 Min-Ye Zhang
Min-Ye Zhang Hong Jiang
Hong Jiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 28 September 2021
Sec. Theoretical and Computational Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.747972
This article is part of the Research Topic Many-Body Green’s Functions and the Bethe-Salpeter Equation in Chemistry: From Single Molecules to Complex Systems View all 10 articles
The pyrite and marcasite polymorphs of FeS2 have attracted considerable interests for their potential applications in optoelectronic devices because of their appropriate electronic and optical properties. Controversies regarding their fundamental band gaps remain in both experimental and theoretical materials research of FeS2. In this work, we present a systematic theoretical investigation into the electronic band structures of the two polymorphs by using many-body perturbation theory with the GW approximation implemented in the full-potential linearized augmented plane waves (FP-LAPW) framework. By comparing the quasi-particle (QP) band structures computed with the conventional LAPW basis and the one extended by high-energy local orbitals (HLOs), denoted as LAPW + HLOs, we find that one-shot or partially self-consistent GW (G0W0 and GW0, respectively) on top of the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation with a converged LAPW + HLOs basis is able to remedy the artifact reported in the previous GW calculations, and leads to overall good agreement with experiment for the fundamental band gaps of the two polymorphs. Density of states calculated from G0W0@PBE with the converged LAPW + HLOs basis agrees well with the energy distribution curves from photo-electron spectroscopy for pyrite. We have also investigated the performances of several hybrid functionals, which were previously shown to be able to predict band gaps of many insulating systems with accuracy close or comparable to GW. It is shown that the hybrid functionals considered in general fail badly to describe the band structures of FeS2 polymorphs. This work indicates that accurate prediction of electronic band structure of FeS2 poses a stringent test on state-of-the-art first-principles approaches, and the G0W0 method based on semi-local approximation performs well for this difficult system if it is practiced with well-converged numerical accuracy.
Iron disulfide FeS2 was studied extensively in the last century in the desire of understanding the structural and electronic properties of transition metal dichalcogenides (TMDC) featuring localized or band-like d electrons (Hulliger and Mooser, 1965a; Hulliger and Mooser, 1965b; Goodenough, 1972; Wilson, 1972; Li et al., 1974; Schlegel and Wachter, 1976; Folkerts et al., 1987). Since 1980s (Chatzitheodorou et al., 1986; Ennaoui et al., 1986), increasing practical interest has been drawn to pyrite FeS2 for its potential as a cheap and competitive candidate material for efficient solar energy conversion (Wadia et al., 2009) because of its natural abundance, non-toxicity, suitable optical gap and extraordinarily large absorption coefficient (Ferrer et al., 1990; Ennaoui et al., 1993). This has led to new solutions under various optoelectronic scenarios, including photovoltaics (Khalid et al., 2018), photo-catalysis (Tian et al., 2015; Barawi et al., 2016), solid-state photo-capacitors (Gong et al., 2013a) and photo-detectors (Wang et al., 2012; Gong et al., 2013b). However, practical application of FeS2-based optoelectronic devices is seriously hampered by its unexpected low efficiency due to a loss of open-circuit voltage VOC (Cabán-Acevedo et al., 2014). A number of factors possibly responsible for the low VOC have been suggested and examined, including the intrinsic and defect surface states (Bronold et al., 1994; Sun et al., 2011; Herbert et al., 2013; Lazić et al., 2013; Cabán-Acevedo et al., 2014; Limpinsel et al., 2014; Walter et al., 2017), bulk sulfur deficiency (Birkholz et al., 1991; Cabán-Acevedo et al., 2014; Shukla et al., 2016) and presence of the metastable marcasite phase as a small-gap impurity (Spagnoli et al., 2010; Sun et al., 2011; Schena et al., 2013).
Despite progress towards understanding the origin of the low VOC in pyrite FeS2 (Rahman et al., 2020), consensus is still not reached on the fundamental band gaps of the two FeS2 phases. Experimentally, values varying from 0.6 to 2.6 eV have been reported for pyrite, primarily due to differences in sample preparation, measuring technique, and analytical model of spectra used in experimental studies (Ferrer et al., 1990; Ennaoui et al., 1993). Measurements of the pyrite band gap are generally carried out through optical absorption spectroscopy (Schlegel and Wachter, 1976; Kou and Seehra, 1978; Ennaoui et al., 1993), which features the neutral excitation (exciton) instead of the charged one as in the photo-electron spectroscopy (PES). Therefore, the measured excitation energies are in fact coupled to the electron-hole binding. Careful investigation by absorption spectroscopy for the marcasite phase is done only recently and gives an optical gap similar to pyrite, which essentially precludes the possibility of marcasite being the culprit for the low VOC of FeS2 photovoltaics (Sánchez et al., 2016; Wu et al., 2016). Furthermore, even though PES measurements of pyrite FeS2 have been conducted (Ohsawa et al., 1974; van der Heide et al., 1980; Folkerts et al., 1987; Mamiya et al., 1997; Ollonqvist et al., 1997; Nesbitt et al., 2003), combined studies of direct and inverse PES (IPS) for regions near the Fermi level are rare. Reported relevant works (Folkerts et al., 1987; Mamiya et al., 1997) were done more than 20 years ago and the spectra were not resolved enough to identify a well-defined fundamental band gap.
Difficulties in characterizing band structures of FeS2 polymorphs are also encountered from the perspective of first-principles calculations. Within the framework of density functional theory (DFT) (Hohenberg and Kohn, 1964), calculations with Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA) (Perdew et al., 1996a) predict pyrite to have a band gap of about 0.3 eV smaller than the experimental value of 0.95 eV as generally accepted (Ennaoui et al., 1993; Schena et al., 2013; Kolb and Kolpak, 2013; Li et al., 2015). Considering the well-known band gap problem of local density approximation (LDA) or GGA (Perdew et al., 1982), orbital-dependent functionals in spirit of generalized Kohn-Sham (GKS) DFT (Seidl et al., 1996; Perdew et al., 2017; Zhang et al., 2020) are also employed to tackle the problem, e.g. PBE plus the Hubbard-U correction (DFT + U) and hybrid functionals (Becke, 1993a; Becke, 1993b; Perdew et al., 1996b). Using an ad hocU of 2 eV, the PBE + U method is able to reproduce the experimental band gap (Sun et al., 2011; Hu et al., 2012; Li et al., 2018) but meanwhile deteriorates the simulated optical spectra compared to PBE (Choi et al., 2012; Schena et al., 2013). Furthermore, despite the good performance in predicting band gaps for typical semiconducting materials (Heyd et al., 2005; Paier et al., 2006b,a; Marsman et al., 2008), hybrid functionals such as Heyd-Scuseria-Ernzerhof (HSE) method (Heyd et al., 2003, 2006) have been shown to give large band gaps for pyrite of over 2 eV (Muscat et al., 2002; Sun et al., 2011; Choi et al., 2012; Hu et al., 2012; Schena et al., 2013; Liu et al., 2019). There are also works using beyond-DFT methods, particularly, the GW method based on many-body perturbation theory (MBPT) (Hedin, 1965). However, the GW results for the pyrite phase are rather scattered, ranging from 0.3 to 1.1 eV (Choi et al., 2012; Lehner et al., 2012; Kolb and Kolpak, 2013; Schena et al., 2013). It is worth noting that Schena and coworkers conducted the state-of-the-art all-electron G0W0 calculations with the linearized augmented plane-wave (LAPW) basis for both pyrite and marcasite, and report a pyrite band gap only about 0.3 eV (Schena et al., 2013). The GW gap value is smaller than that from PBE, which is rarely observed in GW practices and hence deserves closer investigation.
