bertrand et al., 2016). Two recent examples have paired tris(bipyridine)ruthenium and a Ru–porphyrin with therapeutics developed around a Au center (Bertrand et al., 2016). The range of investigations that have been conducted into the creation and use of heterodimetallic systems has highlighted the myriad of potential advantages afforded by employing compounds with different metal centers.
Herein, we report the synthesis and characterization of heterodimetallic complexes based on a symmetric ditopic 2-pyridylimine ligand. The stabilities of these complexes in DMSO and aqueous solutions were investigated and the cytotoxicities of the complexes in human cancer cells were determined.
2 Results and Discussion
Recently, we reported the preparation of homodimetallic compounds of a symmetric, ditopic 2-pyridylimine-based ligand featuring a 1,4-diaminobenzene spacer (Figure 1, LDAB), which is known to form stable coordination complexes (Steel et al., 2021). The formation of these di-Ru, -Os, -Rh and -Ir compounds with two different ditopic ligands highlighted the ability to reliably obtain homodimetallic compounds in good yield (Steel et al., 2021). The complexes also demonstrated a range of structural characteristics and cytotoxic activity (Steel et al., 2021). In the case of those homodimetallic complexes, the ligand was prepared before it was decorated with metal centers. This was feasible due to the symmetric nature of the target complexes. To prepare low-symmetry heterodimetallic compounds based on the same ligand the synthetic strategy had to be adapted, as the previously reported route would likely lead to a mixture of compounds. Accordingly, different pathways were explored (Scheme 1). Initially, inspired by our recent work generating heterometallic PdPt caged systems (Lisboa et al., 2020), we investigated a two-step process, in which the mononuclear precursor 1a would be formed by reaction from mono-topic 2-pyridylimine ligand 1 with dimeric [Ru(cym)Cl2]2 a (cym = η6-p-cymene), followed by conversion with the preformed second metal fragment [Os(cym)(2-pyridine carboxaldehyde)Cl2]2 to target compound 2a. Indeed, 1a was prepared in very good yield (92%), however, the subsequent reaction with [Os(cym)(2-pyridine carboxaldehyde)Cl2]2 only provided 2a in trace amounts (Scheme 1). In an alternative approach, 1a was first treated with 2-pyridine carboxaldehyde to generate the second 2-pyridylimine binding site in situ, and the subsequent reaction with [Os(cym)Cl2]2 afforded 2a in excellent yield (84%).
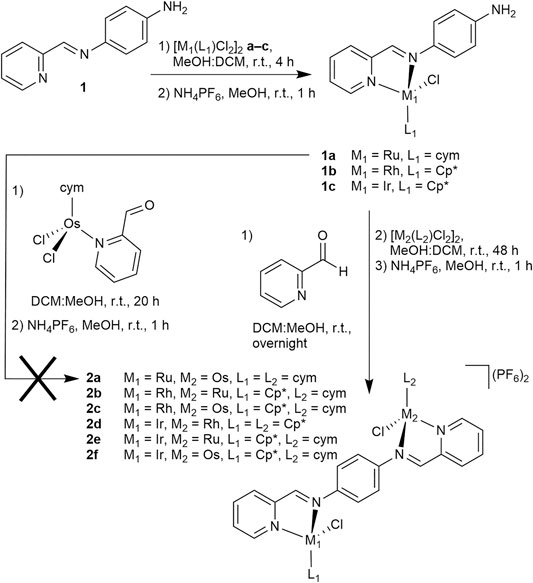
SCHEME 1. Synthetic strategy towards heterodimetallic complexes 2a–2f.
In the 1H NMR spectrum of complex 2a peaks characteristic of π-coordinated cym were observed, with the expected integrals relative to the ligand proton signals. Notably, in contrast to the symmetric dimetallic complexes (Ru2 and Os2, Figure 2), the phenylene protons resonated as two distinct doublets due to the lower molecular symmetry, whereas the corresponding protons in the higher symmetry Ru2 and Os2 complexes were singlets (Steel et al., 2021). The incorporation of two different metals and the resulting loss of symmetry within the molecule also resulted in more complex sets of signals for the remaining protons (Figure 2 and Supplementary Figure S19). For example, the 1H NMR spectrum of 2a features two sets of resonances for the α-pyridyl and imine protons of the coordinating 2-pyridylimine units (Figure 2). The chemical shift values for one set of the α-pyridyl and imine resonances match well with those observed for the homodimetallic Ru2, whilst the second set of peaks display chemical shift values that are very similar to those observed in the Os2 complexes (Figure 2). These data are consistent with the presence of two distinct 2-pyridylimine units in 2a, one coordinated to a [Ru(cym)Cl] fragment and the second binding to an [Os(cym)Cl] unit. In the electrospray ionization (ESI) mass spectrum of 2a, the pseudomolecular ion [M – 2PF6]2+ was observed as the base peak at m/z 459.0743 (m/zcalc 459.0710; Supplementary Table S1) with the isotope pattern matching that predicted for the heterodimetallic complex, in addition to [M – PF6]+ at m/z 1,063.1156 (m/zcalc 1,063.1067).
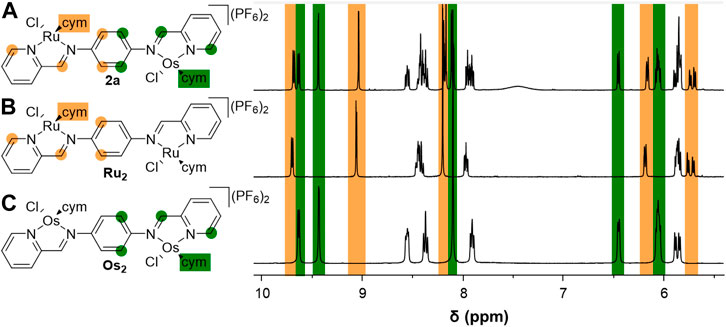
FIGURE 2. Comparison of the 1H NMR spectra for the heterodimetallic Ru-Os complex 2a (A), to those of the symmetric Ru2 (B) and Os2 (C) derivatives.
The heterodimetallic compounds 2b–2f were also synthesized in good yields (Scheme 1; 42–67%) from the mononuclear precursors 1b (for 2b and 2c) and 1c (for 2d–2f) using the same strategy as for 2a and the purity was confirmed by elemental analysis. We considered the relative labilities of the metal centers in the synthetic strategy to optimize the purity, as substitution of the metal center may occur in situ and result in the formation of undesired homodimetallic species. To mitigate the possibility of this occurring, the relatively inert Ir precursor 1c was selected for use in the first reaction step when preparing 2d–2f. 1H NMR spectra of 2b–2f were all indicative of formation of the heterodimetallic complexes (Supplementary Figure S19). By comparison with the equivalent homodimetallic complexes, in particular the protons adjacent to the pyridine and imine nitrogen atoms shifted characteristically depending on the nature of the metal center. ESI-mass spectrometry (MS) was used to characterize the complexes further and the mass spectra of 2b and 2d–2f featured signals assignable to the [M – PF6]+ and [M – 2PF6]2+ pseudomolecular ions (Supplementary Table S1), with the latter usually being present in higher abundance. Each of these peaks showed clearly the isotope pattern expected for the heterodimetallic complex (compare Figure 3 for 2f). In addition to these ions, we often detected, and in particular for 2c, some single imine hydrolysis of the ligand, resulting in the loss of a pyridyl fragment, one of the metal centers and the respective π-bound ligand moiety, giving peaks corresponding to the mononuclear species [M – C6H5N – M(L)Cl – 2PF6]+. It should be noted that usually we detected ions that could be assigned to both metal moieties of the heterodimetallic complexes.
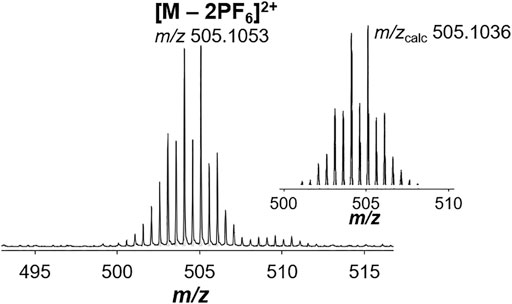
FIGURE 3. ESI-mass spectrometry data for the Os-Ir complex 2f compared to the calculated isotope pattern for the [M – 2PF6]2+ cation.
