
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem., 30 November 2021
Sec. Organic Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.779765
This article is part of the Research TopicAdvances and Future Trends in Bioactive Natural Products: Insights into Total Synthesis and Their Biological and Biomedical ApplicationsView all 4 articles
 Liyan Yang1
Liyan Yang1 Zhonglei Wang2,3,4*
Zhonglei Wang2,3,4*Abstract: Aflatoxins, which are produced by Aspergillus flavus, Aspergillus nomius, and Aspergillus parasiticus, are a group of pentacyclic natural products with difuran and coumarin skeletons. They mainly include aflatoxin B1, B2, G1, G2, M1, and M2. Biologically, aflatoxins are of concern to human health as they can be present as contaminants in food products. The unique skeletons of aflatoxins and their risk to human health have led to the publication of nine remarkable total syntheses (including three asymmetric syntheses) and ten formal total syntheses (including four asymmetric formal syntheses) of aflatoxins in the past 55 years. To better understand the mechanism of the biological activity of aflatoxins and their presence in samples from the food industry, this review summarizes progress in the total synthesis of aflatoxins.
Aflatoxins (Figure 1) are a group of potent hepatocarcinogenic polyketide natural products produced by the fungi Aspergillus flavus and Aspergillus parasiticus (Groopman et al., 2008; Bräse et al., 2009; Wu, 2014). Aflatoxin B1 1) and G1 5) were first isolated together with aflatoxin B2 2) and G2 6) in 1963 (Hartley et al., 1963), and their structures were revealed in 1963 (G1 and B1) (Asao et al., 1963) and 1965 (G2 and B2) (Asao et al., 1965) by the group of Büchi. The absolute stereochemistry of the above four aflatoxins (G1, B1, G2, and B2) was determined by chemical degradation by Büchi’s group (Brechbühler-Bader et al., 1967). Aflatoxin M1 3) and M2 4) are hydroxylated metabolites of aflatoxin B1 and B2 (Bianco et al., 2012; Lee et al., 2017). Biologically, aflatoxins present a significant risk to human health as they can be present as contaminants in food products (Roze et al., 2013; Bashiry et al., 2021; Ismail et al., 2021; Mollayusefian et al., 2021). Aflatoxins are classified as hepatocarcinogens; however, their effects on tissues other than the liver are mainly unclear (Wang et al., 2015; Schrenk et al., 2020). Thus, there is a need to obtain analytical samples from the food industry and understand the mechanism underlying the biological activity of aflatoxins.
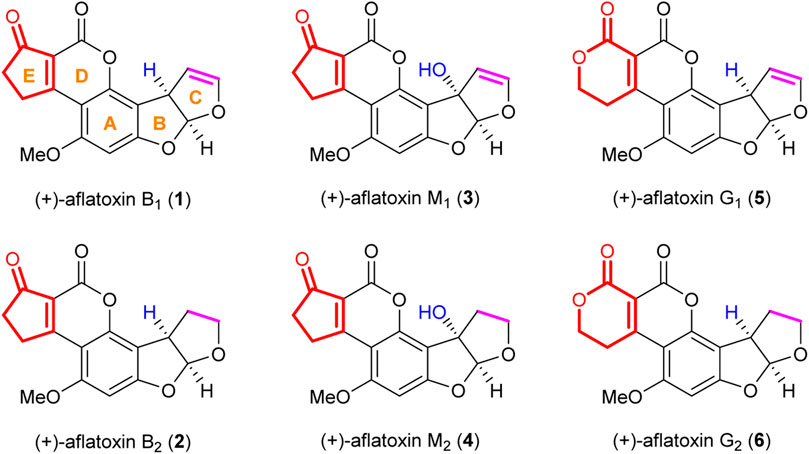
FIGURE 1. Structures of representative aflatoxins.
Given the broad public health implications of aflatoxins, considerable progress has been made in the chemical synthesis of aflatoxins since the first synthesis of racemic aflatoxins by the groups of Büchi and Roberts in the 1960s (Büchi et al., 1966). The mechanisms of the biological activities of many complex natural products, including aflatoxins, remain unknown due to the impracticality of isolating the products from their natural sources; the only alternative to obtaining the natural products is practical total synthesis. Thus, to better understand the effects of aflatoxins, this article reviews important developments in the organic total synthesis of aflatoxins during the past 55 years.
The Büchi group has made outstanding contributions to the chemical total synthesis of aflatoxins. This group has completed several total syntheses of challenging molecules in the aflatoxin family. These syntheses are characterized by Pechmann condensation and cascade reduction rearrangement.
As early as 1966, Büchi’s group (Büchi et al., 1966; Büchi et al., 1967) completed the first total synthesis of aflatoxin B1, as shown in Figure 2. The aldehyde 8 was obtained from acetyl benzene 7 through five steps: non-selective acylation, methylation, deacylation, selective benzylation, veticilienylation, and allyl oxidation. In the presence of Zn/AcOH, the tricyclic skeleton 12 was efficiently constructed. The cascade reaction proceeded in three steps: 1) the reduction of the double bond of coumarin 8 in the presence of Zn/AcOH; 2) the ring opening of the lactone under the action of glacial acetic acid; and 3) the formation of a hemiacetal between the free phenol and aldehyde groups. The construction of the tricyclic framework was then completed via esterification reaction followed by the removal of the benzyl protecting group to realize the tricyclic intermediate 13.
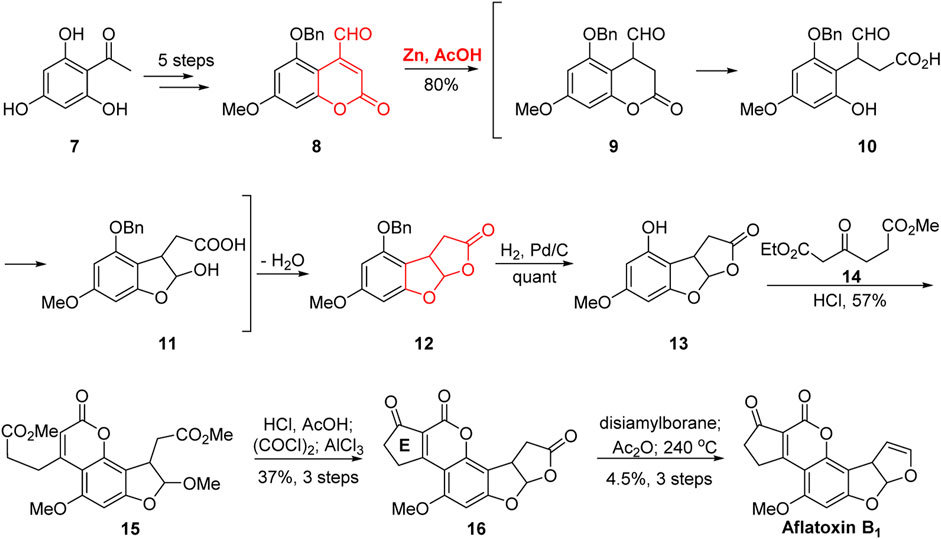
FIGURE 2. First total synthesis of (±)-aflatoxin B1 by Büchi group.
Next, in the presence of hydrochloric acid in methanol, the D-ring was constructed via Pechmann condensation reaction with the β-keto ester 14. Notably, the C-ring was opened in the presence of hydrochloric acid in methanol. Subsequently, under the action of hydrochloric acid and acetic acid, the two ester groups underwent acetal methyl hydrolysis, leading to the re-cyclization of the C-ring. After the activation of the carboxyl group, the E-ring 16 was constructed via Friedel–Crafts reaction catalyzed by AlCl3. Aflatoxin B1 was synthesized by the selective reduction of the C-ring, acylation of the hemiacetal hydroxyl, and pyrolysis at 240°C. The first total synthesis of aflatoxin B1 was completed in 13 steps with a 0.9% total yield.
