 Pietro Franceschi1†
Pietro Franceschi1† Catia Nicoletti1†
Catia Nicoletti1† Ruggero Bonetto1
Ruggero Bonetto1 Marcella Bonchio1
Marcella Bonchio1 Mirco Natali2
Mirco Natali2 Luca Dell’Amico1
Luca Dell’Amico1 Andrea Sartorel1*
Andrea Sartorel1*- 1Nano and Molecular Catalysis Laboratory, Department of Chemical Sciences, University of Padova, Padova, Italy
- 2Department of Chemical, Pharmaceutical and Agricultural Sciences (DOCPAS), University of Ferrara, and Centro Interuniversitario per La Conversione Chimica Dell’Energia Solare (SOLARCHEM), Ferrara, Italy
The utilization of carbon dioxide as a raw material represents nowadays an appealing strategy in the renewable energy, organic synthesis, and green chemistry fields. Besides reduction strategies, carbon dioxide can be exploited as a single-carbon-atom building block through its fixation into organic scaffolds with the formation of new C-C bonds (carboxylation processes). In this case, activation of the organic substrate is commonly required, upon formation of a carbanion C−, being sufficiently reactive toward the addition of CO2. However, the prediction of the reactivity of C− with CO2 is often problematic with the process being possibly associated with unfavorable thermodynamics. In this contribution, we present a thermodynamic analysis combined with density functional theory calculations on 50 organic molecules enabling the achievement of a linear correlation of the standard free energy (ΔG0) of the carboxylation reaction with the basicity of the carbanion C−, expressed as the pKa of the CH/C− couple. The analysis identifies a threshold pKa of ca 36 (in CH3CN) for the CH/C− couple, above which the ΔG0 of the carboxylation reaction is negative and indicative of a favorable process. We then apply the model to a real case involving electrochemical carboxylation of flavone and chalcone as model compounds of α,β-unsaturated ketones. Carboxylation occurs in the β-position from the doubly reduced dianion intermediates of flavone and chalcone (calculated ΔG0 of carboxylation in β = −12.8 and −20.0 Kcalmol-1 for flavone and chalcone, respectively, associated with pKa values for the conjugate acids of 50.6 and 51.8, respectively). Conversely, the one-electron reduced radical anions are not reactive toward carboxylation (ΔG0 > +20 Kcalmol-1 for both substrates, in either α or β position, consistent with pKa of the conjugate acids < 18.5). For all the possible intermediates, the plot of calculated ΔG0 of carboxylation vs. pKa is consistent with the linear correlation model developed. The application of the ΔG0 vs. pKa correlation is finally discussed for alternative reaction mechanisms and for carboxylation of other C=C and C=O double bonds. These results offer a new mechanistic tool for the interpretation of the reactivity of CO2 with organic intermediates.
Introduction
The activation and transformation of small molecules are pillars of artificial photosynthesis. In particular, carbon dioxide is an appealing target substrate because it is the product of combustion of organic compounds, and its levels in the atmosphere are continuously rising due to anthropogenic emissions while contributing to the greenhouse effect and global warming. Activation of CO2 can be accomplished through reduction routes (Francke et al., 2018; Melchionna et al., 2021) in which desirable products are carbon monoxide, formic acid, methanol, methane, or > C2 species (Albero et al., 2020). Alternatively, carbon dioxide can be exploited in cyclic carbonates or heterocycle formation (North et al., 2010; Fiorani et al., 2015; Yu and He, 2015; Guo et al., 2021; Vieira et al., 2018, 2019; Faria et al., 2021) or as a single-carbon-atom building block for its fixation into organic compounds (Liu et al., 2015; Cao et al., 2018; Cherubini-Celli et al., 2018; Tlili and Lakhdar, 2020; Zhang et al., 2020; Sahoo et al., 2021; Yuan et al., 2021; He et al., 2020) upon creation of new C-C or C-heteroatom bonds. Mechanistically, these processes can be accomplished through 1) the reduction of carbon dioxide to its radical anion, followed by its reaction with the organic scaffold (in dimethylformamide, E0(CO2/CO2•–) = −2.21 V vs. saturated calomel electrode, SCE, corresponding to −1.97 V vs. standard hydrogen electrode) (Lamy et al., 1977; Otero et al., 2006; Berto et al., 2015) or 2) upon the formation of reduced intermediates of the organic substrate accomplished through chemical, electrochemical, or photochemical routes and their subsequent reactivity with CO2 (Yuan et al., 2021).
This second possibility includes reductive activation of C-LG bonds (LG– is a leaving group, often a halide ion) (Meng et al., 2017; Isse et al., 1998; Isse et al., 2002; Isse and Gennaro, 2002; Scialdone et al., 2008; Durante et al., 2013) of C=C or C=N double bonds (Seo et al., 2017; Fan et al., 2018; Chen et al., 2020; Fan et al., 2018; Schmalzbauer et al., 2020) and of C-H bonds (Gui et al., 2017; Seo et al., 2017; He et al., 2020). Recent examples include activation of substituted olefins (Alkayal et al., 2020), of diverse carbonyl compounds (Okumura and Uozumi, 2021) including α-ketoamides and α-ketoesters (Cao et al., 2021), α,β-unsaturated esters (Sheta et al., 2021) and ketones (Chen et al., 2020), and of aldimines generated in situ for α-aminoacid synthesis (Naito et al., 2021).
In all cases, a carbanion (hereafter generally indicated as C−) is postulated to be the key intermediate that reacts with CO2 although the nature of the reactive species and the mechanistic comprehension of the reactivity often remain elusive.
As reported by Mayr and coworkers (Li et al., 2020), the prediction of the reactivity of carbon-based nucleophiles with CO2 is problematic using linear-free energy relationships based on nucleophilicity and electrophilicity parameters (Li et al., 2020; Orlandi et al., 2021); the failure to observe carboxylation products with a variety of nucleophilic carbanions may be caused by unfavorable thermodynamics of the reaction (Li et al., 2020).
Therefore, we aimed at developing a general tool to predict the thermodynamics of a carboxylation reaction involving a carbon-based anion C− by exploiting the basicity of C− as a thermodynamic parameter. We propose a thermodynamic analysis supported by density functional theory calculations on 50 small organic molecules that enable the to correlate the standard free energy of the carboxylation reaction with the basicity of the carbanion C−, expressed in terms of the pKa of the C-H/C−couple. We then apply the model to a real case involving electrochemical carboxylation of α,β-unsaturated carbonyls as the selected model substrates, and finally discuss alternative reaction mechanisms for the carboxylation of C=C and C=O double bonds.
Results and Discussion
Thermodynamic and DFT Analysis of Carbanions Reactivity with CO2
We employed a thermodynamic analysis to correlate the standard free energy of carboxylation of C− (
We then considered Eqs. 3, 4, for which the ΔG0 in acetonitrile is reported (Waldie et al., 2018), with the goal of expressing the
Eq. 5 derives from the sum of Eqs. 1–4:
and, rearranging,
Eq. 7, thus, predicts that the difference between
In order to evaluate the separate dependence of
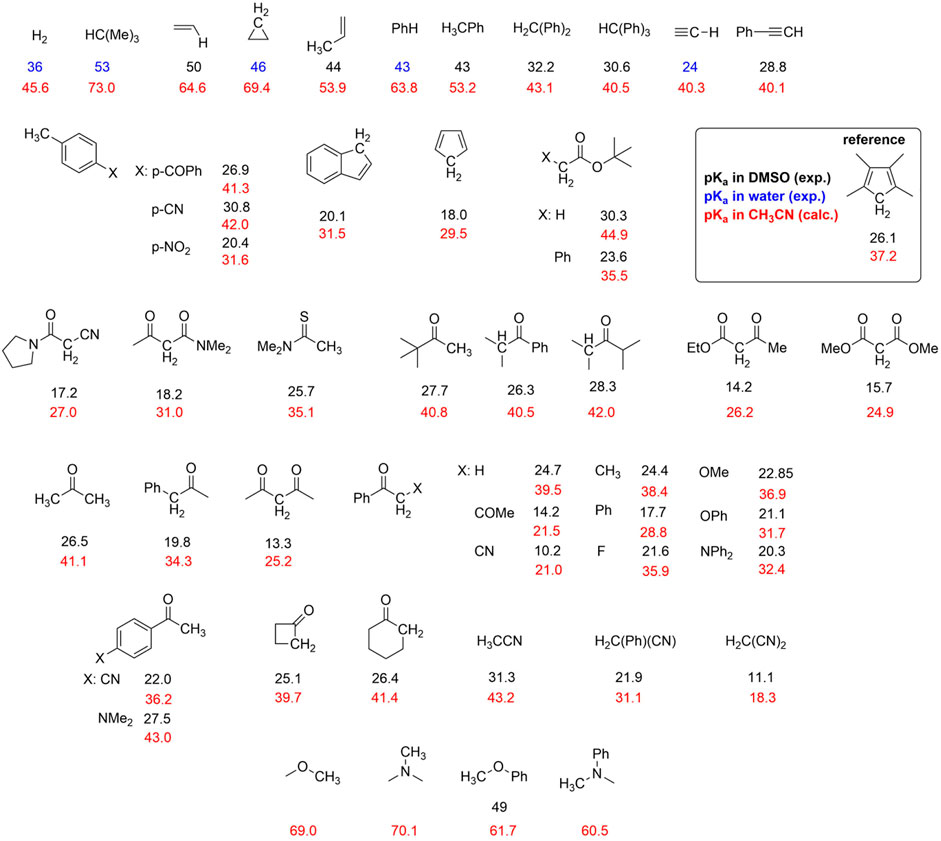
CHART 1. Organic molecules considered in the calculations with experimental and computed pKa values. The experimental pKa values are reported from the Reich database https://organicchemistrydata.org/hansreich/resources/pka/#pka_dmso_compilation (black and blue values refer to DMSO and water solvent, respectively). Computed pKa values (red) were evaluated by DFT calculations using the relative determination method (Ding et al., 2009), by selecting 1,2,3,4-tetramethylcyclopentadiene as the reference (pKa = 37.2).
