 Walter Orellana1
Walter Orellana1 Ricardo Pino-Rios2
Ricardo Pino-Rios2 Osvaldo Yañez3,4
Osvaldo Yañez3,4 Alejandro Vásquez-Espinal5
Alejandro Vásquez-Espinal5 Francesca Peccati6
Francesca Peccati6 Julia Contreras-García7
Julia Contreras-García7 Carlos Cardenas8,9
Carlos Cardenas8,9 William Tiznado5*
William Tiznado5*- 1Departamento de Ciencias Físicas, Universidad Andres Bello, Santiago, Chile
- 2Laboratorio de Química Teórica, Facultad de Química y Biología, Universidad de Santiago de Chile (USACH), Santiago, Chile
- 3Center of New Drugs for Hypertension (CENDHY), Santiago, Chile
- 4Department of Pharmaceutical Science and Technology, School of Chemical and Pharmaceutical Sciences, Universidad de Chile, Santiago, Chile
- 5Departamento de Ciencias Químicas, Computational and Theoretical Chemistry Group, Facultad de Ciencias Exactas, Universidad Andres Bello, Santiago, Chile
- 6Center for Cooperative Research in Biosciences (CIC bioGUNE), Basque Research and Technology Alliance (BRTA), Bizkaia Technology Park, Derio, Spain
- 7Sorbonne Universités, UPMC and CNRS, Laboratoire de Chimie Théorique (LCT), 75005, Paris, France
- 8Departamento de Física, Facultad de Ciencias, Universidad de Chile, Santiago, Chile
- 9Centro para el Desarrollo de la Nanociencias y Nanotecnologia, CEDENNA, Avenida Ecuador, Santiago, Chile
We computationally explore an alternative to stabilize one-dimensional (1D) silicon-lithium nanowires (NWs). The Li12Si9 Zintl phase exhibits the NW
Introduction
The insertion of inorganic materials into single-walled carbon nanotubes (SWCNTs), hereinafter identified simply as CNT, enables the encapsulation of extreme nanowires (NWs) with diameters comparable to a unit cell of the parent material (Green, 1998; Sloan et al., 2002; Spencer et al., 2014). Although NWs of similar diameter can be produced using several templates, such as zeolites (Derouane, 1998), mesoporous phases (Alba-Simionesco et al., 2006; Ke et al., 2009), and metal-organic framework (MOF) (Lu et al., 2012) type materials, CNTs present many advantages as templates; they are atomically smooth, electron transparent, readily available, and can be filled by bulk infiltration to create milligram quantities of encapsulated nanowires, at least on a laboratory scale. Thus, encapsulated NW-CNT are scientifically interesting not only on their own but also as precursors to a wide range of other extreme nanowire materials.
In 2016, Ivanov et al. published a theoretical prediction of helix-shaped lithium-phosphorus nanowires encapsulated into single-walled carbon nanotubes (LiP@CNTs) (Ivanov et al., 2016). Note that helix-shaped LinPn clusters (n = 5–9) had previously been reported as global minimum structures (Ivanov et al., 2012). Some solid phases consist of structural motifs like atomic clusters, i.e., in Zintl phases. This connection brings consistency to the use of models based on stable clusters to generate NWs inside nanotubes, as proposed in Ivanov’s work (Ivanov et al., 2012). The study of these clusters inside CNTs can provide relevant information about these hybrid materials, for example, about their viability (stability analysis), their structural characteristics (geometry analysis), their physical and chemical properties (analysis of their electronic structure).
Due to its excellent energy storage capacity, Si has been extensively studied experimentally as a negative electrode material for Li-ion batteries (Gao et al., 2001; Ryu et al., 2004; Li et al., 2006, Li et al., 2008, Li et al., 2009; Obrovac et al., 2007; Song et al., 2014; Shin et al., 2020). Hence, Si lithifies at high temperature (415°C) in a LiCl-KCl melt, identifying potential plateaus evidencing the crystalline phases Li12Si7, Li7Si3, Li13Si4, and Li22Si5 (Wen and Huggins, 1981). In particular, the binary (non-paramagnetic) Zintl-type Li12Si7 silicide contains semi-infinite sandwich-like
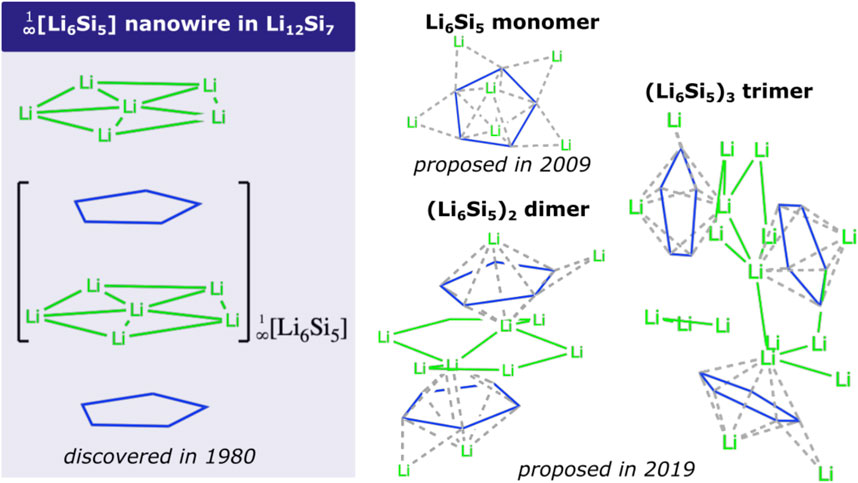
Scheme 1. Structures of
To build new class of materials with desirable properties, using atomic clusters instead of atoms as building blocks, is a remarkable possibility. However, it requires that atomic clusters must retain their identity when assembled, as Khanna and Jena first outlined when they coined the word “cluster-assembled materials” (CAMs) (Khanna and Jena, 1992). These authors argued that the clusters’ coupling would have a unique effect on both the assembled material’s electronic structure and mechanical properties, which is not possible when the assembly blocks are atoms (Khanna and Jena, 1995; Jena et al., 1996; Jena and Khanna, 1996; Claridge et al., 2009; Jena and Sun, 2018). For a more detailed and timely overview of advances in the assembly of materials from clusters, please refer to the following reviews: (Chakraborty and Pradeep, 2017; Jena and Sun, 2018; Pinkard et al., 2018; Zheng et al., 2019; Doud et al., 2020).
