 Miroslav Menšík1
Miroslav Menšík1 David Rais1
David Rais1 Muhammed Arshad Thottappali1,2
Muhammed Arshad Thottappali1,2 Pinar Güloğlu1
Pinar Güloğlu1 Petr Toman1Jiří Vohlídal3
Petr Toman1Jiří Vohlídal3 Jiří Pfleger1*
Jiří Pfleger1*- 1Institute of Macromolecular Chemistry, Czech Academy of Sciences, Prague, Czechia
- 2Faculty of Mathematics and Physics, Charles University, Prague, Czechia
- 3Department of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Prague, Czechia
Managing the excited-state decay by a supramolecular structure is a crucial issue for organic photovoltaics. We show that in thin films of metallo-supramolecular polymers made of bis(terpyridine-4′-yl)terthiophenes and
Introduction
Metallo-supramolecular polymers (MSPs) are composed of defined simple or oligomeric organic molecules capped with chelating end-groups (referred to as unimers) that spontaneously assemble into polymer chains by coordination to metal ions (dubbed ion couplers) (Ciferri, 2002). The ideal MSP should exhibit high thermodynamic stability under operational conditions, but it should be kinetically labile (should significantly dissociate) in solutions and/or at elevated temperature under processing conditions (Constable et al., 1994). Kinetic lability gives to MSPs processing advantages, easier control of the morphology of thin films, and multilayered structures and opens up new possibilities of post-synthesis modifications and tailoring their properties. Polymers showing this so-called constitutional dynamics are referred to as dynamers (Lehn, 2005). Dynamics of MSP chains also allow their structure healing by exchanging and/or reshuffling of their unimeric constituents and/or ion couplers.
During the last decade, MSPs underwent tremendous development because of their unique electronic, photonic, magnetic, and catalytic properties (Hissler, 2019). MSPs containing π-conjugated building blocks gained interest as materials for optoelectronic devices, covering light to electricity conversion, and for polymer light-emitting diodes (PLEDs) (Hissler, 2019). Recent finding of an efficient singlet fission process in thin films of MSP based on α,ω−bis(tpy)terthiophene unimers assembled with Zn2+ ion couplers (Rais et al., 2017b) shows that some MSPs could also be potentially exploited for better light harvesting in photovoltaics. The supramolecular approach to polymer chemistry evidently meets the actual needs of new functional materials for advanced electronics. Linear conjugated MSPs described in the literature mostly comprise α,ω−bis(tpy) unimers (tpy stands for 2,2′,6′,2″-terpyridin-4′-yl end-group) with oligo (p-arylenevinylene) (El-Ghayoury et al., 2002), oligo (p-aryleneethynylene) (Burnworth et al., 2008), oligo(fluorene) (Chen and Lin, 2007; Hrma et al., 2017), oligo (phenylene) (Li et al., 2016; Winter and Schubert, 2016), or co-oligomeric (Chiper et al., 2008; Zhang et al., 2019) central units linked into MSP chains via coordination to Zn2+, Co2+, Fe2+, La3+, or Eu3+ ion couplers (Hissler, 2019). Specific facial-meridian coordination of tridentate terpyridine end-groups to ion couplers (Zhang et al., 2019) gives MSPs well-defined stereochemistry, which is important for reproducible preparation of functional materials. This feature is absent in many other MSPs (Burnworth et al., 2007).