Several experimental pKa values of C-H moieties are reported in DMSO; however, pKa is solvent-dependent (Daasbjerg, 1995; Workentin et al., 1995; Izutzu, 1990), and in acetonitrile, it can be rescaled according to pKa (CH3CN) = 11.6 + 0.98⋅pKa (DMSO); (Ding et al., 2009; Roszak et al., 2019).
When experimental values are not available (as in the case of some intermediates discussed in this work, vide infra), pKa can be predicted computationally. Thus, the pKa values of the C-H groups of the 50 species in Chart 1 were calculated by DFT, using the relative determination method (Ding et al., 2009; Kadiyala et al., 2013; Fu et al., 2005) by employing 1,2,3,4-tetramethylcyclopentadiene reference (pKa value in CH3CN of 37.2 derived from an experimental pKa = 26.1 in DMSO).
As shown in the top panel of Figure 1, the plot of calculated vs. experimental (derived values in CH3CN) pKa values show a linear correlation with a slope of 1.17 ± 0.04, an intercept of -4.7 ± 1.6, and an R-square of 0.95; except for one case, all the points stand within the 95% confidence interval of the linear correlation; the major deviations are observed for species with experimental pKa values > 35, for which the available data are more limited and subject to uncertainties.
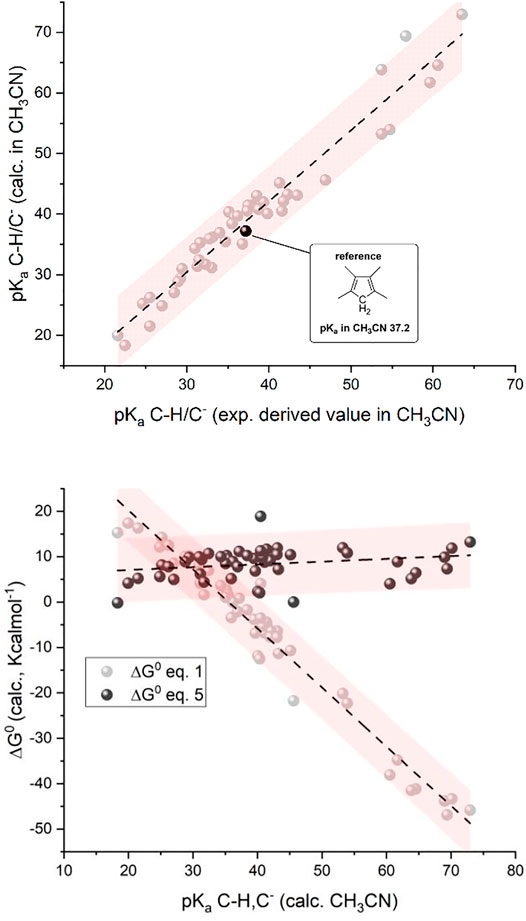
FIGURE 1. Top: Plot of calculated vs. experimental pKa for the 50 organic substances considered; see Chart 1 (CH/C− couples; 1,2,3,4-tetramethylcyclopentadiene as the reference; see the black dot with pKa = 37.2 in CH3CN). Bottom: Plot of calculated standard free energy of carboxylation (
We then determined the ΔG0 of reactions 1 and 5 (by the difference between the calculated free energy of products and reactants) for the 50 organic substrates and plotted the calculated
considering
In short, the presented analysis supports a linear correlation of the standard free energy of carboxylation of C− (
Clearly, the
Evaluation of the Model for Electrochemical Carboxylation of α,β-Unsaturated Carbonyls
We then examined the consistency of the predictive model with the experimental electrochemical carboxylation of flavone and chalcone as representatives of α,β-unsaturated carbonyl scaffolds retaining significant biological interest (Zhuang et al., 2017; Pietta, 2000). Moreover, under cathodic conditions, these substrates lead to the formation of multiple reduced intermediates, thus providing an ideal platform to assess their reactivity with carbon dioxide: the electrochemical methodology is indeed suitable to selectively generate the desired intermediate by tuning the operating potential.
Carboxylation of Flavone
Cyclic voltammetry of flavone F under cathodic scan shows a first, quasi-reversible wave at E1/2 = −2.09 V vs Fc+/Fc (ΔE = 120 mV) due to the one-electron reduction of F to the flavone radical anion, F(RA) (see Figure 2); scanning the CV analysis toward more negative potentials, a second, irreversible wave is observed peaking at E = −2.71 V vs Fc+/Fc, associated with the formation of the dianion F(DA) and its further reduction (see Figure 2); previous polarographic evidence suggests the occurrence of a two-electron process for this second wave due to a further irreversible reduction of F(DA) at this potential (Vakulskaya et al., 2011). Under CO2 saturation, the first wave becomes completely irreversible, and the cathodic peak shifts toward less negative potentials by 50 mV (Figure 2). A major change is instead associated with the second wave, suggesting reactivity of F(DA) with CO2; the decrease of the current suggests that the presence of CO2 inhibits the further reduction of the F(DA) with the latter likely involved in a different reaction pathway with CO2.
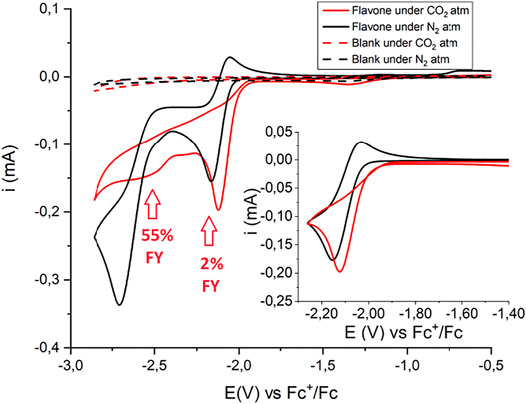
FIGURE 2. CV of 5 mM Flavone in CH3CN with 0.1 M tetrabutylammonium hexafluorophosphate supporting electrolyte under N2 (black traces) and CO2 (red traces). The inset shows the scan conducted in a narrow potential range and limited to the first reduction process. Glassy carbon working electrode (GC, 0.3 cm diameter, 0.07 cm2 geometric area), Pt counter electrode, Ag/AgCl reference electrode, scan rate 0.1 V s−1. Potentials were then converted to Fc+/Fc registering a CV scan of a ferrocene solution under the same conditions. The red arrows and the corresponding FY values refer to the carboxylation process and to the production of flavanone-2-carboxylic acid methyl ester. In principle, the operating potentials could be compatible with the production of the carbon dioxide radical anion, CO2•–: the E0(CO2/CO2•–) = −2.21 V vs SCE corresponding to −2.63 V vs. Fc+/Fc (Lamy et al., 1977; Berto et al., 2015; Christensen et al., 1990). However, only a slight increase of the CV traces below −2.7 V is observed passing from N2 to CO2 in the control experiments in the absence of flavone (dashed black and red traces, respectively). With GC electrodes, reduction of CO2 suffers indeed of an additional overpotential, and gives CO with almost quantitative Faradaic yield, accompanied by formation of CO32- (Berto et al., 2015; Christensen et al., 1990). Considering the ca 15-fold larger current observed in the presence of flavone (5 mM) and the higher concentrations of flavone used in CPE experiments (20 mM), the reduction of flavone is envisaged as the predominant route under these conditions.
