 Martin Obermeier1
Martin Obermeier1 Raoul S. Schaer
Raoul S. Schaer Matthias Schwalbe
Matthias Schwalbe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 30 September 2021
Sec. Catalysis and Photocatalysis
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.751716
This article is part of the Research Topic Light-Assisted Molecular and Hybrid Systems for Artificial Photosynthesis View all 6 articles
Complexes based on nitrogen and sulfur containing ligands involving 3d metal centers are known for the electrocatalytic reduction of CO2. However, photocatalytical activation has rarely been investigated. We herein present results on the light-driven CO2 reduction using either Ir(dFppy)3 [Ir, dFppy = 2-(4,6-difluorophenyl)pyridine] or [Cu(xant)(bcp)]+, (Cu, xant = xantphos, bcp = bathocuproine) as photosensitizer in combination with TEA (triethylamine) as sacrificial electron donor. The 3d metal catalysts have either dptacn (dipicolyl-triazacyclononane, LN3) or dpdatcn (dipicolyl-diazathiocyclononane, LN2S) as ligand framework and Fe3+, Co3+ or Ni2+ as central metal ion. It turned out that the choice of ligand, metal center and solvent composition influences the selectivity for product formation, which means that the gaseous reduction products can be solely CO or H2 or a mixture of both. The ratio between these two products can be controlled by the right choice of reaction conditions. With using Cu as photosensitizer, we could introduce an intermolecular system that is based solely on 3d metal compounds being able to reduce CO2.
Due to the fact that the global energy demand is projected to increase, there will still be a high necessity of usage of fossil fuels over the next couple of decades, as the development of renewable energy sources cannot adjust with the same speed (Sönnchen, 2020). This mismatch will not only cause problems due to an increase in the amount of greenhouse gases in the atmosphere, which are highly responsible for the climate change (e.g. methane, CO2), but also raises the need to search for other alternative energy resources (Aresta, 2010; Ringsmuth et al., 2016). Therefore one of the key answers to face both problems is the conversion of CO2 to liquid fuels (Benson et al., 2009; Ma et al., 2009). This process is forecast to have a positive impact on the global greenhouse gas balance by recycling prior emitted CO2. Due to the inertness of CO2 - both thermodynamically and kinetically - different strategies need to be developed to optimize the conversion to more useful C1 building blocks (Schrag, 2007). One option is using homogeneous catalysts instead of heterogeneous catalysts (Kumar et al., 2016) as often done by industry. One major advantage is the higher variety of spectroscopic techniques that can be used to understand the mechanistic features and, hence, assist in finding the correlation between structure and catalytic activity (Francke et al., 2018). Significant progress has been made for the CO2 to CO reduction with catalytic systems based on heavier transition metals such as rhenium or ruthenium (Tamaki et al., 2013; Tamaki et al., 2015; Gotico et al., 2018). However, these metals are expensive and rare, which leads to the need of finding complexes based on earth-abundant metals to perform the activation and conversion of CO2 (Takeda et al., 2017; Dalle et al., 2019).
As often in modern chemistry, inspiration can be obtained from nature. 3d metal-based enzymes with complex frameworks are known to activate stable small molecules. One of the best studied enzymes is carbon dioxide dehydrogenase (CODH) – capable of oxidizing CO to CO2 and vice versa. The catalytically active C-cluster is composed of a bimetallic Ni-Fe center in a nitrogen/sulfur rich environment (Lubitz et al., 2014; Esselborn et al., 2016; Benvenuti et al., 2020; Ghosh et al., 2020). Beside molecular Ni and Fe complexes, systems containing Co were found to show high electro- as well as photo-catalytic activity towards CO2 reduction by several groups (Tamaki and Ishitani, 2017; Liu et al., 2018). Light-activated processes have the charm, that the necessary activation energy can be harvested from the sun as a limit less power source, which makes the process economical and chemical ultimate efficient. By adding a photosensitizer (PS), which transforms photonic energy into chemical energy, sunlight can be used directly to provide the necessary energy for the CO2 reduction (Thoi et al., 2013; Chan et al., 2015; Guo et al., 2016). Photochemical CO2 reduction can thus be considered a crucial part of artificial photosynthesis.
Apart from the nature of the metal, the ligand environment is also of great importance. For example, Ni(II) complexes – when having π-donor atoms such as S – are more likely to show activity towards CO2 reduction, due to the less negative NiII/I redox potential compared to Ni complexes with just N-donor atoms (Chen et al., 2014). Interestingly, studies on ligands with sulfur donor atoms are rare. The Ni dithiacyclam complex N2S2Ni (N2S2 = 1,8-Dithia-4,11-diazacyclotetradecane) was the first of its kind to be investigated in the electrocatalytic CO2 activation. Although better catalytic properties than the [Ni(cyclam)]2+ congener were observed (cyclam = 1,4,8,11-tetraazacyclotetradecane), it was revealed that the complex seems to be instable after prolonged electrolysis time (Gerschel et al., 2019; Iffland et al., 2020).
Kojima et al. reported about a heterodinuclear nickel magnesium complex, in which the Ni ion resides in a S2N2 ligand sphere and two noncoordinating pyridine substituents can bind to a Lewis-acidic metal ion. The heterodinuclear Ni-Mg complex was successfully applied in photocatalytic CO2 to CO transformation using [Ru (bpy)3]2+ (bpy = 2,2′-bipyridine) as photosensitizer (Hong et al., 2017; Hong et al., 2019). Further examples investigating the photocatalytic properties of 3d metal complexes with sulfur donor atoms in the ligand sphere are missing. Hence, we started out to synthesize a series of complexes with earth abundant transition metals (Fe, Co and Ni) and either a macrocyclic sulfur free or sulfur containing ligand (Scheme 1). Herein we present their photocatalytic activity in the presence of different photosensitizers, Ir ([Ir(dFppy)3], dFppy = 2-(4,6-difluorophenyl)pyridine) and Cu ([Cu(xant)(bcp)]PF6, xant = xantphos, bcp = bathocuproine).