Given the above background, here we evaluated, in silico, the stability of the isolated
Computational Methods
In the finite models (clusters), geometry optimizations and frequency calculations were performed at the PBE0 (Adamo and Barone, 1999)/Def2TZVP(Weigend and Ahlrichs, 2005) level with the Gaussian 16 program (M. J. Frisch, G. W. Trucks, H. B. Schlegel et al., 2016).
For the solid-state study, we performed first-principles calculations based on DFT (Sham and Kohn, 1966; Kohn et al., 1996) as implemented in the Vienna Ab Initio Simulation Package (VASP) (Kresse and Furthmüller, 1996). The exchange-correlation energies were calculated at PBE-D3 level (Ernzerhof and Perdew, 1998; Grimme et al., 2010). Plane-wave basis set with a kinetic energy cutoff of 400 eV, and the projector augmented-wave method for the core-valence interaction was employed (Blöchl et al., 1994). The
Results and Discussion
Finite Model Tests to Estimate the Optimal Width of SWNTS
The first question that arises is which is the optimal SWCNT width to favor the
In the case of small [n]cyclacenes (n = 13–15), Li7Si5+ cluster undergoes noticeable changes in the optimization process due to the confinement effects. In contrast, when [n]cyclocenes with n = 16–20 are used, the Li7Si5+ cluster maintains its structure at the end of the optimization process, leading to the best interaction energy, [Eint = E (Li7Si5+at[n]cyclacene)-(E (Li7Si5)+E ([n]cyclacene))], with [16]cyclacene (-70.1 kcal mol−1 at PBE0/Def2TZVP level). Since this analysis is only a reference for estimating the most suitable nanotube diameters to explore in the periodic calculations, we have not included basis set superposition error (BSSE) corrections. The most stable structures, as well as the Eint, are shown in Figure 1. The structures for the other complexes are shown in Supplementary Figure S1 and their Cartesian coordinates in Supplementary Table S1. These results guided us to use CNTs with diameters in the range of 11–14 Å in next steps of our research.
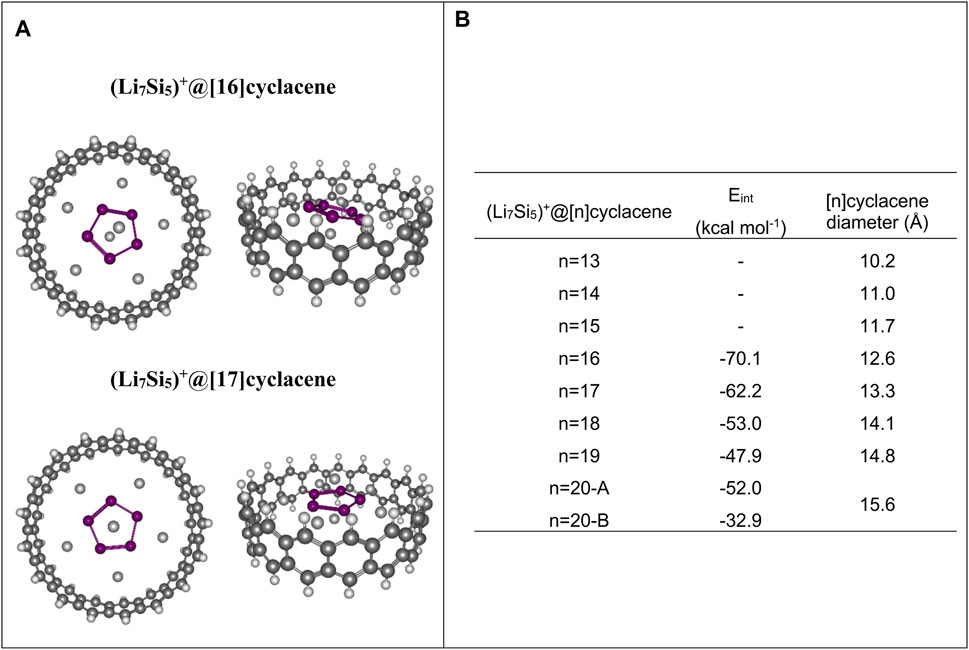
FIGURE 1. (A) Top-and side-views of optimized structures of Li7Si5+ inside both [16]cyclacene and [17]cyclacene at PBE0/def2-TZVP level. (B) Li7Si5+ and [n]cyclacene interaction energy (Eint), number of hexagonal rings and diameter (in Å) of the [n]cyclacene.
Insights on the stability of free
We first studied the stability of periodically repeated Li6Si5 units (Li6Si5-NW), which are stacked along the z direction, forming a one-dimensional structure, as shown in Figure 2.