The kinetics of photoexcited states in α,ω−bis(tpy)terthiophenes (unimer T) and their MSPs with Zn2+ ion couplers (PT) were measured using pump-probe transient absorption spectroscopy. In solutions, their photophysical properties were found to be strongly influenced by chain dynamics. Relaxation processes running in photoexcited molecules of these unimers and MSPs were identified and characterized, and the impact of disturbed coplanarity of adjacent rings (dihedral angles between planes of rings due to the attached hexyl side groups) on these processes was shown (Rais et al., 2015). Two different physical processes were found in thin films of PT depending on the excitation wavelength (Rais et al., 2017b). Using the wavelength 332 nm, the system showed a fast singlet fission (SF) process with a time constant
Experimental
The material chemistry, thin film preparation, X-ray diffractograms (XRDs) of PT films, and transient absorption (TA) spectroscopy experiment were described in details by Rais et al. (2017b) and the references therein. Here, we just briefly mention the following: 1) The procedure of synthesis of compounds of α,ω-bis(tpy)terthiophene (T) and its zinc-bridged supramolecular polymer PT (to keep the same notation as given by Rais et al. (2017b) (cf. Figure 1) in a powder form was described by Svoboda et al. (2011), Bláhová et al. (2014). 2) PT films deposited on 4 × 5 cm rectangular quartz substrates were prepared from a hexafluoroisopropanol solution of an equimolar mixture of T and zinc acetate by spin-casting at 3,000 rpm. 3) The XRD of the as-prepared PT powder samples of PT showed a dominant peak at about
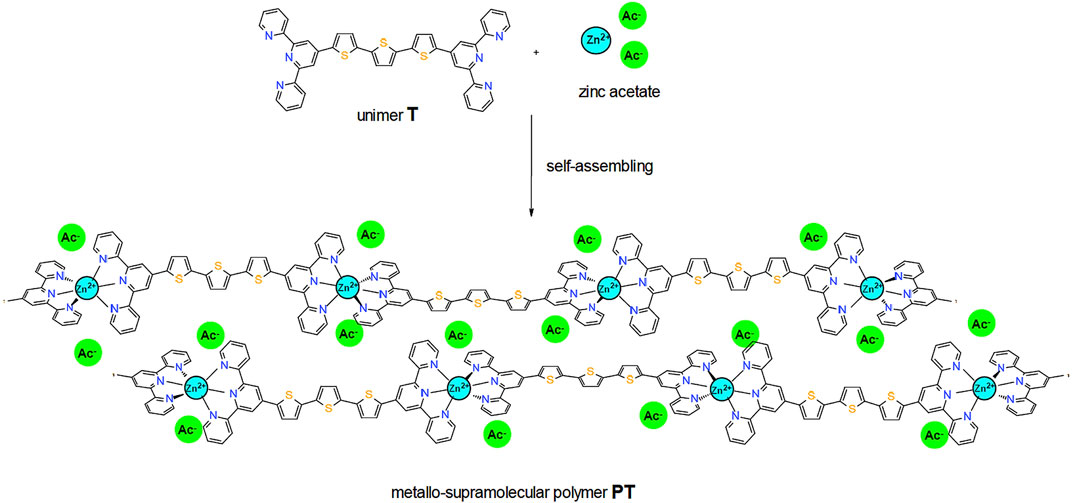
FIGURE 1. Formation of supramolecular structure of thin films of the metallo-supramolecular polymer PT.
The transient differential absorption spectra
Here,
Singlet Exciton Population Kinetics
In Figure 2, we show the evolutions of the populations of the singlet excitons in the PT thin film after excitation by the pump pulse at the wavelength 440 nm, as obtained from Eq. 1. Note that the singlet population equals the ground-state bleach immediately after the excitation because only singlets are directly created by the pump pulse. It is known that the probe at 500 nm provides a nearly exclusively ground-state bleach (GSB) signal, that is, the sum of singlet and triplet excitons with equal weights, without an overlap with the positive ESA signal, which contributes to the spectrum from ca 550–800 nm. Therefore, the measured intensities of the GSB signal can be considered directly proportional to the sum of the concentrations of singlet and triplet excitons. Considering that a singlet exciton is located on a single unimeric unit due to relatively weak mutual interaction of the frontier orbitals of neighboring units, the ratio of the initial TA signal at 500 nm and the steady-state absorption at 500 nm directly shows the fraction of excited unimers in the sample. The absolute values of the singlet populations were obtained from their relative values in Eq. 1, assuming that exclusively singlet excitons contributed to the GSB signal immediately after the photoexcitation.
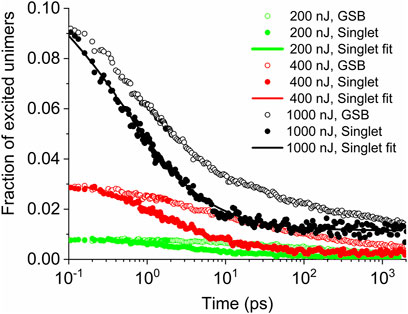
FIGURE 2. Time evolution of the singlet exciton population (full symbols) and of the ground-state bleach (open symbols) for different pump fluences indicated in the legend. Solid lines represent the best fits according to Eq. 2.
The obtained time dependences of the fraction of singlet excited unimers
where time
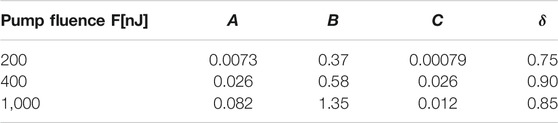
TABLE 1. Parameters from Eq. 2 obtained by fitting experimental data for different pump fluences.