Aflatoxin B1 also attracted the attention of Roberts’ group because of its unique chemical structure, although Büchi’s group was first to report the total synthesis of aflatoxin B1. Roberts’ group (Roberts et al., 1968) then switched their focus to the total synthesis of aflatoxin B2. In 1967, the total synthesis of aflatoxin B2 in 10 steps was reported for the first time, as shown in Figure 3A.
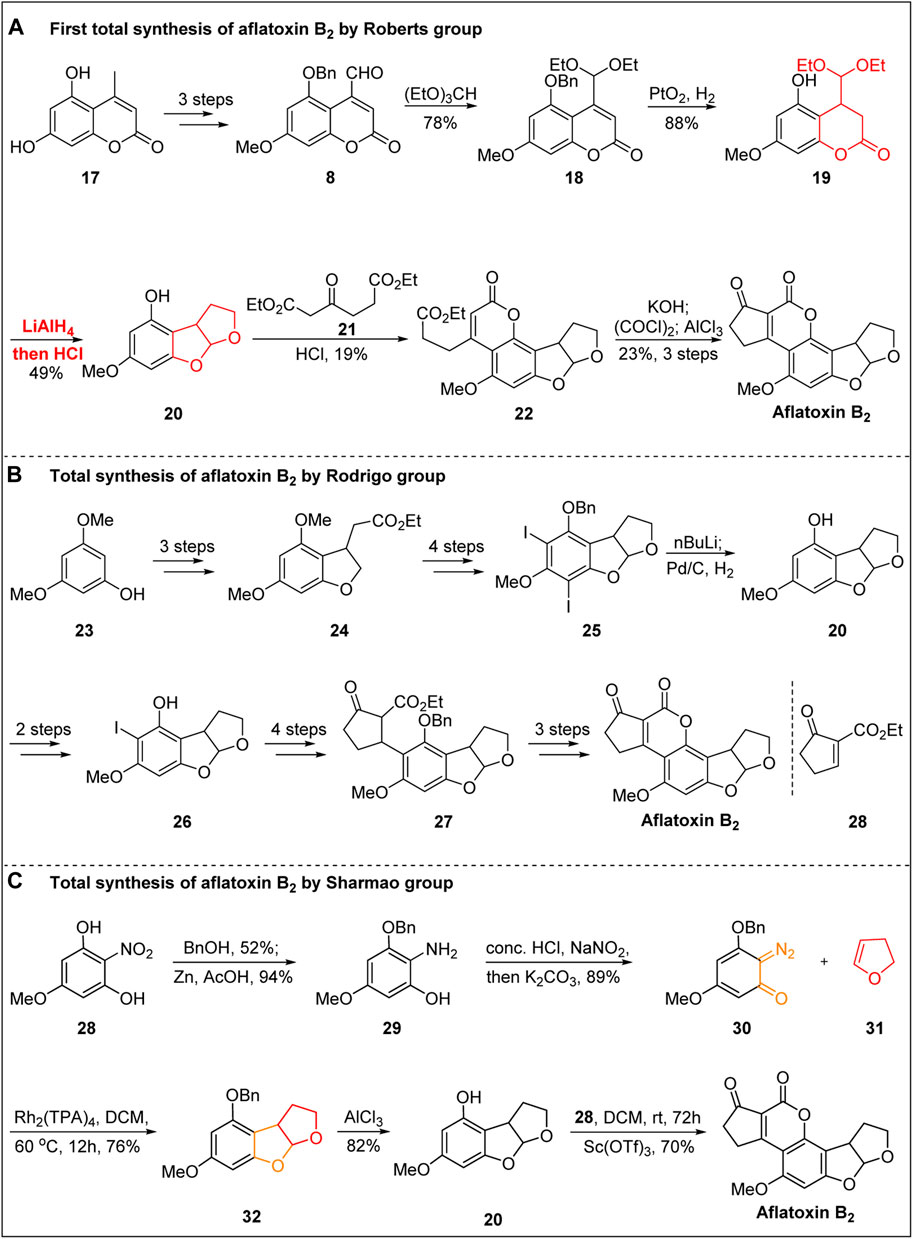
FIGURE 3. Total synthesis of (±)-aflatoxin B2.
Using the same strategy reported by Büchi’s group, starting from the coumarin 17, the coumarin intermediate 8 containing an aldehyde group was obtained by selective methylation, benzylation, and allyl oxidation under the action of SeO2. After the aldehyde group was protected, the benzyl protecting group was removed, and the double bond was hydrogenated under the action of Adam’s catalyst. Subsequently, under the action of LiAlH4, the ester group was reduced to an alcohol, and the aldehyde group was released in the presence of hydrochloric acid. The intramolecular acetal was spontaneously generated, and the synthesis of 20 was achieved. The first synthesis of aflatoxin B2 was then achieved by Pechmann condensation and Friedel–Crafts acylation using the same strategy reported by Büchi’s group.
In 1988, Rodrigo’s group (Weeratunga et al., 1988) started to the total synthesis of (±)-aflatoxin B2 starting from 3,5-dimethoxyphenol 23 and realized the construction of the B-ring through three steps: iodine substitution, alkylation, and intramolecular addition. The C-ring skeleton was then realized by reduction, selective demethylation, iodination, benzyl protection, and cyclization. After the deiodination and debenzyl reaction, the intermediate 20 was synthesized in 4% total yield.
In 1990, Rodrigo’s group (Horne et al., 1990) reported the second total synthesis of aflatoxin B2 after two years of trying and failing (Figure 3B). From the advanced intermediate 20, the key precursor 26 was obtained through diiodization, selective deiodization, benzyl protection of phenolic OH, lithium halide exchange, and transfer metallization. The second synthesis of aflatoxin B2 was then completed via the 1,4-addition of unsaturated cyclopentanone 28, removal of the benzyl protective group, hydrolysis of the ester group under acidic conditions, spontaneous esterification, and DDQ oxidation. The total yield of aflatoxin B2 in the above nine linear steps was 2.5%.
In 2021, an efficient approach for the total synthesis of aflatoxin B2 was described by Sharmao’s group (Paymode and Sharma, 2021) (Figure 3C). The key step involved in this synthesis is the Rh-catalyzed [3 + 2]-annulation of ortho-diazoquinone with enol ether. The key diazoquinone precursor 30 was obtained after mono-benzylation, the reduction of the nitro group, and diazotization. The key Rh-catalyzed [3 + 2]-annulation of diazoquinone 30 with enol ether 31 was then carried out in the presence of Rh2(OAc)4 in DCM, resulting in the annulation product 32. The advanced intermediate 20 was then obtained after deprotecting the benzyl group using AlCl3. Finally, in the presence of the Lewis acid Sc(OTf)3 in DCM, as reported by Zu’s group (Wang and Zu, 2019), the total synthesis of aflatoxin B2 was completed via Pechmann-type annulation and aerobic oxidation.
In 1969, Büchi’s group (Büchi and Weinreb, 1969) reported the first chemical total synthesis of aflatoxin M1, as shown in Figure 4. Based on the structural characteristic of aflatoxin M1, the dihydroxybenzofuranone 37 with a B-ring structure was used as the starting material, which created the conditions for the introduction of hydroxyl group and avoided the problem of constructing the B-ring. The hydroxyl-protected benzo furanone 38 was obtained after dimethylation, selective demethylation, and benzylation. The aldehyde 40 was then obtained via bromination (α-carbonyl group), substitution of benzyl alcohol, addition of allyl magnesium bromide to the ketone carbonyl group, and the oxidative fracturing of the double bond using osmium tetroxide. Two benzyl protection groups were removed by hydrogenation followed by the acylation of the phenol and hemiacetal hydroxyl groups and high-temperature pyrolysis (450°C), resulting in the construction of the C-ring double bond.