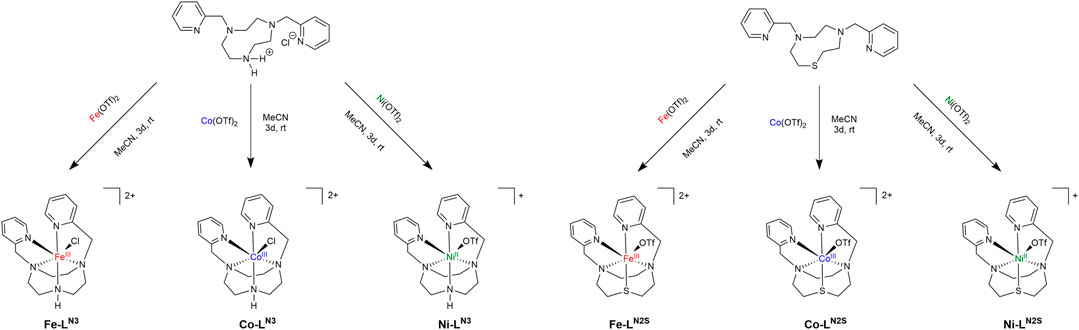
SCHEME 1. Reaction scheme and picture of the complexes M-LN3 (with M = Fe, Co, Ni; left) and M-LN2S (with M = Fe, Co, Ni; right) with LN3 being dipicolyl-triazacyclononane and LN2S being dipicolyl-diazathiacyclononane.
The complexes were prepared by first synthesizing the corresponding ligands 1,4-di(picolyl)-1,4,7-triazacyclononane LN3 and 1-thia-4,7-di(picolyl)-diazacyclononane LN2S. We followed published procedures with slight adjustments (see also the Supporting Information). Briefly, LN3 was prepared beginning with the tosylation of diethylenetriamine and glycol (Friscourt et al., 2012). The next step was a Cs2CO3 catalyzed macrocyclization using the tosylated compounds to form tri-tosyl-triazacyclononane (ts3-tacn) (Cao et al., 2010). Partial detosylation of the formed macrocycle using HBr in acetic acid resulted in the formation of ts-tacn (Cao et al., 2010), which was transformed into dipicolyl-tosyl-tacn using two equivalents of 2-picolylchloride hydrochloride in a base-catalyzed SN2 reaction (Stavila et al., 2008). To obtain the desired ligand LN3 the remaining tosyl protecting group was cleaved using conc. H2SO4 (Luk’yanenko et al., 1990; Stavila et al., 2008). LN2S was prepared in a similar fashion: First N,N’-bis-tosyl-bis(2-aminoethyl)sulfide was synthesized via sodium ethoxide driven SN2 reaction of cysteamine hydrochloride and 2-chloroethylamine hydrochloride (Wilson, 2007), followed by tosylation as described earlier for LN3 (Friscourt et al., 2012). The macrocyclization was started with LiOH (2.5% in water) in toluene and using NaBu4Br as phase-transfer catalyst (Wilson, 2007). To obtain LN2S a detosylation and substitution with 2-picolylchloride hydrochloride was performed (Chak et al., 1994; Wilson, 2007). Both proligands were used immediately to obtain the complexes, due to sensitivity towards oxidation. Hence it was not possible to store the proligands for longer time periods.
For both compounds, LN3 and LN2S, the analytical data nonetheless agreed with those published earlier. Metal complexation could be achieved by reaction of the corresponding ligand with an equimolar amount of simple metal salt, e.g., metal triflates M(OTf)2 (M = Fe, Co, Ni). Work-up of the reaction mixture was done under air atmosphere leading to oxidation of the iron and cobalt compounds. Purification of the crude product was obtained via washing with CH2Cl2 to remove excess of ligand and washing with toluene (Fe(OTf)2 and Co(OTf)2) or THF (Ni(OTf)2) to remove unreacted metal salt. The desired complexes were obtained as solids, which were redissolved in a small amount of MeCN and precipitated using Et2O. Since LN3 was synthesized as hydrochloride salt (stemming from column chromatographic purification using dichloromethane), Fe-LN3 and Co-LN3 were obtained as complexes with one chloride ligand and two uncoordinated triflate counter anions, which could be verified by mass spectrometry and X-ray diffractometry. Ni-LN3 was isolated as a chloride free complex by precipitating AgCl using Ag(OTf). The complexes M-LN2S were prepared in the same fashion giving yields in the range of 44–50%. All complexation reactions should be done soon after preparation of the proligands, due to sensitivity against oxidation of the compounds LN3 and LN2S.
The successful complex formation was supported by high resolution MS revealing characteristic peaks at m/z 516.0974 (Fe-LN3), 519.0975 (Co-LN3), 518.1018 (Ni-LN3), as well as 533.0584 (Fe-LN2S), 536.0568 (Co-LN2S) and 535.0590 (Ni-LN2S) for the corresponding [M + OTf]+ fragments (Supplementary Figures S13–S18). To verify the oxidation state of +3 for both the Fe and the Co complexes, different approaches have been made. Since Co-LN3 and Co-LN2S represent diamagnetic compounds, i.e., they are in the low-spin d6 form, NMR spectroscopy could be applied. Thereby, Co-LN3 gives a nicely resolved 1H NMR spectrum (Supplementary Figure S11) where all signals can be assigned. The ligand conformation leads to an asymmetry in the complex in contrast to the symmetric nature of the ligand itself (see also crystal structure in Figure 1). Thus, the pyridyl substituents give rise to eight individual signals in the aromatic region, ranging from 9.5 to 7.2 ppm. The methylene groups arise at 5.1, 4.6, and 4.5 ppm and the macrocyclic protons from 4.2 to 2.0 ppm. In case of Co-LN2S the 1H NMR spectrum shows broad signals in the region from 2 to 9 ppm. It seems that the S-donor atom in the ligand macrocycle is less strongly bound leading to a higher mobility in the system and hence stronger dynamics.
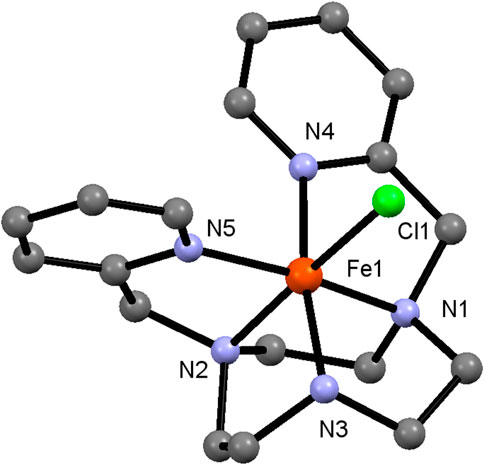
FIGURE 1. Crystal Structure of Fe-LN3. Hydrogen atoms and counter anions are omitted for clarity.
On the other hand, Fe-LN3 and Fe-LN2S are paramagnetic compounds, which give rise to non-interpretable 1H NMR spectra. The magnetic properties indicate a +3 oxidation state in octahedral environment with a strong ligand field, as for +2 oxidation state a d6 low-spin state, and thus a diamagnetic compound, would be expected. The +3 oxidation state was confirmed by using EPR spectroscopy for both compounds giving a FeIII typical spectrum with signals having a g value of around 4.0 and 2.0 (Supplementary Figure S23) (Srinivasan and Gralla, 2002).