It is seen from Figure 2 that the term
Solution of which is a function
In Eqs 3, 4,
Eq. 5 can be also expressed in terms of the singlet exciton population
Substituting
The Förster transfer is controlled by the long-range dipole–dipole interaction of two, in this case both excited, unimers. Within the discussed recombination process one of them becomes de-excited, while the other one is excited to a higher excited state followed by the subsequent fast Kasha de-excitation from higher to lower excited state or, alternatively, by singlet fission. As a result, there is either only one unimer remaining in the singlet state or two unimers in the triplet state. Such a process can be formally expected due to a significant overlap between the luminescence spectrum and the excited state absorption (ESA) spectrum at wavelengths between 600 and 800 nm (cf. Figure 4-4 of Rais et al. (2017b)).
The diffusion (hopping) proceeds, in turn, by subsequent exciton hopping between the nearest neighbor molecules, facilitated by the overlap of their electronic orbitals. It is, in principle, supported by certain regularity of the arrangement of PT units in thin films characterized by the inter-planar distance of 3.4 Å. It allows getting excitations to the nearest distance where they can recombine.
The dependences
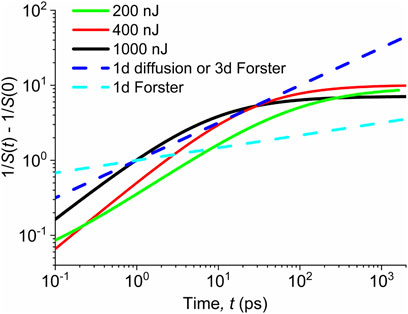
FIGURE 3. Solid lines—evolution of the inverse of the singlet exciton population estimated from the TA data measured with various pump fluences indicated in the legend. Dashed lines—power-law profiles of the exciton–exciton annihilation controlled by either 1d diffusion with the time-independent diffusion coefficient or 3d Förster transfer (light blue) or 1d Förster transfer (dark blue). The initial singlet populations are normalized to unity (independent on the pre-factors in Eqs 5, 6) for all pump fluences.
Interpretation of the Singlet Exciton Annihilation by the Förster Transfer
If the singlet annihilation is interpreted within the Förster transfer model, the slopes of the dependences shown in Figure 3 suggest that for times up to ca 10 ps, the process is rather of 3d nature, while it takes a 1d character at longer times. This is not in contradiction with the physical intuition. Namely, respective transition dipole moments for the absorption or luminescence are probably aligned parallel (or antiparallel) in the anisotropic 1d structure. If the vector
Interpretation of the Singlet Exciton Annihilation by Diffusion
On the microscopic level, the diffusion of singlet excitons is controlled by hopping probability between nearest neighbor unimers. Due to face-to-face molecular stacking, such a process is dominantly of 1d nature. In Figure 3, we see that the data can be well-interpreted within the concept of the 1d model with the diffusion coefficient gradually decreasing in time, but the 1d model with the time-independent diffusion coefficient fails. For this case, we can relate the term
or equivalently
In Eq. 7,
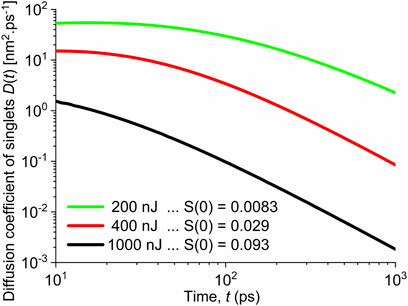
FIGURE 4. Diffusion coefficient of singlet excitons assuming the singlet–singlet annihilation is fully controlled by the singlet exciton diffusion. Shown for different pump fluences and corresponding initial excitation populations marked in the legend.
We conjecture from the abovementioned discussion that the long-range Förster energy transfer process will be dominant. The diffusion-controlled annihilation participates only as a secondary additive process, but becomes important when the initial excitation population is high.