For GW implementations involving explicit summation of states, it is established recently by a number of works (Friedrich et al., 2006; Friedrich et al., 2011a; Friedrich et al., 2011b; Klimes et al., 2014; Jiang and Blaha, 2016; Nabok et al., 2016; Jiang, 2018; Zhang and Jiang, 2019; Ren et al., 2021) that an accurate description of high-lying empty states is essential to give accurate correlation self-energy operator and consequent QP band structure. In the pseudo-potential framework, one can improve the accuracy by using a norm-conserving potential with specifically tailored projectors at high energies (Klimes et al., 2014; van Setten et al., 2018). In all-electron calculations with the LAPW basis set, local orbitals with large energy parameters (usually 101∼2 Ry higher than the Fermi level) are introduced as additional basis functions to remove the linearization error in unoccupied states up high in the conduction band regime (Friedrich et al., 2006, 2011a,b; Jiang and Blaha, 2016; Nabok et al., 2016). The LAPW basis extended by these high-energy local orbitals (HLOs), termed as LAPW + HLOs, has succeeded in helping produce accurate QP band structures in good agreement with experiment for a variety of semiconductors (Jiang and Blaha, 2016) including the conventionally challenging systems such as ZnO (Friedrich et al., 2011a; Friedrich et al., 2011b; Stankovski et al., 2011; Jiang and Blaha, 2016; Nabok et al., 2016), d/f-electron oxides (Jiang, 2018) and cuprous and silver halides (Zhang and Jiang, 2019). Particularly, the effects of including HLOs on the QP correction have been demonstrated quantitatively to be larger for states with stronger metal-d characters (Zhang and Jiang, 2019). For the FeS2 polymorphs with states of significant Fe-3d characters in both valence and low-energy conduction band regimes, GW with LAPW + HLOs is likely to give better description of the QP energies and dispersion relation than that with the standard LAPW basis.
A competitive alternative in the DFT framework to GW for band structure prediction is the doubly screened hybrid (DSH) functional method (Cui et al., 2018) in the category of hybrid functionals with system-dependent parameters (Zhang et al., 2020). Derived from a model dielectric function (Cappellini et al., 1993; Shimazaki and Yoshihiro, 2008), the exchange-correlation potential in DSH can be regarded as a further approximation to the Coulomb hole and screened exchange (COHSEX) approximation to the GW self-energy, and is able to capture both dielectric and metallic screening in the exchange interaction (Cui et al., 2018). It is shown that the DSH can evaluate band gaps of typical sp semiconductors with accuracy comparable to GW with the LAPW + HLOs basis while only at modest computational cost (Cui et al., 2018). Furthermore, the one-shot variant DSH0 can outperform fixed-parameter hybrid functionals for band gap predictions in a wide range of materials including narrow-gap semiconductors and transition metal mono-oxides (Cui et al., 2018; Liu et al., 2020). Hence we consider DSH as a hopeful approach to solve the FeS2 band gap puzzle within the GKS framework of DFT.
In the present work, we investigate the electronic band structures of the pyrite and marcasite polymorphs of FeS2 by applying the state-of-the-art all-electron GW method with the LAPW + HLOs basis. For comparison, we examine the results from GW with the standard LAPW basis as well. We also investigate the performances of several hybrid functionals, including PBE0 (Perdew et al., 1996b), HSE06 (Heyd et al., 2003, Heyd et al., 2006), screened-exchange-PBE hybrid functional (SX-PBE) (Bylander and Kleinman, 1990; Seidl et al., 1996) and DSH (Cui et al., 2018), in attempt to obtain insights into the failure of the conventional fixed-parameter functionals in predicting the band gap of FeS2.
The central task of the GW method is to solve the quasi-particle (QP) equation with the self-energy operator Σ in the frequency domain expressed as (Hedin, 1965)
where G is the time-ordered Green’s function
With ψnk and εnk being the wave function and energy of the single-particle state
where v (r, r′) = 1/|r − r′| is the bare Coulomb interaction and ε(r, r′; ω) is the microscopic dielectric function calculated at the level of random phase approximation (RPA). In principle, Eqs 1–3 have to be solved self-consistently along with the Dyson equation for the Green’s function (Hedin, 1965). However, due to the computational cost and generally unsatisfactory results of the fully self-consistent GW for solids [e.g. Grumet et al. (2018)], one usually turns to the non-self-consistent variant G0W0. Considering the resemblance of KS and QP wave functions in weakly correlated systems (Hybertsen and Louie, 1986), the self-energy or QP energy
where Vxc is the KS exchange-correlation potential and Znk a renormalization factor. One can further perform the so-called energy-only self-consistent GW0 calculations, where QP energies
In the all-electron framework with LAPW, KS wave functions are expanded by the LAPW basis (Andersen, 1975; Singh and Nordström, 2006; Blaha et al., 2020)
where Vα is the region enclosed by the muffin-tin (MT) sphere of atom α centered at rα with radius
where
HLOs fall into the category of LOs with
In the current implementation, HLOs are generated systematically by following the way described by Laskowski and Blaha (2012). The quality of LAPW + HLOs is controlled by two parameters besides those for the LAPW basis, namely, the additional number of nodes in the radial function of highest energy local orbital with respect to that of the LAPW function with the same angular quantum number and the maximum angular quantum number of used HLOs, denoted as nLO and
Hybrid functionals have been widely used in first-principles simulations of condensed matter for their good balance between performance and computational cost, and have been actively developed to further exploit the potential of its particular functional form. Readers interested in detailed description on the current status of hybrid functional development are directed to several recent reviews (Kümmel and Kronik, 2008; Baer et al., 2010; Maier et al., 2019; Zhang et al., 2020). Here we briefly introduce the general formalism of the range-separated hybrid functionals and the variants relevant to the current study.
The essential ingredient in hybrid functional methods is the exchange-correlation energy Exc or potential Vxc composed of non-local orbital-dependent (screened) Hartree-Fock (HF) exchange terms. In the present work, we focus on hybrid functionals with Vxc in the range-separated form as (Zhang et al., 2020)
where
In Eq. 8vsr (r, r′; μ) denotes the short-ranged Coulomb interaction of a certain form characterized by screening parameter μ (Zhang et al., 2020). x denotes collectively the spatial and spin coordinates of an electron, x ≡ (r, σ).