The molecular structures of the complexes 2a, 2d and 2e were determined by single crystal X-ray diffraction studies (Figure 4). The single crystals were grown via the slow diffusion of toluene into a saturated solution of the respective complex in acetonitrile. All complexes have a triclinic crystal system and crystallized in the P-1 space group. While 2a co-crystallized with an acetonitrile molecule, the structure of 2d featured a disordered toluene molecule which was found sandwiched between the Cp* ligands of two neighboring molecules of 2d (Figure 5A). The molecular structure of 2e also contains a strongly disordered toluene molecule, which was excluded from the final refinement (Supplementary Figure S20, Supplementary Table S2). The structure features a co-crystallized water molecule, resulting in hydrogen bonding with the chlorido ligand coordinated to the Ru center, [d(Cl2–O) = 3.205 Å], and one of the PF6− counterions (d(F10–O) = 2.929 Å). Furthermore, in all three structures, the PF6− counterions were extensively involved in H bonding networks with several neighboring complex molecules (Figure 5B for that of 2a). In the cases of 2a (with cym ligands coordinated to both Ru and Os) and 2d (with Cp* ligands coordinated to both Rh and Ir), the metal centers were statistically distributed between the two positions in the crystal lattice; hence, it was not possible to distinguish between the two M(cym) or M(Cp*) moieties in the molecular structures. In contrast, in the structure of 2e, the Ru and Ir centers could be unambiguously distinguished based on their different π-bound ligands, i.e., cym and Cp*, respectively. For 2a and 2d, two independent molecules with R,R and S,S configurations at the metal centers were found in the unit cell, whereas in 2e the two molecules had R,S and S,R configurations. For 2d and 2e the metal centers were arranged on opposite faces of the ligand whereas in 2a the M(cym) moieties were found on the same face of the phenylene spacer (Table 1). In contrast to 2e, the statistical distribution of the metal centers in 2a and 2d over the two sites, makes it impossible to determine accurately the M–donor atom bond lengths (Table 1).
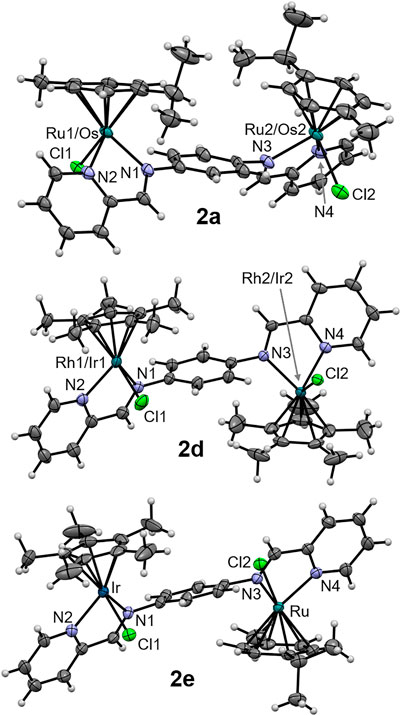
FIGURE 4. ORTEP representation of one of the two enantiomers of complexes 2a, 2d and 2e drawn at 50% probability level. Any co-crystallized solvent molecules and hexafluorophosphate counterions have been omitted for clarity.
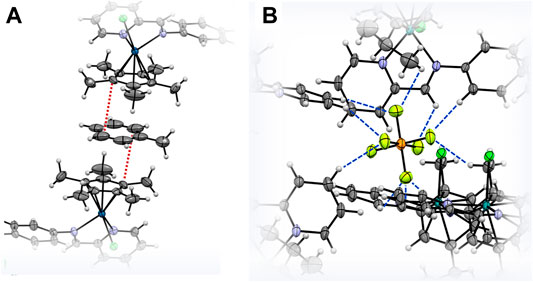
FIGURE 5. ORTEP representation of (A) co-crystallized toluene π-bonding with the Cp* ligands of two adjacent molecules of 2d, and (B) H-bonding network around a PF6− counteranion and 2a molecules. The shortest C–C distances at 3.604 Å in (A) are highlighted by red dashed lines while the H bonds in (B) are indicated in blue.
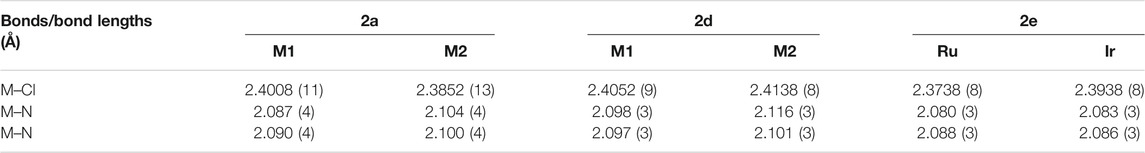
TABLE 1. Key bond lengths (Å) in the molecular structures of 2a, 2d and 2e.
Imines can hydrolyze in aqueous solution into the respective amine and aldehyde building blocks, and metal complexes may undergo ligand exchange reactions, especially in the presence of coordinating solvents like DMSO which is often used for the preparation of stock solutions for biological assays. Therefore, the stabilities of the representative complexes Os-Rh 2c and Os-Ir 2f were analyzed by 1H NMR spectroscopy over a 72 h time period in DMSO-d6 and 10% DMSO-d6/D2O. When 2c was dissolved in DMSO-d6, a second set of peaks appeared over a 72 h period suggesting coordination of DMSO to the Os center (Supplementary Figure S21). In the case of the Os-Ir compound 2f, minor changes were observed, but these were not as pronounced as those observed for 2c (Supplementary Figure S22). Interestingly, for the Cp* signals in the spectra of both compounds, we observed changes in the aliphatic region which require further investigations (Banerjee et al., 2018; Lee B. Y. T. et al., 2021; Lee B. et al., 2021). In 10% DMSO-d6/D2O, the spectra of 2f changed over time which was suppressed by the addition of 100 mM NaCl, to approximate the standard chloride concentration in blood. In contrast, treatment with AgNO3 resulted in precipitation of AgCl and significant alteration of the spectra, most likely owing to the formation of the aqua complex after abstraction of the chlorido ligand (Figure 6). An analogous pattern was observed in the case of 2c, with the addition of AgNO3 causing a precipitate of AgCl and a noticeable change in the shifts of the aromatic protons (Supplementary Figure S23). Note that in both experiments, precipitation occurred over the 72 h time period which resulted in spectra with lower signal-to-noise ratios which made the interpretation of the spectra more difficult.
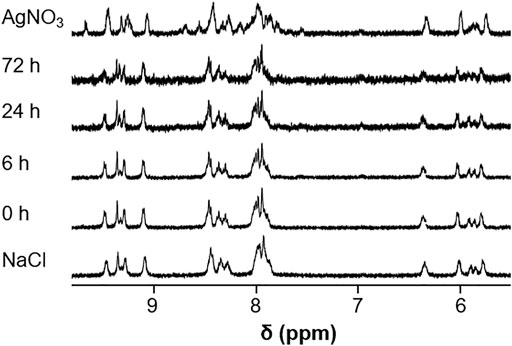
FIGURE 6. 1H NMR spectra of complex 2f recorded in 10% DMSO-d6/D2O over a period of 72 h as well as after addition of AgNO3 (2 equiv.) or NaCl (100 mM).
All complexes were investigated for their cytotoxic activity in HCT116 (human colon cancer cell line), SW480 (human colon adenocarcinoma cell line), NCI-H460 (human non-small-cell lung cancer cell line) and SiHa (human cervical cancer cell line) cancer cell lines with the sulforhodamine B assay. Compounds that gave mean growth inhibitory concentrations (IC50) > 100 µM were deemed to be inactive in the respective cell line. The IC50 values obtained for each complex have been summarized in Table 2 and compared to the bidentate pyridyl-imine-based ligand 2 and the analogous homodimetallic complexes in the same cell lines and under the same conditions (Steel et al., 2021). Ligand 2 demonstrated only very moderate activity in HCT116, NCI-H460 and SiHa cells, while it was inactive in SW480. The formation of coordination compounds of 2 unfortunately did not significantly alter the overall picture, and only for Os-Rh compound 2c IC50 values could be determined, which only showed moderate cytotoxicity. This reflects to a large extent the observations we made for the analogous homodiatomic complexes Ru2, Os2, Rh2 and Ir2 (Steel et al., 2021). While we and others have reported significant improvements in terms of cytotoxic effects when two or more metal centers are linked in a single molecule (Mendoza-Ferri et al., 2009), it is apparent that the incorporation of multiple metal centers in a single molecule does not necessarily result in high antiproliferative activity. The biological effect is significantly influenced by the ligand system used. This may in part be because of the structure of the ligand but may also be related to the stability of the formed complexes, with possibly even the ligands being the cytotoxic species upon cleavage of the metal centers from the coordinating motif. In any case, cellular accumulation of the active species is a prerequisite to observe biological activity which may be the reason for the low activity of this compound type (Jakupec et al., 2005). However, the low antiproliferative activity of the investigated complexes does not warrant further biological investigations.