Triplet Exciton Population Kinetics
Experimental data of the triplet population decay for different pump fluences are shown in Figure 5. For the sake of clarity, we show them as two independent sets: 1) derived from Figure 2 by subtracting the ground-state bleached population
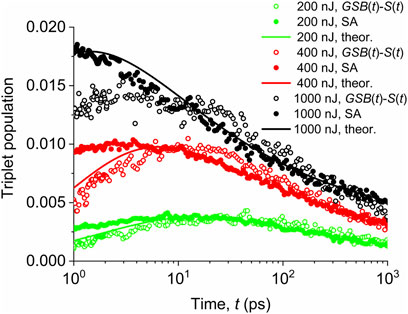
FIGURE 5. Evolution of the triplet exciton population for different pump fluences indicated in the legend. Empty symbols—values obtained by subtracting experimental data of ground-state bleach and singlet populations. Full symbols—data from the spectral analysis (SA) of transient absorption reported by Rais et al. (2017b). Solid lines—data calculated using a theoretical model based on the triplet generation by the singlet–singlet annihilation and the triplet population decay controlled by the triplet–triplet annihilation.
In the second case, the triplet population kinetics was directly obtained from Eq. 1, where it is proportional to
We see in Figure 5 that both methods provide identical profiles at longer times, when the excited-state manifold is already thermalized, while they differ for short times, when the hot-state kinetics of the excited-state manifold can play a role.
We also see that for low pump fluence of 200 nJ (initial singlet exciton population
We should note that the power-law decay is typically observed for diffusion processes (see discussion below). The rate of the triplet population decay depends on the pump pulse fluence, which also determines the initial density of triplet state population.
In the study by Rais et al. (2017b), it was shown that the SF process occurs after the excitation to the S2 state and not to the S1 state because the energy of the triplet pair is higher than the energy of S1. In reverse, the triplet–triplet annihilation can also yield partly S1 singlets. It would also explain the decreasing rate of the singlet population decay at very long times.
Based on the abovementioned discussion, the triplet population kinetics can be described by the following equation
Here,
In the second term in Eq. 9, the term
which asymptotically describes the early time “triplet pumping” when the triplet–triplet annihilation could be neglected due to their low populations.
For longer time limit, the experimental data approach the asymptote
which follows from Eq. 9 if the “pumping rate”
The abovementioned two asymptotes describe the evolution at early and long-time limits, respectively. At the same time, it is evident that
We can define the point
We could see that the rate of triplet population generation at time t1 is already lower than the mean rate of triplet generation in the interval (0, t1). Thus, near the maximum of the triplet population also the “pumping rate” of triplet states decreases. It also proves that the triplet generation is directly bound to singlet exciton population decay.
The fact that near the point of the maximum of triplet population, the function
Taking into account the function used for fitting experimental data for the triplet annihilation rate
We see that the second derivative
The experimental data show that not only the position of the maximum of the triplet population but also the transition time between the concavity and convexity of the triplet population time course shifts to shorter time with increasing pump fluence. It means that also the saturation of the “pumping rate integral”
Triplet Exciton Formation by Inter-System Crossing