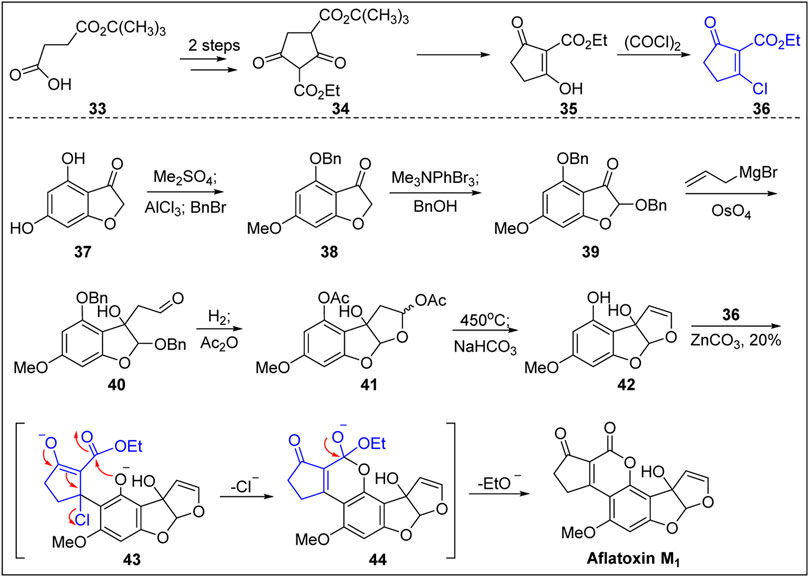
FIGURE 4. First total synthesis of (±)-aflatoxin M1 by Büchi group.
The free phenolic hydroxyl group in the A-ring was released via hydrolysis in the presence of weak base. The free phenolic compound 42 was then reacted with chlorinated unsaturated cyclopentanone 36 under the action of ZnCO3 to efficiently construct a D,E-bicyclic compound. The specific reaction process was as follows. First, under the catalysis of Zn ion, the 1,4-addition reaction took place smoothly between the phenolic OH group in the ortho-position and the unsaturated carbonyl substrate 36. Next, the chloride anion as the leaving group left to form the unsaturated carbonyl intermediate. Finally, under the action of ZnCO3, the phenolic OH group underwent transesterification to produce ethyl ester, thereby completing the first total synthesis of aflatoxin M1. This procedure represents a new solution for the synthesis of aflatoxins.
In 1971, Büchi’s group (Büchi and Weinreb, 1971) optimized the synthesis of aflatoxin B1 and completed the first total synthesis of aflatoxin G1 via a sequence of 1,4-addition, elimination, and transesterification reactions, as shown in Figure 5.

FIGURE 5. First total synthesis of (±)-aflatoxin G1 by Büchi group.
The synthesis of the advanced intermediate 46 was carried out from intermediate 12 through six steps: DIBAL-H reduction, acylation, hydrogenation of the benzyl group, acylation of the phenolic OH group, pyrolysis at high temperature (400°C), and deacylation. Finally, using the more active bromo-unsaturated cyclopentanone 45, the D,E-bicyclic compound was formed in one step under reflux with ZnCO3 (130 equiv.). This method achieved the second-generation synthesis of aflatoxin B1 and provided an important reference for Corey’s group to synthesize aflatoxin B2.
In addition, Büchi’s group reported the bromo-unsaturated cyclopentanone 45 to the bromo-unsaturated caprolactone 48. Based on this strategy, Büchi’s group achieved the chemical total synthesis of aflatoxin G2 for the first time, although the operation is complicated, and the yield was only 14%. This marks an important breakthrough in the synthesis of the G class of aflatoxins.
In 2003, Trost’s group (Trost and Toste, 2003) achieved the first chemical synthesis of aflatoxin B1 with high enantioselectivity via Pd-catalyzed dynamic kinetic asymmetric transformation (DYKAT), as shown in Figure 6.
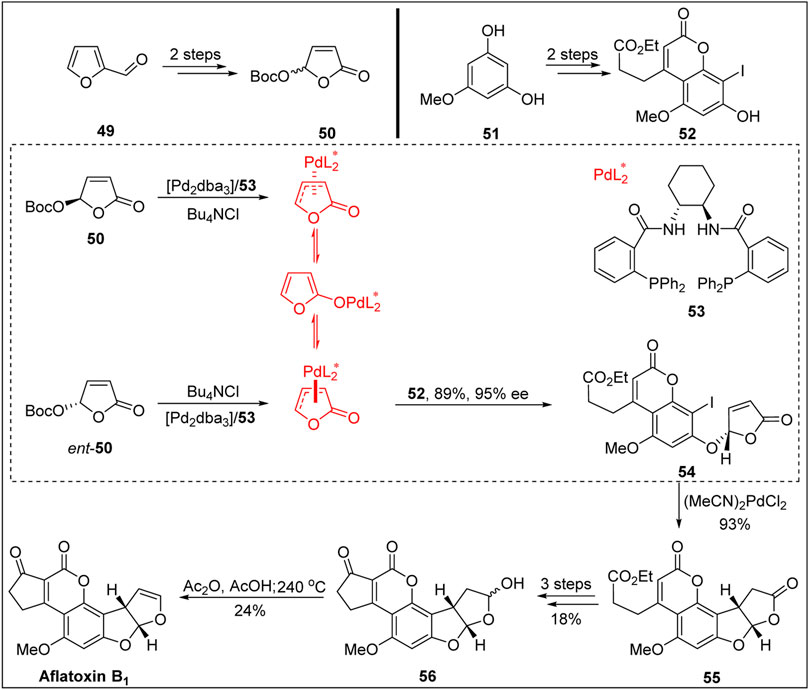
FIGURE 6. First asymmetric total synthesis of aflatoxin B1 by Trost group.
Based on the work of the Buchi and Roberts groups, the coumarin precursor was constructed via Pechmann reaction from 5-methoxy-m-catechol 51 and β-keto ester. The key precursor 52 was then obtained via regionally selective iodination in the presence of ICl. Next, Trost’s group used their previously developed method (namely, the dynamic asymmetric transformation reaction of iodide and lactone under palladium catalysis) to construct the chiral center of the B-ring. The coupling product, which was obtained in high yield and with an excellent ee value, was subjected to intramolecular reduction via Heck reaction under standard conditions. The chiral center of the BC-ring was constructed with a high ee value, and the coumarin product 55 with an ABCD four-ring skeleton was obtained. In a later work, the Büchi group achieved the construction of the E-ring through a related transformation and then completed the chiral synthesis of aflatoxin B2a 56) via the selective reduction of the C-ring. Finally, aflatoxin B1 was synthesized via the acylation of the hemiacetal hydroxyl group followed by pyrolysis at 240 °C. The total yield of aflatoxin B1 in nine linear steps was 1.6%.
In 2005, inspired by the excellent work of the Büchi and Noland groups (Büchi and Weinreb, 1971; Noland and Kedrowski, 2000), Corey’s group (Zhou and Corey, 2005) reported the first asymmetric synthesis of aflatoxin B2 based on the use of several advanced intermediates, as shown in Figure 7. The key intermediate 58 was obtained with high efficiency from dihydrofuran 31 and 1,4-benzoquinone 57 based on a highly enantioselective [3 + 2] cycloaddition catalyzed by an organoboron reagent. The highly efficient formation of the ABC ring system was achieved in only one step.