Electrochemical and spectroscopic properties were determined via cyclic voltammetry (CV) and UV/vis measurements. The UV/vis studies in DMF solution (Table 1; Supplementary Figures S19–S21) reveal a strong ligand-based absorption peaking at 268 nm for all compounds, which can be assigned to be situated at the aromatic picolyl substituents. These maxima are outside the visible spectrum and do not have an impact on the color of the complexes. For the two Fe complexes the dark brown color derives from a broad absorption feature ranging from 600 to 350 nm with a shoulder at around 432 nm (Fe-LN3) or 502 nm (Fe-LN2S). The Ni complex Ni-LN3 only has an additional absorption maximum at 308 nm and hence is colorless. As the Ni-LN2S absorbs in a broader area from 500 to 300 nm it is isolated as light brown solid. The most striking difference is revealed for the Co complexes where d-d transitions are observed at 362 nm (ε = 320 dm3/mol*cm) for Co-LN3 and 490 nm (ε = 390 dm3/mol*cm) for Co-LN2S leading to a light red/pink color.
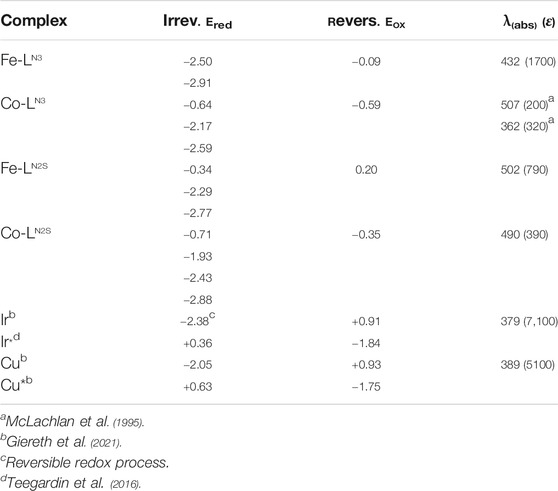
TABLE 1. Redox potentials [V], excited state redox potentials [V] (marked with *) and MLCT absorption maxima [nm] with corresponding molar attenuation coefficients ɛ [dm3 mol−1cm−1] of the complexes. Peak potentials are given vs. Fc/Fc+.
In the CV measurements of the complexes Fe-LN3 and Fe-LN2S a reversible redox event is observed at a half-wave potential of −0.09 and 0.20 V (referenced to Fc/Fc+), respectively, representing the FeII/FeIII redox couple (Table 1; Supplementary Figure S22). In addition, there are irreversible reduction events at −2.50 V and −2.91 V for Fe-LN3 and −2.29 V and −2.77 V for Fe-LN2S, which can tentatively be assigned to the FeII/FeI reduction and the formal FeI/Fe0 reduction. However, there is certainly the possibility that the true electronic distribution is different and ligand-based reduction cannot be neglected without further experiments.
Interestingly, Fe-LN2S is thus easier to reduce (by about 200 mV) than the sulfur free analogue. This result can be attributed to the electron donating property of the sulfur atom, and a similar trend is also observed for the related cobalt complexes. It is important to note, though, that for Fe-LN2S an additional reduction process occurs at −0.34 V, which we tentatively relate to the sulfur donor atom. A similar behavior is observed for Co-LN2S that shows and additional reduction process at −0.71 V, which is absent for Co-LN3.
In case of Co-LN3 a reversible redox event at −0.59 V is indicative for the CoII/CoIII redox couple. This event is accompanied by two irreversible reduction processes at −2.17 V and −2.59 V resulting in the formation of CoI and finally the Co0 state – most likely being a CoI(LN3−) species. In accordance with the observations for Co-LN3, Co-LN2S shows one reversible reduction at −0.35 V and three irreversible reduction events at more negative potentials at −1.93 V, −2.43 V and −2.88 V that are shifted to higher voltages when compared to Co-LN3. An assignment of these redox processes is not straightforward and currently not possible. The Ni complexes Ni-LN3 and Ni-LN2S do not show any redox processes in the investigated solvent window.
X-ray quality crystals of Co-LN3 and Fe-LN3 were obtained by slow vapor diffusion of Et2O to a high concentrated MeCN solution. Both complexes are isostructural (Figure 1; Supplementary Figure S24; Supplementary Tables S1, S2) with all five nitrogen atoms acting as donor atoms. The octahedral coordination sphere is completed by coordination of one chloride ion. The crystal structure shows a hydrogen atom at the secondary amine which confirms the +3 oxidation state (two triflate counter ions were also found per cation).
In case of Co-LN3 shorter Co-N bond lengths (1.94–1.97 Å) are observed, which is in line with Co3+ being smaller than Fe3+. The Co-Cl bond distance, on the other hand, is almost identical to the Fe-Cl bond with ∼2.23 Å (Table 2; Supplementary Table S2). For Co-LN3 an isostructural complex [CoLN3(OH2)][CIO4]3•H2O was published earlier (McLachlan et al., 1995) having similar bond angles and distances, but a shorter Co-O bond since H2O is a smaller ligand than the chloride ion. Furthermore, there are a few crystal structures of similar Fe compounds with the LN3 ligand described in the literature. Two prominent examples are the complexes [FeLN3(Cl)](PF6) and [FeLN3(NCS)](PF6), which were obtained via reaction under inert conditions and, hence, possess an oxidation state for Fe of +2 (Spiccia et al., 1998). Both complexes nonetheless show a similar ligand geometry, with Fe in an octahedral coordination environment surrounded by five nitrogen and one (pseudo-)halide donor atom.

TABLE 2. Selected interatomic distances (Å) and bond angles (°) for Fe-LN3.
In case of the LN2S complexes we were not able to obtain crystals so far. Nonetheless, crystal structures have been reported for similar FeII and NiII complexes (Wasielewski and Mattes, 1993; Zhang et al., 1998).
The photocatalytic activity of the prepared complexes was determined using Ir(dFppy)3 and triethylamine (TEA) as sacrificial electron donor. In earlier studies we successfully applied the following conditions (Giereth et al., 2021): 5 × 10−5 M catalyst concentration in 5 ml DMF with 5 × 10−5 M Ir as photosensitizer (PS) and 5 Vol% TEA as sacrificial electron donor (SR). The catalysis mixture is placed in front of a 200 W Hg-lamp equipped with a 400 nm low-wavelength cut-off filter to exclude UV light. Ir was selected because of its ability to still absorb solar photons in the region of 400–460 nm (Becker et al., 2020) and its potentially well suited (excited state) redox properties (Table 1) (Koike and Akita, 2014; Lee and Han, 2020; Giereth et al., 2021). For this work we just focused on the gaseous reduction products formed and did not investigate the formation of liquid products. Further information is given in the supplementary data.