Here, we assume that the spin-orbital coupling will allow the singlet-to-triplet transition via inter-system crossing (Sun et al., 2021). The rate of the formation of the triplet population
where α is a constant independent on the exciton population, and thus also the initial pump fluence. After integration over time, we get
so that the rate of the depletion of the “pumping rate” generated by the inter-system crossing can be obtained by analyzing the time evolution of
Triplet Formation by the Singlet–Singlet Annihilation
The possibility of this process can be justified by the following reasons. In the study by Rais et al. (2017b), it was shown that the maximum absorption from the ground state takes place at 2.53 eV, while the maximum of the emission takes place at 1.80 eV. From the Stokes shift energy 0.73 eV, we can estimate the energy of
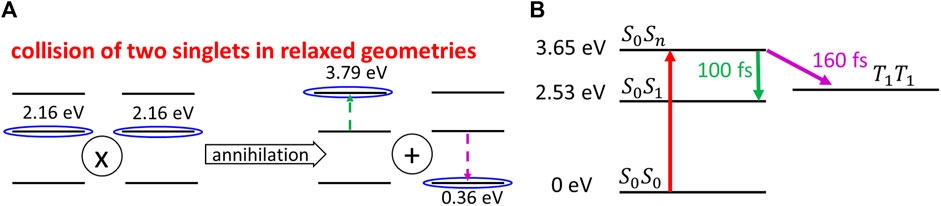
FIGURE 6. (A) Scheme of the singlet–singlet annihilation process, which can be described as a simultaneous virtual ESA (indicated by the green dashed arrow) and the virtual luminescence (shown by the violet dashed arrow). (B) Internal conversion to the
Assuming singlet–singlet annihilation, we can derive the rate
and
Thus, the decrease in the generation rate of triplets by the singlet–singlet annihilation can be obtained by analyzing the experimental data of the time evolution of
As observed from the previous two paragraphs, by comparing the rates of saturation of
The dependences of
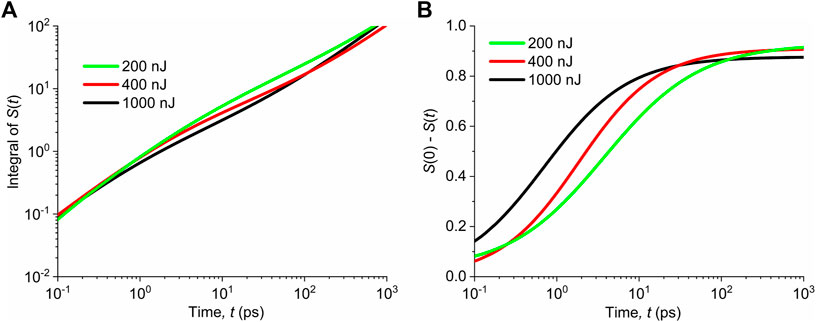
FIGURE 7. (A) Calculated dependences of
It indicates that at the initial rise times (rate of formation), the triplet states are not governed by the singlet-to-triplet inter-system crossing.
The time evolutions of
It should be noted that the dependences in Figure 7 are obtained by numerical fitting of the experimental data. The saturation of
We can also compare the different mechanisms of the triplet formation looking at the explicit dependence of the rise time (or maximum position) of the triplet population on the pump fluence F, that is, on the initial singlet population
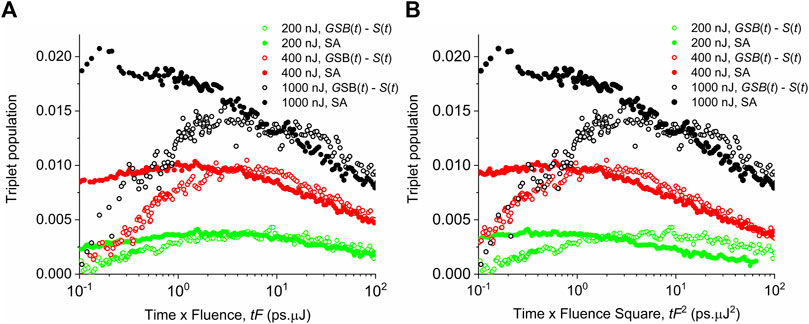
FIGURE 8. Experimental data of the triplet exciton population evolution for different pump fluences F shown in the legend. Empty symbols—data obtained by subtracting the ground-state bleach and the singlet populations. Full symbols—data from the spectral analysis (SA) of transient absorption reported by Rais et al., 2017b. In x-axis, time is rescaled as
Based on the analysis of Figures 7, 8, we conjecture that triplets are created by the singlet–singlet annihilation rather than by inter-system crossing. Hot individual triplet excitons migrate by diffusion and gradually thermalize until they finally collide and annihilate. The whole process of singlet–singlet annihilation, triplet pair formation and dissociation, and triplet diffusion and annihilation is schematically drawn in Figure 9. The triplet–triplet annihilation in long times can also potentially contribute to the formation of singlet states
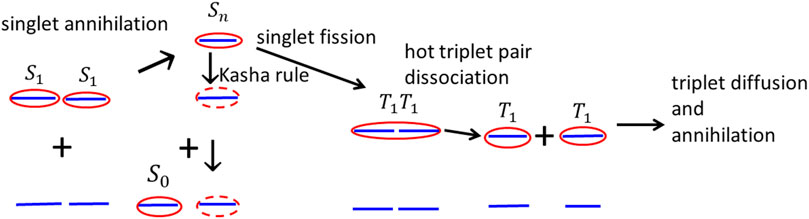
FIGURE 9. Schematic figure of subsequent processes of mutual singlet exciton annihilation, formation of intermediate hot high excited singlet exciton, formation of the hot intermediate triplet exciton pair, its dissociation to two triplet states, triplet state diffusion, and triplet exciton–exciton annihilation.