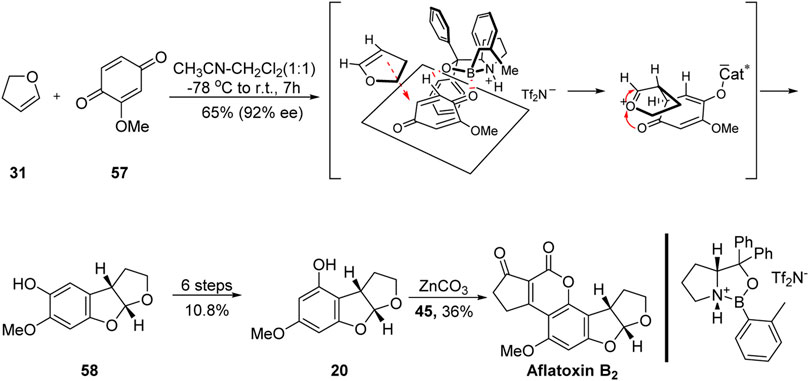
FIGURE 7. First asymmetric total synthesis of aflatoxin B2 by Corey group.
It should be noted that the key precursor 58 obtained using this method was not a good match for the A-ring of aflatoxin B2. The authors then followed the synthetic strategy of Noland’s group (Noland and Kedrowski, 2000) (that is, Friedel–Crafts acylation, hydroxyl protection, 1,2-addition, DMP oxidation, oxygen insertion, saponification, and reduction) to obtain the advanced precursor 20. Subsequently, they synthesized the DE-ring of aflatoxin B2 using the reaction conditions employed by Büchi’s group (Büchi and Weinreb, 1971). The first total synthesis of (+)-aflatoxin B2 was achieved in 2.5% yield through eight linear steps.
In recent years, green and facile single-pot reactions have received considerable attention in the field of chemistry because they can give rise to complex structures in few synthetic steps and with simple starting materials (Newhouse et al., 2009; Trost, 1991; István T. Horváth and Anastas, 2007; Wender et al., 2008; Hayashi, 2016). In 2019, an efficient Sc(OTf)3-promoted green and facile single-pot reaction involving 1,4-addition, intramolecular lactonization, and spontaneous aerobic oxidation was developed by Zu’s group (Wang and Zu, 2019) to synthesize the DE-ring system of aflatoxin B2 (Figure 8).
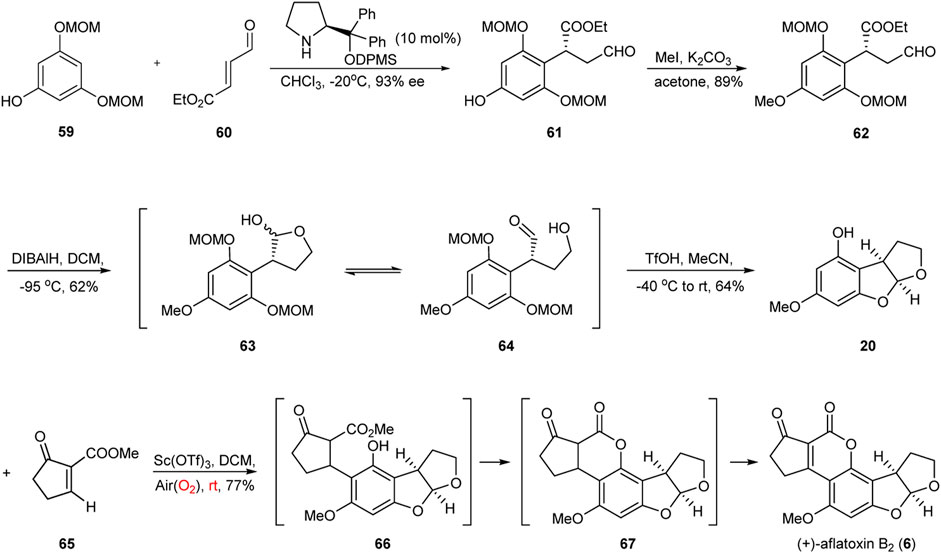
FIGURE 8. Asymmetric total synthesis of aflatoxin B2 by Zu group.
The synthesis started from the known phloroglucinol derivative 59 and the α,β-unsaturated aldehyde 60. The enantioselective Friedel–Crafts alkylation of 59 with 60 provided the alkylation product 61, which was methylated with MeI in acetone to provide intermediate 62. It was gratifying to find that the partial reduction of the intermediate 62 with DIBALH in DCM at −95 °C generated hemiacetal 63 and hydroxyl aldehyde 64 as an inseparable mixture in 62% yield. In the presence of TfOH in MeCN, the tricycle 20 was obtained via the cleavage of the MOM groups followed by intramolecular cyclization. Having successfully assembled tricycle 20, the group turned toward the final stage of the total synthesis of aflatoxin B2: the Sc(OTf)3-promoted one-pot sequential reaction. Finally, they successfully completed the required conversion in 77% yield using the Lewis acid Sc(OTf)3, thereby achieving the asymmetric chemical total synthesis of (+)-aflatoxin B2 with excellent atom-, redox-, and step-economy. This work also demonstrates that the application potential of the new developed strategy for the construction of benzyl chiral centers in the synthesis of complex molecules.
In 1988, Snieckus’ group (Sloan et al., 1988) reported the formal total synthesis of aflatoxin B1 based on radical cyclization, as shown in Figure 9. The radical precursor 70 was obtained via substitution reaction from o-bromophenol 68 and bromobutenolactone 69. The B-ring skeleton was then successfully constructed by intramolecular 1,4-addition mediated by free radicals. Finally, the MOM protecting group was removed to obtain the advanced intermediate 13. According to Büchi’s strategy, the aflatoxin B1 was successfully synthesized.
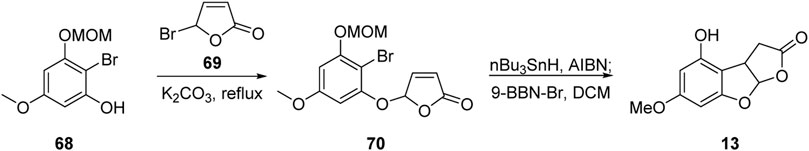
FIGURE 9. Formal total synthesis of aflatoxin B1 by Snieckus group.
In 1986, Rapoport’s group (Castellino and Rapoport, 1986; Civitello and Rapoport, 1994) formally synthesized aflatoxin B2 via Oxaza–Cope rearrangement, as shown in Figure 10A. Hydroxylamine 71 and aldehyde 72 were condensed to obtain oxime compounds, which were refluxed in a sealing tube containing 3.9 M HCl in tetrahydrofuran for 24 h followed by Oxaza–Cope rearrangement, imine hydrolysis, tetrahydrofuran cleavage by chlorine, and spontaneous addition. Subsequently, one methyl sulfonyl group was removed by lithium hydroxide hydrolysis, and the C-ring was constructed under the catalysis of p-toluenesulfonic acid. The isomers 80 and 20 were then obtained in a 16:1 ratio. The formal total synthesis of aflatoxin B2 was achieved in six steps with a total yield of 2.9%.