It can be observed, that not only the ligand but also the metal centre do have an impact on both the formed products and the catalytic activity (Figure 2; Table 3). While Fe-LN3 shows a slightly higher TON for H2 than for CO formation after 24 h reaction time, Co-LN3 shows no H2 formation and the highest TONco for the here investigated complexes. This result might be due to the fact that Fe tends to form hydride intermediates (Drosou et al., 2020). When a sulfur atom is added to the ligand framework, considerable H2 formation is observed. It is known, that sulfur donor atoms can take part in hydrogen evolution reaction by forming a sulfur-hydride species (Darmon et al., 2014; Drosou et al., 2020). On a side note, the illumination of the reaction solution without any catalysts results in no CO or H2 release, indicating that the photosensitizer is stable and its (photo) decomposition products are not responsible for the formation of the gaseous products.
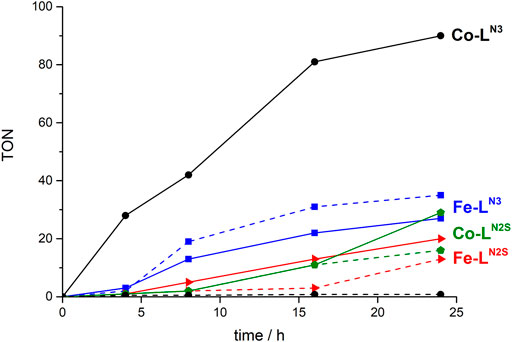
FIGURE 2. Comparison of catalytic activity of the Fe and Co based catalysts (5 × 10−5 M), in DMF applying a λ > 400 nm longpass filter, with 5% TEA as SR, Ir(dFppy)3 and a 200 W Hg-lamp. Dotted line: H2 formation and solid line: CO formation.

TABLE 3. Turn over number (TON) for CO and H2 formation for the respective complexes after 24 h of illumination. The catalytic reaction was performed with a catalyst concentration of 5 × 10-5 M in DMF with 5% TEA as sacrificial electron donor applying a λ > 400 nm longpass filter and a 200 W Xe-lamp (—no product determined by GC).
Further evidence for molecular catalysis could be obtained by adding mercury to a catalysis solution with Co-LN3. Similar TONs were obtained indicating that Co colloids did not form. In case of Co-LN2S the ratio of CO:H2 changes to about 2:1 (i.e., 66 % selectivity for CO formation) in comparison to no H2 formation for Co-LN3. In case of Fe-LN2S the CO:H2 ratio stays very similar to the one of Co-LN2S, but the activity drops by about one third, and about half in comparison to Fe-LN3. For the Ni complexes the electrochemical data predicted no (photo)catalytic activity, because they do not show a reduction process in the solvent window investigated. This could be confirmed for Ni-LN3, whereas Ni-LN2S shows a moderate TONH2, which again is probably induced by the sulfur atom in the ligand system.
In order to investigate if the product ratio or the activity can be adjusted by adding an additional proton source, water was added to a Co-LN2S catalytic mixture in different concentrations. Thereby a shift to higher TONH2 is observable, while TONCO is decreasing (Supplementary Table S4). When adding 1 Vol.% water a ratio of CO:H2 18:24 was observed, while the addition of 5 Vol.% water led to a ratio of CO:H2 5:54. This result strikingly demonstrates that the product selectivity can be manipulated to a large amount by the addition of a proton source.
To investigate the influence of the type of PS, catalytic experiments were performed using Cu. This PS was chosen due to its similar absorption and redox features as compared to Ir (Table 1) (Giereth et al., 2021). Unexpectedly, the combination of Cu and Co-LN3 shows only a TONCO of 13 after 24 h of illumination, i.e., only a seventh part compared to the reaction using Ir. The much lower overall TONCO can be attributed to the rather fast photodecomposition of Cu (Giereth et al., 2021). In the first 2 hours of illumination, Cu actually shows a better performance than Ir (Supplementary Figure S25; TONCO of 9 vs. 3). However, there is almost no further increase in CO evolution for Cu indicating that most of the photosensitizer is decomposed already after a few hours of reaction time. The initial better performance of Cuvs.Ir is tentatively assigned to the more suitable excited state oxidation potential of Cu (Table 1) and, hence, more efficient oxidative quenching of the excited state by the catalyst (see next section).
Luminescence quenching experiments were performed to obtain some insight into the reaction mechanism. The excited state of the (Ir) photosensitizer can be quenched oxidatively by the catalyst Co-LN3 (Supplementary Figure S26; Table 1). Specifically, Stern-Volmer luminescence quenching experiments in acetonitrile yield a quenching rate constant kq = 1.1·109 M−1s−1 (for Ir), which is roughly a factor of seven (Montalti et al., 2006) below the diffusion limit. Under the catalytically relevant reaction conditions, 5·10−5 M Co-LN3 was present, and given a rate constant of 1.1·109 M−1 s−1 for oxidative Ir luminescence quenching, this leads to a pseudo first-order rate constant of 5.5·104 s−1 for photoinduced electron transfer from Ir to Co-LN3. Given an inherent excited-state decay of 5·105 s−1 (lifetime of Ir of about 2 μs) (Giereth et al., 2021) this implies that 11 in 100 photo-excitations (5.5·104 s−1/5·105 s−1 = 0.11) will lead to electron transfer from Ir to Co-LN3 under the catalytically relevant conditions with 5·10-5 M Co-LN3.
Similar bimolecular rate constants were determined by Chan and co-workers (Chan et al., 2015), who investigated the photocatalytic CO2 reduction in acetonitrile with [CoII(tpa)Cl]Cl (tpa = tris(2-pyridylmethyl)amine) and using Ir(ppy)3 (ppy = 2-phenylpyridine anion) as photocatalyst. Stern–Volmer analysis revealed quenching rate constants for the photosensitizer of 5.29 × 109 M−1s−1 for [CoII(tpa)Cl]Cl and 4.14 × 104 M−1s−1 for TEA, respectively. It was further noted that reductive quenching of excited [Ir(ppy)3] is not feasible from thermodynamic consideration (E1/2 [(Ir(ppy)3)*/(Ir(ppy)3)−] = +0.31 V vs. SCE) and, hence, oxidative quenching of the PS is the more favourable reaction path. These findings are also in line with previous studies by some of us (Giereth et al., 2021), in which we used Ir in the presence of a large excess of TEA and a dirhenium catalyst. In the photocatalytic experiments, Ir (E1/2[(Ir(dFppy)3)*/(Ir(dFppy)3)−] = +0.36 V vs. Fc/Fc+) (Teegardin et al., 2016) seemingly had no influence on the catalytic performance, because the dirhenium compound was itself an efficient photocatalyst and because of very inefficient reductive quenching of Ir by TEA. It is interesting to note that smaller quantities of a stronger reductant (such as BIH, 1,3-dimethyl-2-phenylbenzimidazoline) gave significantly greater turnover numbers and led to very fast CO2 transformation.