Conventionally, the parameters are determined by either theoretical analysis or fitting against some dataset of particular properties, and then applied to other systems as fixed. Famous examples of the fixed-parameter hybrid functionals include PBE0 αsr = αlr = 1/4 (Perdew et al., 1996b) and the HSE series αsr = 1/4, αlr = 0, μ = 0.2–0.3 Å-1 (Heyd et al., 2003; Heyd et al., 2006). Recently, hybrid functionals with system-dependent parameters are developed by several groups (Shimazaki and Yoshihiro, 2008; Marques et al., 2011; Kronik et al., 2012; Koller et al., 2013; Skone et al., 2014; Chen et al., 2018; Cui et al., 2018). Among different methods, the doubly screened hybrid (DSH) functional has been demonstrated as a competitive candidate for accurate description of band structures of both wide- and narrow-gap semiconductors (Cui et al., 2018). The underlying idea of DSH is to approximate the screening effect in solids by employing the Bechstedt model dielectric function (Bechstedt et al., 1992)
where ɛM is the macroscopic dielectric constant, qTF the Thomas-Fermi wave vector and α an empirical parameter chosen for semiconductors (Cappellini et al., 1993). A screened Coulomb interaction can be derived from this model to take both dielectric and metallic screening into account, leading to parameters in Eq. 7 as
The corresponding short-ranged Coulomb interaction in Eq. 8 is
where erfc is the complementary error function. In practice, an initial ɛM is required, which can be obtained from the PBE calculation or experimental measurements, to construct the DSH potential and solve the GKS equation. The resulting single-particle states act as the inputs to compute a new ɛM, which is in turn used to update the DSH potential. The self-consistent loop stops when ɛM is converged. Alternatively, one can break after solving the GKS equation with the initial ɛM, leading to the one-shot scheme denoted as DSH0.
The unit cells of pyrite and marcasite FeS2 used in our calculations are shown in the left panel of Figure 1. The crystal structure of pyrite FeS2 (Figure 1A) can be viewed as a faced-centered cubic cell of Fe atoms with S2 dumbbells occupying the octahedral interstitials and pointing to different <111> crystallographic axes. The anion coordination octahedra (FeS6) are connected only through sharing vertices. In the orthorhombic marcasite phase (Figure 1C), (FeS6) are connected by sharing edges with the two neighbors along c-axis and linked together through sharing vertices on the aOb plane. In terms of lattice parameters, we use a = 5.418 Å, u = 0.3850 for pyrite (space group
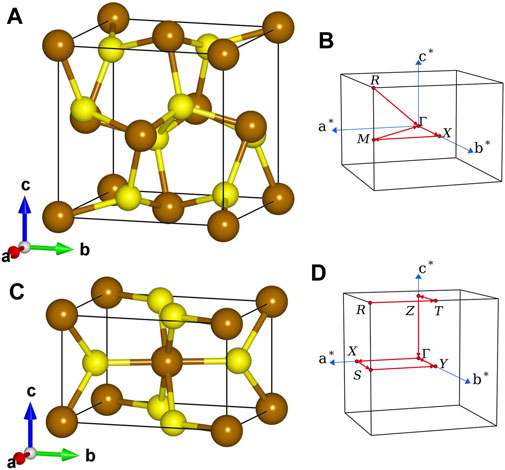
FIGURE 1. Lattice structures (A,C) and first Brillouin zones (B,D) of pyrite (upper) and marcasite (lower) phases of FeS2. Brown and yellow spheres represent Fe and S atoms, respectively.
The present all-electron GW calculations are performed by the GW facilities in the GAP2 program (Jiang et al., 2013; Jiang and Blaha, 2016) interfaced to WIEN2k (Blaha et al., 2001, 2020). Results in both G0W0 and GW0 schemes are presented, where KS orbital energies and wave functions calculated with the PBE (Perdew et al., 1996a) GGA are used as the input to construct one-body Green’s function and screened Coulomb interaction. The KS states are obtained by using charge density pre-converged under self-consistent field (SCF) calculation with PBE and the standard LAPW basis. The energy criterion for convergence of SCF iterations is set to 10–8 Rydberg (Ry). 64 (4 × 4 × 4) and 120 (5 × 4 × 6) k points are sampled in the first Brillouin zones of pyrite and marcasite FeS2, respectively. All available unoccupied states are considered in the summation of states for screened Coulomb interaction W and self-energy Σ. Mixed product basis is used to describe the wave function products in the two-point functions, e.g. W and Σ (Aryasetiawan and Gunnarsson, 1994; Kotani and van Schilfgaarde, 2002). We choose Q = 0.75 and
In terms of the LAPW basis, the usual or standard LAPW basis set is created automatically in the recent version of WIEN2k (Blaha et al., 2001), which is actually a mixture of the APW + lo basis for the valence states (Madsen et al., 2001), the ordinary LAPW basis for higher l channels up to lmax = 10 and additional local orbitals (LOs) for semi-core Fe-3s and Fe-3p states (Blaha et al., 2001). The convergence with respect to the two HLOs parameters nLO and
For hybrid functional calculations, we consider PBE0 (Perdew et al., 1996b), HSE06 (Heyd et al., 2003, 2006) and screened exchange SX-PBE (Bylander and Kleinman, 1990) methods as well as DSH. All hybrid functional calculations are performed with the projector augmented waves (PAW) method (Blöchl, 1994) implemented in the Vienna ab-initio Simulation Package (VASP) (Kresse and Furthmüller, 1996). The static dielectric function is calculated from the average of diagonal elements of macroscopic dielectric tensor computed by using density functional perturbation theory (DFPT) with local field effect included (Baroni et al., 2001). Apart from 3d and 4s, the 3s and 3p electrons of Fe are also treated explicitly in the valence region. The Thomas-Fermi wave vectors are 2.57 and 2.56 Å-1 for pyrite and marcasite FeS2, respectively. The cut-off energy of plane-wave basis for wave function expansion is chosen as 400 eV, which is sufficient to converge
In this part, we present the electronic band structures of pyrite and marcasite FeS2 computed by the all-electron GW method. In particular, we analyse the effect of high-energy local-orbitals (HLOs) by comparing the results from GW with the standard LAPW and LAPW + HLOs basis.