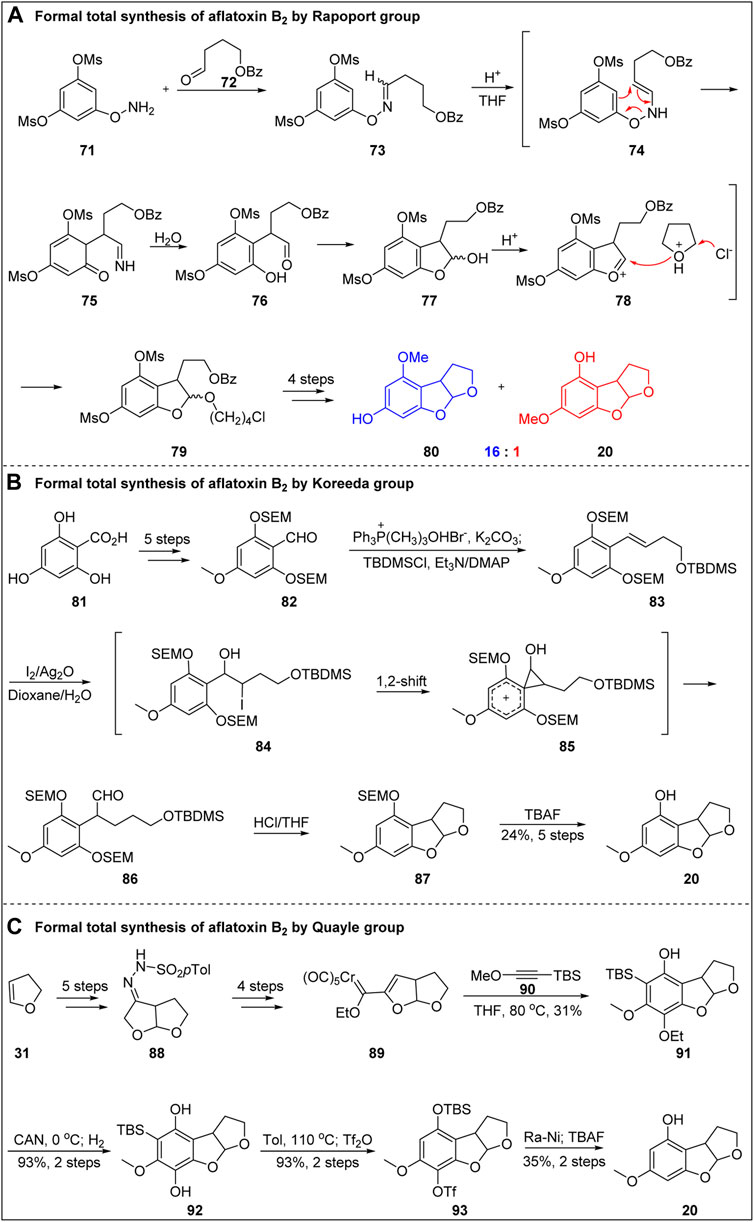
FIGURE 10. Formal total synthesis of aflatoxin B2.
In 1993, Koreeda’s group (Koreeda et al., 1993) formally synthesized aflatoxin B2 through Kikuchi rearrangement, as shown in Figure 10B. Starting from benzoic acid 81, aldehyde 82 was obtained by esterification of carboxylic acid and selective methylation of phenol, followed by protection of phenol OH with SEM, reduction of ester group and subsequent oxidation. Then, they introduced side chain compound 83 containing double bond by Wittig reaction. The key precursor 83 was subjected to Kikuchi rearrangement in iodine, silver oxide, and dioxane/water system, resulting in chain-branching 1,2-migration product 86. After the removal of protective group and tandem cyclization reaction, the construction of advanced intermediate 20 was completed. It is worth to mention that Kikuchi rearrangement, as a newly developed method, plays an important role in the formal synthesis of aflatoxin B2, which confirms that the advance of the method has important promoting value to the total synthesis.
In 2006, Quayle’s group (Eastham et al., 2006; Eastham et al., 2008) reported an efficient method for the formal total synthesis of aflatoxin B2 via Wulff-Dötz reaction, as shown in Figure 10C. Starting from the C-ring compound dihydrofuran 31, the B-ring skeleton was constructed by cobalt-mediated cyclization, and then 88 underwent a series of functional group transformations, such as ozone breaking and hydrazine to form a hydrazone, to obtain the key precursor of wulff-Dötz reaction. Then wulff-Dötz reaction of 89 with alkyneen 90 in THF was performed to obtain the construction of A-ring. Finally, the formal synthesis of aflatoxin B2 was successfully completed through simple transformation of 91.
In 1999, Kraus’ group (Kraus and Wang, 1999) reported the first formal synthesis of aflatoxin M2 via the 1,2-addition of dichloromethyl lithium to carbonyl, as shown in Figure 11. The Friedel–Crafts acylation product 96 was obtained by the reaction of isotrimethoxylbenzene 94 with propanolactone 95 under the catalysis of AlCl3. Subsequently, 96 reacted with dichloromethyl lithium to obtain the key intermediate triol 97 via 1,2-addition. The hemiacetal intermediate 98 was hydrolyzed and cyclized under the action of potassium carbonate in isopropyl solution.
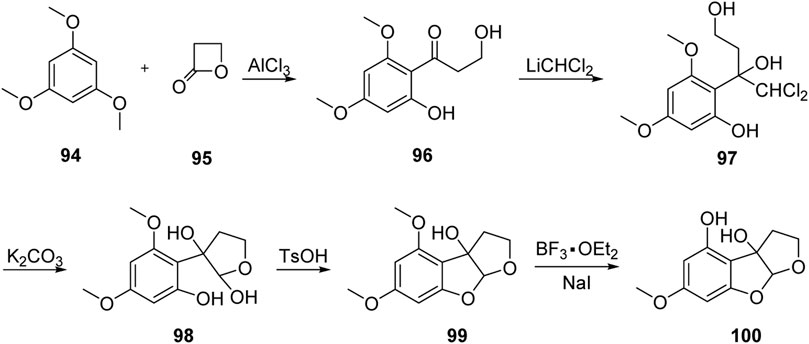
FIGURE 11. Formal total synthesis of aflatoxin M2 by Kraus group.
The B-ring was then constructed under the catalysis of TsOH. Finally, 99 was subjected to selective demethylation under the action of BF3•Et2O, and the advanced intermediate 100 was synthesized in five steps with a total yield of 26%. Later, the chemical synthesis of aflatoxin M2 was achieved based on Büchi’s method for the total synthesis of aflatoxin M1.
In 1993, the Marino group (Marino, 1993; Marino et al., 2011) described an efficient and stereoselective approach for the formal total synthesis of aflatoxin B1 with 80% ee. This approach is characterized by the [3,3]-σ rearrangement of chiral vinyl sulfoxide 103 and dichloroethylene ketone 104, as shown in Figure 12.
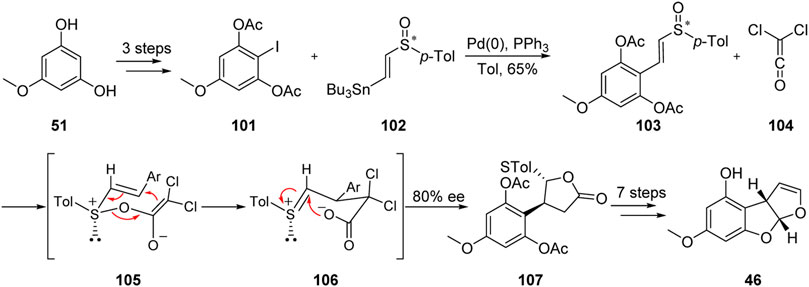
FIGURE 12. Stereoselective formal total synthesis of aflatoxin B1 by Marino group.
The key sulfoxide precursor 101 was obtained from diphenol 51 by acylation, iodization, and Stille coupling. The C-ring was then successfully constructed by [3,3]-σ rearrangement. Finally, the formal synthesis of aflatoxin B1 was successfully achieved through eight steps including deacylation and cyclization.
In 1997, Shishido’s group (Bando and Shishido, 1997) used the lipase-mediated asymmetric acetylation of prochiral diol compounds as the key strategy to accomplish the asymmetric formal synthesis of aflatoxin B2, as shown in Figure 13A. The coupling product was obtained from iodide 108 via Heck reaction. After ozonation and NaBH4 reduction, the diol 110 was obtained. After screening with a large number of lipases, the authors found that the lipase AL mediated the transfer of the ester group from Achromobacter sp., resulting in high yield with an ee of 89%. Compound 111 was then transformed into cyanide 112 by the introduction of a methyl sulfonyl group, cyanogen substitution, and deacylation. Subsequently, the B-ring was constructed via the oxidation of alcohol, the deprotection of phenol, and tandem cyclization. The benzylation of phenolic OH group, homeopathic reduction to alcohol after hydrolysis of cyanide. Next, under the action of TsOH, the C-ring was obtained through intramolecular cyclization. Finally, the asymmetric formal synthesis of aflatoxin B2 was realized via debenzylation.