Though the fluorinated Ir complex is a somewhat stronger photo-oxidant than Ir(ppy)3 (+0.36 V vs SCE compared to +0.31 V vs SCE, see above) one may assume that the rate constant for bimolecular quenching of photoexcited Ir by TEA is on a similar order of magnitude, roughly 4.14·104 M−1s−1 (see above). Under the catalytically relevant reaction conditions in which 0.36 M TEA is present, this leads to a pseudo-first order rate constant of 1.5·104 s−1. Co-LN3 is present at 5·10-5 M concentration under the catalytically relevant conditions, and given a rate constant of 1.1·109 M−1s−1 for bimolecular electron transfer from photoexcited Ir to Co-LN3 (see above), one obtains a pseudo-first order rate constant of 5.5·104 s−1. Thus, under the assumption that reductive excited-state quenching of Ir by TEA is not more rapid than reductive quenching of [Ir(ppy)3] despite its 0.05 V higher oxidative power (see above), oxidative quenching of Ir by Co-LN3 is a factor of 3.6 (= 5.5·104 s−1/1.5·104 s−1) faster than reductive quenching by TEA.
Therefore we speculate that the reaction mechanism is as illustrated in Scheme 2. Following the absorption of light by the photosensitizer, the excited PS* transfers an electron to the catalyst. Subsequently, PS+ is reduced by TEA to reform PS, which then restarts the cycle (Pellegrin and Odobel, 2017). The reduced catalyst cat− is probably only able to interact with protons, derived from TEA decomposition, to form a hydride intermediate. Consecutive electron and proton transfer results in hydrogen formation (Dempsey et al., 2009; Darmon et al., 2014; Wiedner and Bullock, 2016). In order to be able to reduce CO2 to CO, the catalyst very likely needs to be present in the double reduced form cat2−. The double reduced species could form in a dark reaction by further reduction of cat− by the radical cation TEA•+, which is a potent reductant (Shimoda et al., 2018). Alternatively, cat2− could result from a disproportionation reaction (indicated in Scheme 2) as has been proposed for other 3d transition metal compounds (Takeda et al., 2017; Dalle et al., 2019). Such disproportionation reactions have likewise been demonstrated to play a key role in photoinduced charge accumulation (Skaisgirski et al., 2017). In another alternative, the attack of two single reduced catalyst species on CO2 could be a possible reaction path, as has been suggested for rhenium complexes (Morris et al., 2009), though this requires the formation of a ternary encounter complex in solution. Further investigations to elucidate the reaction mechanism by characterization and isolation of intermediates are currently performed.
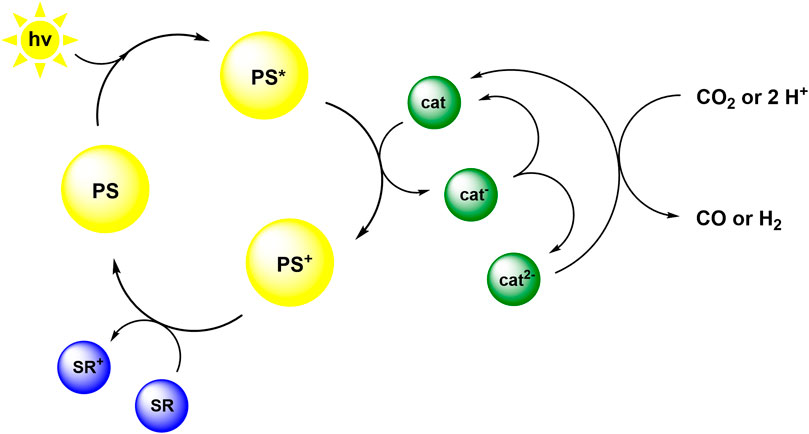
SCHEME 2. Proposed reaction mechanism based on oxidative quenching of an excited PS and disproportionation of the mono-reduced catalyst.
General procedure for the synthesis of complexes Fe-LN3 and Co-LN3. Macrocycle LN3 (1 eq.) was dissolved in MeCN (5 ml) and M(OTf)2 (1. eq) was added. After stirring for 3 days at room temperature, the solvent was removed. The resulting oil was washed with CH2Cl2 and toluene, dissolved in MeCN and precipitated using Et2O.
After purification Fe-LN3 was obtained as brownish red solid (38 mg, 54%).
ESI-MS: m/z: calc. for [M + OTf-]+, 516.0974; found, 516.0974.
UV/vis: λ 268, 312, 432 nm.
After purification Co-LN3 was obtained as pink solid (49 mg, 38%).
1H NMR (400 MHz, CD3CN): δ 9.24 (d, J = 5.8 Hz, 1H), 8.33 (td, J = 1.5, 7.8 Hz, 1H), 8.13 (td, J = 1.5, 7.8 Hz, 1Hpico), 7.86 (m, 2Hpico), 7.72 (d, J = 7.9 Hz, 1Hpico), 7.47 (t, J = 7.4, 1Hpico), 7.21 (d, J = 5.9 Hz, 1Hpico), 6.67 (s, NH), 5.09 (d, J = 16.5 Hz, 1Halkyl), 4.64 (d, J = 16.5 Hz, 1Halkyl), 4.46 (m, 2Halkyl), 4.10 (m, 2Hmacro), 3.79 (m, 3Hmacro), 3.45 (m, 1Hmacro), 3.31 (dd, J = 6.6, 15.2 Hz, 1Hmacro), 3.23 (dd, 5.8, 12.5 Hz, 1H), 3.01 (dd, 5.3, 13.7 Hz, 1Hmacro), 2.77 (td, 5.9, 13.7 Hz, 1Hmacro), 2.54 (dd, 5.9, 13.7 Hz, 1Hmacro), 2.01(m, 1Hmacro).13C NMR (400 MHz, CD3CN): δ 164.83 (Cquart), 162.48 (Cquart), 153.65 (Cpico), 149.45 (Cpico), 141.83 (CPico), 140.83 (CPico), 126.69 (CPico), 126.61 (CPico), 125.51 (CPico), 129.94 (CPico), 69.43 (Calkyl), 67.27 (Calkyl), 63.92 (Cmacro), 62.37 (Cmacro), 61.53 (Cmacro), 60.16 (Cmacro), 54.23 (Cmacro), 52.93 (Cmacro). 19F NMR (300 MHz, CD3CN): δ -76.95 (Ftriflat).