Controlled potential electrolysis (CPE) experiments were then performed to assess the reactivity of both the F(RA) and F(DA) species by applying a suitable operating potential to a glassy carbon rod working electrode. Electrolysis was conducted in a cell with two compartments separated by a ceramic frit. To evaluate the Faradaic yield of formation of the carboxylation product(s), an esterification procedure was performed involving treatment of the electrolyzed solution with H2SO4 in methanol for 1 h under microwave heating at 80°C (Scheme 1).
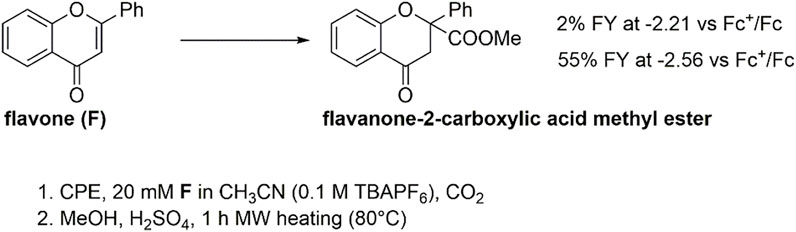
SCHEME 1. Electrochemical carboxylation of flavone F and formation of flavanone-2-carboxylic acid methyl ester (from carboxylation in β-position to the carbonyl group) after esterification of the electrolysis solution.
Reactivity of flavone radical anion, F(RA): In the presence of CO2, a CPE held at −2.21 V (a potential associated with the electrogeneration of F(RA)), led to the production of flavanone-2-carboxylic acid methyl ester in a 2% Faradaic yield (Scheme 1, see also the red arrow in Figure 2; in CPE, a 20 mM concentration of flavone was used, fourfold higher with respect to CV conditions), and the electrolysis led mainly to the formation of 2,2-biflavanone (racemate and meso forms) dimerization by-products (see Supporting Information) (Sisa et al., 2010). This result suggests that F(RA) is not an intermediate favorably reacting with CO2 along a carboxylation reaction.
The unfavorable reactivity of F(RA) with CO2 is supported by DFT analysis. F(RA) was optimized as a doublet, displaying spin density mainly at the carbon in β to the carbonyl group (0.28 spin density) at the ortho and para positions of the phenyl ring in β (0.16–0.22 spin density) and at the carbonyl group (0.12 and 0.13 spin density at the carbon and oxygen atoms, respectively); no significant spin density is observed at the carbon in α to the carbonyl (see Supplementary Figure S1).
Calculations on the conjugate acids of F(RA) were performed by considering protonation of F(RA) in α or β positions; the computed free energies allowed to determine the calculated pKa of the C-H/C− couples according to the abovementioned procedure, resulting in pKa values of 18.6 and 11.8 for the α and β positions, respectively. Both these values fall above the previously discussed threshold to reach a favorable carboxylation process. Consistently, a calculated ΔG0 of +19.6 kcalmol−1 was found for the carboxylation of F(RA) in α position, and the carboxylation product of F(RA) in the β position was unstable during the optimization process, decomposing into CO2 and F(RA); see Scheme 2 and Figure 3.
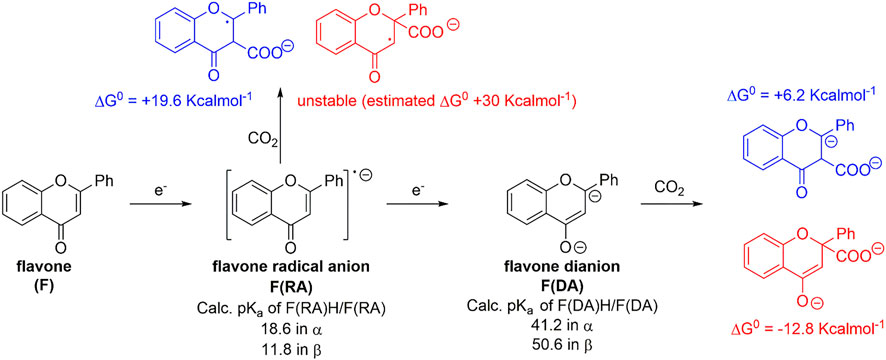
SCHEME 2. Generation of radical anion F(RA) and dianion F(DA) of flavone F, calculated pKa values of their conjugate acids, and their predicted reactivity with CO2 in terms of calculated ΔG0 values. Blue and red structures refer to carboxylation in α and β positions, respectively.
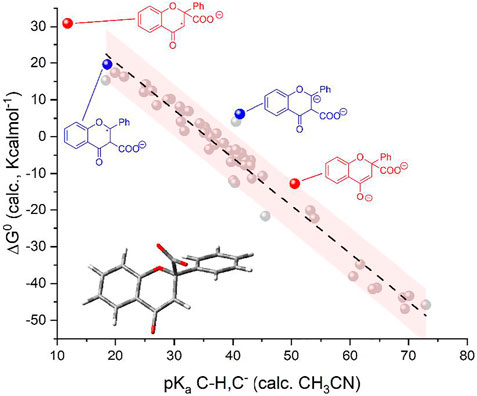
FIGURE 3. Plot of calculated standard free energy of carboxylation (
Reactivity of flavone dianion, F(DA): CPE at -2.56 V vs Fc+/Fc (Scheme 1, see also the red arrow in Figure 2) leads to the β-C carboxylation of flavone with 55% Faradaic yield upon isolation of the flavanone-2-carboxylic acid methyl ester. This evidence indicates a reaction of F(DA) with CO2 in the β-position (Senboku et al., 2011; Senboku et al., 2012). The reactivity of F(DA) with CO2 was supported by DFT calculations. F(DA) was optimized as a singlet state (Supplementary Figure S2), and similarly to the case of F(RA) previously discussed, calculations on the conjugate acids of the F(DA) were performed by considering protonation of F(DA) in the α and β positions to determine the calculated pKa of the C-H/C− couples: pKa values of 41.2 and 50.6 were obtained for the conjugate acids of F(DA) in the α and β positions, respectively (Scheme 2 and Figure 3). Concerning the carboxylation upon reaction of F(DA) with CO2, calculated ΔG0 of +6.2 kcalmol−1 and of −12.8 Kcalmol-1 were found for the carboxylation of F(DA) in the α and β positions, respectively (Scheme 2 and Figure 3), thus supporting the preferred carboxylation in β-position (Senboku et al., 2011; Senboku et al., 2012).
Importantly, the calculated ΔG0 vs pKa values for F(RA) (in the α position) and F(DA) (in both α and β positions) were observed to be consistent with the trend predicted in Figure 1 right for the 50 organic substrates (Scheme 2 and Figure 3).
Carboxylation of Trans-Chalcone
An analogous scenario was reached exploring the carboxylation of trans-chalcone (C). The CV analysis under cathodic scan and N2 atmosphere shows the presence of a first irreversible wave peaking at −1.9 V vs Fc+/Fc, attributed to the formation of the chalcone radical anion C(RA), Supplementary Figure S3. This is followed by a second wave, composed of two contributions at E1/2 = −2.3 V vs Fc+/Fc and E1/2 = −2.45 V vs Fc+/Fc, likely associated with the formation of the chalcone dianion C(DA) (Chen et al., 2020); the splitting of the wave into two contributions with ca halved intensity with respect to the first one could be ascribed to the rotation of the C-C bond in C(RA), leading to two cis/trans isomeric forms that are further reduced to C(DA) at slightly different potentials. Upon addition of CO2, the first wave is almost unaffected, and the second one shows marked changes with the formation of a single irreversible wave peaking at Epc = −2.5 V vs. Fc+/Fc and, thus, suggesting reactivity of C(DA) with CO2. This is confirmed by CPE experiments, that allowed the isolation of methyl-4-oxo-2,4-diphenylbutanoate with 41% FY upon electrolysis at −2.7 V vs Fc+/Fc followed by esterification of the carboxylate product, consistent with effective carboxylation in the β position to the carbonyl group (Scheme 3).