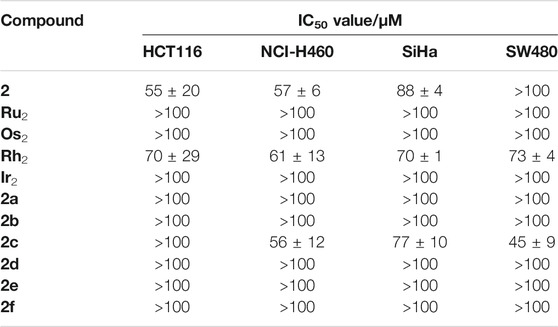
TABLE 2. IC50 values (µM) complexes 2a–2f in HCT116, NCI-H460, SiHa and SW480 cancer cell lines expressed as mean ± standard error (n = 3), in comparison to ligand 2 and the homodimetallic analogs Ru2, Os2, Rh2 and Ir2 (Steel et al., 2021).
3 Conclusion
In an attempt to improve the anticancer activity of organometallic anticancer agents, we designed and prepared heterodimetallic compounds based on a ditopic symmetric ligand. The synthetic strategy was developed by taking into consideration the labilities of the intermediate complexes to facilitate the isolation of the target heterodimetallic compounds in pure form. 1H NMR spectroscopy in acetone-d6 enabled the ligand components coordinated to the respective metal center to be distinguished, while ESI-MS confirmed the formation of the target compounds. Single crystal X-ray diffraction analysis showed statistical distribution of the metal centers in the heterometallic Ru/Os(cym) and Rh/Ir(Cp*) compounds 2a and 2d, while in case of complex 2e the Ru(cym) and Ir(Cp*) moieties were easily distinguishable based on the π-bound ligand. Compounds 2c and 2f were investigated for stability in solution and both were found to undergo ligand exchange reactions, although at different rates. In assays to investigate antiproliferative activity, only 2c showed very moderate potency. While the antiproliferative activity of the current complexes was only modest, the robust, facile and modular nature of the method to form these heterodimetallic compounds means that a wide range of systems could be synthesized and examined for biological activity. Furthermore, this method to heterodimetallic complexes may also find applications in the development of new catalysts (Mata et al., 2014; Gaston et al., 2021; Patra and Maity, 2021).
4 Experimental Section
4.1 Materials and Methods
All air and moisture-sensitive reactions were carried out under a nitrogen (N2) atmosphere, and light sensitive reactions were protected from photolytic degradation by covering the apparatus in aluminum foil. Chemicals and solvents purchased from commercial suppliers were used without further purification. Solvents were dried prior to use when necessary. Solvents were evaporated under reduced pressure using a rotary evaporator. The precursor complexes [Ru(cym)Cl2]2 a (Bennett and Smith, 1974), [Rh(Cp*)Cl2]2 b (Vogt et al., 2005; Nejman et al., 2015), [Ir(Cp*)Cl2]2 c (Vogt et al., 2005; Jakoobi et al., 2017) and [Os(cym)Cl2]2 d (Peacock et al., 2007) were prepared according to literature procedures.
1D [1H, and 13C{1H} DEPT-Q, 31P{1H}] and 2D (1H-1H COSY, 1H-1H NOESY, 1H-13C HSQC, 1H-13C HMBC) NMR spectra were recorded on Bruker DRX 400 MHz NMR spectrometers at 25 °C. The measurement frequencies for 1H, 13C{1H}, and 31P{1H} NMR spectra were 399.89, 100.55 and 161.85 MHz, respectively. Deuterated chloroform, acetone-d6, D2O, and DMSO-d6 were used as NMR solvents and the chemical shifts are reported relative to the residual solvent peaks. The mass spectra were recorded on a Bruker micrOTOF-Q II ESI-MS in positive ion mode. X-ray diffraction measurements of single crystals were conducted on a Rigaku Oxford Diffraction XtaLABSynergy-S single-crystal diffractometer with a PILATUS 200 K hybrid pixel array detector using Cu Kα radiation (λ = 1.54184 Å). The structure solution and refinements were performed with the SHELXS-97, SHELXL-2016 (Sheldrick, 2008) and Olex2 program packages (Dolomanov et al., 2009; Bourhis et al., 2015). Molecular structures were visualized using Mercury 4.0.0. Elemental analyses were carried out on the vario EL cube CHNOS Elemental analyzer at the University of Auckland for Ru, Rh, and Ir complexes, and at the Campbell Microanalytical Laboratory, the University of Otago for Os complexes.
4.2 Syntheses
4.2.1 General Procedure for the Synthesis of Mononuclear Complexes 1a, 1c and 1d
Pyridyl-imine ligand 1 (1.0 eq) was added to a solution of dimeric precursor a, c or d (0.5 eq) in DCM:MeOH (15 ml, 1:1) and stirred at r. t. for 4 h. The solvent was then removed under reduced pressure. A solution of ammonium hexafluorophosphate (NH4PF6, 20 eq) in MeOH (20 ml) was added to a solution of the crude product in MeOH (20 ml) and the resulting mixture was stirred for 1 h. The solution was concentrated under reduced pressure and diethyl ether was added. The resulting precipitate was collected by filtration, washed with cold MeOH and dried. The crude product was dissolved in DCM (80 ml), filtered and the solvent was removed from the filtrate under reduced pressure to yield the products (Figure 7 for the NMR numbering scheme) after drying the residue in vacuo.
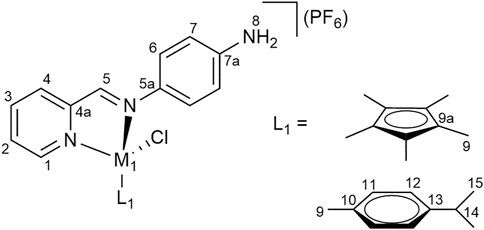
FIGURE 7. NMR numbering scheme for complexes 1a–1c.
[Chlorido(η6-p-cymene)(4-((pyridine-2-ylmethylene)amino)aniline)ruthenium(II)] hexafluorophosphate 1a.
1 (161 mg, 0.10 mmol), [Ru(cym)Cl2]2 a (250 mg, 0.05 mmol) and NH4PF6 (1.33 g, 1.02 mmol) to afford 1a as a dark red powder (461 mg, 92%). m. p.: 104.8–118.2°C (decomposition). 1H NMR (acetone-d6): δ 9.55 (d, 3J = 2 Hz, 1H, H-1), 8.73 (s, 1H, H-5), 8.32–8.24 (m, 2H, H-2,3), 7.84–7.79 (m, 1H, H-4), 7.69–7.64 (m, 2H, H-6), 6.87–6.81 (m, 2H, H-7), 6.11 (d, 3J = 3 Hz, 1H, H-11), 5.77 (d, 3J = 3 Hz, 1H, H-12), 5.72 (d, 3J = 3 Hz, 1H, H-11), 5.66 (d, 3J = 3 Hz, 1H, H-12), 5.45 (s, 2H, H-8), 2.68 (sept, 3J = 7 Hz, 1H, H-10), 2.30 (s, 3H, H-13), 1.15–1.07 ppm (m, 6H, H-9). 13C{1H} DEPT-Q (acetone-d6): δ 162.6 (C-5), 156.6 (C-1), 156.4 (C-4a), 151.9 (C-7a), 142.6 (C-5a), 140.6 (C-2,3), 129.6 (C-2,3), 128.9 (C-4), 125.2 (C-6), 114.6 (C-7), 106.8 (C-13), 104.8 (C-10), 87.9 (C-12), 87.3 (C-12), 86.7 (C-11), 85.9 (C-11), 31.9 (C-14), 22.2 (C-15), 18.7 ppm (C-9). 31P{1H} NMR (acetone-d6): δ –144.29 ppm (sept, 2J = 711 Hz, PF6−). MS (ESI+): m/z 468.0788 [M – PF6]+ (mcalc = 468.0775).
[Chlorido(η5-pentamethylcyclopentadienyl)(4-((pyridine-2-ylmethylene)amino)aniline)rhodium(III)] hexafluorophosphate 1b.