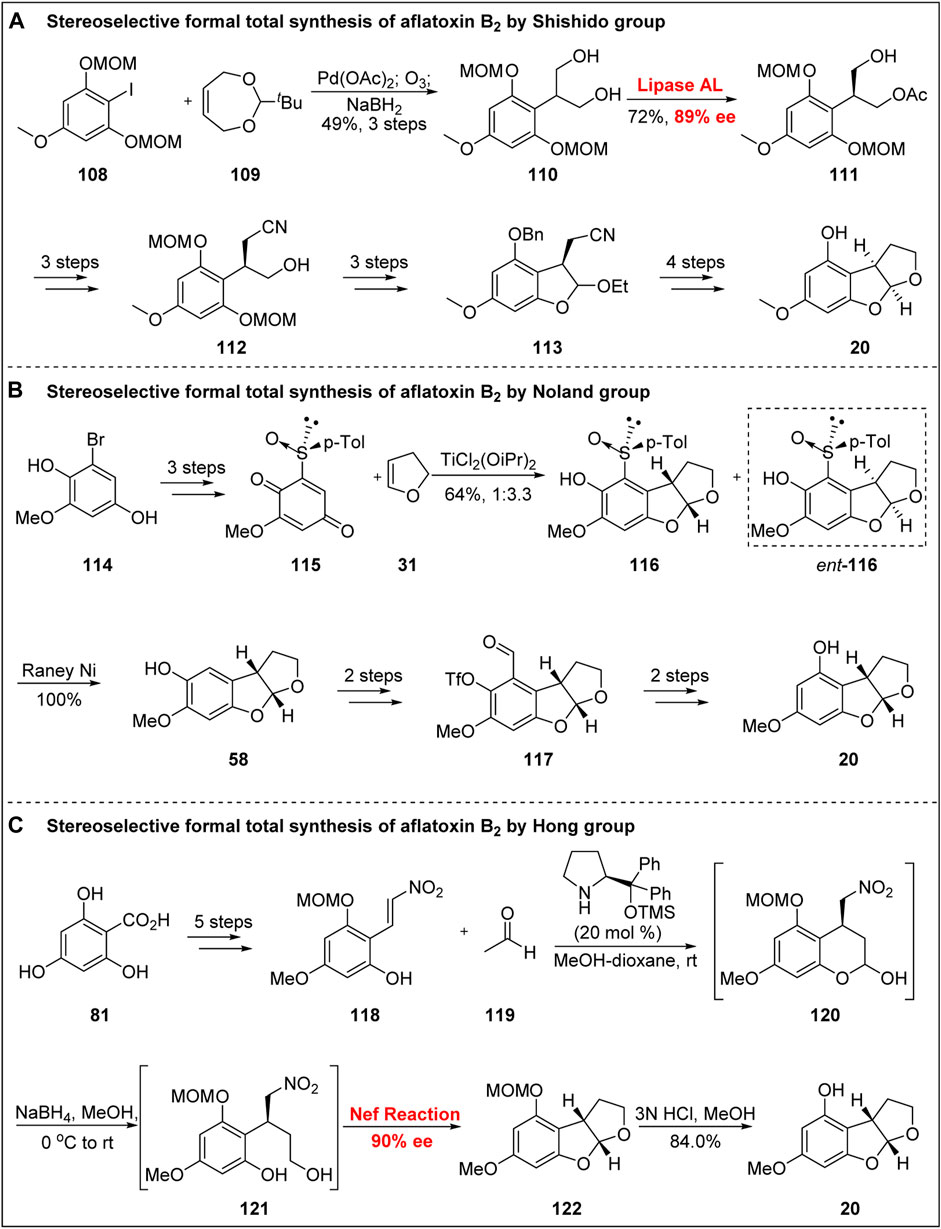
FIGURE 13. Stereoselective formal total synthesis of aflatoxin B2.
In 2000, Noland’s group (Noland and Kedrowski, 2000) reported the asymmetric formal synthesis of aflatoxin B2 using chiral sulfoxide, as shown in Figure 13B. Based on the method developed by Andersen’s group (Andersen et al., 1964), a compound containing a sulfoxide subsidiary was synthesized from the known intermediate 114 after MOM protection. Under the action of CAN, the quinone sulfoxide intermediate 115 was obtained by oxidative demethylation. Under the catalysis of the Lewis acid TiCl2(OiPr)2, compounds 116 and ent-116 were obtained in a ratio of 1:3.3, and the construction of the ABC tricyclic skeleton was realized. The sulfoxide auxiliary group was then removed by Laney nickel. After Duff reaction, the formylation product was obtained, and the phenol was esterified.
The functional group transformation of 117 was performed by Baeyer–Villiger oxidation, saponification, and reduction under the action of Laney nickel to obtain the precursor 20. Notably, their synthetic strategy and particularly the late functional group transformation provided important ideas and inspiration for the asymmetric total synthesis of aflatoxin B2 by Corey’s group (Zhou and Corey, 2005).
In 2017, Hong’s group (Huang et al., 2017) reported the concise formal total synthesis of (-)-aflatoxin B2 based on the synthesis of an advanced intermediate of 20 in seven steps using an organic-catalyzed tandem one-pot reaction (Figure 13C). Starting from benzoic acid 81, the key precursor 118 was obtained by using acetone to protect the carboxyl group and its o-phenol OH followed by selective methylation, MOM protection, reductive deprotection using DIBAL-H, and aldol reaction. Subsequently, using the organic catalytic tandem one-pot reaction developed by their own group, Hong’s group obtained the BC double-ring skeleton with excellent enantioselectivity. The specific sequence of the tandem reaction was as follows: first, under the action of Jørgensen catalyst and in the presence of acetic acid, the hemiacetal 120 was obtained with high ee; second, under the action of NaBH4, diol 121 was generated; finally, the BC double ring was constructed by Nef reaction. Subsequently, the asymmetric synthesis of 20 was completed by the removal of MOM.
Given the broad implications that aflatoxins have for public health, considerable progress has been made in the total synthesis of aflatoxins since the 1960s. During the past 55 years, three enantioselective total syntheses have been described, including the first asymmetric total synthesis of aflatoxin B1 and B2 by the groups of Trost and Corey, and the second asymmetric total synthesis of aflatoxin B2 by the group of Zu. These works represent wonderful progress in the total synthesis of aflatoxins in terms of elegance, efficiency, and environmental friendliness.
Most reported studies have focused on four types of methods. Taking the preparation of the DE ring system as an example (Figure 14), in 1966, the group of Büchi assembled the DE ring system via Pechmann condensation and Friedel–Crafts acylation in four consecutive steps with an extremely low overall yield (4.4% yield). Later, in 1971, Büchi’s group used a brominated five-membered cyclic ketone or six-membered lactone in the presence of ZnCO3 (130 equiv.) and NaHCO3 (138 equiv.) to successfully obtain the DE ring system in one step. However, it should be noted that the preparation of the brominated ketone or lactone requires four or five steps along with CCl4 and benzene as solvents, which is not ideal. In 1990, the group of Rodrigo assembled the DE ring system using a very different strategy from Büchi’s. Rodrigo’s approach involved transmetalation and addition with nine tedious consecutive steps, and both low temperature (−100°C) and high pressure (200 psi) were required. In 2019, the group of Zu developed a new Sc(OTf)3-promoted, green, facile, and economic single-pot strategy to synthesize the DE ring system of aflatoxins.