ESI-MS: m/z: calc. for [M + OTf-]+, 519.0956; found, 519.0975.
UV/Vis: 268, 362, 507 nm.
General procedure for the synthesis of Ni-LN3, Fe-LN2S, Co-LN2S and Ni-LN2S. The macrocycles LN3 and LN2S were dissolved in dry MeCN (5 ml) and M(OTf)2 (1 eq.) was added. The suspension was stirred for 3 days at room temperature and the solvent was removed subsequently. The resulting oil was washed with CH2Cl2 and toluene [Fe(OTf)2 and Co(OTf)2] or THF [Ni(OTf)2], dissolved in MeCN and the remaining chloride ions were precipitated using an excess of Ag(OTf). The filtrate was evaporated and afterwards the residue was taken up in a small amount of MeCN and precipitated using Et2O.
After purification Ni-LN3 was obtained as beige solid (39 mg, 30%).
ESI-MS: m/z: calc. for [M + OTf-]+, 518.0984; found, 518.1018.
UV/Vis: 268, 308 nm.
After purification Fe-LN2S was obtained as dark brown solid (34 mg, 46%).
ESI-MS: m/z: calc. for [M + OTf-]+, 533.0586; found, 533.0584.
UV/vis: 268, 490, 502 nm.
After purification Co-LN2S was obtained as red solid (31 mg, 50%).
ESI-MS: m/z: calc. for [M + OTf-]+, 536.0571; found, 536.0568.
UV/vis: 268, 490 nm.
After purification Ni-LN2S was obtained as light brown solid (28 mg, 44%).
ESI-MS: m/z: calc. for [M + OTf-]+, 535.0590; found, 535.0590.
UV/vis: 268 nm.
We herein describe the synthesis of two series of macrocyclic complexes M-LN3 and M-LN2S containing Fe, Co and Ni, which can be applied in the photocatalytic activation of CO2. Initially, an Ir photosensitizer was used and the combination of Ir and Co-LN3 showed the highest TONCO (90 after 24 h of illumination) of all investigated complexes. The ratio of the gaseous products CO and H2 can be varied by the choice of metal ion, macrocyclic ligand and solvent composition. The product selectivity can even be adjusted to almost solely formation of CO (in case of Co-LN3) or H2 (in case of Ni-LN2S). In addition, we were able to demonstrate that a Cu photosensitizer can be used for the catalytic reaction as well, making the whole system solely 3d metal based. The initially higher catalytic activity compared to Ir (TONCO of 9 vs. 3 after 2 h reaction time) can tentatively be assigned to the more positive excited state oxidation potential of Cu making the oxidative quenching process by the catalyst more efficient. On the contrary, the long-term stability of the Cu photosensitizer is clearly inferior compared to the Ir photosensitizer with a TONCO after 24 h of illumination of 13 vs. 90.
First mechanistic investigations confirm that an oxidative quenching mechanism is more likely than a reductive quenching process. Further experiments are on the way to confirm possible intermediates involved, such as a hydride species, and the influence of further reaction parameters on the outcome of the photocatalytic reaction, such as changing the sacrificial electron donor. The latter might also influence the quenching process and thus the overall TON and product selectivity.
The raw data supporting the conclusion of this article are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
MO was carrying out the synthetical and experimental work and wrote the manuscript, FB was measuring and solving the crystallographic data, RSS was executing the photophysical quenching studies with OSW supervising the photophysical experiments and editing the manuscript, MS was supervising the work and editing the manuscript.
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) via the priority program 2102 Light-controlled reactivity of metal complexes (DFG SCHW1454/9-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the Humboldt Universität zu Berlin for the generous support of this project. We further thank the SPP 2102 (see Funding) for financial support. Furthermore, the authors want to thank Beatrice Cula for supervising the X-ray diffractometric analysis and Beatrice Battistella for performing the EPR measurements.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.751716/full#supplementary-material
Aresta, M. (2010). Carbon Dioxide as Chemical Feedstock. Weinheim, Germany: Wiley‐Vch Verlag Gmbh & Co. Kgaa.
Becker, M. R., Wearing, E. R., and Schindler, C. S. (2020). Synthesis of Azetidines via Visible-Light-Mediated Intermolecular [2+2] Photocycloadditions. Nat. Chem. 12 (10), 898–905. doi:10.1038/s41557-020-0541-1
Benson, E. E., Kubiak, C. P., Sathrum, A. J., and Smieja, J. M. (2009). Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 38 (1), 89–99. doi:10.1039/b804323j
Benvenuti, M., Meneghello, M., Guendon, C., Jacq-Bailly, A., Jeoung, J.-H., Dobbek, H., et al. (2020). The Two CO-dehydrogenases of Thermococcus Sp. AM4. Biochim. Biophys. Acta (Bba) - Bioenerg. 1861 (7), 148188. doi:10.1016/j.bbabio.2020.148188
Cao, R., Müller, P., and Lippard, S. J. (2010). Tripodal Tris-Tacn and Tris-Dpa Platforms for Assembling Phosphate-Templated Trimetallic Centers. J. Am. Chem. Soc. 132 (49), 17366–17369. doi:10.1021/ja108212v
Chak, B., McAuley, A., and Whitcombe, T. W. (1994). Crystal and Solution Structure of a Pendant-Armed Macrocyclic Complex of Palladium(II). Can. J. Chem. 72 (6), 1525–1532. doi:10.1139/v94-189
Chan, S. L.-F., Lam, T. L., Yang, C., Yan, S.-C., and Cheng, N. M. (2015). A Robust and Efficient Cobalt Molecular Catalyst for CO2 Reduction. Chem. Commun. 51 (37), 7799–7801. doi:10.1039/c5cc00566c