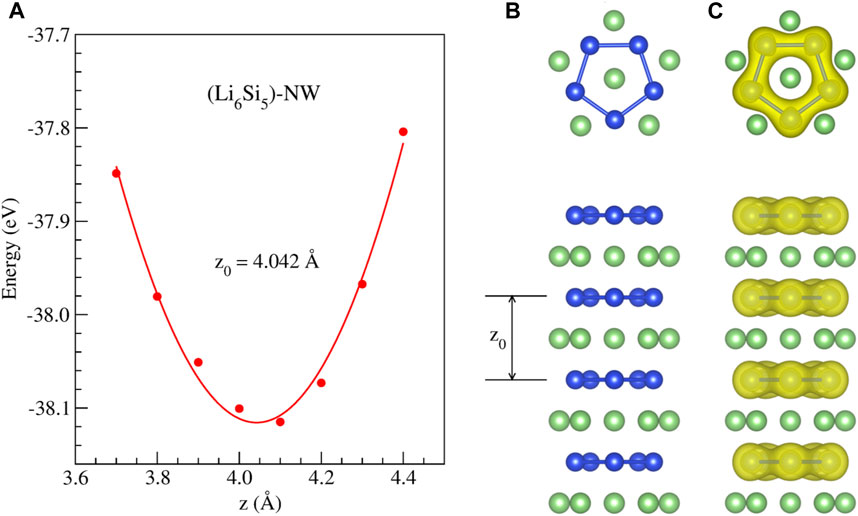
FIGURE 2. Stability of the
We found a stable structure with an equilibrium distance between Si5 rings of 4.04 Å which is the periodicity of the Li6Si5 unit cell. In the equilibrium geometry, the distance between Li atoms of the border is 3.33 Å while the distance with respect to the center one is 2.83 Å. The Si-Si distance between neighboring atoms is 2.37 Å, very close to the ones in Li6Si5 monomer (between 2.30 and 2.35 Å). Our computations of the electronic band structure of the Li6Si5-NW in the primitive unit cell suggest a metallic character (see Figure 3). Note that the bandgaps of Li12Si7 was reported from conductivity-temperature experimental measurements and found to be 0.6 eV (Nesper et al., 1986b).
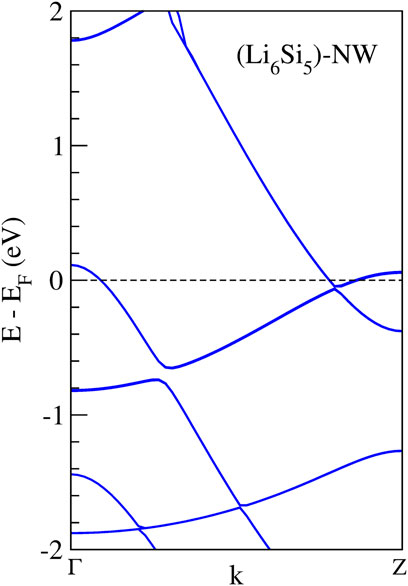
FIGURE 3. Band structures calculation of the Li6Si5-NW in the unit cell. The dashed line indicates the Fermi energy.
The stability of the Li6Si5-NW was also verified by BO-AIMD simulations at 300 K and 500K, during a simulation time of 10 ps. The simulation was performed by considering four Li6Si5 units in the periodic unit cell, as shown in Figure 4. We observe that at 500 K the Si5Li6 NW preserves its stability, showing energy fluctuations of around 2 eV. Supporting information contains short movies extracted from the BO-AIMD simulations.
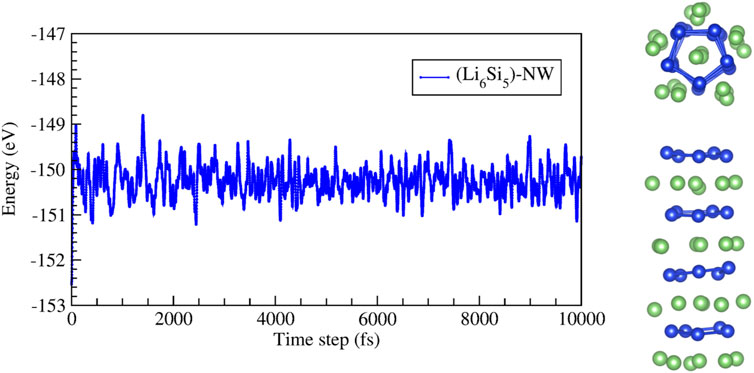
FIGURE 4. Energy as a function of time for the molecular dynamic simulations at 500 K of the infinite Si5Li6 nanowire. The unit cell for this simulation considers four Si5Li6 units. The right images show top and side views of a snapshot taken at 10,000 fs
Stability of the Li6Si5-NW Inside the CNTs
Next, we studied a finite Li6Si5 structure (f-Li6Si5) in the free space and encapsulated it inside both armchair and zigzag carbon nanotubes (f-Li6Si5@CNT). For the f-Li6Si5 structure, we consider four Si5 rings surrounded by five Li6 moieties. BO-AIMD simulations provide insights on the stability of the f-Li6Si5 system in the free space at 300 K and 500 K. We find that at 300 K, the f-Li6Si5 structure preserves its stability. Still, at 500 K, it tends to form Si-Si bonds between adjacent Si rings without losing its one-dimensional array, as shown in Figure 5. However, it is important to note that this model does not have the exact stoichiometry of NW because, to maintain symmetry, an extra Li6 unit group is added, i.e., [(Li6)5(Si5)4].