Triplet Diffusion and Annihilation
We can, thus, assume that the evolution of the triplet population is controlled simultaneously by their generation from singlet states and by the triplet–triplet annihilation. The first process dominates at early stages, as it was discussed above, while the triplet–triplet annihilation becomes dominant at longer times. As a supporting argument, we can point to the sensitivity of the triplet population decay rate to the pump fluence at longer times, seen in Figure 5, which is in accordance with the mutual collisions of triplet excitons. These long-time tails of the triplet population decay show the power-law dependence, which is typical for the bi-molecular collisions, too. The kinetics of the triplet population can, thus, be described by the equation
The triplet–triplet annihilation rate
The time dependences of the normalized 1d diffusion coefficient
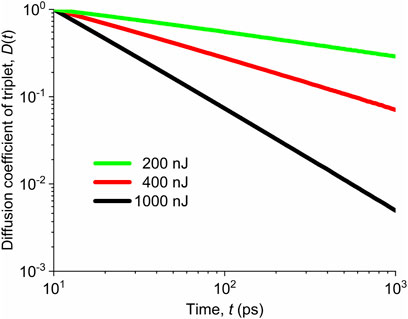
FIGURE 10. Normalized values of the diffusion coefficient of triplets for different pump fluences marked in the legend.
We did not show the diffusion coefficient
For the pump fluences around 200 nJ, the diffusion coefficient

TABLE 2. Dependence of the exponent
The correlation between the power-law time dependence of the exciton population and the time-dependent diffusion coefficient was proved theoretically and experimentally documented on thin films of α,ω-bis(tpy)terthiophene (T) unimers without metal couplers under strong excitation conditions (Menšík et al., 2019). The power-law diffusion kinetics can generally occur in systems with an increased structural and dynamic disorder (see Devižis et al., 2009; Král and Menšík, 2016; Toman et al., 2017; Menšík et al., 2018b and references therein). In the system discussed in this article, the structure can be disturbed by both the electronic excitations and heat dissipation at stronger pump fluence which, subsequently, can affect triplet diffusivity.
Conclusion
The detailed analysis of the kinetics of the photoexcited states in thin films of metallo-supramolecular polymers with ditopic thiophene-bridged terpyridine ligands showed that singlet excitons
The reported model is generally applicable to systems in which the energy of triplet pairs falls within the energy levels of S1 and S2 states. In systems where the triplet diffusion is slower, the excitation energy can be temporarily stored in these triplets and can be restored as S1 energy later, which is manifested by prolonged lifetime of S1 excitons, observable, for example, as delayed fluorescence.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
Conceptualization: MM and JP; methodology: MM and PT; theoretical calculations: MM and PT, data processing: MT and PG; formal analysis DR and MT; validation: JP; writing—original draft preparation, MM and PT; writing—reviewing and editing: MM and JP; contributions to the interpretation of data and overall revision of the text: JV; project resources: JP. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the Ministry of Education, Youth and Sports of the Czech Republic—Program INTEREXCELLENCE, No. LTAUSA19066 for financial support.
References
Bláhová, P., Zedník, J., Šloufová, I., Vohlídal, J., and Svoboda, J. (2014). Synthesis and Photophysical Properties of New α,Ω-Bis(Tpy)Oligothiophenes and Their Metallo-Supramolecular Polymers with Zn2+Ion Couplers. Soft Mater. 12, 214–229. doi:10.1080/1539445X.2013.858358
Burnworth, M., Knapton, D., Rowan, S. J., and Weder, C. (2007). Metallo-supramolecular Polymerization: A Route to Easy-To-Process Organic/inorganic Hybrid Materials. J. Inorg. Organomet. Polym. 17, 91–103. doi:10.1007/s10904-006-9075-2