To achieve a balance between the computational cost and numerical accuracy of the LAPW + HLOs based GW method, we have to decide an optimized HLOs setup for the FeS2 polymorphs of interest. That is to say, certain convergence with respect to the two HLOs parameters, namely nLO and ΔlLO, must be achieved for the QP band structures of both polymorphs, while the number of basis functions should be kept as few as possible. To simplify the notation, we denote the setup of HLOs by (nLO, ΔlLO) so that (1, 1) indicates a set of HLOs with nLO = 1 and ΔlLO = 1, for example. Since we are most interested in the band gaps (direct and indirect) of the systems, we choose the indirect band gap from the X point to the Γ point,
Before discussing the results, we briefly illustrate the appropriateness of this choice. First of all,
Figure 2 summarizes the results of convergence study for the G0W0@PBE method.
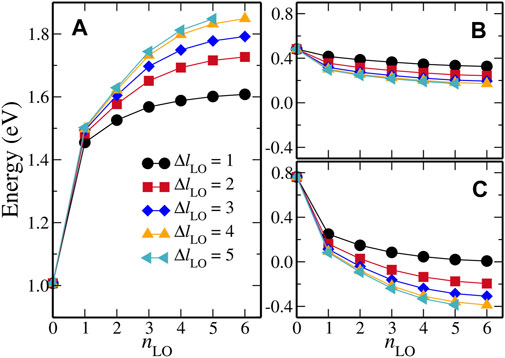
FIGURE 2. Dependence on HLOs parameters of G0W0@PBE calculated (A) the indirect band gap between X and Γ,
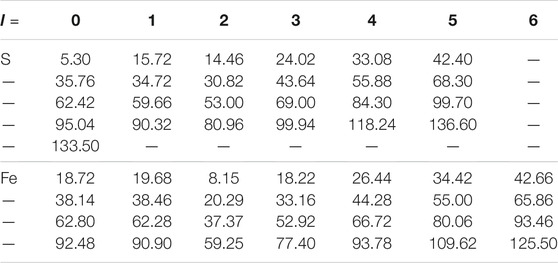
TABLE 1. Energy parameters (unit: Rydberg) of high-energy local orbitals (HLOs) used in pyrite FeS2 corresponding to HLOs parameters nLO = 4, ΔlLO = 4 for Fe and S. Those for marcasite are essentially the same with difference by 0.02 Ry at most in each element and l channel.
It is worth noting that the effect of including HLOs on the QP correction to
After having obtained the optimized HLOs, we perform the PBE-based GW calculations for pyrite and marcasite FeS2 with the LAPW + HLOs basis set, and compare with the PBE method and GW with the standard LAPW basis.
The band gaps of pyrite and marcasite FeS2 calculated by PBE and GW methods are presented in Table 2. The fundamental band gaps are obtained by computing the band energies along the k-point paths indicated in the right panel of Figure 1. In the PBE reference, pyrite and marcasite are predicted to have indirect fundamental band gaps of 0.70 and 0.83 eV, respectively. Our PBE results are consistent with those from previous all-electron LAPW study (Schena et al., 2013) and close to the recently reported optical band gaps obtained from diffuse reflectance spectroscopy (Sánchez et al., 2016). However, our PBE band gap for pyrite is slightly larger than several reported PBE results (Sun et al., 2011; Kolb and Kolpak, 2013; Lazić et al., 2013; Li et al., 2015; Zhang et al., 2018). This can be attributed to the use of different lattice structures in those studies (Eyert et al., 1998; Lazić et al., 2013; Schena et al., 2013) from the current work. Particularly, geometry optimization by PBE (Eyert et al., 1998; Schena et al., 2013) generally gives a longer S-S dimer, which leads to smaller splitting between bonding and anti-bonding S-3pσ orbitals and a consequent shrink in the band gap.
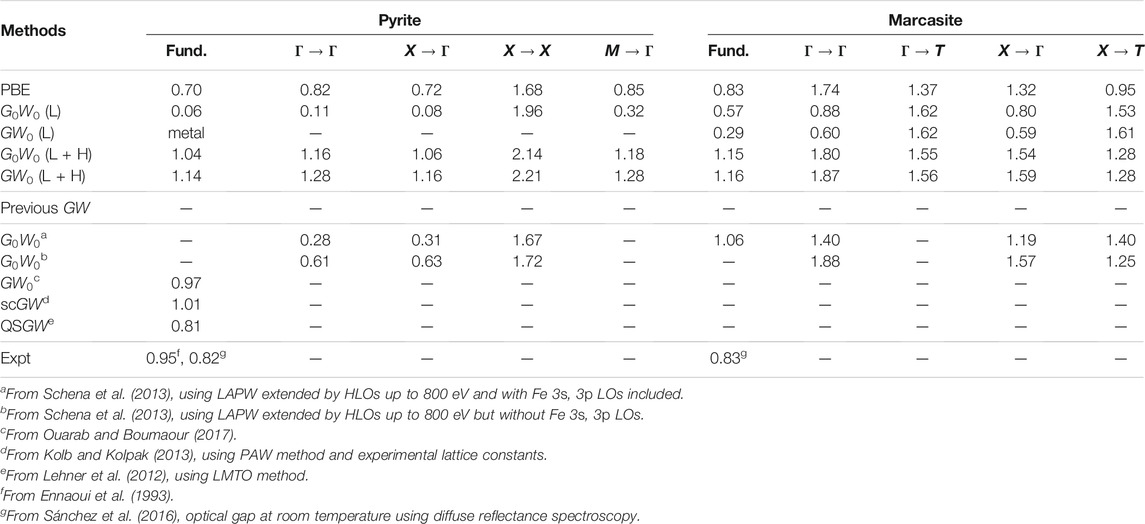
TABLE 2. Fundamental band gap (indicated by “fund.”) and other direct and indirect band gaps (unit: eV) for pyrite and marcasite FeS2 calculated by PBE and GW methods. Results from previous GW studies and experimental measurements are presented for comparison. To simplify the notation, we use “L” and “L + H” to denote the standard LAPW and LAPW + HLOs basis sets, respectively. PBE is used as the starting point for G0W0 and GW0 calculations unless stated otherwise.
For GW calculations with the standard LAPW basis, the QP fundamental band gaps by G0W0@PBE are smaller than the PBE counterparts in both FeS2 polymorphs. Pyrite FeS2 is predicted to have a band gap of only 0.06 eV, which is 0.64 eV smaller than that by PBE. The negative QP correction for pyrite band gap has been reported by Schena et al. (2013). The QP fundamental band gap for marcasite predicted by G0W0@PBE (LAPW) is also smaller than PBE, while the change (0.26 eV) is less dramatic than that for pyrite. Such negative QP corrections to LDA/GGA band gaps are uncommon in GW studies for closed-shell systems (Klimes et al., 2014; Jiang and Blaha, 2016; van Setten et al., 2017; Zhang and Jiang, 2019) as well as open-shell d/f-electron semiconductors (Jiang, 2018). Switching on self-consistency of the Green’s function by GW0@PBE further reduces the fundamental band gaps of FeS2. In particular, pyrite is predicted to be metallic by GW0@PBE, which disagrees qualitatively with its semiconducting nature in experiment (Ennaoui et al., 1993). For other direct and indirect band gaps, those for Γ → Γ and X → Γ in pyrite and marcasite and M → Γ in marcasite are decreased from PBE to G0W0@PBE (LAPW). The decrease is largest for the Γ → Γ gap in the two phases, 0.71 and 0.86 eV for pyrite and marcasite, respectively. On the other hand, the gaps for X → X in pyrite, Γ → T and X → T in marcasite are increased by 0.28, 0.25, and 0.58 eV, respectively. However, it should be noted that the distinction in signs of corrections to the QP gaps in different channels should not be considered as intrinsic for FeS2. Instead, it is an artifact as a result of the incomplete basis, which we will discuss in details below.