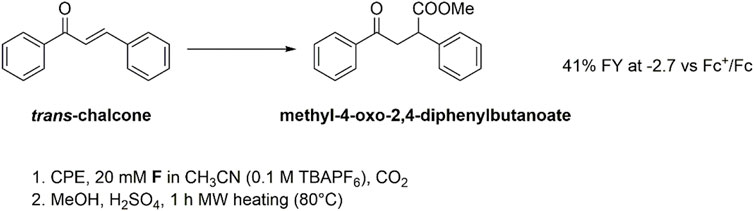
SCHEME 3. Electrochemical carboxylation of trans-chalcone C and formation of ethyl-4-oxo-2,4-diphenylbutanoate after esterification in methanol of the electrolysis solution. The Faradaic yield results lower with respect to galvanostatic conditions performed in a single compartment cell and employing aluminum anodes (Chen et al., 2020); this can be due to a stabilizing effect of the carboxylate product by electrogenerated Al(III) ions.
Similar to what previously discussed in the case of flavone, the reactivity trend of chalcone reduced species C(RA) and C(DA) toward CO2 was supported by DFT calculations (Scheme 4 and Figure 4). C(RA), optimized as a doublet, shows a planar structure and a spin density localized mainly on the carbon in β to the carbonyl (0.33 spin density) and on the carbonyl group (0.18 and 0.19 spin density for C and O, respectively); see Supplementary Figure S4. Calculations on the conjugate acids of C(RA) by considering a protonation in the α and β positions to the carbonyl, lead to the determination of pKa values of 18.5 and 18.2 in the α and β positions, respectively; these pKa values are below the predicted threshold of reactivity with CO2, and consistently, positive ΔG0 of +22.0 and of +20.2 kcalmol−1 were obtained for the carboxylation of C(RA) in the α and β positions, respectively, see Scheme 4 and Figure 4.
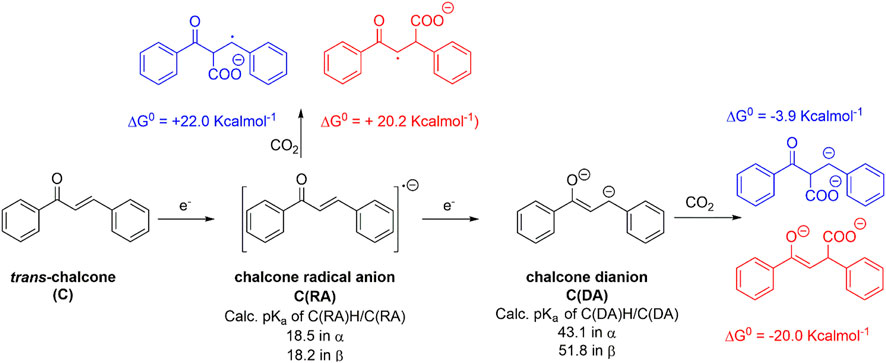
SCHEME 4. Generation of radical anion C(RA) and dianion C(DA) from trans-chalcone C, calculated pKa values of their conjugate acids and their predicted reactivity with CO2 in terms of calculated ΔG0 values. Blue and red structures refer to carboxylation in α and β positions, respectively.
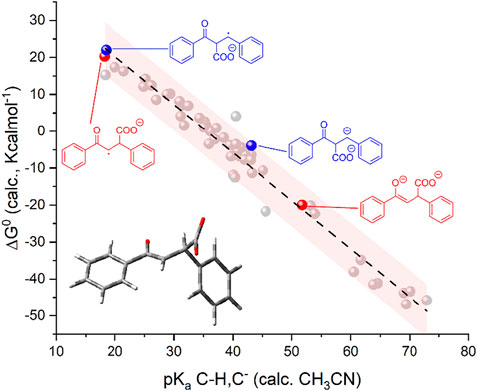
FIGURE 4. Plot of calculated standard free energy of carboxylation (
Optimization of C(DA) as a singlet led to a planar structure (Supplementary Figure S5); calculations on the conjugate acids of C(DA) by inserting a proton in the α and β positions, led to the determination of pKa of 43.1 and 51.8 for C-H groups in the α and β positions, respectively. Consistently, negative ΔG0 for the carboxylation reaction involving C(DA) were determined of −3.9 and −20.0 Kcalmol-1 for the α and β positions, respectively (Scheme 4 and Figure 4).
Again, the calculated ΔG0 vs. pKa values for C(RA) and C(DA) in both α and β positions were observed to be consistent with the linear trend predicted in Figure 1 for the 50 organic substrates (Figure 4) with all the points standing within the 95% confidence interval, supporting the need to generate the dianion of chalcone to achieve the carboxylation in the β-position as experimentally observed.
Alternative Mechanisms and Perspectives in the Carboxylation of C=C and C=O Bonds
Based on the above discussion, some considerations of general relevance can be finally addressed and focused in particular on 1) the reactivity of radical anions, generated from a one-electron reduction of the parent C=C bond; this type of intermediate is often envisaged in photochemical carboxylation processes (Nikolaitchik et al., 1996; Seo et al., 2017); 2) the reactivity of carbanions generated from an activation of C=O bonds, via an umpolung strategy (Juhl and Lee, 2018; Juhl et al., 2019; Cao et al., 2021).
In the case of α,β-unsaturated carbonyl compounds, a positive, unfavorable ΔG0 in the reactivity of the radical anion toward CO2 seems to be a general feature as predicted by the DFT calculations on other α,β-unsaturated carbonyl scaffolds summarized in Supplementary Table S1. However, other reaction pathways that are alternative to the generation of a further reduced dianion intermediate can be envisaged to achieve carboxylation of this class of substrates. One possibility exploits hydrogen atom transfer (HAT) from a suitable HAT donor (Capaldo and Ravelli, 2017; Costas and Bietti, 2018). Still considering the representative case of flavone and chalcone, a HAT to F(RA) and C(RA) occurs preferentially in β positions to generate the corresponding flavone and chalcone anions, F(A) and C(A) (Scheme 5; these are 12.8 and 11.9 Kcalmol-1 more stable with respect to the isomeric species generated by a HAT in the α position to flavone and chalcone, respectively). Because the HAT is more favorable in the β position, the possible reactivity of F(A) and of C(A) with CO2 should occur in the α position.
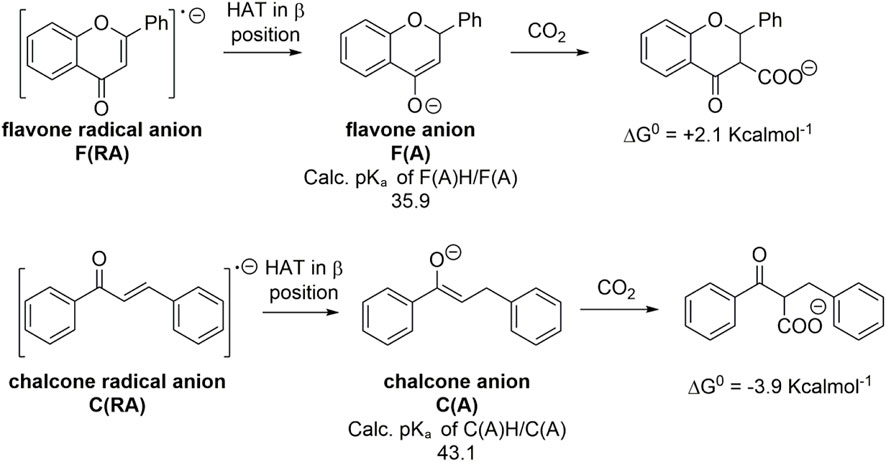
SCHEME 5. Generation of flavone and chalcone anions, F(A) and C(A) from a HAT to the flavone and chalcone radical anions F(RA) and C(RA), calculated pKa values of their conjugate acids, and their predicted reactivity with CO2 in terms of calculated ΔG0 values of the carboxylation reaction.
Calculations predict pKa values of 35.9 and 43.1 for the conjugate acids of F(A) and C(A), respectively, and the ΔG0 for the carboxylation are +2.1 and -3.9 Kcalmol-1 starting from F(A) and C(A), respectively (Scheme 5); the pKa and ΔG0 values fit well with the model previously developed, standing within the 95% confidence interval (Figure 5). Therefore, the basicity of F(A) and C(A) is greatly enhanced with respect to the corresponding radical anions F(RA) and C(RA) with differences in the pKa values of 17.3 and 24.6, respectively; the enhancement of basicity leads to a favorable gain in the ΔG0 of carboxylation in the α position of 17.5 and 25.9 Kcalmol-1 for flavone and chalcone, respectively, when passing from the radical anions to the anions. The use of a HAT donor additive can, thus, be considered in the carboxylation processes although the regioselectivity should be properly evaluated.