Burnworth, M., Mendez, J. D., Schroeter, M., Rowan, S. J., and Weder, C. (2008). Decoupling Optical Properties in Metallo-Supramolecular Poly(p-Phenylene Ethynylene)s. Macromolecules 41, 2157–2163. doi:10.1021/ma702712e
Chen, Y.-Y., and Lin, H.-C. (2007). Metallo-homopolymer and Metallo-Copolymers Containing Light-Emitting poly(fluorene/ethynylene/(terpyridyl)Zinc(II)) Backbones and 1,3,4-oxadiazole (OXD) Pendants. Polymer 48, 5268–5278. doi:10.1016/j.polymer.2007.06.040
Chiper, M., Meier, M. A. R., Wouters, D., Hoeppener, S., Fustin, C.-A., Gohy, J.-F., et al. (2008). Supramolecular Self-Assembled Ni(II), Fe(II), and Co(II) ABA Triblock Copolymers. Macromolecules 41, 2771–2777. doi:10.1021/ma0718954
Ciferri, A. (2002). Supramolecular Polymerizations. Macromol. Rapid Commun. 23, 511–529. doi:10.1002/1521-3927(20020601)23:9<511:aid-marc511>3.0.co;2-f
Constable, E. C., Edwards, A. J., Marcos, M. D., Raithby, P. R., Martínez-Máñez, R., and Tendero, M. J. L. (1994). A New Functionalised Oligopyridine Ligand Containing Ferrocene as a ball-bearing Spacer for Metallosupramolecular Chemistry. Inorg. Chim. Acta 224, 11–14. doi:10.1016/0020-1693(94)04147-4
Devižis, A., Serbenta, A., Meerholz, K., Hertel, D., and Gulbinas, V. (2009). Ultrafast Dynamics of Carrier Mobility in a Conjugated Polymer Probed at Molecular and Microscopic Length Scales. Phys. Rev. Lett. 103, 027404. doi:10.1103/PhysRevLett.103.027404
El-Ghayoury, A., Schenning, A. P. H. J., and Meijer, E. W. (2002). Synthesis of ?-conjugated Oligomer that Can Form Metallo Polymers. J. Polym. Sci. A. Polym. Chem. 40, 4020–4023. doi:10.1002/pola.10477
Engel, E., Leo, K., and Hoffmann, M. (2006). Ultrafast Relaxation and Exciton-Exciton Annihilation in PTCDA Thin Films at High Excitation Densities. Chem. Phys. 325, 170–177. doi:10.1016/j.chemphys.2005.09.004
Fleming, G. R., Morris, J. M., and Robinson, G. W. (1976). Direct Observation of Rotational Diffusion by Picosecond Spectroscopy. Chem. Phys. 17, 91–100. doi:10.1016/0301-0104(76)85009-4
Hissler, M. (2019). “Metallo-Supramolecular Polymers J Vohlídal and M Hissler,” in Smart Inorganic Polymers: Synthesis, Properties, and Emerging Applications in Materials and Life Sciences. Editors E. Hey-Hawkins, and M. Hissler, 141–162.
Hrma, M., Šichová, K., Svoboda, J., and Vohlídal, J. (2017). Assembling of Bis(tpy)fluorenes with Zn2+ and Fe2+ Ions into Metallo-Supramolecular Polymers with Highly Efficient white-light Emission. Polymer 122, 22–33. doi:10.1016/j.polymer.2017.06.037
Král, K., and Menšík, M. (2016). Transfer of Energy or Charge between Quasi-Zero-Dimensional Nanostructures. J. Comput. Theor. Transport 45, 243–255. doi:10.1080/23324309.2016.1157492
Lehn, J.-M. (2005). Dynamers: Dynamic Molecular and Supramolecular Polymers. Prog. Polym. Sci. 30, 814–831. doi:10.1016/j.progpolymsci.2005.06.002
Li, H.-Q., Ding, Z.-Y., Pan, Y., Liu, C.-H., and Zhu, Y.-Y. (2016). Fluorescence Tuning of Zn(II)-based Metallo-Supramolecular Coordination Polymers and Their Application for Picric Acid Detection. Inorg. Chem. Front. 3, 1363–1375. doi:10.1039/c6qi00267f
Marciniak, H., Li, X.-Q., Würthner, F., and Lochbrunner, S. (2011). One-dimensional Exciton Diffusion in Perylene Bisimide Aggregates. J. Phys. Chem. A. 115, 648–654. doi:10.1021/jp107407p
Menšík, M., Pfleger, J., and Toman, P. (2018a). Analysis of Collision-Controlled Time Dependence of Diffusion Coefficient of Polaron Pairs from Transient Absorption Spectra of Conducting Polymers. Chem. Pap. 72, 1729–1734. doi:10.1007/s11696-018-0439-1
Menšík, M., Rais, D., Pfleger, J., and Toman, P. (2019). Evolution of Diffusion Coefficient of Photoexcited Species in Excimer Forming Organic Thin Films. J. Phys. Chem. C 124, 52–59. doi:10.1021/acs.jpcc.9b08953