The synthesis was performed according to the general procedure using 1 (128 mg, 0.65 mmol), [Rh(Cp*)Cl2]2 b (201 mg, 0.32 mmol), and NH4PF6 (1.1 g, 6.5 mmol) to afford 1b as a bright orange powder (335 mg, 84%). m. p.: 147 °C (clear point). 1H NMR (DMSO-d6): δ 8.98 (d, 3J = 6 Hz, 1H, H-1), 8.76 (d, 3J = 2 Hz, 1H, H-5), 8.31 (td, 3J = 8 Hz, 4J = 2 Hz, 1H, H-3), 8.20 (d, 3J = 8 Hz, 1H, H-4), 7.91–7.86 (m, 1H, H-2), 7.49–7.43 (m, 2H, H-6), 6.72–6.70 (m, 2H, H-7), 5.79 (s, 2H, H-8), 1.45 ppm (s, 15H, H9). 13C{1H} DEPT-Q (DMSO-d6): δ 162.3 (C-5), 154.4 (C-4a), 152.6 (C-1), 150.6 (C-5a), 140.3 (C-3), 137.2 (C-7a), 128.9 (C-2), 128.6 (C-4), 124.1 (C-6), 113.2 (C-7), 97.0 (C-9a), 8.3 ppm (C-9). 31P{1H} NMR (DMSO-d6): δ –144.7 ppm (sept, 2J = 711 Hz, PF6−). MS (ESI+): m/z 470.0899 [M – PF6]+ (mcalc = 470.0870).
[Chlorido(η5-pentamethylcyclopentadienyl)(4-((pyridine-2-ylmethylene)amino)aniline)iridium(III)] hexafluorophosphate 1c.
The synthesis was performed according to the general procedure using 1 (282 mg, 1.40 mmol), [Ir(Cp*)Cl2]2 c (569 mg, 0.71 mmol), and NH4PF6 (3.5 g, 29 mmol) to afford 1c as a dark orange powder (652 mg, 82%). m. p.: 176°C (clear point). 1H NMR (DMSO-d6): δ 9.14 (s, 1H, H-5), 8.97 (d, 3J = 6 Hz, 1H, H-1), 8.33–8.25 (m, 2H, H-3/4), 7.89–7.83 (m, 1H, H-2), 7.41 (d, 3J = 9 Hz, 2H, H-6), 6.68 (d, 3J = 9 Hz, 2H, H-7), 5.80 (s, 2H, H-8), 1.44 ppm (s, 15H, H9). 13C{1H} DEPT-Q (DMSO-d6): δ 163.8 (C-5), 156.0 (C-4a), 152.1 (C-1), 150.8 (C-5a), 140.5 (C-3/4), 137.6 (C-7a), 129.5 (C-2), 128.7 (C-3/4), 124.2 (C-6), 113.1 (C-7), 89.6 (C-9a), 8.0 ppm (C-9). 31P{1H} NMR (DMSO-d6): δ –144.7 ppm (sept, 2J = 711 Hz, PF6−). MS (ESI+): m/z 560.1440 [M – PF6]+ (mcalc = 560.1444).
4.2.2 General Procedure for the Syntheses of Heterodimetallic Complexes 2a–2f
2-Pyridine carboxaldehyde (1 eq) was added to a solution of precursor complexes 1a–1c (1 eq) in DCM:MeOH (30 ml, 1:1) and stirred at r. t. overnight. Dimeric precursor a, b or d (0.5 eq) was added and the resulting mixture was stirred for ca. 48 h. The solvent was removed under reduced pressure. A solution of NH4PF6 (20 eq) in MeOH (20 ml) was added to the solution of the crude product in MeOH (20 ml) and the resulting mixture was stirred for 1 h. The solution was concentrated under reduced pressure and diethyl ether was added. The precipitate was filtered, washed with cold MeOH and dried. The crude product was dissolved in acetone (40 ml), filtered and the solvent was removed from the filtrate under reduced pressure, yielding pure complexes (Figure 8 for the NMR numbering scheme) after drying the residue in vacuo.
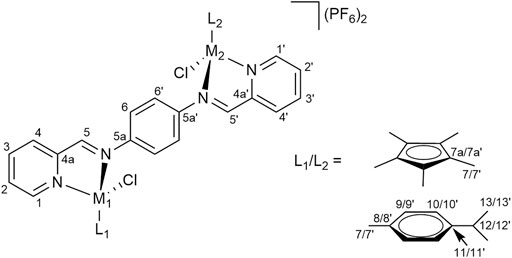
FIGURE 8. NMR numbering scheme for complexes 2a–2f.
[Chlorido(η6-p-cymene)osmium(II)](N,N'-(1,4-phenylene)(bis(1-(pyridin-2-yl)(methanimine)-κ2N,N')[chlorido(η6-p-cymene)ruthenium(II)] hexafluorophosphate 2a.
The synthesis was performed according to the general procedure using 1a (240 mg, 0.4 mmol), 2-pyridine carboxaldehyde (37 µL, 0.39 mmol), [Os(cym)Cl2]2 (158 mg, 0.20 mmol) and NH4PF6 (1.30 g, 8.0 mmol) to afford 2a as a dark red-brown powder (397 mg, 84%). m. p.: 161.2–173.5°C (decomp.). 1H NMR (acetone-d6): δ 9.71 (d, 3J = 2 Hz, 1H, H-1), 9.66 (d, 3J = 3 Hz, 1H, H-1'), 9.46 (s, 1H, H-5'), 9.06 (s, 1H, H-5), 8.60–8.51 (m, 1H, H-3'), 8.48–8.36 (m, 3H, H-3,4', 4), 8.23–8.18 (m, 2H, H-6), 8.15–8.10 (m, 2H, H-6'), 8.00–7.91 (m, 2H, H-2,2'), 6.47 (d, 3J = 4 Hz, 1H, H-9'), 6.19 (d, 3J = 4 Hz, 1H, H-9), 6.10–6.02 (m, 2H, H-9'/10'), 5.91–5.83 (m, 3H, H-9, 10', 10), 5.71 (dd, 3J = 8 Hz, 4J = 3 Hz, 1H, H-10), 2.78 (m, 1H, H-12), 2.64 (sept, 3J = 7 Hz 1H, H-12'), 2.40 (d, 3J = 3 Hz, 3H, H-7), 2.33 (d, 3J = 3 Hz, 3H, H-7'), 1.18–1.12 (m, 6H, H-13), 1.11–1.06 ppm (m, 6H, H-13'). 13C{1H} DEPT-Q (acetone-d6): δ 170.0 (C-5'), 169.1 (C-5), 157.1 (C-1), 156.7 (C-1'), 155.8 (C-4a'), 154.2 (C-5a'), 154.0 (C-5a), 153.9 (C-4a), 141.2 (C-4,4'), 141.0 (C-4,4'), 131.5 (C-3), 131.3 (C-3'), 131.1 (C-2'), 130.2 (C-2), 125.4 (C-6'), 124.9 (C-6), 107.9 (C-11), 104.9 (C-8), 99.6 (C-11'), 98.8 (C-8'), 87.8 (C-9), 86.8 (C-9,10), 86.4 (C-10), 79.7 (C-9'), 79.1 (C-9'/10'), 77.3 (C-9'/10'), 76.9 (C-10'), 32.2 (C-12), 32.0 (C-12'), 22.6 (C-13'), 22.2 (C-13), 18.9 ppm (C-7,7'). 31P{1H} NMR (acetone-d6): δ –144.37 ppm (sept, 2J = 707 Hz, PF6−). MS (ESI+): m/z 459.0721 [M – 2PF6]2+ (mcalc = 459.0723). EA calculated for C38H42Cl2F12N4P2RuOs·0.67NH4PF6: C 34.69%, H 3.42%, N 4.97%. Found: C 35.03%, H 3.50%, N 5.07%.
[Chlorido(η5-pentamethylcyclopentadienyl)rhodium(III)](N,N'-(1,4-phenylene)(bis(1-(pyridin-2-yl)(methanimine)-κ2N,N')[chlorido(η6-p-cymene)ruthenium(II)] hexafluorophosphate 2b.