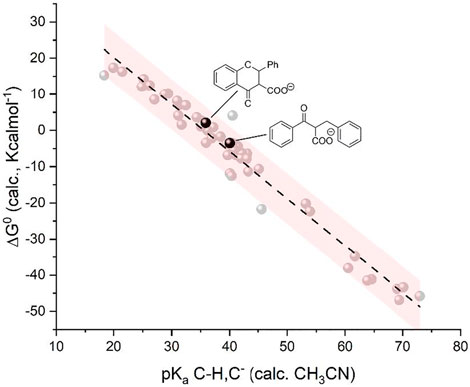
FIGURE 5. Plot of calculated standard free energy of carboxylation (
Although unfavorable in the case of α,β-unsaturated carbonyls, the negligible reactivity of radical anion intermediates toward carboxylation should not be considered as a general feature of C=C double bonds. We performed DFT calculations on the radical anions of selected alkenes, such as ethylene, 2-butene, and diphenylethylene isomers as well as of phenanthrene as representative of a fully aromatic scaffold to calculate the corresponding basicity and the ΔG0 of the carboxylation reaction involving these radical anions. The results are summarized in Supplementary Table S1 and plotted in the ΔG0 vs. pKa graph of Figure 6. A nice match is observed between these data and those of the 50 organic substrates previously employed in the construction of the linear trend. Interestingly, these calculations predict that 1) radical anions of alkenes can be sufficiently basic to achieve a favorable ΔG0 for carboxylation when hydrogen or alkyl groups are bound to the C=C double bond; 2) phenyl groups bound to the carbon atoms of the C=C bond reduce the basicity of the radical anion and tend to disfavor the carboxylation (differences of ca 30 pKa units and of 40 Kcalmol-1 in ΔG0 of carboxylation are observed by comparing trans-2-butene and trans-1,2-diphenylethylene); 3) when phenyl groups are present, reactivity is expected to be favorable if one of the carbon atoms of the C=C bond does not bear phenyl substituents as in the case of 1,1-diphenylethylene; and 4) radical anions of C=C bonds in aromatic scaffolds show unfavorable basicity and carboxylation reactivity as in the case of phenanthrene. Further investigations on structure-reactivity analysis on this kind of substrate are ongoing.
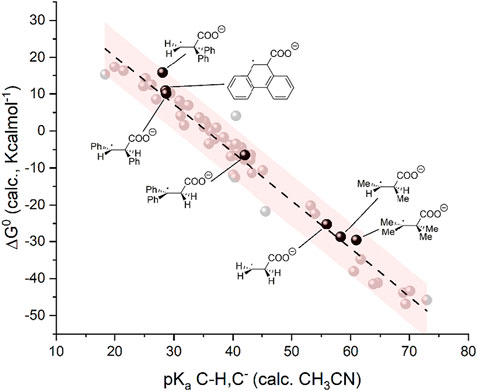
FIGURE 6. Plot of calculated standard free energy of carboxylation (
We finally verified if the model is suitable for the prediction of carboxylation of carbanions generated by activation of C=O groups through an umpolung strategy. In particular, we considered a carbanion of 4-fluorobenzaldehyde activated via a cyanohydrin intermediate (Juhl and Lee, 2018) and the carbanions of alkyl aryl ketones, α-ketoesters, and aryl aldehydes generated through a photochemical process combining a trimethylsilyl (in the case of the alkyl aryl ketones and of α-ketoesters) or triphenylsilyl (in the case of aryl aldehydes) activating/protecting group, see Scheme 6 (Cao et al., 2021). Gratifyingly, the calculations predict a negative ΔG0 for the carboxylation of such intermediates (Juhl et al., 2019), thus supporting the experimental outcome (see the yields of carboxylation in Scheme 6) (Juhl and Lee, 2018; Cao et al., 2021). In addition, the pKa and ΔG0 values fit well with the model previously developed (Figure 7).
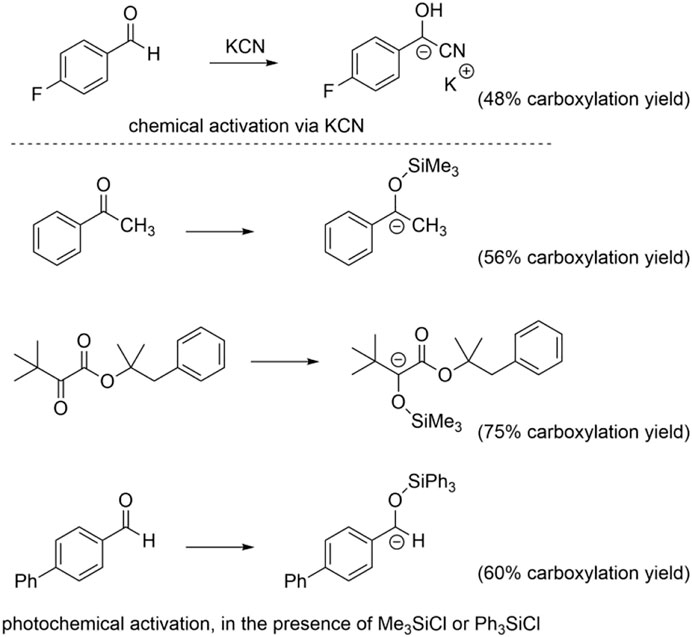
SCHEME 6. Formation of carbanions capable of carboxylation reactions via Umpolung activation of C=O bonds. See references for further experimental conditions (Juhl and Lee, 2018; Juhl et al., 2019; Cao et al., 2021).
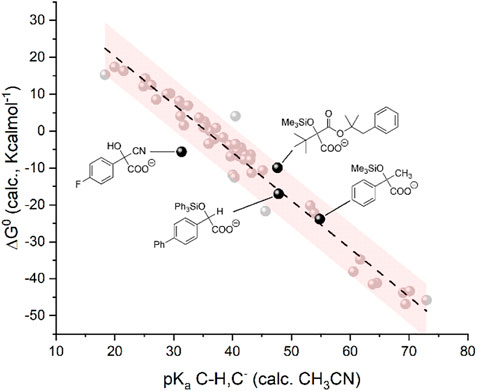
FIGURE 7. Plot of calculated standard free energy of carboxylation (ΔG0) vs. calculated pKa of the CH/C− couples. The black dots refer to the products upon carboxylation of anions generated from activation of C=O groups. The light gray dots indicate the same 50 organic molecules reported in Chart 1 and plotted in Figure 1.
Conclusion and Perspectives
We present a thermodynamic analysis combined with density functional theory calculations that enable linearly correlating the standard free energy ΔG0 of the carboxylation reaction of a carbanion C− with its basicity expressed as the pKa of the CH/C− couple. This offers a new mechanistic tool for the interpretation of the reactivity of CO2 with organic intermediates. The analysis identifies a threshold pKa of ca 36 (in CH3CN) for the CH/C− couple, above which the ΔG0 of the carboxylation reaction is negative and, thus, indicative of a thermodynamically favorable process. Because fast tools are nowadays available for the estimation of pKa of C-H groups (Roszak et al., 2019), the pKa vs ΔG0 correlation enables a fast analysis and prediction of the thermodynamics of the carboxylation reaction.
Application of the model to the electrochemical carboxylation of flavone and chalcone as representative compounds of α,β-unsaturated ketones allows the prediction of the carboxylation occurring in the β-position from the doubly reduced dianion intermediates of the starting compounds (ΔG0 of carboxylation in β = −12.8 and −20.0 Kcalmol-1 for flavone and chalcone, respectively, associated to pKa values for the conjugate acid of 50.6 and 51.8, respectively). The one-electron reduced radical anions are instead not reactive toward carboxylation (ΔG0 > +20 Kcalmol-1 for both substrates in either α or β position, consistent with pKa of the conjugate acid < 18.5). In all cases, the calculated pKa and ΔG0 of carboxylation are consistent with the linear correlation model developed, thus supporting its application also to more complex organic scaffolds.
The analysis was extended to alternative carboxylation mechanisms and to other organic substrates that were already employed in carboxylation reactions in previous literature studies.