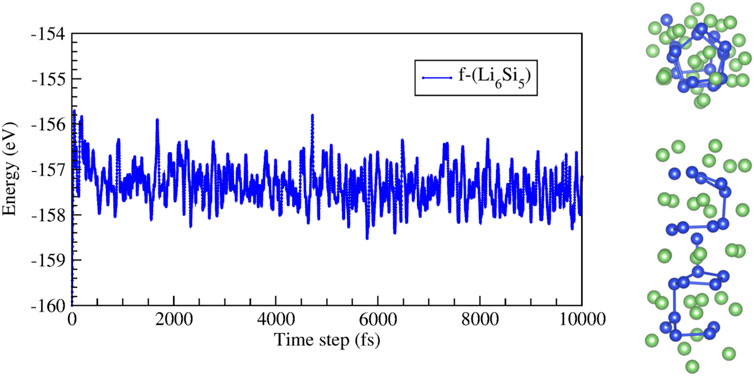
FIGURE 5. Energy as a function of time for molecular dynamic simulations at 500 K for the finite Li6Si5 structure (f-Li6Si5). The right images show top and side views of a snapshot taken at 10,000 fs
To study the f-Li6Si5 structure inside the CNTs, we consider three armchair CNTs with chiral indexes of (8,8), (9,9), and (10,10), and three zigzag CNTs with chiral indexes of (14,0), (15,0), and (16,0). With this choice, we seek to find the CNT size that best accommodates the Li6Si5-NW inside. Note that we selected these CNTs according to our preliminary findings from the Li7Si5+@[n]cyclacene model, suggesting diameters between 12 and 15 Å. The CNTs were simulated with periodic boundary conditions along its axis with a periodicity of z0 = 30 Å. The latter allows a vacuum region for the encapsulated f-Li6Si5 structure of 14 Å, allowing the atomic movement inside the CNT. Next, we calculate the
where
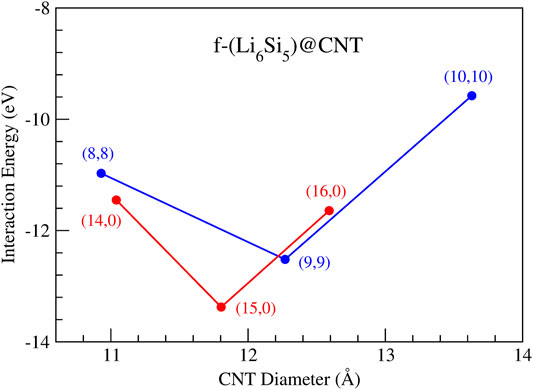
FIGURE 6. Interaction energy as a function of the nanotube diameters for the finite Li6Si5 structures inside armchair CNTs with chiral indexes (8,8), (9,9), and (10,10) and zigzag CNTs with chiral indexes (14,0), (15,0), and (16,0).
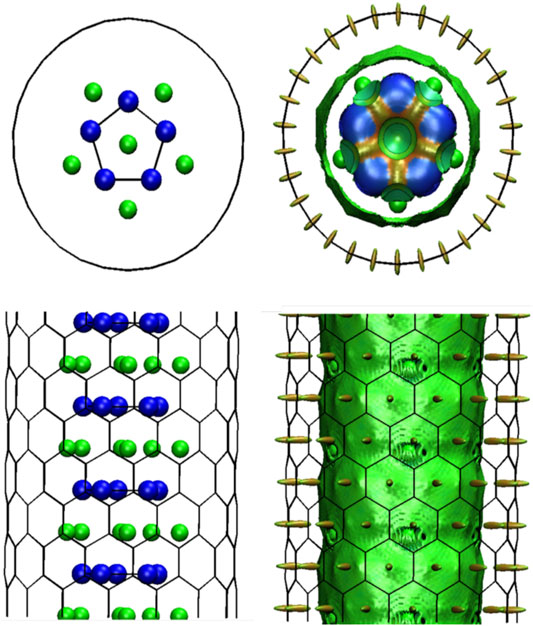
FIGURE 7. f-Li6Si5@NTC optimized structure, for zigzag CNTs with chiral indexes (15,0), with NCI surfaces (s = 0.3, color range: −0.03 to 0.03 a.u.). Geometries and electron density were taken from solid state computations.
We also calculate the band structure of the Li6Si5-NW inside the zigzag (15,0) CNT. It is important to note that the unit cells of the Li6Si5-NW and the CNT have a mismatch of 5.7%, which means that the Li6Si5-NW is not in its equilibrium geometry in the Li6Si5-NW@CNT unit cell, where the distance between the Si5 rings increases by 0.23 Å. However, this mismatch is relatively small and should not affect the electronic properties of the system. For the isolated CNT we find a small bandgap of 0.02 eV as shown in Figure 8A, which is in good agreement with the measured value of 0.029 ± 0.004 eV (Ouyang et al., 2001). Whereas the Li6Si5-NW@CNT system exhibits metallic properties as shown Figure 8B, suggesting that the Li6Si5-NW would preserve its electronic properties inside the CNT as can be compared with Figure 3.