The synthesis was performed according to the general procedure using 1b (165 mg, 0.27 mmol), 2-pyridine carboxaldehyde (26 µL, 0.27 mmol), [Ru(cym)Cl2]2 (82 mg, 0.14 mmol) and NH4PF6 (872 mg, 5.35 mmol) to afford 2b as a yellow powder (152 mg, 51%). m. p.: 141°C (clear point). 1H NMR (acetone-d6): δ 9.69 (d, 3J = 6 Hz, 1H, H-1'), 9.27–9.23 (m, 1H, H-1), 9.10–9.04 (m, 2H, H-5/5'), 8.49–8.37 (m, 4H, H-3/3'/4/4'), 8.25–8.18 (m, 1H, H-6'), 8.14–8.05 (m, 3H, H-2/6), 7.98–7.93 (m, 1H, H-2'), 6.21–6.16 (m, 1H, H-9'), 5.89–5.69 (m, 3H, H-8'/9'), 5.70 (d, 3J = 6 Hz, 1H, H-8'), 2.74 (m, 1H, H-10'), 2.34–2.30 (m, 3H, H-7'), 1.69–1.63 (m, 15H, H-7), 1.20–1.12 ppm (m, 6H, H-11'). 13C{1H} DEPT-Q (acetone-d6): δ 169.1 (C-5/5'), 157.0 (C-1'), 153.9 (C-1), 153.7 (C-4a/4a'/5a), 150.7 (C-4a/4a'), 150.6 (C-5a'), 141.6 (C-3/3'), 141.0 (C-3/3'), 131.5 (C-4/4'), 131.4 (C-4/4'), 131.2 (C-2), 130.2 (C-2'), 125.1 (C-6), 125.0 (C-6'), 107.8 (C-11'), 105.0 (C-8'), 98.7 (C-7a), 87.7–86.2 (C-9'/10'), 32.0 (C-12'), 22.3 (C-12'), 19.0 (C-7'), 9.0 ppm (C-7). 31P{1H} NMR (acetone-d6): δ –144.3 ppm (sept, 2J = 708 Hz, PF6−). MS (ESI+): m/z 470.0870 [M – 2PF6 – C6NH5 – RuCl(p-cym)]+ (mcalc = 470.0870). EA calculated for C38H43Cl2F12RhN4P2Ru·0.2NH4PF6: C 39.57%, H 3.84%, N 5.10%. Found: C 39.67%, H 4.20%, N 4.80%.
[Chlorido(η6-p-cymene)osmium(II)](N,N'-(1,4-phenylene)(bis(1-(pyridin-2-yl)(methanimine)-κ2N,N')[chlorido(η5-pentamethylcyclopentadienyl)rhodium(III)] hexafluorophosphate 2c.
The synthesis was performed according to the general procedure using 1b (153 mg, 0.25 mmol), 2-pyridine carboxaldehyde (24 µL, 0.25 mmol), [Os(cym)Cl2]2 (98 mg, 0.13 mmol) and NH4PF6 (808 mg, 4.96 mmol) to afford 2c as an orange powder (127 mg, 42%). m. p.: 154°C (clear point). 1H NMR (acetone-d6): δ 9.64 (d, 3J = 6 Hz, 1H, H-1'), 9.50 (s, 1H, H-5'), 9.26 (d, 3J = 6 Hz, 1H, H-1), 9.06 (m, 1H, 1H, H-5), 8.55 (m, 1H, H-4'), 8.47–8.43 (m, 2H, H-3/4), 8.37 (m, 1H, H-3'), 8.16–8.07 (m, 5H, H-2/6/6'), 7.93–7.89 (m, 1H, H-2'), 6.49–6.45 (m, 1H, H-8'), 6.09–5.98 (m, 2H, H-8'/9'), 5.92–5.84 (m, 1H, H-9'), 2.64 (sept, 3J = 7 Hz, 1H, H-10'), 2.41–2.37 (m, 3H, H-7'), 1.68–1.64 (m, 15H, H-7), 1.27–1.06 ppm (m, 6H, H-11'). 13C{1H} DEPT-Q (acetone-d6): δ 170.0 (C-5'), 169.2 (C-5), 157.2 (C-4a'), 156.7 (C-1'), 153.9 (C-1), 153.7 (C-5a/5a'), 150.9 (C-5a/5a'), 141.6 (C-3), 141.1 (C-3'), 131.4 (C-4), 131.3 (C-4') 131.2 (C-2), 131.1 (C-2'), 125.5 (C-6/6'), 124.9 (C-6/6'), 99.5 (C-11'), 99.3 (C-8'), 98.6 (C-7a), 79.6–76.7 (C-9'/10'), 32.2 (C-12'), 22.6 (C-13'), 18.9 (C-7'), 9.0 ppm (C-7). 31P{1H} NMR (acetone-d6): δ –144.3 ppm (sept, 2J = 708 Hz, PF6−). MS (ESI+): m/z 470.0817 [M – 2PF6 – C6NH5 – OsCl(p-cym)]+ (mcalc = 470.0870). EA calculated for C38H43Cl2F12RhN4P2Os: C 37.73%, H 3.58%, N 4.63%. Found: C 37.77%, H 3.39%, N 4.44%.
[Chlorido(η5-pentamethylcyclopentadienyl)iridium(III)](N,N'-(1,4-phenylene)(bis(1-(pyridin-2-yl)(methanimine)- κ2N,N')[chlorido(η5-pentamethylcyclopentadienyl)rhodium(III)] hexafluorophosphate 2d.
The synthesis was performed according to the general procedure using 1c (174 mg, 0.25 mmol), 2-pyridine carboxaldehyde (24 µL, 0.25 mmol), [Rh(Cp*)Cl2]2 (76 mg, 0.13 mmol) and NH4PF6 (806 mg, 5.0 mmol) to afford 2d as a dark yellow powder (170 mg, 58%). Single crystals suitable for X-ray diffraction analysis were grown by slow diffusion of toluene into a saturated solution of the complex in acetonitrile. m. p.: 135 °C (clear point). 1H NMR (acetone-d6): δ 9.53–9.50 (m, 1H, H-5), 9.27–9.23 (m, 2H, H-1/1'), 9.10 (dd, 3J = 6 Hz, 4J = 3 Hz, 1H, H-5'), 8.59–8.55 (m, 1H, H-4), 8.49–8.41 (m, 3H, H-3/3'/4'), 8.16–8.05 (m, 6H, H-2/2'/6/6'), 1.66–1.65 (m, 15H, H-7') 1.64–1.63 ppm (m, 15H, H-7). 13C{1H} DEPT-Q (acetone-d6): δ 170.6 (C-5), 169.3 (C-5'), 153.8 (C-1/1'), 153.4 (C-1/1'), 150.7 (C-4a), 150.6 (C-4a'), 141.7 (C-4/4'), 141.5 (C-3/3'), 131.8 (C-2/2'), 131.4 (C-2/2'), 131.3 (C-4') 131.2 (C-4), 125.2 (C-6/6'), 124.9 (C-6/6'), 98.7 (C-7a'), 91.4 (C-7a), 9.0 (C-7'), 8.8 ppm (C-7). 31P{1H} NMR (acetone-d6): δ –144.3 ppm (sept, 2J = 707 Hz, PF6−). MS (ESI+): m/z 461.0794 [M – 2PF6]2+ (mcalc = 461.0814). 461.0794. EA calculated for C38H44Cl2F12IrN4P2Rh·0.7H2O: C 37.24%, H 3.74%, N 4.57%. Found: C 36.84%, H 4.11%, N 4.39%.
[Chlorido(η5-pentamethylcyclopentadienyl)iridium(III)](N,N'-(1,4-phenylene)(bis(1-(pyridin-2-yl)(methanimine)-κ2N,N')[chlorido(η6-p-cymene)ruthenium(II)] hexafluorophosphate 2e.
The synthesis was performed according to the general procedure using 1c (175 mg, 0.25 mmol), 2-pyridine carboxaldehyde (24 µL, 0.25 mmol), [Ru(cym)Cl2]2 (76 mg, 0.13 mmol) and NH4PF6 (808 mg, 5.0 mmol) to afford 2e as a dark brown powder (151 mg, 50%). Single crystals suitable for X-ray diffraction analysis were grown by slow diffusion of toluene into a saturated solution of the complex in acetonitrile. m. p.: 169°C (clear point). 1H NMR (acetone-d6): δ 9.68 (d, 3J = 6 Hz, 1H, H-1'), 9.48 (s, 1H, H-5), 9.25 (d, 3J = 6 Hz, 1H, H-1), 9.08 (s, 1H, H-5'), 8.54–8.45 (m, 1H, H-4), 8.46–8.37 (m, 3H, H-3/3'/4'), 8.25–8.18 (m, 2H, H-6') 8.10–8.04 (m, 3H, H-2/6), 7.97–7.92 (m, 1H, H-2') 6.21–6.15 (m, 1H, H-9'), 5.87–5.73 (m, 2H, H-8'/9'), 5.69 (d 3J = 6 Hz, 1H, H-8'), 2.75 (m, 1H, H-10'), 2.32 (s, 3H, H-7'), 1.65–1.62 (m, 15H, H-7), 1.19–1.13 ppm (m, 6H, H-11'). 13C{1H} DEPT-Q (acetone-d6): δ 170.5 (C-5), 169.2 (C-5'), 157.0 (C-1'), 156.5 (C-4a'), 155.7 (C-4a), 153.9 (C-5a), 153.4 (C-1), 150.9 (C-5a'), 141.7 (C-3/3') 141.0 (C-3/3'), 131.8 (C-2), 131.5 (C-4), 131.3 (C-4'), 130.2 (C-2'), 125.1 (C-6'), 125.0 (C-6), 107.8 (C-11'), 105.1 (C-8'), 91.4 (C-7a), 87.8–86.2 (C-9'/10'), 32.0 (C-12'), 22.3 (C-13'), 18.9 (C-7'), 8.7 ppm (C-7). 31P{1H} NMR (acetone-d6): δ –144.3 ppm (sept, 2J = 708 Hz, PF6−). MS (ESI+): m/z 460.0785 [M – 2PF6]2+ (mcalc = 460.0769). EA calculated for C38H43Cl2F12IrN4P2Ru·0.9H2O: C 37.22%, H 3.68%, N 4.57%. Found: C 36.87%, H 3.72%, N 4.95%.