Further tuning of the model could consider possible specific stabilization of the species involved and, in particular, of the charged ones by the nature of the solvent or by the presence of additives. As discussed by (Pletcher and Slevin, 1996), Mg2+ ions are known to stabilize reduced intermediates and carboxylate species (Corbin et al., 2021) and are proposed to play a key role in the electrochemical carboxylation of benzalacetone (Mg2+ are typically generated under electrochemical conditions, when employing Mg sacrificial anodes) (Wang et al., 2017; Bhasha Sayyed and Sakaki, 2014).
Experimental
The cyclic voltammetry (CV) characterizations were carried out with a three-electrode system controlled by a BASi EC Epsilon potentiostat-galvanostat. The working electrode was a glassy carbon disk electrode (BioLogic, nominal diameter 3 mm), the auxiliary electrode was a platinum electrode (BASi), and the reference electrode was an Ag/AgCl (NaCl 3 M) electrode; potentials were then referenced to the ferrocenium/ferrocene (Fc+/Fc) couple upon addition, at the end of each experiment session, of ferrocene to the analyte solutions as internal standard; 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) was used as a supporting electrolyte.
Constant potential electrolysis experiments were performed with a Metrohm Autolab PGSTAT204 potentiostat-galvanostat in combination with the NOVA 2.1.4 software (https://www.metrohm-autolab.com/Products/Echem/Software/Nova.html).
The cell generally employed for preparative electrolysis was a custom-made, six-necked, two-compartment glass cell with the two compartments being separated by a porous glass frit.
Quantitative gas chromatographic (GC) analysis were performed on a Shimadzu GC-2010 Pro gas chromatograph equipped with a flame ionization detector (FID). Every measurement was performed by automatic injection of 1 μL of the sample solution. Quantification of the starting material and ester products was achieved by internal calibration of the instrument upon the construction of a calibration curve by the injection of known volumes of reagents and mesitylene as a standard. The response factor of the initial substrate was used also to quantify the ester product because the presence of one -COOCH3 additional group with respect to the initial substrate is expected to have a minor effect in the FID response.
1H NMR spectra were recorded on a Bruker 300 Advance spectrometer equipped with BBO probe head 5 mm. NMR spectra were processed using MestReNova software.
EI-MS spectra were registered using an Agilent 6,850 Network GC system equipped with a 5975 Series MSD detector. ESI-MS spectra were acquired with an Agilent Technology LC/MSD Trap SL, interfaced to an Agilent 1100 binary pump.
Esterification procedures were done by a CEM Discover microwave reactor (300 W maximum power) setting the bulk temperature at 80°C for 1 h.
For all species, geometry optimizations and frequency calculations were done to give the best suited Gibbs energies by DFT calculations performed at the b3lyp/6–311 + g(d,p) level of theory with Gaussian16 and GaussView 6 software packages (Frisch et al., 2016). The self-consistent reaction field was used with DFT energies, optimizations, and frequency calculations to model systems in acetonitrile solution. The convergence criteria for interatomic force minimization (geometry optimization) were the standard ones of the Gaussian16 software.
Further details are reported in Supplementary Information.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author Contributions
The laboratory work was carried out by PF, CN, and RB. PF, CN, RB, MB, MN, LD’A, and AS (all authors) contributed to the analysis, review and interpretation of data. LD’A and AS supervised the work. AS designed the study and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Fondazione Cariparo, project Synergy, within the call Ricerca Scientifica d’Eccellenza 2018 (AS), and by the University of Padova P-DiSC#11BIRD2020-UNIPD (LD’A).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Prof. Abdirisak Ahmed Isse, Prof. Flavio Maran, Prof. Cristiano Zonta, Manuel Orlandi (University of Padova) and Giacomo Saielli (Italian C.N.R., Consiglio Nazionale delle Ricerche) for fruitful discussions. We thank Chiara Conti (University of Padova) and Elena Rossin (University of Ferrara) for preliminary experiments. We thank Mauro Meneghetti and Stefano Mercanzin (technical services at the University of Padova) for the irreplaceable contribution in the construction of the electrochemical cell and for their technical assistance. CN acknowledges Ministero dell’Università e della Ricerca for a Ph.D. scholarship (Programma Operativo Nazionale, PON, Ricerca e Innovazione, dottorati su tematiche green).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.783993/full#supplementary-material
References
Albero, J., Peng, Y., and García, H. (2020). Photocatalytic CO2 Reduction to C2+ Products. ACS Catal. 10, 5734–5749. doi:10.1021/acscatal.0c00478
Alkayal, A., Tabas, V., Montanaro, S., Wright, I. A., Malkov, A. V., and Buckley, B. R. (2020). Harnessing Applied Potential: Selective β-Hydrocarboxylation of Substituted Olefins. J. Am. Chem. Soc. 142, 1780–1785. doi:10.1021/jacs.9b13305
Azcarate, I., Costentin, C., Robert, M., and Savéant, J.-M. (2016). Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 138, 16639–16644. doi:10.1021/jacs.6b07014
Berto, T. C., Zhang, L., Hamers, R. J., and Berry, J. F. (2015). Electrolyte Dependence of CO2 Electroreduction: Tetraalkylammonium Ions Are Not Electrocatalysts. ACS Catal. 5, 703–707. doi:10.1021/cs501641z
Blanksby, S. J., and Ellison, G. B. (2003). Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 36, 255–263. doi:10.1021/ar020230d
Cao, G.-M., Hu, X.-L., Liao, L.-L., Yan, S.-S., Song, L., Chruma, J. J., et al. (2021). Visible-light Photoredox-Catalyzed Umpolung Carboxylation of Carbonyl Compounds with CO2. Nat. Commun. 12, 2–11. doi:10.1038/s41467-021-23447-8
Cao, Y., He, X., Wang, N., Li, H.-R., and He, L.-N. (2018). Photochemical and Electrochemical Carbon Dioxide Utilization with Organic Compounds. Chin. J. Chem. 36, 644–659. doi:10.1002/cjoc.201700742
Capaldo, L., and Ravelli, D. (2017). Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2056–2071. doi:10.1002/ejoc.201601485
Chen, R., Tian, K., He, D., Gao, T., Yang, G., Xu, J., et al. (2020). Carboxylation of α,β-Unsaturated Ketones by CO2 Fixation through Photoelectro-Chemistry. ACS Appl. Energ. Mater. 3, 5813–5818. doi:10.1021/acsaem.0c00728
Cherubini-Celli, A., Mateos, J., Bonchio, M., Dell'Amico, L., and Companyó, X. (2018). Transition Metal-free CO2 Fixation into New Carbon-Carbon Bonds. ChemSusChem 11, 3056–3070. doi:10.1002/cssc.201801063
Chiba, K., Tagaya, H., Karasu, M., Ishizuka, M., and Sugo, T. (1994). Carboxylation of Active Methylene Compounds Using Anilide, Potassium Carbonate, and Carbon Dioxide. Bcsj 67, 452–454. doi:10.1246/bcsj.67.452
Chiba, K., Tagaya, H., Miura, S., and Karasu, M. (1992). The Carboxylation of Active Methylene Compounds with Carbon Dioxide in the Presence of 18-Crown-6 and Potassium Carbonate. Chem. Lett. 21, 923–926. doi:10.1246/cl.1992.923
Christensen, P. A., Hamnett, A., Muir, A. V. G., and Freeman, N. A. (1990). CO2 Reduction at Platinum, Gold and Glassy Carbon Electrodes in Acetonitrile. J. Electroanalytical Chem. Interfacial Electrochemistry 288, 197–215. doi:10.1016/0022-0728(90)80035-5
Corbin, N., Yang, D.-T., Lazouski, N., Steinberg, K., and Manthiram, K. (2021). Suppressing Carboxylate Nucleophilicity with Inorganic Salts Enables Selective Electrocarboxylation without Sacrificial Anodes. Chem. Sci. 12, 12365–12376. doi:10.1039/d1sc02413b
Costas, M., and Bietti, M. (2018). Uncovering the Complexity of the Simplest Atom Transfer Reaction. Acc. Chem. Res. 51, 2601–2602. doi:10.1021/acs.accounts.8b00525
Daasbjerg, K., Oslob, J. D., Åkermark, B., and Norrby, P.-O. (1995). Estimation of the pKa for Some Hydrocarbons and Aldehydes and Solvation Energies of the Corresponding Anions. Acta Chem. Scand. 49, 878–887. doi:10.3891/acta.chem.scand.49-0878
Destro, G., Horkka, K., Loreau, O., Buisson, D. A., Kingston, L., Del Vecchio, A., et al. (2020). Transition‐Metal‐Free Carbon Isotope Exchange of Phenyl Acetic Acids. Angew. Chem. Int. Ed. 59, 13490–13495. doi:10.1002/anie.202002341
Ding, F., Smith, J. M., and Wang, H. (2009). First-Principles Calculation of pKa Values for Organic Acids in Nonaqueous Solution. J. Org. Chem. 74, 2679–2691. doi:10.1021/jo802641r
Durante, C., Isse, A. A., Todesco, F., and Gennaro, A. (2013). Electrocatalytic Activation of Aromatic Carbon-Bromine Bonds toward Carboxylation at Silver and Copper Cathodes. J. Electrochem. Soc. 160, G3073–G3079. doi:10.1149/2.008307jes
Fan, X., Gong, X., Ma, M., Wang, R., and Walsh, P. J. (2018). Visible Light-Promoted CO2 Fixation with Imines to Synthesize Diaryl α-amino Acids. Nat. Commun. 9, 1–8. doi:10.1038/s41467-018-07351-2
Faria, D. J., dos Santos, L. M., Bernard, F. L., Pinto, I. S., Romero, I. P., Chaban, V. V., et al. (2021). Performance of Supported Metal Catalysts in the Dimethyl Carbonate Production by Direct Synthesis Using CO2 and Methanol. J. CO2 Utilization 53, 101721. doi:10.1016/j.jcou.2021.101721
Fiorani, G., Guo, W., and Kleij, A. W. (2015). Sustainable Conversion of Carbon Dioxide: The Advent of Organocatalysis. Green. Chem. 17, 1375–1389. doi:10.1039/c4gc01959h
Francke, R., Schille, B., and Roemelt, M. (2018). Homogeneously Catalyzed Electroreduction of Carbon Dioxide-Methods, Mechanisms, and Catalysts. Chem. Rev. 118, 4631–4701. doi:10.1021/acs.chemrev.7b00459
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16. Wallingford CT: Gaussian, Inc.