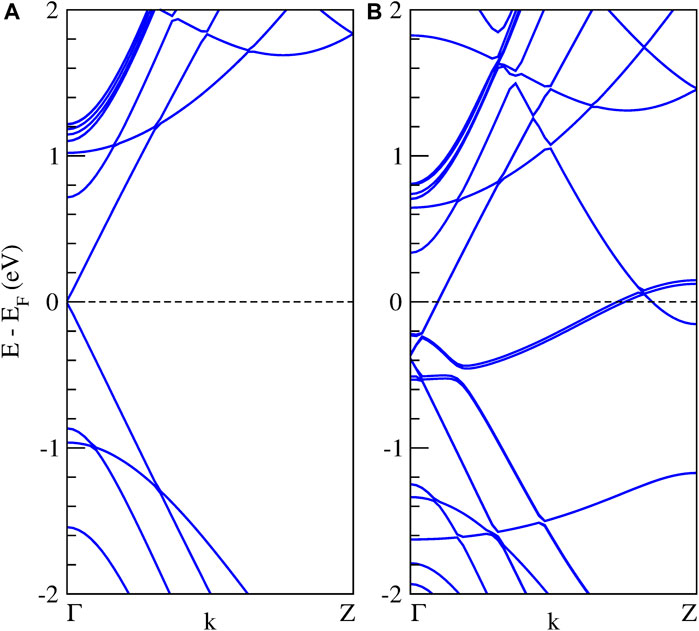
FIGURE 8. Band structure calculations of the infinite Li6Si5-NW inside the zigzag carbon nanotubes, (A) the isolated (15,0) CNT, and (B) the Li6Si5-NW@CNT system. The dashed line indicates the Fermi energy.
Finally, we study the stability of the f-Li6Si5 structure inside both zigzag (15,0) and armchair (9,9) CNTs by performing BO-AIMD simulations. Figure 9A shows the equilibrium geometry of the f-Si5Li6 structure inside the (15,0) CNT. We find that the structure remains almost unchanged with respect to f-Si5Li6 in the free space, showing that the CNT would have a small influence in the Li6Si5 NW stability. We only note a small displacement of the Li ions at the extreme of the f-Li6Si5 structure which move toward the CNT wall. The integrity of the f-Li6Si5 structure inside the (15,0) and (9,9) CNTs was investigated by BO-AIMD simulations at 300 K. We find that the f-Si5Li6 structure preserves its stability where the Li ions move around the Si5 ring without detaching. Similar results are found for the f-Li6Si5 structure inside the armchair (9,9) CNT, indicating that the formation and stability of the Li6Si5 NW inside the CNTs is independent of its chirality. This result suggests that Li6 Si5-NW are likely to form inside CNTs in a compact form, which would allow efficient storage of Li ions into CNTs mediated by Si5 rings. Supplementary Figure S2 shows the variation of the total energy for the BO-AIMD simulation of f-Li6Si5 inside the (15,0) and (9,9) CNTs at 300 K. We observe energy fluctuation of around 5 eV in both CNTs, preserving the stability of the f-Li6Si5 structure.
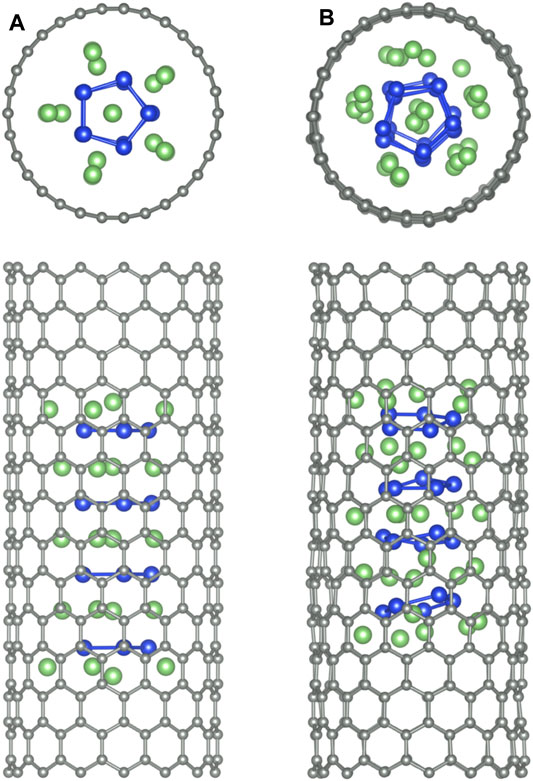
FIGURE 9. The f-Li6Si5 structure inside a zigzag (15,0) CNT. (A) Top and side views of the equilibrium geometry at 0 K. (B) Top and side views of a snapshot taken at 2000 fs of BO-AIMD simulation at 300 K. The periodicity of the CNT along its axis is z0 = 30 Å.