[Chlorido(η5-pentamethylcyclopentadienyl)iridium(III)](N,N'-(1,4-phenylene)(bis(1-(pyridin-2-yl)(methanimine)-κ2N,N')[chlorido(η6-p-cymene)osmium(II)] hexafluorophosphate 2f.
The synthesis was performed according to the general procedure using 1c (163 mg, 0.23 mmol), 2-pyridine carboxaldehyde (22 µL, 0.23 mmol), [Os(cym)Cl2]2 (91 mg, 0.12 mmol) and NH4PF6 (753 mg, 4.6 mmol) to afford 2f as a dark red powder (201 mg, 67%). m. p.: 147°C (clear point). 1H NMR acetone-d6): δ 9.64 (d, 3J = 6 Hz, 1H, H-1'), 9.49–9.47 (m, 2H, H-5/5'), 9.25 (d, 3J = 6 Hz, 1H, H-1), 8.59–8.54 (m, 2H, H-4/4'), 8.44 (td, 3J = 8 Hz, 3J = 1 Hz, 1H, H-3), 8.38 (m, 1H, H-3'), 8.16–8.12 (m, 2H, H-6'), 8.11–8.04 (m, 3H, H-2/6), 7.94–7.89 (m, 1H, H-2'), 6.50–6.44 (m, 1H, H-9'), 6.08–5.98 (m, 2H, H-8'/9'), 5.87 (dd, 3J = 20.6 Hz, 4J = 5.8 Hz, 1H, H8'), 2.63 (sept, 3J = 7 Hz, 1H, H-10'), 2.39 (d, 3J = 7.2 Hz, 3H, H-7'), 1.67–1.62 (m, 15H, H-7), 1.12–1.07 ppm (m, 6H, H-11'). 13C{1H} DEPT-Q (acetone-d6): δ 170.7 (C-5/5'), 170.1 (C-5/5'), 157.1 (C-4a'), 156.5 (C-4a) 156.7 (C-1'), 154.0 (C-5a), 153.4 (C-1), 151.0 (C-5a'), 141.7 (C-4), 141.1 (C-4'), 131.8 (C-2), 131.4 (C-3/3'), 131.1 (C-2'), 125.4 (C-6/6'), 125.15 (C-6/6'), 99.6 (C-11'), 99.4 (C-8'), 91.4 (C-7a), 79.6–76.7 (C-9'/10'), 32.2 (C-12'), 22.6 (C-13'), 19.0 (C-7'), 8.7 ppm (C-7). 31P{1H} NMR (acetone-d6): δ –144.3 ppm (sept, 2J = 708 Hz, PF6−). MS (ESI+): m/z 505.1053 [M – 2PF6]2+ (mcalc = 505.1055). EA calculated for C38H43Cl2F12IrN4P2Os·0.6H2O: C 34.84%, H 3.41%, N 4.28%. Found: C 34.44%, H 3.29%, N 4.04%.
4.3 DMSO and Aqueous Stability Studies
Stability studies in DMSO were conducted for 2c and 2f by dissolving ca. 1 mg of the complex in DMSO-d6 (ca. 0.5 ml). 1H NMR spectra were recorded at t = 0, 2, 6, 24, 48, and 72 h.
The stability studies in aqueous solution were conducted by dissolving ca. 1 mg of 2c or 2f in DMSO-d6 (0.05 ml) and diluting it with D2O (0.45 ml) to form a 10% DMSO-d6/D2O solution. 1H NMR spectra were recorded at t = 0, 2, 6, 24, 48, and 72 h. The compounds were investigated via the same procedure in a solution of 100 mM NaCl in D2O (0.45 ml) added to a solution of ca. 1 mg of the complex in DMSO-d6 (0.05 ml). In addition, a solution of the hydrolyzed product was prepared by the addition of AgNO3 (2 eq) to a suspension of ca. 1 mg of the complex (1 eq) in 10% DMSO-d6/D2O. After vigorous shaking, the formed AgCl was removed by filtration and a 1H NMR spectrum of the filtrate was recorded.
4.4 Cell Cytotoxicity Studies
The antiproliferative activity of compounds 2a–2f was investigated in HCT116, SW480, SiHa and NCI-H460 cells as described elsewhere (Movassaghi et al., 2018). In brief, the cells were grown in α-MEM supplemented with 5% fetal calf serum at 37°C in a humidified incubator with 5% CO2 after seeding them at 750 (HCT116, NCI-H460), 4,000 (SiHa) and 5,000 (SW480) cells per well in 96-well plates. The complexes were added to the plates in a series of 3-fold dilutions in 0.5% DMSO at the highest concentration for 72 h before the assay was terminated and the cells were stained with 0.4% sulforhodamine B (Sigma-Aldrich). The IC50 values were calculated with SigmaPlot 14.0 (Systat Software Inc.) using a three-parameter logistic sigmoidal dose-response curve between the calculated growth inhibition and the compound concentration. The presented IC50 values are the mean of at least three independent experiments, where 10 concentrations were tested in duplicate for each compound.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
LG and TRS synthesized the compounds under supervision of MH and CH, TS refined the molecular structures, and LG and MR determined the cytotoxicity supervised by SJ. The project was conceptualized by LW, JC and CH, and the manuscript was drafted by LG and CH and finalized with input from all co-authors.