Now we turn to the LAPW + HLOs-based GW calculations. With the G0W0@PBE method, including HLOs increases the QP fundamental gap by 0.98 eV for pyrite and 0.58 eV for marcasite. The resulting G0W0@PBE band gaps are 1.04 and 1.15 eV for pyrite and marcasite, respectively. In contrast, all band gaps investigated are increased by G0W0 with LAPW + HLOs compared to their PBE counterparts. We note that HLOs have distinct effects among band gaps for different channels. Once the HLOs are included, band gaps for channels with the conduction state at the Γ point are increased by about 1 eV. On the other hand, the QP correction to the X → X band gap in pyrite increases by only 0.18 eV. Moreover, the gaps for Γ → T and X → T in marcasite even decrease. With the LAPW + HLOs basis, using GW0 to switch on partial self-consistency further increases the band gaps, but the change is moderate and no more than 0.1 eV.
As explained at the beginning, the fundamental band gap of FeS2 has been controversial in the recent decades, partly due to the widely varying experimental values (Ennaoui et al., 1993). In the present study, the GW0@PBE method with the LAPW + HLOs predicts that pyrite and marcasite have indirect fundamental band gaps of 1.14 and 1.16 eV, respectively. The GW0 gap of pyrite is slightly larger than the generally accepted experimental value of 0.95 eV (Ennaoui et al., 1993). Furthermore, the fact that the two polymorphs have almost identical band gaps is consistent with the optical measurements by Sánchez et al. (2016), although our predicted band gaps are about 0.3 eV larger. However, it should be noted that one must take exciton binding energy EB into account for a meaningful comparison between the QP fundamental band gap and experimentally measured optical gap. The difference between the fundamental and optical gaps can be significant when the exciton is localized, i.e. of Frenkel type (Fox, 2010). On the other hand, while it is more straightforward to compare the QP gap with spectral data from direct and inverse PES (Folkerts et al., 1987; Mamiya et al., 1997), the resolutions of available measurements for pyrite FeS2 are too low to extract a meaningful gap value for comparison. Moreover, to the best knowledge of the authors, no data of combined PES/IPS measurements are available for marcasite. Therefore, further experimental studies are required to determine and verify the band gaps of the FeS2 polymorphs.
To close this part, we highlight that the present work resolve two issues reported in previous GW studies in terms of QP band structures of FeS2. First, Schena et al. (2013) performed a G0W0@PBE study on pyrite and marcasite FeS2 with similar HLOs-extended LAPW basis. The fundamental band gap of pyrite was estimated as about 0.3 eV, by which the authors claimed to explain the low VOC encountered in the pyrite solar cell. However, according to our convergence study, such a small band gap is likely to result from inadequate convergence with respect to HLOs. More specifically, the largest angular momentum of HLOs
To further illustrate the significance of HLOs in applying the GW methods to FeS2, we present the QP band structures of pyrite and marcasite FeS2 calculated from the G0W0@PBE method with the LAPW + HLOs basis set, and compare the results to those with the standard LAPW basis.
Figure 3 shows the electronic band structures of the two FeS2 phases from different methods. Note that the bands are aligned to the CBM at the Γ point for a better view of QP correction to the valence states. With PBE, pyrite (Figure 3A) is found to be an indirect band gap material with the CBM located at the Γ point and the VBM near the X point along Γ–X. The top valence bands within 1 eV below the VBM are dominated by the localized Fe-3d states, also manifested by their flat dispersion. The dispersive bands about 2 eV below the VBM are mainly composed of S-3p states and well separated from the Fe-3d (t2g) valence bands. In the conduction band region, the lowermost conduction bands are also largely composed of Fe-3d (eg), except for the states close to the Γ point with predominant S-3p characters. Particularly, the CBM Γc state is exclusively formed by the σ anti-bonding overlapping of S-3p orbitals in the S-S dimer (see projected bands in Figure 4A). Valence and conduction bands with strong Fe-3d characters are separated by about 2 eV. For marcasite, an indirect band gap is also observed, with the CBM located at the T point (Tc) and the VBM along Γ–X (Δv). Both states at the VBM and CBM of marcasite are of dominant Fe-3d characters (Figure 4D), in contrast to pyrite where CBM is of pure S-3p characters. The wider Fe-3d valence bands overlap with the S-3p bands near about 1.5 eV below the VBM, which indicates stronger covalent bonding between Fe and S in marcasite than in pyrite.
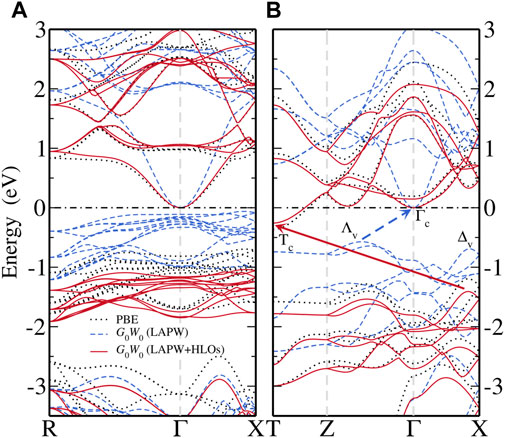
FIGURE 3. Comparison of band structures computed from PBE (black dotted), G0W0@PBE with the standard LAPW basis (blue dashed) and G0W0@PBE with the LAPW basis extended by optimized HLOs (LAPW + HLOs, red solid) for (A) pyrite and (B) marcasite FeS2. The conduction band minimum is aligned as the energy zero and marked by the black dash-dotted line.