Conclusion
Using periodic DFT calculations and Born-Oppenheimer ab initio molecular dynamic simulations, we have shown that Li6Si5 units can be stacked one above the other, forming a one-dimensional structure linked together by Coulomb interactions. This study complements previous findings, where we demonstrated that Li6Si5, (Li6Si5)2, and (Li6Si5)3 lowest energy structures contain one, two, and three Si56- aromatic rings stabilized by Li+ counterions. Additionally, the
We also found that finite (Li6Si5)4 systems are stable inside both armchair and zigzag carbon nanotubes of around 12 Å in diameter, preserving its stability at room temperature, supporting the viable formation of Li6Si5-NW inside the CNTs. Interestingly, the Li6Si5-NW@CNTs hybrid nanocomposite maintains the metallic character. Finally, in the Li6Si5-NW, the Li6Si5 units are connected by strong electrostatic interactions (Si56- aromatic pentagons intercalated with the Li66+ moiety) in agreement with the Zintl ion concept. In the [Li6Si5-NW]@CNTs, NCI predicts that Li6Si5-NW interacts with the CNT walls by van der Waals interactions Frisch et al., 2016.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
We thank the financial support of National Agency for Research and Development (ANID) through ECOS170045, FONDECYT projects 1211128 (W.T.), 1181121 (C.C.), and 1170480 (W.O.). FONDECYT Postdoctorado 3180119 (R.P.-R.), ANID/PIA ACT192144 (O.Y.). Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02) of the Universidad de Chile. Computational resources for periodic DFT calculations and BO-AIMD simulations were provided by Fenix HCP of the Universidad Andres Bello. C.C. acknowledges Center for the Development of Nanoscience and Nanotechnology CEDENNA AFB180001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.767421/full#supplementary-material
References
Adamo, C., and Barone, V. (1999). Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 110, 6158–6170. doi:10.1063/1.478522
Alba-Simionesco, C., Coasne, B., Dosseh, G., Dudziak, G., Gubbins, K. E., Radhakrishnan, R., et al. (2006). Effects of Confinement on Freezing and Melting. J. Phys. Condens. Matter 18, R15–R68. doi:10.1088/0953-8984/18/6/r01
Blöchl, P. E., Jepsen, O., and Andersen, O. K. (1994). Improved Tetrahedron Method for Brillouin-Zone Integrations. Phys. Rev. B 49, 16223–16233. doi:10.1103/physrevb.49.16223
Chakraborty, I., and Pradeep, T. (2017). Atomically Precise Clusters of noble Metals: Emerging Link between Atoms and Nanoparticles. Chem. Rev. 117, 8208–8271. doi:10.1021/acs.chemrev.6b00769
Chevrier, V. L., Zwanziger, J. W., and Dahn, J. R. (2010). First Principles Study of Li-Si Crystalline Phases: Charge Transfer, Electronic Structure, and Lattice Vibrations. J. Alloys Compd. 496, 25–36. doi:10.1016/j.jallcom.2010.01.142
Claridge, S. A., Castleman, A. W., Khanna, S. N., Murray, C. B., Sen, A., and Weiss, P. S. (2009). Cluster-assembled Materials. ACS Nano 3, 244–255. doi:10.1021/nn800820e
Contreras, M., Osorio, E., Ferraro, F., Puga, G., Donald, K. J., Harrison, J. G., et al. (2013). Isomerization Energy Decomposition Analysis for Highly Ionic Systems: Case Study of Starlike E5Li7+Clusters. Chem. Eur. J. 19, 2305–2310. doi:10.1002/chem.201203329
Contreras-García, J., Johnson, E. R., Keinan, S., Chaudret, R., Piquemal, J.-P., Beratan, D. N., et al. (2011). NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theor. Comput. 7, 625–632. doi:10.1021/ct100641a
Derouane, E. G. (1998). Zeolites as Solid solvents1Paper Presented at the International Symposium `Organic Chemistry and Catalysis' on the Occasion of the 65th Birthday of Prof. H. Van Bekkum, Delft, Netherlands, 2-3 October 1997.1. J. Mol. Catal. A: Chem. 134, 29–45. doi:10.1016/s1381-1169(98)00021-1
Doud, E. A., Voevodin, A., Hochuli, T. J., Champsaur, A. M., Nuckolls, C., and Roy, X. (2020). Superatoms in Materials Science. Nat. Rev. Mater. 5, 371–387. doi:10.1038/s41578-019-0175-3
Ernzerhof, M., and Perdew, J. P. (1998). Generalized Gradient Approximation to the Angle- and System-Averaged Exchange Hole. J. Chem. Phys. 109, 3313–3320. doi:10.1063/1.476928
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scalmani, G., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, Inc.Revis. B.01. (Wallingford CT: Gaussian).
Gao, B., Sinha, S., Fleming, L., and Zhou, O. (2001). Alloy Formation in Nanostructured Silicon. Adv. Mater. 13, 816–819. doi:10.1002/1521-4095(200106)13:11<816:aid-adma816>3.0.co;2-p
Green, M. H. (1998). The Opening and Filling of Single Walled Carbon Nanotubes (SWTs). Chem. Commun. (3), 347–348. doi:10.1039/a707632k
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Hückel, E. (1931a). Quanstentheoretische Beitrage Zum Benzolproblem. Z. Physik 72, 310–337. doi:10.1007/BF01341953
Hückel, E. (1931b). Quantentheoretische Beitrage Zum Benzolproblem. Z. Physik 70, 204–286. doi:10.1007/BF01339530
Hückel, E. (1930). Zur Quantentheorie der Doppelbindung. Z. Physik 60, 423–456. doi:10.1007/BF01341254
Ivanov, A. S., Kar, T., and Boldyrev, A. I. (2016). Nanoscale Stabilization of Zintl Compounds: 1D Ionic Li-P Double helix Confined inside a Carbon Nanotube. Nanoscale 8, 3454–3460. doi:10.1039/c5nr07713c