Funding
The project was funded through the Marsden Fund Council, managed by the Royal Society Te Apārangi. MH is supported by a Sir Charles Hercus Fellowship of the Health Research Council of New Zealand.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the University of Auckland for Doctoral Scholarships to LG and TS and a Faculty Research Development Fund grant of the Faculty of Science to MH. We are grateful to Tanya Groutso for collecting the X-ray diffraction, and to Tony Chen and Mansa Nair for collecting the MS data. MR would like to thank Eva and Henry Frænkels Minde fond, Knud Højgaards Fond, Dagmar Marshalls Fond, Carl og Ellen Hertz’ legat til Dansk Læge-og Naturvidenskab, Viet-Jacobsen Fonden, Christian og Ottilia Brorsons Rejselegat for yngre videnskabsmænd—og kvinder, and Direktør Jacob Madsens og Hustru Olga Madsens Fond for financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.786367/full#supplementary-material
References
Arango, D., Wilson, A. J., Shi, Q., Corner, G. A., Arañes, M. J., Nicholas, C., et al. (2004). Molecular Mechanisms of Action and Prediction of Response to Oxaliplatin in Colorectal Cancer Cells. Br. J. Cancer. 91, 1931–1946. doi:10.1038/sj.bjc.6602215
Banerjee, S., Soldevila-Barreda, J. J., Wolny, J. A., Wootton, C. A., Habtemariam, A., Romero-Canelón, I., et al. (2018). New Activation Mechanism for Half-Sandwich Organometallic Anticancer Complexes. Chem. Sci. 9, 3177–3185. doi:10.1039/c7sc05058e
Batchelor, L. K., Ortiz, D., and Dyson, P. J. (2019). Histidine Targeting Heterobimetallic Ruthenium(II)-Gold(I) Complexes. Inorg. Chem. 58, 2501–2513. doi:10.1021/acs.inorgchem.8b03069
Benjamin Garbutcheon-Singh, K., Grant, M., Harper, B., Krause-Heuer, A., Manohar, M., Orkey, N., et al. (2011). Transition Metal Based Anticancer Drugs. Curr. Top. Med. Chem. 11, 521–542. doi:10.2174/156802611794785226
Bennett, M. A., and Smith, A. K. (1974). Arene Ruthenium(II) Complexes Formed by Dehydrogenation of Cyclohexadienes With Ruthenium(III) Trichloride. J. Chem. Soc. Dalton Trans., 233–241. doi:10.1039/dt9740000233
Bertrand, B., Doulain, P.-E., Goze, C., and Bodio, E. (2016). Development of Trackable Metal-Based Drugs: New Generation of Therapeutic Agents. Dalton Trans. 45, 13005–13011. doi:10.1039/c5dt04275e
Bertrand, B., Passador, K., Goze, C., Denat, F., Bodio, E., and Salmain, M. (2018). Metal-Based BODIPY Derivatives as Multimodal Tools for Life Sciences. Coord. Chem. Rev. 358, 108–124. doi:10.1016/j.ccr.2017.12.007
Boros, E., Dyson, P. J., and Gasser, G. (2020). Classification of Metal-Based Drugs According to Their Mechanisms of Action. Chem. 6, 41–60. doi:10.1016/j.chempr.2019.10.013
Bourhis, L. J., Dolomanov, O. V., Gildea, R. J., Howard, J. A. K., and Puschmann, H. (2015). The Anatomy of a Comprehensive Constrained, Restrained Refinement Program for the Modern Computing Environment -Olex2 dissected. Acta Cryst. Sect. A. 71, 59–75. doi:10.1107/s2053273314022207
Buccella, D., Lim, M. H., and Morrow, J. R. (2019). Metals in Biology: From Metallomics to Trafficking. Inorg. Chem. 58, 13505–13508. doi:10.1021/acs.inorgchem.9b02965
Burgoyne, A. R., Kaschula, C. H., Parker, M. I., and Smith, G. S. (2016). In Vitro Cytotoxicity of Half-Sandwich Platinum Group Metal Complexes of a Cationic Alkylated Phosphaadamantane Ligand. Eur. J. Inorg. Chem. 2016, 1267–1273. doi:10.1002/ejic.201501458
Curado, N., Giménez, N., Miachin, K., Aliaga‐Lavrijsen, M., Cornejo, M. A., Jarzecki, A. A., et al. (2019). Preparation of Titanocene-Gold Compounds Based on Highly Active Gold(I)-N-Heterocyclic Carbene Anticancer Agents: Preliminary In Vitro Studies in Renal and Prostate Cancer Cell Lines. ChemMedChem. 14, 1086–1095. doi:10.1002/cmdc.201800796
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K., and Puschmann, H. (2009). OLEX2: a Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 42, 339–341. doi:10.1107/s0021889808042726
Gaston, A. J., Greindl, Z., Morrison, C. A., and Garden, J. A. (2021). Cooperative Heterometallic Catalysts for Lactide Ring-Opening Polymerization: Combining Aluminum With Divalent Metals. Inorg. Chem. 60, 2294–2303. doi:10.1021/acs.inorgchem.0c03145
Geldmacher, Y., Oleszak, M., and Sheldrick, W. S. (2012). Rhodium(III) and Iridium(III) Complexes as Anticancer Agents. Inorg. Chim. Acta 393, 84–102. doi:10.1016/j.ica.2012.06.046
Ghosh, A. C., Duboc, C., and Gennari, M. (2021). Synergy Between Metals for Small Molecule Activation: Enzymes and Bio-Inspired Complexes. Coord. Chem. Rev. 428, 213606. doi:10.1016/j.ccr.2020.213606
Gonchar, M. R., Matnurov, E. M., Burdina, T. A., Zava, O., Ridel, T., Milaeva, E. R., et al. (2020). Ruthenium(II)-Arene and Triruthenium-Carbonyl Cluster Complexes With New Water-Soluble Phopsphites Based on Glucose: Synthesis, Characterization and Antiproliferative Activity. J. Organomet. Chem. 919, 121312. doi:10.1016/j.jorganchem.2020.121312
Gorle, A. K., Haselhorst, T., Katner, S. J., Everest‐Dass, A. V., Hampton, J. D., Peterson, E. J., et al. (2021). Conformational Modulation of Iduronic Acid‐Containing Sulfated Glycosaminoglycans by a Polynuclear Platinum Compound and Implications for Development of Antimetastatic Platinum Drugs. Angew. Chem. 133, 3320–3326. doi:10.1002/ange.202013749
Gorle, A. K., Katner, S. J., Johnson, W. E., Lee, D. E., Daniel, A. G., Ginsburg, E. P., et al. (2018). Substitution‐Inert Polynuclear Platinum Complexes as Metalloshielding Agents for Heparan Sulfate. Chem. Eur. J. 24, 6606–6616. doi:10.1002/chem.201706030
Haas, K. L., and Franz, K. J. (2009). Application of Metal Coordination Chemistry to Explore and Manipulate Cell Biology. Chem. Rev. 109, 4921–4960. doi:10.1021/cr900134a
Hartinger, C. G., Jakupec, M. A., Zorbas-Seifried, S., Groessl, M., Egger, A., Berger, W., et al. (2008). KP1019, A New Redox-Active Anticancer Agent - Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodiversity 5, 2140–2155. doi:10.1002/cbdv.200890195
Heffeter, P., Böck, K., Atil, B., Reza Hoda, M. A., Körner, W., Bartel, C., et al. (2010). Intracellular Protein Binding Patterns of the Anticancer Ruthenium Drugs KP1019 and KP1339. J. Biol. Inorg. Chem. 15, 737–748. doi:10.1007/s00775-010-0642-1
Herry, B., Batchelor, L. K., Roufosse, B., Romano, D., Baumgartner, J., Borzova, M., et al. (2019). Heterobimetallic Ru(μ-dppm)Fe and Homobimetallic Ru(μ-dppm)Ru Complexes as Potential Anti-Cancer Agents. J. Organomet. Chem. 901, 120934. doi:10.1016/j.jorganchem.2019.120934
Jain, A. (2019). Multifunctional, Heterometallic Ruthenium-Platinum Complexes With Medicinal Applications. Coord. Chem. Rev. 401, 213067. doi:10.1016/j.ccr.2019.213067
Jakoobi, M., Halcovitch, N., Whitehead, G. F. S., and Sergeev, A. G. (2017). Selective Arene Cleavage by Direct Insertion of Iridium Into the Aromatic Ring. Angew. Chem. 129, 3314–3317. doi:10.1002/ange.201611409
Jakupec, M. A., Galanski, M., Arion, V. B., Hartinger, C. G., and Keppler, B. K. (2008). Antitumour Metal Compounds: More Than Theme and Variations. Dalton Trans., 183–194. doi:10.1039/b712656p
Jakupec, M. A., Reisner, E., Eichinger, A., Pongratz, M., Arion, V. B., Galanski, M., et al. (2005). Redox-Active Antineoplastic Ruthenium Complexes With Indazole: Correlation of In Vitro Potency and Reduction Potential. J. Med. Chem. 48, 2831–2837. doi:10.1021/jm0490742
Johnstone, T. C., Park, G. Y., and Lippard, S. J. (2014). Understanding and Improving Platinum Anticancer Drugs-Pphenanthriplatin. Anticancer Res. 34, 471–476.
Kenny, R. G., and Marmion, C. J. (2019). Toward Multi-Targeted Platinum and Ruthenium Drugs-A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 119, 1058–1137. doi:10.1021/acs.chemrev.8b00271
Lee, B. Y. T., Phillips, A. D., Hanif, M., Tong, K. K. H., Söhnel, T., and Hartinger, C. G. (2021a). Heptadentate, Octadentate, or Even Nonadentate? Denticity in the Unexpected Formation of an All-Carbon Donor-Atom Ligand in RhIII(Cp*)(Anthracenyl-NHC) Complexes. Inorg. Chem. 60, 8734–8741. doi:10.1021/acs.inorgchem.1c00711
Lee, B. Y. T., Sullivan, M. P., Yano, E., Tong, K. K. H., Hanif, M., Kawakubo-Yasukochi, T., et al. (2021b). Anthracenyl Functionalization of Half-Sandwich Carbene Complexes: In Vitro Anticancer Activity and Reactions With Biomolecules. Inorg. Chem. 60, 19, 14636–14644. doi:10.1021/acs.inorgchem.1c01675
Lisboa, L. S., Findlay, J. A., Wright, L. J., Hartinger, C. G., and Crowley, J. D. (2020). A Reduced‐Symmetry Heterobimetallic [PdPtL4]4+ Cage: Assembly, Guest Binding, and Stimulus‐Induced Switching. Angew. Chem. Int. Ed. 59, 11101–11107. doi:10.1002/anie.202003220
Lisboa, L. S., Riisom, M., Vasdev, R. A. S., Jamieson, S. M. F., Wright, L. J., Hartinger, C. G., et al. (2021). Cavity-Containing [Fe2L3]4+ Helicates: An Examination of Host-Guest Chemistry and Cytotoxicity. Front. Chem. 9, 697684. doi:10.3389/fchem.2021.739785
Maillet, A., Yadav, S., Loo, Y. L., Sachaphibulkij, K., and Pervaiz, S. (2013). A Novel Osmium-Based Compound Targets the Mitochondria and Triggers ROS-Dependent Apoptosis in Colon Carcinoma. Cell Death Dis. 4, e653. doi:10.1038/cddis.2013.185
Manzotti, C., Pratesi, G., Menta, E., Di Domenico, R., Cavalletti, E., Fiebig, H. H., et al. (2000). BBR 3464: a Novel Triplatinum Complex, Exhibiting a Preclinical Profile of Antitumor Efficacy Different From Cisplatin. Clin. Cancer Res. 6, 2626–2634.