Menšík, M., Toman, P., Bielecka, U., Bartkowiak, W., Pfleger, J., and Paruzel, B. (2018b). On the Methodology of the Determination of Charge Concentration Dependent Mobility from Organic Field-Effect Transistor Characteristics. Phys. Chem. Chem. Phys. 20, 2308–2319. doi:10.1039/C7CP06423C
Rais, D., Menšík, M., Paruzel, B., Kurunthu, D., and Pfleger, J. (2017a). Phonons Spreading from Laser-Heated Gold Nanoparticle Array Accelerate Diffusion of Excitons in an Underlying Polythiophene Thin Film. Phys. Chem. Chem. Phys. 19, 10562–10570. doi:10.1039/c7cp00286f
Rais, D., Menšík, M., Paruzel, B., Toman, P., and Pfleger, J. (2018). Concept of the Time-dependent Diffusion Coefficient of Polarons in Organic Semiconductors and its Determination from Time-Resolved Spectroscopy. J. Phys. Chem. C 122, 22876–22883. doi:10.1021/acs.jpcc.8b07395
Rais, D., Menšík, M., Štenclová-Bláhová, P., Svoboda, J., Vohlídal, J., and Pfleger, J. (2015). Time-Resolved Transient Optical Absorption Study of Bis(terpyridyl)oligothiophenes and Their Metallo-Supramolecular Polymers with Zn(II) Ion Couplers. J. Phys. Chem. A. 119, 6203–6214. doi:10.1021/jp512213s
Rais, D., Pfleger, J., Menšík, M., Zhigunov, A., Štenclová, P., Svoboda, J., et al. (2017b). Singlet Fission in Thin Films of Metallo-Supramolecular Polymers with Ditopic Thiophene-Bridged Terpyridine Ligands. J. Mater. Chem. C 5, 8041–8051. doi:10.1039/c7tc00484b
Stern, H. L., Cheminal, A., Yost, S. R., Broch, K., Bayliss, S. L., Chen, K., et al. (2017). Vibronically Coherent Ultrafast Triplet-Pair Formation and Subsequent Thermally Activated Dissociation Control Efficient Endothermic Singlet Fission. Nat. Chem 9, 1205–1212. doi:10.1038/nchem.2856
Sun, X., Gong, H., Zhang, Y., Tian, Y., Zhang, H., Bai, F., et al. (2021). Investigating Phosphorescence Capability of Halogen-Substituted Metal-free Organic Molecules: A Theoretical Study. Spectrochimica Acta A: Mol. Biomol. Spectrosc. 255, 119642. doi:10.1016/j.saa.2021.119642
Svoboda, J., Štenclová, P., Uhlík, F., Zedník, J., and Vohlídal, J. (2011). Synthesis and Photophysical Properties of α,ω-bis(terpyridine)oligothiophenes. Tetrahedron 67, 75–79. doi:10.1016/j.tet.2010.11.039
Toman, P., Menšík, M., Bartkowiak, W., and Pfleger, J. (2017). Modelling of the Charge Carrier Mobility in Disordered Linear Polymer Materials. Phys. Chem. Chem. Phys. 19, 7760–7771. doi:10.1039/c6cp07789g
Winter, A., and Schubert, U. S. (2016). Synthesis and Characterization of Metallo-Supramolecular Polymers. Chem. Soc. Rev. 45, 5311–5357. doi:10.1039/C6CS00182C
Keywords: metallo-supramolecular polymers, transient absorption spectroscopy, singlet and triplet excitons, time-dependent diffusion coefficient, polythiophene
Citation: Menšík M, Rais D, Thottappali MA, Güloğlu P, Toman P, Vohlídal J and Pfleger J (2022) Kinetics of the Photoexcited States in Thin Films of Metallo-Supramolecular Polymers With Ditopic Thiophene-Bridged Terpyridine Ligands. Front. Chem. 9:766121. doi: 10.3389/fchem.2021.766121
Received: 28 August 2021; Accepted: 21 December 2021;
Published: 20 January 2022.
Edited by:
Tomasz Marszalek, Max Planck Institute for Polymer Research, GermanyReviewed by:
Charusheela Ramanan, VU Amsterdam, NetherlandsZdenek Remes, Institute of Physics (ASCR), Czechia
Krzysztof Gibasiewicz, Adam Mickiewicz University, Poland
Vaclav Spicka, Academy of Sciences of the Czech Republic, Czechia
Copyright © 2022 Menšík, Rais, Thottappali, Güloğlu, Toman, Vohlídal and Pfleger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiří Pfleger, cGZsZWdlckBpbWMuY2FzLmN6