Ivanov, A. S., Morris, A. J., Bozhenko, K. V., Pickard, C. J., and Boldyrev, A. I. (2012). Inorganic Double-Helix Structures of Unusually Simple Lithium-Phosphorus Species. Angew. Chem. Int. Ed. 51, 8330–8333. doi:10.1002/anie.201201843
Jena, P., and Khanna, S. N. (1996). Physics of Cluster Assembled Materials. Mater. Sci. Eng. A 217-218, 218–222. doi:10.1016/s0921-5093(96)10361-0
Jena, P. K., Khanna, S. N., and Rao, B. K. (1996). Stability and Electronic Structure of Cluster Assembled Materials. Msf 232, 1–26. doi:10.4028/www.scientific.net/msf.232.1
Jena, P., and Sun, Q. (2018). Super Atomic Clusters: Design Rules and Potential for Building Blocks of Materials. Chem. Rev. 118, 5755–5870. doi:10.1021/acs.chemrev.7b00524
Johnson, E. R., Keinan, S., Mori-Sánchez, P., Contreras-García, J., Cohen, A. J., and Yang, W. (2010). Revealing Noncovalent Interactions. J. Am. Chem. Soc. 132, 6498–6506. doi:10.1021/ja100936w
Ke, J., Su, W., Howdle, S. M., George, M. W., Cook, D., Perdjon-Abel, M., et al. (2009). Electrodeposition of Metals from Supercritical Fluids. Proc. Natl. Acad. Sci. 106, 14768–14772. doi:10.1073/pnas.0901986106
Khanna, S. N., and Jena, P. (1992). Assembling Crystals from Clusters. Phys. Rev. Lett. 69, 1664–1667. doi:10.1103/physrevlett.69.1664
Khanna, S. N., and Jena, P. (1995). Atomic Clusters: Building Blocks for a Class of Solids. Phys. Rev. B 51, 13705–13716. doi:10.1103/physrevb.51.13705
Kohn, W., Becke, A. D., and Parr, R. G. (1996). Density Functional Theory of Electronic Structure. J. Phys. Chem. 100, 12974–12980. doi:10.1021/jp960669l
Köster, T. K.-J., Salager, E., Morris, A. J., Key, B., Seznec, V., Morcrette, M., et al. (2011). Resolving the Different Silicon Clusters in Li12Si7 by 29Si and 6,7Li Solid-State NMR Spectroscopy. Angew. Chem. Int. Ed. 50, 12591–12594. doi:10.1002/anie.201105998
Kresse, G., and Furthmüller, J. (1996). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186. doi:10.1103/physrevb.54.11169
Kuhn, A., Sreeraj, P., Pöttgen, R., Wiemhöfer, H.-D., Wilkening, M., and Heitjans, P. (2011a). Li Ion Diffusion in the Anode Material Li12Si7: Ultrafast quasi-1D Diffusion and Two Distinct Fast 3D Jump Processes Separately Revealed by 7Li NMR Relaxometry. J. Am. Chem. Soc. 133, 11018–11021. doi:10.1021/ja2020108
Kuhn, A., Sreeraj, P., Pöttgen, R., Wiemhöfer, H.-D., Wilkening, M., and Heitjans, P. (2011b). Li NMR Spectroscopy on Crystalline Li12Si7: Experimental Evidence for the Aromaticity of the Planar Cyclopentadienyl-Analogous Si56− Rings. Angew. Chem. Int. Ed. 50, 12099–12102. doi:10.1002/anie.201105081
Li, J., Christensen, L., Obrovac, M. N., Hewitt, K. C., and Dahn, J. R. (2008). Effect of Heat Treatment on Si Electrodes Using Polyvinylidene Fluoride Binder. J. Electrochem. Soc. 155, A234. doi:10.1149/1.2830545
Li, J., Lewis, R. B., and Dahn, J. R. (2006). Sodium Carboxymethyl Cellulose: A Potential Binder for Si Negative Electrodes for Li-Ion Batteries. Electrochem. Solid State. Lett. 10, A17. doi:10.1149/1.2398725
Li, J., Smith, A., Sanderson, R. J., Hatchard, T. D., Dunlap, R. A., and Dahn, J. R. (2009). In Situ [sup 119]Sn Mössbauer Effect Study of the Reaction of Lithium with Si Using a Sn Probe. J. Electrochem. Soc. 156, A283. doi:10.1149/1.3073879
Lu, G., Li, S., Guo, Z., Farha, O. K., Hauser, B. G., Qi, X., et al. (2012). Imparting Functionality to a Metal-Organic Framework Material by Controlled Nanoparticle Encapsulation. Nat. Chem 4, 310–316. doi:10.1038/nchem.1272
Manrique-de-la-Cuba, M. F., Leyva‐Parra, L., Inostroza, D., Gomez, B., Vásquez‐Espinal, A., Garza, J., et al. (2021). Li8Si8, Li10Si9, and Li12Si10: Assemblies of Lithium‐Silicon Aromatic Units. ChemPhysChem 22 (10), 906–910. doi:10.1002/cphc.202001051
Monkhorst, H. J., and Pack, J. D. (1976). Special Points for Brillouin-Zone Integrations. Phys. Rev. B 13, 5188–5192. doi:10.1103/physrevb.13.5188
Nesper, R., Curda, J., and Von Schnering, H. G. (1986a). Li8MgSi6, a Novel Zintl Compound Containing Quasi-Aromatic Si5 Rings. J. Solid State. Chem. 62, 199–206. doi:10.1016/0022-4596(86)90232-x
Nesper, R. (1990). Structure and Chemical Bonding in Zintl-Phases Containing Lithium. Prog. Solid State. Chem. 20, 1–45. doi:10.1016/0079-6786(90)90006-2
Nesper, R., von Schnering, H. G., and Curda, J. (1986b). Li12Si7, eine Verbindung mit trigonal-planaren Si4-Clustern und isometrischen Si5-Ringen. Chem. Ber. 119, 3576–3590. doi:10.1002/cber.19861191207
Obrovac, M. N., Christensen, L., Le, D. B., and Dahn, J. R. (2007). Alloy Design for Lithium-Ion Battery Anodes. J. Electrochem. Soc. 154, A849. doi:10.1149/1.2752985
Ouyang, M., Huang, J.-L., Cheung, C. L., and Lieber, C. M. (2001). Energy Gaps in "Metallic" Single-Walled Carbon Nanotubes. Science 292, 702–705. doi:10.1126/science.1058853
Perez-Peralta, N., Contreras, M., Tiznado, W., Stewart, J., Donald, K. J., and Merino, G. (2011). Stabilizing Carbon-Lithium Stars. Phys. Chem. Chem. Phys. 13, 12975–12980. doi:10.1039/C1CP21061K
Pinkard, A., Champsaur, A. M., and Roy, X. (2018). Molecular Clusters: Nanoscale Building Blocks for Solid-State Materials. Acc. Chem. Res. 51, 919–929. doi:10.1021/acs.accounts.8b00016
Robles-Navarro, A., Fuentealba, P., Muñoz, F., and Cardenas, C. (2020). Electronic Structure of First and Second Row Atoms under Harmonic Confinement. Int. J. Quan. Chem. 120, e26132.