Fu, Y., Liu, L., Yu, H.-Z., Wang, Y.-M., and Guo, Q.-X. (2005). Quantum-chemical Predictions of Absolute Standard Redox Potentials of Diverse Organic Molecules and Free Radicals in Acetonitrile. J. Am. Chem. Soc. 127, 7227–7234. doi:10.1021/ja0421856
Gui, Y.-Y., Zhou, W.-J., Ye, J.-H., and Yu, D.-G. (2017). Photochemical Carboxylation of Activated C(sp3)−H Bonds with CO2. ChemSusChem 10, 1337–1340. doi:10.1002/cssc.201700205
Guo, L., Lamb, K. J., and North, M. (2021). Recent Developments in Organocatalysed Transformations of Epoxides and Carbon Dioxide into Cyclic Carbonates. Green. Chem. 23, 77–118. doi:10.1039/d0gc03465g
He, X., Qiu, L.-Q., Wang, W.-J., Chen, K.-H., and He, L.-N. (2020). Photocarboxylation with CO2: an Appealing and Sustainable Strategy for CO2 Fixation. Green. Chem. 22, 7301–7320. doi:10.1039/d0gc02743j
Isse, A. A., Galia, A., Belfiore, C., Silvestri, G., and Gennaro, A. (2002). Electrochemical Reduction and Carboxylation of Halobenzophenones. J. Electroanalytical Chem. 526, 41–52. doi:10.1016/S0022-0728(02)00815-X
Isse, A. A., and Gennaro, A. (2002). Electrochemical Synthesis of Cyanoacetic Acid from Chloroacetonitrile and Carbon Dioxide. J. Electrochem. Soc. 149, D113. doi:10.1149/1.1490358
Isse, A. A., Gennaro, A., and Vianello, E. (1998). Mechanism of the Electrochemical Reduction of Benzyl Chlorides Catalysed by Co(salen). J. Electroanalytical Chem. 444, 241–245. doi:10.1016/S0022-0728(97)00572-X
Izutzu, K. (1990). Acid-base Dissociation Constants in Dipolar Aprotic Solvents. 1990th ed. Oxford: Blackwell Scientific publications.
Juhl, M., Kim, M., Lee, H.-Y., Baik, M.-H., and Lee, J.-W. (2019). Aldehyde Carboxylation: A Concise DFT Mechanistic Study and a Hypothetical Role of CO2 in the Origin of Life. Synlett 30, 987–996. doi:10.1055/s-0037-1611738
Juhl, M., and Lee, J.-W. (2018). Umpolung Reactivity of Aldehydes toward Carbon Dioxide. Angew. Chem. Int. Ed. 57, 12318–12322. doi:10.1002/anie.201806569
Kadiyala, R. R., Tilly, D., Nagaradja, E., Roisnel, T., Matulis, V. E., Ivashkevich, O. A., et al. (2013). Computed CH Acidity of Biaryl Compounds and Their Deprotonative Metalation by Using a Mixed lithium/Zinc-TMP Base. Chem. Eur. J. 19, 7944–7960. doi:10.1002/chem.201300552
Kong, D., Moon, P. J., Lui, E. K. J., Bsharat, O., and Lundgren, R. J. (2020). Direct Reversible Decarboxylation from Stable Organic Acids in Dimethylformamide Solution. Science 369, 557–561. doi:10.1126/science.abb4129
Lamy, E., Nadjo, L., and Saveant, J. M. (1977). Standard Potential and Kinetic Parameters of the Electrochemical Reduction of Carbon Dioxide in Dimethylformamide. J. Electroanalytical Chem. Interfacial Electrochemistry 78, 403–407. doi:10.1016/s0022-0728(77)80143-5
Li, Z., Mayer, R. J., Ofial, A. R., and Mayr, H. (2020). From Carbodiimides to Carbon Dioxide: Quantification of the Electrophilic Reactivities of Heteroallenes. J. Am. Chem. Soc. 142, 8383–8402. doi:10.1021/jacs.0c01960
Liu, Q., Wu, L., Jackstell, R., and Beller, M. (2015). Using Carbon Dioxide as a Building Block in Organic Synthesis. Nat. Commun. 6, 1–15. doi:10.1038/ncomms6933
Mateos, J., Rigodanza, F., Vega‐Peñaloza, A., Sartorel, A., Natali, M., Bortolato, T., et al. (2020). Naphthochromenones: Organic Bimodal Photocatalysts Engaging in Both Oxidative and Reductive Quenching Processes. Angew. Chem. Int. Ed. 59, 1302–1312. doi:10.1002/anie.201912455
Melchionna, M., Fornasiero, P., Prato, M., and Bonchio, M. (2021). Electrocatalytic CO2 Reduction: Role of the Cross-Talk at Nano-Carbon Interfaces. Energy Environ. Sci. 14, 5816–5833. doi:10.1039/d1ee00228g
Meng, Q.-Y., Wang, S., and König, B. (2017). Carboxylation of Aromatic and Aliphatic Bromides and Triflates with CO2 by Dual Visible-Light-Nickel Catalysis. Angew. Chem. Int. Ed. 56, 13426–13430. doi:10.1002/anie.201706724
Naito, Y., Nakamura, Y., Shida, N., Senboku, H., Tanaka, K., and Atobe, M. (2021). Integrated Flow Synthesis of α-Amino Acids by In Situ Generation of Aldimines and Subsequent Electrochemical Carboxylation. J. Org. Chem. 1. doi:10.1021/acs.joc.1c00821
Nikolaitchik, A. V., Rodgers, M. A. J., and Neckers, D. C. (1996). Reductive Photocarboxylation of Phenanthrene: A Mechanistic Investigation. J. Org. Chem. 61, 1065–1072. doi:10.1021/jo951702n
North, M., Pasquale, R., and Young, C. (2010). Synthesis of Cyclic Carbonates from Epoxides and CO2. Green. Chem. 12, 1514–1539. doi:10.1039/c0gc00065e
Okumura, S., and Uozumi, Y. (2021). Photocatalytic Carbinol Cation/Anion Umpolung: Direct Addition of Aromatic Aldehydes and Ketones to Carbon Dioxide. Org. Lett. 23, 7194–7198. doi:10.1021/acs.orglett.1c02592
Orlandi, M., Escudero-Casao, M., and Licini, G. (2021). Nucleophilicity Prediction via Multivariate Linear Regression Analysis. J. Org. Chem. 86, 3555–3564. doi:10.1021/acs.joc.0c02952
Otero, M. D., Batanero, B., and Barba, F. (2006). CO2 Anion-Radical in Organic Carboxylations. Tetrahedron Lett. 47, 2171–2173. doi:10.1016/j.tetlet.2006.01.113
Pietta, P.-G. (2000). Flavonoids as Antioxidants. J. Nat. Prod. 63, 1035–1042. doi:10.1021/np9904509
Pletcher, D., and Slevin, L. (1996). Influence of Magnesium(II) Ions on Cathodic Reactions in Aprotic Solvents-Carboxylation of Methyl Aryl Ketones. J. Chem. Soc. Perkin Trans 2, 217–220. doi:10.1039/p29960000217
Roszak, R., Beker, W., Molga, K., and Grzybowski, B. A. (2019). Rapid and Accurate Prediction of pKa Values of C-H Acids Using Graph Convolutional Neural Networks. J. Am. Chem. Soc. 141, 17142–17149. doi:10.1021/jacs.9b05895