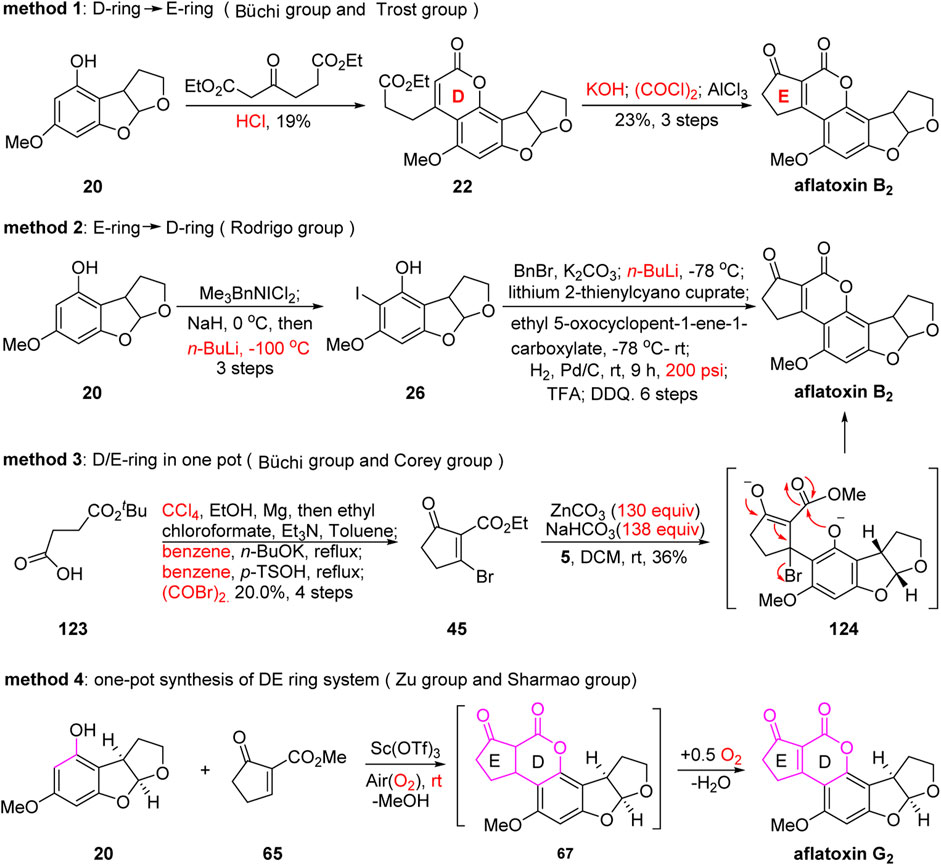
FIGURE 14. Four types of methods for the preparation of the DE ring system.
In brief, economic, facile, and green processes are being advanced as effective ways to not only form the key structural motifs of aflatoxins but also to circumvent tedious purification steps and promote environmental sustainability. Given the vital importance of this topic, the organic chemistry community should continue to invest efforts in developing new methods for the efficient total synthesis of aflatoxins.
LY conceived the review. LY collected the literatures. ZW and LY wrote the manuscript. ZW edited the manuscript. All authors read and approved the final version of the manuscript.
This work was supported by the project of the PhD research start-up funds of Qufu Normal University, China (Grant Nos. 614901, and 615201).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors, therefore, gratefully acknowledge the Qufu Normal University for the financial supports.
Andersen, K. K., Gaffield, W., Papanikolaou, N. E., Foley, J. W., and Perkins, R. I. (1964). Optically Active Sulfoxides. The Synthesis and Rotatory Dispersion of Some Diaryl Sulfoxides2. J. Am. Chem. Soc. 86, 5637–5646. doi:10.1021/ja01078a047
Asao, T., Büchi, G., Abdel-Kader, M. M., Chang, S. B., Wick, E. L., and Wogan, G. N. (1963). Aflatoxins B and G. J. Am. Chem. Soc. 85, 1706–1707. doi:10.1021/ja00894a050
Asao, T., Büchi, G., Abdel-Kader, M. M., Chang, S. B., Wick, E. L., and Wogan, G. N. (1965). The Structures of Aflatoxins B and G1. J. Am. Chem. Soc. 87, 882–886. doi:10.1021/ja01082a031
Bando, T., and Shishido, K. (1997). Enantioselective Access to the Mycotoxin, Aflatoxin B2. Synlett 1997, 665–666. doi:10.1055/s-1997-3266
Bashiry, M., Javanmardi, F., Sadeghi, E., Shokri, S., Hossieni, H., Oliveira, C. A. F., et al. (2021). The Prevalence of Aflatoxins in Commercial Baby Food Products: A Global Systematic Review, Meta-Analysis, and Risk Assessment Study. Trends Food Sci. Tech. 114, 100–115. doi:10.1016/j.tifs.2021.05.014
Bianco, G., Russo, R., Marzocco, S., Velotto, S., Autore, G., and Severino, L. (2012). Modulation of Macrophage Activity by Aflatoxins B1 and B2 and Their Metabolites Aflatoxins M1 and M2. Toxicon 59, 644–650. doi:10.1016/j.toxicon.2012.02.010
Bräse, S., Encinas, A., Keck, J., and Nising, C. F. (2009). Chemistry and Biology of Mycotoxins and Related Fungal Metabolites. Chem. Rev. 109, 3903–3990. doi:10.1021/cr050001f
Brechbühler-Bader, S., Büchi, G., and Milne, G. (1967).The Absolute Configuration of the Aflatoxins. J. Org. Chem. 32, 2641–2642. doi:10.1021/jo01283a068
Büchi, G., Foulkes, D. M., Kurono, M., Mitchell, G. F., and Schneider, R. S. (1967). Total Synthesis of Racemic Aflatoxin B1. J. Am. Chem. Soc. 89, 6745–6753. doi:10.1021/ja01001a062
Büchi, G., Foulkes, D. M., Kurono, M., and Mitchell, G. F. (1966). The Total Synthesis O Racemic Aflatoxin B1. J. Am. Chem. Soc. 88, 4534–4536. doi:10.1021/ja00971a061
Büchi, G., and Weinreb, S. M. (1969). Total Synthesis of Racemic Aflatoxin-M1 (Milk Toxin). J. Am. Chem. Soc. 91, 5408–5409. doi:10.1021/ja01047a052
Büchi, G., and Weinreb, S. M. (1971). Total Syntheses of Aflatoxins M1 and G1 and an Improved Synthesis of Aflatoxin B1. J. Am. Chem. Soc. 93, 746–752. doi:10.1021/ja00732a032
Castellino, A. J., and Rapoport, H. (1986). Syntheses of Tetrahydrofuro[2,3-B]benzofurans: a Synthesis of (.+-.)-aflatoxin B2. J. Org. Chem. 51, 1006–1011. doi:10.1021/jo00357a011
Civitello, E. R., and Rapoport, H. (1994). Synthesis of the Enantiomeric Furobenzofurans, Late Precursors for the Synthesis of (+)- and (-)-Aflatoxins B1, B2, G1, and G2. J. Org. Chem. 59, 3775–3782. doi:10.1021/jo00093a008
Eastham, S. A., Ingham, S. P., Hallett, M. R., Herbert, J., Modi, A., Morley, T., et al. (2008). Towards Aflatoxins: a Formal Synthesis of Aflatoxin B2. Tetrahedron 64, 936–948. doi:10.1016/j.tet.2007.10.114
Eastham, S. A., Ingham, S. P., Hallett, M. R., Herbert, J., Quayle, P., and Raftery, J. (2006). A Formal Synthesis of Aflatoxin B2: a Dötz Benzannulation Approach. Tetrahedron Lett. 47, 2299–2304. doi:10.1016/j.tetlet.2006.02.024
Groopman, J. D., Kensler, T. W., and Wild, C. P. (2008). Protective Interventions to Prevent Aflatoxin-Induced Carcinogenesis in Developing Countries. Annu. Rev. Public Health 29, 187–203. doi:10.1146/annurev.publhealth.29.020907.090859
Hartley, R. D., O'Kelly, J., and Nesbitt, F. (1963). Toxic Metabolites of Aspergillus Flavus. Nature 198, 1056–1058. doi:10.1038/1981056a0
Hayashi, Y. (2016). Pot Economy and One-Pot Synthesis. Chem. Sci. 7, 866–880. doi:10.1039/c5sc02913a
Horne, S., Weeratunga, G., and Rodrigo, R. (1990). The Regiospecific P-Deiodination of 2,4-Di-Iodo Phenols; a New Synthesis of Aflatoxin B2. J. Chem. Soc. Chem. Commun., 39–41. doi:10.1039/c39900000039