Ryu, J. H., Kim, J. W., Sung, Y.-E., and Oh, S. M. (2004). Failure Modes of Silicon Powder Negative Electrode in Lithium Secondary Batteries. Electrochem. Solid-state Lett. 7, A306. doi:10.1149/1.1792242
Sham, L. J., and Kohn, W. (1966). One-particle Properties of an Inhomogeneous Interacting Electron Gas. Phys. Rev. 145, 561–567. doi:10.1103/physrev.145.561
Shin, H.-J., Hwang, J.-Y., Kwon, H. J., Kwak, W.-J., Kim, S.-O., Kim, H.-S., et al. (2020). Sustainable Encapsulation Strategy of Silicon Nanoparticles in Microcarbon Sphere for High-Performance Lithium-Ion Battery Anode. ACS Sustain. Chem. Eng. 8, 14150–14158. doi:10.1021/acssuschemeng.0c04828
Sloan, J., Kirkland, A. I., Hutchison, J. L., and Green, M. L. H. (2002). Integral Atomic Layer Architectures of 1D Crystals Inserted into Single Walled Carbon Nanotubes. Chem. Commun. 2010, 1319–1332. doi:10.1039/b200537a
Song, T., Hu, L., and Paik, U. (2014). One-dimensional Silicon Nanostructures for Li Ion Batteries. J. Phys. Chem. Lett. 5, 720–731. doi:10.1021/jz4027979
Spencer, J. H., Nesbitt, J. M., Trewhitt, H., Kashtiban, R. J., Bell, G., Ivanov, V. G., et al. (2014). Raman Spectroscopy of Optical Transitions and Vibrational Energies of ∼1 Nm HgTe Extreme Nanowires within Single Walled Carbon Nanotubes. ACS Nano 8, 9044–9052. doi:10.1021/nn5023632
Tiznado, W., Perez-Peralta, N., Islas, R., Toro-Labbe, A., Ugalde, J. M., and Merino, G. (2009). Designing 3-D Molecular Stars. J. Am. Chem. Soc. 131, 9426–9431. doi:10.1021/ja903694d
Vásquez-Espinal, A., Palacio‐Rodríguez, K., Ravell, E., Orozco‐Ic, M., Barroso, J., Pan, S., et al. (2018). E5M7+ (E= C–Pb, M= Li–Cs): A Source of Viable Star‐Shaped Clusters. Chem. Asian J. 13, 1751–1755. doi:10.1002/asia.201800654
Weigend, F., and Ahlrichs, R. (2005). Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/b508541a
Wen, C. J., and Huggins, R. A. (1981). Chemical Diffusion in Intermediate Phases in the Lithium-Silicon System. J. Solid State. Chem. 37, 271–278. doi:10.1016/0022-4596(81)90487-4
Yañez, O., Báez-Grez, R., Inostroza, D., Rabanal-León, W. A., Pino-Rios, R., Garza, J., et al. (2019a). AUTOMATON: a Program that Combines a Probabilistic Cellular Automata and a Genetic Algorithm for Global Minimum Search of Clusters and Molecules. J. Chem. Theor. Comput. 15, 1463–1475. doi:10.1021/acs.jctc.8b00772
Yañez, O., Garcia, V., Garza, J., Orellana, W., Vásquez‐Espinal, A., and Tiznado, W. (2019b). (Li 6 Si 5 ) 2-5 : The Smallest Cluster‐Assembled Materials Based on Aromatic Si 5 6− Rings. Chem. Eur. J. 25, 2467–2471. doi:10.1002/chem.201805677
Yañez, O., Inostroza, D., Usuga-Acevedo, B., Vásquez-Espinal, A., Pino-Rios, R., Tabilo-Sepulveda, M., et al. (2020). Evaluation of Restricted Probabilistic Cellular Automata on the Exploration of the Potential Energy Surface of Be6B11−. Theor. Chem. Acc. 139, 41. doi:10.1007/s00214-020-2548-5
Zhao, L., Grande-Aztatzi, R., Foroutan-Nejad, C., Ugalde, J. M., and Frenking, G. (2017). Aromaticity, the Hückel 4 N+2 Rule and Magnetic Current. ChemistrySelect 2, 863–870. doi:10.1002/slct.201602080
Keywords: nanowire, density functional theory, silicon-lithium clusters, carbon nanotube, metallic character
Citation: Orellana W, Pino-Rios R, Yañez O, Vásquez-Espinal A, Peccati F, Contreras-García J, Cardenas C and Tiznado W (2021) Cluster Assembled Silicon-Lithium Nanostructures: A Nanowire Confined Inside a Carbon Nanotube. Front. Chem. 9:767421. doi: 10.3389/fchem.2021.767421
Received: 30 August 2021; Accepted: 01 October 2021;
Published: 12 November 2021.
Edited by:
Iwona Anusiewicz, University of Gdansk, PolandReviewed by:
Fengyu Li, Inner Mongolia University, ChinaTanmoy Chakraborty, Sharda University, India
Copyright © 2021 Orellana, Pino-Rios, Yañez, Vásquez-Espinal, Peccati, Contreras-García, Cardenas and Tiznado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William Tiznado, d3Rpem5hZG9AdW5hYi5jbA==