Chen, L., Chen, G., Leung, C.-F., Yiu, S.-M., Ko, C.-C., Anxolabéhère-Mallart, E., et al. (2014). Dual Homogeneous and Heterogeneous Pathways in Photo- and Electrocatalytic Hydrogen Evolution with Nickel(II) Catalysts Bearing Tetradentate Macrocyclic Ligands. ACS Catal. 5 (1), 356–364. doi:10.1021/cs501534h
Dalle, K. E., Warnan, J., Leung, J. J., Reuillard, B., Karmel, I. S., and Reisner, E. (2019). Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 119 (4), 2752–2875. doi:10.1021/acs.chemrev.8b00392
Darmon, J. M., Raugei, S., Liu, T., Hulley, E. B., Weiss, C. J., Bullock, R. M., et al. (2014). Iron Complexes for the Electrocatalytic Oxidation of Hydrogen: Tuning Primary and Secondary Coordination Spheres. ACS Catal. 4 (4), 1246–1260. doi:10.1021/cs500290w
Dempsey, J. L., Brunschwig, B. S., Winkler, J. R., and Gray, H. B. (2009). Hydrogen Evolution Catalyzed by Cobaloximes. Acc. Chem. Res. 42 (12), 1995–2004. doi:10.1021/ar900253e
Drosou, M., Kamatsos, F., and Mitsopoulou, C. A. (2020). Recent Advances in the Mechanisms of the Hydrogen Evolution Reaction by Non-innocent Sulfur-Coordinating Metal Complexes. Inorg. Chem. Front. 7 (1), 37–71. doi:10.1039/c9qi01113g
Esselborn, J., Muraki, N., Klein, K., Engelbrecht, V., Metzler-Nolte, N., Apfel, U.-P., et al. (2016). A Structural View of Synthetic Cofactor Integration into [FeFe]-Hydrogenases. Chem. Sci. 7 (2), 959–968. doi:10.1039/c5sc03397g
Francke, R., Schille, B., and Roemelt, M. (2018). Homogeneously Catalyzed Electroreduction of Carbon Dioxide-Methods, Mechanisms, and Catalysts. Chem. Rev. 118 (9), 4631–4701. doi:10.1021/acs.chemrev.7b00459
Friscourt, F., Fahrni, C. J., and Boons, G.-J. (2012). A Fluorogenic Probe for the Catalyst-free Detection of Azide-Tagged Molecules. J. Am. Chem. Soc. 134 (45), 18809–18815. doi:10.1021/ja309000s
Gerschel, P., Warm, K., Farquhar, E. R., Englert, U., Reback, M. L., Siegmund, D., et al. (2019). Apfel, U. P.Sulfur Substitution in a Ni(cyclam) Derivative Results in Lower Overpotential for CO2 Reduction and Enhanced Proton Reduction. Dalton Trans. 48 (18), 5923–5932. doi:10.1039/c8dt04740e
Ghosh, D., Sinhababu, S., Santarsiero, B. D., and Mankad, N. P. (2020). A W/Cu Synthetic Model for the Mo/Cu Cofactor of Aerobic CODH Indicates that Biochemical CO Oxidation Requires a Frustrated Lewis Acid/Base Pair. J. Am. Chem. Soc. 142 (29), 12635–12642. doi:10.1021/jacs.0c03343
Giereth, R., Obermeier, M., Forschner, L., Karnahl, M., Schwalbe, M., and Tschierlei, S. (2021). Exploring the Full Potential of Photocatalytic Carbon Dioxide Reduction Using a Dinuclear Re2Cl2 Complex Assisted by Various Photosensitizers. ChemPhotoChem 5, 644–653. doi:10.1002/cptc.202100034
Gotico, P., Del Vecchio, A., Audisio, D., Quaranta, A., Halime, Z., Leibl, W., et al. (2018). Visible-Light-Driven Reduction of CO2 to CO and its Subsequent Valorization in Carbonylation Chemistry and 13C Isotope Labeling. ChemPhotoChem 2 (8), 715–719. doi:10.1002/cptc.201800012
Guo, Z., Cheng, S., Cometto, C., Anxolabéhère-Mallart, E., Ng, S.-M., Ko, C.-C., et al. (2016). Highly Efficient and Selective Photocatalytic CO2 Reduction by Iron and Cobalt Quaterpyridine Complexes. J. Am. Chem. Soc. 138 (30), 9413–9416. doi:10.1021/jacs.6b06002
Hong, D., Kawanishi, T., Tsukakoshi, Y., Kotani, H., Ishizuka, T., and Kojima, T. (2019). Efficient Photocatalytic CO2 Reduction by a Ni(II) Complex Having Pyridine Pendants through Capturing a Mg2+ Ion as a Lewis-Acid Cocatalyst. J. Am. Chem. Soc. 141 (51), 20309–20317. doi:10.1021/jacs.9b10597
Hong, D., Tsukakoshi, Y., Kotani, H., Ishizuka, T., and Kojima, T. (2017). Visible-Light-Driven Photocatalytic CO2 Reduction by a Ni(II) Complex Bearing a Bioinspired Tetradentate Ligand for Selective CO Production. J. Am. Chem. Soc. 139 (19), 6538–6541. doi:10.1021/jacs.7b01956
Iffland, L., Siegmund, D., and Apfel, U. P. (2020). Electrochemical CO2 and Proton Reduction by a Co(dithiacyclam) Complex. Z. Anorg. Allg. Chem. 646 (13), 746–753. doi:10.1002/zaac.201900356
Koike, T., and Akita, M. (2014). Visible-light Radical Reaction Designed by Ru- and Ir-Based Photoredox Catalysis. Inorg. Chem. Front. 1 (8), 562–576. doi:10.1039/c4qi00053f
Kumar, B., Brian, J. P., Atla, V., Kumari, S., Bertram, K. A., White, R. T., et al. (2016). New Trends in the Development of Heterogeneous Catalysts for Electrochemical CO2 Reduction. Catal. Today 270, 19–30. doi:10.1016/j.cattod.2016.02.006
Lee, S., and Han, W.-S. (2020). Cyclometalated Ir(III) Complexes towards Blue-Emissive Dopant for Organic Light-Emitting Diodes: Fundamentals of Photophysics and Designing Strategies. Inorg. Chem. Front. 7 (12), 2396–2422. doi:10.1039/d0qi00001a
Liu, D.-C., Wang, H.-J., Wang, J.-W., Zhong, D.-C., Jiang, L., and Lu, T.-B. (2018). Highly Efficient and Selective Visible-Light Driven CO2-to-CO Conversion by a Co-based Cryptate in H2O/CH3CN Solution. Chem. Commun. 54 (80), 11308–11311. doi:10.1039/c8cc04892d
Lubitz, W., Ogata, H., Rüdiger, O., and Reijerse, E. (2014). Hydrogenases. Chem. Rev. 114 (8), 4081–4148. doi:10.1021/cr4005814
Luk’yanenko, N. G., Basok, S. S., Filonova, L. K., Kulikov, N. V., and Pastushok, V. N. (1990). Macroheterocycles. 51. Synthesis of Macrocyclic Polyamines in a Biphasic System. Chem. Heterocycl. Comp. 26, 346–349. doi:10.1007/BF00472559
Ma, J., Sun, N., Zhang, X., Zhao, N., Xiao, F., Wei, W., et al. (2009). A Short Review of Catalysis for CO2 Conversion. Catal. Today 148 (3-4), 221–231. doi:10.1016/j.cattod.2009.08.015