Huang, W.-L., Raja, A., Hong, B.-C., and Lee, G.-H. (2017). Organocatalytic Enantioselective Michael-Acetalization-Reduction-Nef Reaction for a One-Pot Entry to the Functionalized Aflatoxin System. Total Synthesis of (−)- Dihydroaflatoxin D2 and (−)- and (+)-Microminutinin. Org. Lett. 19, 3494–3497. doi:10.1021/acs.orglett.7b01473
Ismail, A., Naeem, I., Gong, Y. Y., Routledge, M. N., Akhtar, S., Riaz, M., et al. (2021). Early Life Exposure to Dietary Aflatoxins, Health Impact and Control Perspectives: A Review. Trends Food Sci. Tech. 112, 212–224. doi:10.1016/j.tifs.2021.04.002
István T. Horváth, I. T., and Anastas, P. T. (2007). Introduction: Green Chemistry. Chem. Rev. 107, 2167–2168. doi:10.1021/cr0783784
Koreeda, M., Dixon, L. A., and Hsi, J. D. (1993). An Efficient Formal Synthesis of Aflatoxin B2 by Use of the Kikuchi Rearrangement Reaction. Synlett 1993, 555–556. doi:10.1055/s-1993-22526
Kraus, G. A., and Wang, X. (1999). A Direct Synthesis of Aflatoxin M2. Tetrahedron Lett. 40, 8513–8514. doi:10.1016/s0040-4039(99)01820-1
Lee, H. S., Lindahl, J., Nguyen-Viet, H., Khong, N. V., Nghia, V. B., Xuan, H. N., et al. (2017). An Investigation into Aflatoxin M 1 in Slaughtered Fattening Pigs and Awareness of Aflatoxins in Vietnam. BMC Vet. Res. 13, 363. doi:10.1186/s12917-017-1297-8
Marino, J. P. (1993). Asymmetric Synthesis via Optically Active Vinyl Sulfoxides. Chem. 65, 667–674. doi:10.1351/pac199365040667
Marino, J. P., Kieler, K. A., and Kim, M.-W. (2011). An Enantioselective Synthesis of (−)-4-Hydroxy-6-Methoxy-3a,8a-Dihydrofuro[2,3-B]benzofuran: an Advanced Intermediate in the Synthesis of (−)-aflatoxin B1 and G1. Tetrahedron 67, 837–841. doi:10.1016/j.tet.2010.10.003
Mollayusefian, I., Ranaei, V., Pilevar, Z., Cabral-Pinto, M. M. S., Rostami, A., Nematolahi, A., et al. (2021). The Concentration of Aflatoxin M1 in Raw and Pasteurized Milk: A Worldwide Systematic Review and Meta-Analysis. Trends Food Sci. Tech. 115, 22–30. doi:10.1016/j.tifs.2021.06.033
Newhouse, T., Baran, P. S., and Hoffmann, R. W. (2009). The Economies of Synthesis. Chem. Soc. Rev. 38, 3010–3021. doi:10.1039/b821200g
Noland, W. E., and Kedrowski, B. L. (2000). Quinone Approaches toward the Synthesis of Aflatoxin B2. Org. Lett. 2, 2109–2111. doi:10.1021/ol006014r
Paymode, D. J., and Sharma, I. (2021). Rhodium‐Catalyzed [3+2]‐Annulation of Ortho‐ Diazoquinones with Enol Ethers: Diversity‐Oriented Total Synthesis of Aflatoxin B2. Eur. J. Org. Chem. 2021, 2034–2040. doi:10.1002/ejoc.202100186
Roberts, J. C., Sheppard, A. H., Knight, J. A., and Roffey, P. (1968). Studies in Mycological Chemistry. Part XXII. Total Synthesis of (±)-Aflatoxin-B2. J. Chem. Soc. C, 22–24. doi:10.1039/j39680000022
Roze, L. V., Hong, S.-Y., and Linz, J. E. (2013). Aflatoxin Biosynthesis: Current Frontiers. Annu. Rev. Food Sci. Technol. 4, 293–311. doi:10.1146/annurev-food-083012-123702
EFSA Panel on Contaminants in the Food Chain (CONTAM) Schrenk, D., Bignami, M., Bodin, L., Chipman, J. K., Mazo, J. D., Grasl-Kraupp, B., et al. (2020). Risk Assessment of Aflatoxins in Food. EFSA J. 18, e06040. doi:10.2903/j.efsa.2020.6040
Sloan, C. P., Cuevas, J. C., Quesnelle, C., and Snieckuss, V. (1988). Aryl Radical-Induced Cyclization Routes to Furo[2,3-B]benzofurans. Abbreviated Formal Syntheses of Aflatoxins B1 and B2. Tetrahedron Lett. 29, 4685–4686. doi:10.1016/s0040-4039(00)80580-8
Trost, B. M., and Toste, F. D. (2003). Palladium Catalyzed Kinetic and Dynamic Kinetic Asymmetric Transformations of γ-Acyloxybutenolides. Enantioselective Total Synthesis of (+)-Aflatoxin B1 and B2a. J. Am. Chem. Soc. 125, 3090–3100. doi:10.1021/ja020988s
Trost, B. (1991). The Atom Economy-A Search for Synthetic Efficiency. Science 254, 1471–1477. doi:10.1126/science.1962206
Wang, Y., Wenjuan, T., Wang, C. C., and Leung, L. K. (2015). Aflatoxin B1 Augments the Synthesis of Corticotropin Releasing Hormone in JEG-3 Placental Cells. Chemico-Biological Interactions 237, 73–79. doi:10.1016/j.cbi.2015.05.015
Wang, Z., and Zu, L. (2019). Organocatalytic Enantioselective Direct Alkylation of Phloroglucinol Derivatives: Asymmetric Total Synthesis of (+)-aflatoxin B2. Chem. Commun. 55, 5171–5174. doi:10.1039/c9cc01833f
Weeratunga, G., Horne, S., and Rodrigo, R. (1988). A Formal Synthesis of Aflatoxin B2. J. Chem. Soc. Chem. Commun., 721–722. doi:10.1039/c39880000721
Wender, P. A., Verma, V. A., Paxton, T. J., and Pillow, T. H. (2008). Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res. 41, 40–49. doi:10.1021/ar700155p
Keywords: aflatoxins, natural products, contaminants, total syntheses, formal syntheses
Citation: Yang L and Wang Z (2021) Advances in the Total Synthesis of Aflatoxins. Front. Chem. 9:779765. doi: 10.3389/fchem.2021.779765
Received: 19 September 2021; Accepted: 02 November 2021;
Published: 30 November 2021.
Edited by:
Essa M. Saied, Humboldt UnivSAersity of Berlin, GermanyReviewed by:
Otniel Freitas-Silva, Embrapa Food agroindustry, BrazilCopyright © 2021 Yang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhonglei Wang, d2FuZ3psMTZAdHNpbmdodWEub3JnLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.