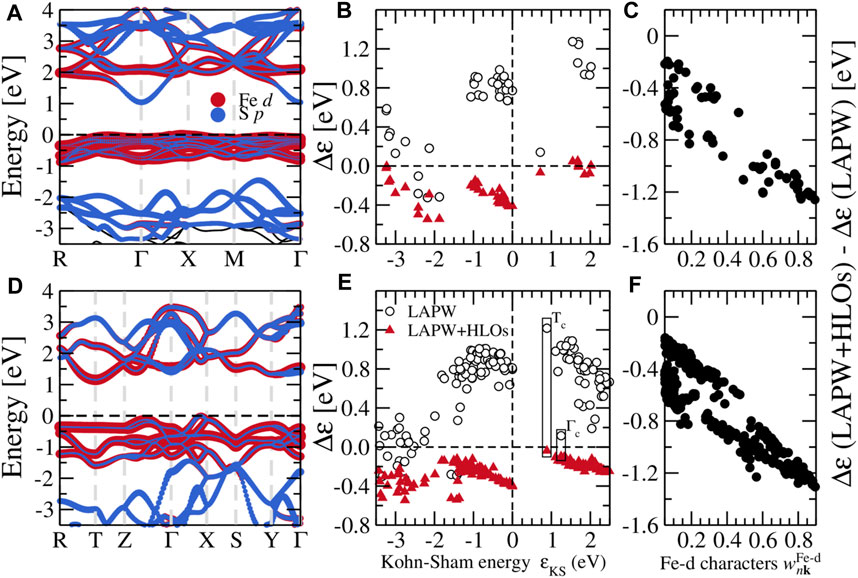
FIGURE 4. (A) Projected band structure from GW with the LAPW + HLOs basis, (B) self-energy corrections Δɛ to Kohn-Sham states, and (C) the difference between Δɛ with LAPW + HLOs and LAPW basis against the weight of Fe-d characters
Then we compare the QP band structures obtained from G0W0@PBE with the LAPW and LAPW + HLOs basis (Figure 3). With the standard LAPW basis, G0W0@PBE predicts pyrite almost as a semimetal with a nearly vanishing band gap (Figure 3A). Dispersion of the conduction band around the Γ point and the separation between the Fe-3d and S-3p valence bands are enhanced compared to the PBE reference. For marcasite (Figure 3B), although a noticeable gap (0.57 eV) is predicted by G0W0@PBE, the band edges are different from those in PBE: the CBM is located at the Γ point (Γc) and the VBM in the middle of the Z–Γ path (Λv). The change in the nature of band edges from semi-local functional to GW method is also observed by Schena et al. (2013). Once HLOs are included in the basis set, QP band gaps of both phases are dramatically enlarged. The fundamental gaps of pyrite and marcasite are 1.04 and 1.15 eV, respectively, which are 0.2 ∼ 0.3 eV larger than the optical gaps from absorption spectra (Sánchez et al., 2016). Band edges of marcasite by GW are also recovered to those by PBE. The comparison indicates that both negative QP corrections to band gaps and change of band edges in GW (LAPW) are indeed artifacts due to the inadequate numerical accuracy of the basis set.
To better understand how the HLOs basis functions influence the QP band structures of FeS2, we scrutinize the QP correction to Kohn-Sham state Δɛ, defined by the difference between the QP energy ɛQP and the KS energy ɛKS, i.e. Δɛ ≡ɛQP − ɛKS. For pyrite, with the standard LAPW basis, Δɛ to the CBM is smaller than those to the valence Fe-3d t2g and conduction Fe-3d eg states as shown in Figure 4B. Particularly, Δɛ for the VBM is about 0.7 eV greater than that for the CBM. This leads to a up-shift of Fe-3d states with respect to the CBM on a whole. Extending LAPW with HLOs reduces Δɛ for all states, but the reduction in Δɛ to the VBM is more than that to the CBM by about 1.0 eV, resulting in the sign change of the QP correction to the band gap. Similar conclusion can be drawn from Δɛ in the marcasite phase (Figure 4E). With the standard LAPW method, Δɛ to Tc exceeds that to Γc by more than 1.1 eV. Consequently, Γc drops down below Tc and becomes the CBM, as we have seen in Figure 3B. Upon including HLOs, Δɛ to Tc is reduced more significantly than Δɛ to Γc such that Tc recovers the conduction band edge as in PBE.
Such biased effects of HLOs are clearly associated with the atomic characteristics of Kohn-Sham states, as we have demonstrated in the GW calculations of cuprous and silver halides (Zhang and Jiang, 2019). In Figures 4C,F, we plot the difference between Δɛ computed by G0W0 with LAPW + HLOs and LAPW against the weight of Fe-d characters of the Kohn-Sham orbitals
where
To end this section, we present the GW calculated density of states (DOS) of FeS2 polymorphs in Figure 5. The results for pyrite FeS2 are shown in Figure 5A. Due to different definitions of the Fermi level in theoretical results and experimental spectral data, we have shifted the experimental data to match up the highest valence peak near the Fermi level. With this alignment, the overall DOS from G0W0 (LAPW + HLOs) agrees well with the energy distribution curves (EDCs) from the PES experiments. The width of the valence Fe-3d band and separation between the Fe-3d and S-3p valence bands are consistent with the UPS experiment by Mamiya et al. (1997) and the XPS experiment by Folkerts et al. (1987). The location of the first peak in the conduction band region is also in good agreement with the BIS data (Folkerts et al., 1987). Interestingly, although G0W0 (LAPW) underestimates the band gap severely, the location of the first peak in the conduction region is almost identical to that by G0W0 (LAPW + HLOs), probably due to the error cancellation between QP corrections to the valence and conduction Fe-3d bands. However, such fortuitous cancellation does not hold in the valence region as inferred by the too deep S-3p band in the G0W0 (LAPW) results.
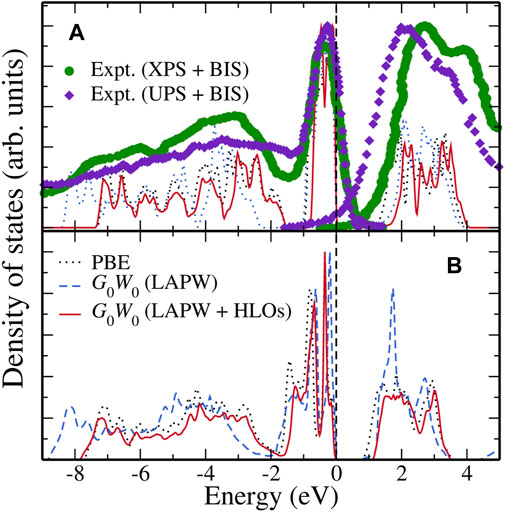
FIGURE 5. Density of states (DOS) computed from PBE (black dotted), G0W0@PBE with the standard LAPW basis (blue dashed) and G0W0@PBE with the LAPW + HLOs basis (red solid) for (A) pyrite and (B) marcasite FeS2. Energy distribution curves (EDCs) of pyrite extracted from photo-electron spectroscopy (PES) are presented for comparison. Each dataset is normalized with respect to its highest peak. Theoretical data are aligned to its valence band maximum as energy zero. The XPS + BIS and UPS + BIS data for pyrite are obtained from Folkerts et al. (1987) and Mamiya et al. (1997), respectively. To take into account the different definitions of the Fermi level in theory and experiment, a rigid shift of 0.60 and 0.40 eV are employed for the EDCs from XPS + BIS and UPS + BIS, respectively, to match the highest valence peaks below the Fermi level.