Mata, J. A., Hahn, F. E., and Peris, E. (2014). Heterometallic Complexes, Tandem Catalysis and Catalytic Cooperativity. Chem. Sci. 5, 1723–1732. doi:10.1039/c3sc53126k
Mendoza-Ferri, M.-G., Hartinger, C. G., Eichinger, R. E., Stolyarova, N., Severin, K., Jakupec, M. A., et al. (2008). Influence of the Spacer Length on the In Vitro Anticancer Activity of Dinuclear Ruthenium−Arene Compounds. Organometallics 27, 2405–2407. doi:10.1021/om800207t
Mendoza-Ferri, M. G., Hartinger, C. G., Mendoza, M. A., Groessl, M., Egger, A. E., Eichinger, R. E., et al. (2009). Transferring the Concept of Multinuclearity to Ruthenium Complexes for Improvement of Anticancer Activity. J. Med. Chem. 52, 916–925. doi:10.1021/jm8013234
Movassaghi, S., Singh, S., Mansur, A., Tong, K. K. H., Hanif, M., Holtkamp, H. U., et al. (2018). (Pyridin-2-yl)-NHC Organoruthenium Complexes: Antiproliferative Properties and Reactivity Toward Biomolecules. Organometallics 37, 1575–1584. doi:10.1021/acs.organomet.8b00153
Nazarov, A. A., Mendoza-Ferri, M.-G., Hanif, M., Keppler, B. K., Dyson, P. J., and Hartinger, C. G. (2018). Understanding the Interactions of Diruthenium Anticancer Agents With Amino Acids. J. Biol. Inorg. Chem. 23, 1159–1164. doi:10.1007/s00775-018-1597-x
Nejman, P. S., Morton-Fernandez, B., Moulding, D. J., Athukorala Arachchige, K. S., Cordes, D. B., Slawin, A. M. Z., et al. (2015). Structural Diversity of Bimetallic Rhodium and Iridium Half Sandwich Dithiolato Complexes. Dalton Trans. 44, 16758–16766. doi:10.1039/c5dt02542g
Nováková, O., Nazarov, A. A., Hartinger, C. G., Keppler, B. K., and Brabec, V. (2009). DNA Interactions of Dinuclear RuII Arene Antitumor Complexes in Cell-Free media. Biochem. Pharmacol. 77, 364–374. doi:10.1016/j.bcp.2008.10.021
Parveen, S., Arjmand, F., and Tabassum, S. (2019a). Development and Future Prospects of Selective Organometallic Compounds as Anticancer Drug Candidates Exhibiting Novel Modes of Action. Eur. J. Med. Chem. 175, 269–286. doi:10.1016/j.ejmech.2019.04.062
Parveen, S., Hanif, M., Leung, E., Tong, K. K. H., Yang, A., Astin, J., et al. (2019b). Anticancer Organorhodium and -iridium Complexes with Low Toxicity In Vivo but High Potency In Vitro: DNA Damage, Reactive Oxygen Species Formation, and Haemolytic Activity. Chem. Commun. 55, 12016–12019. doi:10.1039/c9cc03822a
Patra, S., and Maity, N. (2021). Recent Advances in (Hetero)Dimetallic Systems Towards Tandem Catalysis. Coord. Chem. Rev. 434, 213803. doi:10.1016/j.ccr.2021.213803
Payne, R., Govender, P., Therrien, B., Clavel, C. M., Dyson, P. J., and Smith, G. S. (2013). Neutral and Cationic Multinuclear Half-Sandwich Rhodium and Iridium Complexes Coordinated to Poly(propyleneimine) Dendritic Scaffolds: Synthesis and Cytotoxicity. J. Organomet. Chem. 729, 20–27. doi:10.1016/j.jorganchem.2013.01.009
Peacock, A. F. A., Habtemariam, A., Moggach, S. A., Prescimone, A., Parsons, S., and Sadler, P. J. (2007). Chloro Half-Sandwich Osmium(II) Complexes: Influence of Chelated N,N-ligands on Hydrolysis, Guanine Binding, and Cytotoxicity. Inorg. Chem. 46, 4049–4059. doi:10.1021/ic062350d
Redrado, M., Fernández‐Moreira, V., and Gimeno, M. C. (2021). Theranostics Through the Synergistic Cooperation of Heterometallic Complexes. ChemMedChem. 16, 932–941. doi:10.1002/cmdc.202000833
Ruhayel, R. A., Langner, J. S., Oke, M.-J., Berners-Price, S. J., Zgani, I., and Farrell, N. P. (2012). Chimeric Platinum-Polyamines and DNA Binding. Kinetics of DNA Interstrand Cross-Link Formation by Dinuclear Platinum Complexes With Polyamine Linkers. J. Am. Chem. Soc. 134, 7135–7146. doi:10.1021/ja301397h
Sava, G., Zorzet, S., Turrin, C., Vita, F., Soranzo, M., Zabucchi, G., et al. (2003). Dual Action of NAMI-A in Inhibition of Solid Tumor Metastasis: Selective Targeting of Metastatic Cells and Binding to Collagen. Clin. Cancer Res. 9, 1898–1905.
Sheldrick, G. M. (2008). A Short History of SHELX. Acta Cryst. Sect. A. 64, 112–122. doi:10.1107/s0108767307043930
Simpson, P. V., Desai, N. M., Casari, I., Massi, M., and Falasca, M. (2019). Metal-Based Antitumor Compounds: beyond Cisplatin. Future Med. Chem. 11, 119–135. doi:10.4155/fmc-2018-0248
Steel, T. R., Tong, K. K. H., Söhnel, T., Jamieson, S. M. F., Wright, L. J., Crowley, J. D., et al. (2021). Homodinuclear Organometallics of Ditopic N,N-chelates: Synthesis, Reactivity and In Vitro Anticancer Activity. Inorg. Chim. Acta 518, 120220. doi:10.1016/j.ica.2020.120220
Tremlett, W. D. J., Goodman, D. M., Steel, T. R., Kumar, S., Wieczorek-Błauż, A., Walsh, F. P., et al. (2021). Design Concepts of Half-sandwich Organoruthenium Anticancer Agents Based on Bidentate Bioactive Ligands. Coord. Chem. Rev. 445, 213950. doi:10.1016/j.ccr.2021.213950
Vogt, M., Pons, V., and Heinekey, D. M. (2005). Synthesis and Characterization of a Dicationic Dihydrogen Complex of Iridium With a Bis-Carbene Ligand Set. Organometallics 24, 1832–1836. doi:10.1021/om049045p
Keywords: anticancer activity, structural characterization, ligand exchange reactions, bioorganometallics, heterodimetallic complexes
Citation: Green LPM, Steel TR, Riisom M, Hanif M, Söhnel T, Jamieson SMF, Wright LJ, Crowley JD and Hartinger CG (2021) Synthetic Strategy Towards Heterodimetallic Half-Sandwich Complexes Based on a Symmetric Ditopic Ligand. Front. Chem. 9:786367. doi: 10.3389/fchem.2021.786367
Received: 30 September 2021; Accepted: 02 November 2021;
Published: 03 December 2021.
Edited by:
Dinorah Gambino, Universidad de la República, UruguayReviewed by:
Alzir Azevedo Batista, Federal University of São Carlos, BrazilRoberto Santana Da Silva, University of São Paulo, Brazil
Copyright © 2021 Green, Steel, Riisom, Hanif, Söhnel, Jamieson, Wright, Crowley and Hartinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian G. Hartinger, Yy5oYXJ0aW5nZXJAYXVja2xhbmQuYWMubno=