Sahoo, P. K., Zhang, Y., and Das, S. (2021). CO2-Promoted Reactions: An Emerging Concept for the Synthesis of Fine Chemicals and Pharmaceuticals. ACS Catal. 11, 3414–3442. doi:10.1021/acscatal.0c05681
Sayyed, F. B., and Sakaki, S. (2014). The Crucial Roles of MgCl2 as a Non-innocent Additive in the Ni-Catalyzed Carboxylation of Benzyl Halide with CO2. Chem. Commun. 50, 13026–13029. doi:10.1039/c4cc04962d
Schmalzbauer, M., Svejstrup, T. D., Fricke, F., Brandt, P., Johansson, M. J., Bergonzini, G., et al. (2020). Redox-Neutral Photocatalytic C−H Carboxylation of Arenes and Styrenes with CO2. Chem 6, 2658–2672. doi:10.1016/j.chempr.2020.08.022
Scialdone, O., Galia, A., Filardo, G., Isse, A. A., and Gennaro, A. (2008). Electrocatalytic Carboxylation of Chloroacetonitrile at a Silver Cathode for the Synthesis of Cyanoacetic Acid. Electrochimica Acta 54, 634–642. doi:10.1016/j.electacta.2008.07.012
Senboku, H., Yamauchi, Y., Kobayashi, N., Fukui, A., and Hara, S. (2011). Electrochemical Carboxylation of Flavones: Facile Synthesis of Flavanone-2-Carboxylic Acids. Electrochemistry 79, 862–864. doi:10.5796/electrochemistry.79.862
Senboku, H., Yamauchi, Y., Kobayashi, N., Fukui, A., and Hara, S. (2012). Some Mechanistic Studies on Electrochemical Carboxylation of Flavones to Yield Flavanone-2-Carboxylic Acids. Electrochimica Acta 82, 450–456. doi:10.1016/j.electacta.2012.03.131
Seo, H., Katcher, M. H., and Jamison, T. F. (2017). Photoredox Activation of Carbon Dioxide for Amino Acid Synthesis in Continuous Flow. Nat. Chem 9, 453–456. doi:10.1038/nchem.2690
Sheta, A. M., Alkayal, A., Mashaly, M. A., Said, S. B., Elmorsy, S. S., Malkov, A. V., et al. (2021). Selective Electrosynthetic Hydrocarboxylation of α,β‐Unsaturated Esters with Carbon Dioxide**. Angew. Chem. Int. Ed. 60, 21832–21837. doi:10.1002/anie.202105490
Sisa, M., Bonnet, S. L., Ferreira, D., and Van Der Westhuizen, J. H. (2010). Photochemistry of Flavonoids. Molecules 15, 5196–5245. doi:10.3390/molecules15085196
Tlili, A., and Lakhdar, S. (2021). Acridinium Salts and Cyanoarenes as Powerful Photocatalysts: Opportunities in Organic Synthesis. Angew. Chem. Int. Ed. 60, 19526–19549. doi:10.1002/anie.202102262
Vakulskaya, T. I., Larina, L. I., and Vashchenko, A. V. (2011). Radical Anions of Flavonoids. Magn. Reson. Chem. 49, 508–513. doi:10.1002/mrc.2783
Vieira, M. O., Monteiro, W. F., Neto, B. S., Chaban, V. V., Ligabue, R., and Einloft, S. (2019). Chemical Fixation of CO2: the Influence of Linear Amphiphilic Anions on Surface Active Ionic Liquids (SAILs) as Catalysts for Synthesis of Cyclic Carbonates under Solvent-free Conditions. Reac Kinet Mech. Cat 126, 987–1001. doi:10.1007/s11144-019-01544-6
Vieira, M. O., Monteiro, W. F., Neto, B. S., Ligabue, R., Chaban, V. V., and Einloft, S. (2018). Surface Active Ionic Liquids as Catalyst for CO2 Conversion to Propylene Carbonate. Catal. Lett. 148, 108–118. doi:10.1007/s10562-017-2212-4
Waldie, K. M., Ostericher, A. L., Reineke, M. H., Sasayama, A. F., and Kubiak, C. P. (2018). Hydricity of Transition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction. ACS Catal. 8, 1313–1324. doi:10.1021/acscatal.7b03396
Wang, H., Zhu, H.-W., Guo, R.-R., Hu, Q.-L., Zeng, S., and Lu, J.-X. (2017). Computational and Experimental Study on Electrocarboxylation of Benzalacetone. Asian J. Org. Chem. 6, 1380–1384. doi:10.1002/ajoc.201700233
Wiedner, E. S., Chambers, M. B., Pitman, C. L., Bullock, R. M., Miller, A. J. M., and Appel, A. M. (2016). Thermodynamic Hydricity of Transition Metal Hydrides. Chem. Rev. 116, 8655–8692. doi:10.1021/acs.chemrev.6b00168
Workentin, M. S., Maran, F., and Wayner, D. D. M. (1995). Reduction of Di-tert-butyl Peroxide: Evidence for Nonadiabatic Dissociative Electron Transfer. J. Am. Chem. Soc. 117, 2120–2121. doi:10.1021/ja00112a037
Yu, B., and He, L.-N. (2015). Upgrading Carbon Dioxide by Incorporation into Heterocycles. ChemSusChem 8, 52–62. doi:10.1002/cssc.201402837
Yuan, L., Qi, M. Y., Tang, Z. R., and Xu, Y. J. (2021). Coupling Strategy for CO 2 Valorization Integrated with Organic Synthesis by Heterogeneous Photocatalysis. Angew. Chem. Int. Ed. 60, 21150–21172. doi:10.1002/anie.202101667
Zhang, Z., Ye, J.-H., Ju, T., Liao, L.-L., Huang, H., Gui, Y.-Y., et al. (2020). Visible-Light-Driven Catalytic Reductive Carboxylation with CO2. ACS Catal. 10, 10871–10885. doi:10.1021/acscatal.0c03127
Zhou, S., Wang, Y., and Gao, J. (2021). Solvation Induction of Free Energy Barriers of Decarboxylation Reactions in Aqueous Solution from Dual-Level QM/MM Simulations. JACS Au 1, 233–244. doi:10.1021/jacsau.0c00110
Keywords: carbon dioxide fixation, thermodynamic analysis, DFT calculations, reaction intermediates, unsaturated carbonyl
Citation: Franceschi P, Nicoletti C, Bonetto R, Bonchio M, Natali M, Dell’Amico L and Sartorel A (2021) Basicity as a Thermodynamic Descriptor of Carbanions Reactivity with Carbon Dioxide: Application to the Carboxylation of α,β-Unsaturated Ketones. Front. Chem. 9:783993. doi: 10.3389/fchem.2021.783993
Received: 27 September 2021; Accepted: 28 October 2021;
Published: 24 November 2021.
Edited by:
Simelys Hernández, Politecnico di Torino, ItalyReviewed by:
Vitaly Chaban, Harvard University, United StatesLiang-Nian He, Nankai University, China
Ji-Woong Lee, University of Copenhagen, Denmark
Copyright © 2021 Franceschi, Nicoletti, Bonetto, Bonchio, Natali, Dell’Amico and Sartorel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Sartorel, YW5kcmVhLnNhcnRvcmVsQHVuaXBkLml0
†These authors have contributed equally to this work