Figure 5B shows the calculated DOS for marcasite. Regardless of the theoretical method used, the valence Fe-3d band of marcasite has larger width than that of pyrite, indicating a stronger Fe-S interaction in the marcasite phase. In the conduction region, a sharp peak is observed with the G0W0 (LAPW) method, while only a plateau is found with G0W0 (LAPW + HLOs). However, the sharp peak is actually an artifact of wrongly pushed up Fe-3d conduction bands due to the inaccuracy of the standard LAPW basis as explained above.
As mentioned in the introduction, previous theoretical studies found that various hybrid functionals, which are typically able to describe the band gaps of semiconductors quite accurately, performed badly for FeS2. In this section, we look into this issue and present results by several hybrid schemes including the DSH functional with system-dependent parameters.
Band gaps computed by different hybrid functionals are collected in Table 3. The widely used PBE0 and HSE06 functionals have been reported to predict fundamental gaps of pyrite and marcasite FeS2 larger than 2 eV in the literature (Sun et al., 2011; Choi et al., 2012; Hu et al., 2012; Schena et al., 2013; Liu et al., 2019), which is confirmed by our results. DSH, the hybrid functional with system-tuned parameters, does not improve the prediction over PBE0 and HSE06. This is surprising, given that DSH has been previously shown to outperform several other hybrids in evaluating band structures for wide- and narrow-gap systems (Cui et al., 2018; Liu et al., 2020), including PBE0, HSE06 and the dielectric-dependent hybrid (DDH) functionals (Marques et al., 2011; Skone et al., 2014). SX-PBE screened exchange functional gives band gaps of FeS2 significantly smaller than the hybrids mentioned above, but the gaps are still larger than those from GW with the LAPW + HLOs basis (Table 2) by about 0.5 eV.
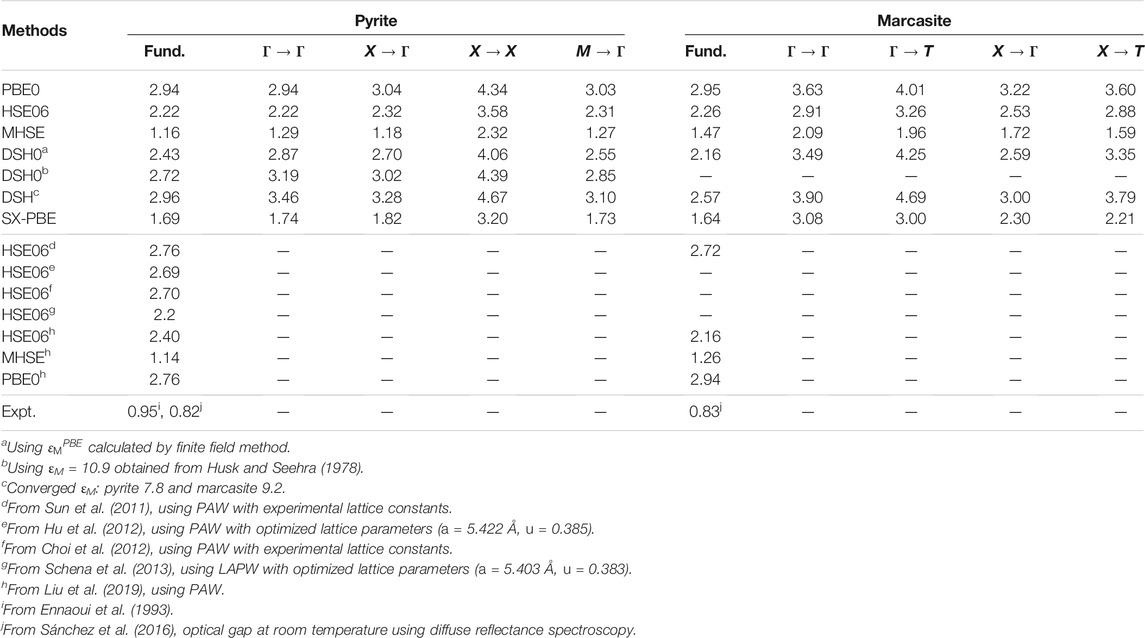
TABLE 3. Fundamental band gap (indicated by “fund.”) and other direct and indirect band gaps (unit: eV) for pyrite and marcasite FeS2 calculated by different hybrid functionals. Results from other theoretical studies and experimental measurements are presented as comparison.
Considering that the one-shot DSH, i.e. DSH0, may outperform the self-consistent scheme in some transition metal compounds (Cui et al., 2018; Liu et al., 2020), we also employ DSH0 to calculate the two FeS2 polymorphs. The macroscopic dielectric constant calculated with PBE
As summarized above, the investigated hybrid functionals except for MHSE fail to give reasonable predictions for the band gaps of pyrite and marcasite FeS2. In this section, we take a close look at the band structures computed from these methods to understand the failure.
The band structures for pyrite calculated from selected hybrid functionals are shown in the upper panel of Figure 6. With PBE0 and HSE06 (Figures 6A,B), the fundamental band gap is a direct one with both VBM and CBM located at the Γ point. An indirect fundamental gap is obtained by SX-PBE and DSH (Figures 6C,D), but the VBM is different from that in PBE or the GW method (Figure 3A). In addition, compared to the GW (LAPW + HLOs) results, the separation between valence Fe-3d and S-3p bands is reduced and the splitting between the valence and conduction Fe-3d bands is significantly increased by the hybrid functionals. We note that both features can be understood tentatively as a result of increased ligand field strength from the perspective of ligand field theory. This indicates an overestimated interaction between the ligand S-3pσ and Fe-3d orbitals in the selected hybrid functionals than that in PBE. The overestimation is most significant in the DSH method (Figure 6D), where the state of predominant S-3pπ characters along the M–Γ path becomes the VBM and conduction Fe-3d bands are raised beyond 6 eV above the Fermi level.