McLachlan, G. A., Brudenell, S. J., Fallon, G. D., Martin, R. L., Spiccia, L., and Tiekink, E. R. T. (1995). Synthesis, Structure and Properties of Cobalt(III) Complexes of Pentadentate Ligands with Pyridyl Pendant Arms. J. Chem. Soc. Dalton Trans. 1995 (1), 439–447. doi:10.1039/DT9950000439
Montalti, M., Credi, A., Prodi, L., and Gandolfi, M. T. (2006). Handbook of Photochemistry. 3rd Edition. Boca Raton, Florida: CRC Press, 664. doi:10.1201/9781420015195
Morris, A. J., Meyer, G. J., and Fujita, E. (2009). Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels. Acc. Chem. Res. 42 (12), 1983–1994. doi:10.1021/ar9001679
Pellegrin, Y., and Odobel, F. (2017). Sacrificial Electron Donor Reagents for Solar Fuel Production. Comptes Rendus Chim. 20 (3), 283–295. doi:10.1016/j.crci.2015.11.026
Ringsmuth, A. K., Landsberg, M. J., and Hankamer, B. (2016). Can Photosynthesis Enable a Global Transition from Fossil Fuels to Solar Fuels, to Mitigate Climate Change and Fuel-Supply Limitations? Renew. Sustain. Energ. Rev. 62, 134–163. doi:10.1016/j.rser.2016.04.016
Schrag, D. P. (2007). Preparing to Capture Carbon. Science 315 (5813), 812–813. doi:10.1126/science.1137632
Shimoda, T., Morishima, T., Kodama, K., Hirose, T., Polyansky, D. E., Manbeck, G. F., et al. (2018). Photocatalytic CO2 Reduction by Trigonal-Bipyramidal Cobalt(II) Polypyridyl Complexes: The Nature of Cobalt(I) and Cobalt(0) Complexes upon Their Reactions with CO2, CO, or Proton. Inorg. Chem. 57 (9), 5486–5498. doi:10.1021/acs.inorgchem.8b00433
Skaisgirski, M., Guo, X., and Wenger, O. S. (2017). Electron Accumulation on Naphthalene Diimide Photosensitized by [Ru(2,2′-Bipyridine)3]2+. Inorg. Chem. 56 (5), 2432–2439. doi:10.1021/acs.inorgchem.6b02446
Spiccia, L., Fallon, G. D., Grannas, M. J., Nichols, P. J., and Tiekink, E. R. T. (1998). Synthesis and Characterisation of Mononuclear and Binuclear Iron(II) Complexes of Pentadentate and Bis(pentadentate) Ligands Derived from 1,4,7-triazacyclononane. Inorg. Chim. Acta 279, 192–199. doi:10.1016/S0020-1693(98)00122-4
Srinivasan, C., and Butler Gralla, E. (2002). Measurement of "Free" or Electron Paramagnetic Resonance-Detectable Iron in Whole Yeast Cells as Indicator of Superoxide Stress. Method. Enzymol. 349, 173–180. doi:10.1016/S0076-6879(02)49333-0
Stavila, V., Allali, M., Canaple, L., Stortz, Y., Franc, C., Maurin, P., et al. (2008). Significant Relaxivity gap between a Low-Spin and a High-Spin Iron(ii) Complex of Structural Similarity: an Attractive Off-On System for the Potential Design of Responsive MRI Probes. New J. Chem. 32 (3), 428–435. doi:10.1039/b715254j
Takeda, H., Cometto, C., Ishitani, O., and Robert, M. (2017). Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 7 (1), 70–88. doi:10.1021/acscatal.6b02181
Tamaki, Y., and Ishitani, O. (2017). Supramolecular Photocatalysts for the Reduction of CO2. ACS Catal. 7 (5), 3394–3409. doi:10.1021/acscatal.7b00440
Tamaki, Y., Koike, K., and Ishitani, O. (2015). Highly Efficient, Selective, and Durable Photocatalytic System for CO2 Reduction to Formic Acid. Chem. Sci. 6 (12), 7213–7221. doi:10.1039/c5sc02018b
Tamaki, Y., Koike, K., Morimoto, T., and Ishitani, O. (2013). Substantial Improvement in the Efficiency and Durability of a Photocatalyst for Carbon Dioxide Reduction Using a Benzoimidazole Derivative as an Electron Donor. J. Catal. 304, 22–28. doi:10.1016/j.jcat.2013.04.002
Teegardin, K., Day, J. I., Chan, J., and Weaver, J. (2016). Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process. Res. Dev. 20 (7), 1156–1163. doi:10.1021/acs.oprd.6b00101
Thoi, V. S., Kornienko, N., Margarit, C. G., Yang, P., and Chang, C. J. (2013). Visible-light Photoredox Catalysis: Selective Reduction of Carbon Dioxide to Carbon Monoxide by a Nickel N-Heterocyclic Carbene-Isoquinoline Complex. J. Am. Chem. Soc. 135 (38), 14413–14424. doi:10.1021/ja4074003
Wasielewski, K., and Mattes, R. (1993). Nickel-, Palladium- und Platinkomplexe funktionalisierter Makrocyclen. Die Kristallstrukturen von [Ni(py2-tasn)(H2O)](ClO4)2, [Pd(py2-tasn)](PF6)2 und [Pt(py2-tasn)](PF6)2. (py2-tasn=4,7-Bis(2-methylpyridyl)-1-thia-4,7-diazacyclononan. Z. Anorg. Allg. Chem. 619, 158–162. doi:10.1002/zaac.19936190126
Wiedner, E. S., and Bullock, R. M. (2016). Electrochemical Detection of Transient Cobalt Hydride Intermediates of Electrocatalytic Hydrogen Production. J. Am. Chem. Soc. 138 (26), 8309–8318. doi:10.1021/jacs.6b04779
Wilson, J. (2007). Synthesis of Biologically Active Heterocyclic Compounds. Great Britain: University of Glasgow. Available at: http://theses.gla.ac.uk/45/.
Keywords: photocatalysis, carbon dioxide reduction, 3d metal complexes, electron transfer, sulfur containing ligand
Citation: Obermeier M, Beckmann F, Schaer RS, Wenger OS and Schwalbe M (2021) Sensitized Photocatalytic CO2 Reduction With Earth Abundant 3d Metal Complexes Possessing Dipicolyl-Triazacyclononane Derivatives. Front. Chem. 9:751716. doi: 10.3389/fchem.2021.751716
Received: 01 August 2021; Accepted: 14 September 2021;
Published: 30 September 2021.
Edited by:
Souvik Roy, University of Lincoln, United KingdomReviewed by:
Jared H. Delcamp, University of Mississippi, United StatesCopyright © 2021 Obermeier, Beckmann, Schaer, Wenger and Schwalbe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Schwalbe, bWF0dGhpYXMuc2Nod2FsYmVAaHUtYmVybGluLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.