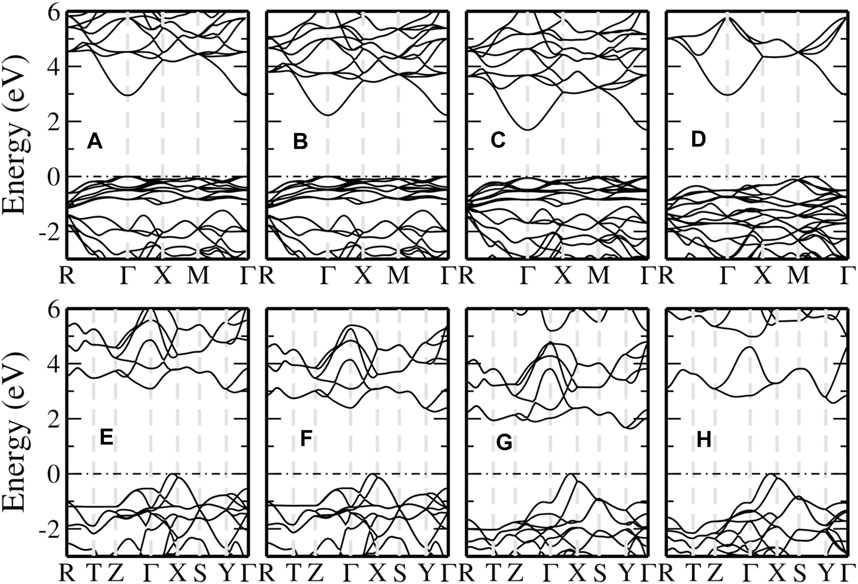
FIGURE 6. Band structures of pyrite (upper panel) and marcasite (lower panel) FeS2 computed from different hybrid functional methods. From left to right, the methods used are PBE0 (A,E), HSE06 (B,F), SX-PBE (C,G) and DSH (D,H), respectively. The valence band maximum is aligned as the energy zero and indicated by the black dash-dotted line.
We can observe similar features in marcasite band structures from hybrid functionals, as shown in the lower panel of Figure 6. In the valence band region, the S-3p bands are pushed up relatively to Fe-3d bands compared with PBE and GW. The increase is so significant that the VBM along Γ–X, which is mainly of Fe-3d in PBE and GW, is now of predominant S-3p characters. This also leads to a considerable overlap between the two sets of bands in the energy window 1 ∼ 3 eV below the Fermi level. The conduction bands are also shifted to higher energies. However, the shifts are larger for the conduction Fe-3d bands than for S-3p. For the DSH method as an extreme case, the Fe-3d bands are raised up too high and even separated from the S-3p bands.
The radical failure of DSH invites a close inspection of feasibility of DSH for FeS2. As a preliminary exploration to the possible cause, we make a direct comparison between the inverse static dielectric function used in the DSH with
We note that Eq. 13 differs from the inverse of Eq. 9 because in the derivation of DSH, the exponential function is replaced by erfc [Cui et al. (2018) for more details]. As shown in Figure 7, while DSH overestimates ɛ−1 and underestimates the screening in the short-wavelength region, i.e. near |G| = 0, DSH0 model dielectric function with
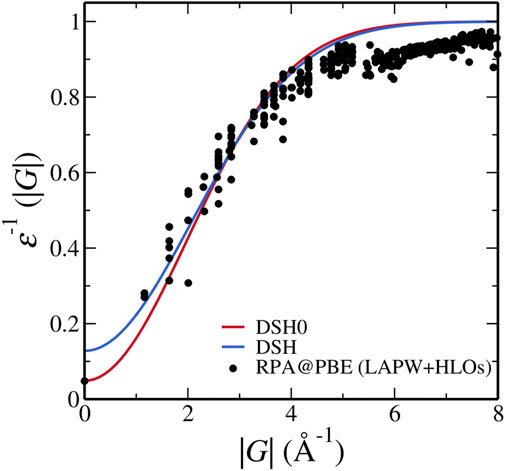
FIGURE 7. Inverse dielectric functions of pyrite used in the DSH model and calculated from RPA@PBE with the LAPW + HLOs basis set. Dielectric constant computed by PBE is used in the one-shot DSH0 method.
In the present study, we have investigated the electronic band structures of two FeS2 polymorphs, namely pyrite and marcasite, by using methods in different frameworks. With the all-electron many-body GW method implemented in the LAPW framework, we find that by using GW0@PBE with the LAPW + HLOs basis, pyrite and marcasite are predicted to have indirect fundamental band gaps of 1.14 and 1.16 eV, respectively. The closeness of band gaps for the two polymorphs agrees with the experimental observation (Sánchez et al., 2016). The pyrite band gap from GW0@PBE with LAPW + HLOs is very close to the generally accepted experimental value (Ennaoui et al., 1993) and the corresponding density of states also agrees well with energy distribution curves obtained from the photoelectron spectroscopy measurements (Folkerts et al., 1987; Mamiya et al., 1997). In contrast, with the standard LAPW basis, PBE-based G0W0 and GW0 both lead to negative QP correction to the PBE fundamental gap, which is rarely observed in LDA/GGA-based G0W0 and GW0 treatments of semiconductors. The splitting between Fe-3d and S-3p valence bands of pyrite is also significantly overestimated compared to experiment. These artifacts exist not only in calculations with the standard LAPW basis, but also in those with LAPW basis extended by an inadequately converged HLOs (Schena et al., 2013). Therefore in order to eliminate such artifacts, it is instrumental to carefully converge the fundamental band gap with respect to the two controlling parameters, namely nLO and ΔlLO. We have further studied electronic band structures of FeS2 polymorphs with different hybrid functionals, including PBE0, HSE06, the screened exchange SX-PBE and the recently developed DSH functional with system-tuned hybridization parameters. We find that all those methods overestimate the band gaps of the two polymorphs by 0.5 ∼ 1.9 eV compared to the results obtained from G0W0 (LAPW + HLOs). The overestimation by PBE0 and HSE06 as reported in the literature is reproduced in this work. Furthermore, either self-consistent or one-shot DSH method fails to improve over the conventional fixed-parameter hybrid functionals. By comparing the model dielectric function used in DSH with that from RPA calculation with LAPW + HLOs in pyrite, we point out that the failure of DSH may not be caused by the insufficiency of the dielectric model used and therefore requires further investigation. Our investigations clearly show that accurate prediction of electronic band structures of FeS2 polymorphs poses a stringent test on the state-of-the-art first-principles approaches, and the GW method based on semi-local density approximation performs well for this difficult system if it is practiced with well-converged numerical accuracy.
Finally, we note that further work in the following aspects can be done to shed more light onto the band gap problem of FeS2 in terms of GW and hybrid functional calculations. For one thing, it is possible to build the screened Coulomb interaction W using the KS states from the LAPW calculations and calculate the self-energy Σ with G from LAPW + HLOs. One can compare it with GW using LAPW to see whether it is the inaccurate band summation in W or G to blame. For another, replacing the PBE with the hybrid functional as starting point will be worthwhile to evaluate the dependence of G0W0/GW0 results on initial input for FeS2. Particularly, considering the severe overestimation of FeS2 band gaps by the hybrid functionals, it is of great interest to see whether G0W0/GW0 can produce a negative QP correction to the gap from hybrid functional calculations such that the experimental gap is approached from above.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
M-YZ performed the calculations, conducted the analysis and wrote the manuscript. HJ revised the manuscript and supervised all the work. All authors listed contributed to the article and approved it for publication.
National Natural Science Foundation of China (grant numbers: 21873005 and 21911530231).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors acknowledge the support by High-performance Computing Platform of Peking University for the computational resources.
Andersen, O. K. (1975). Linear Methods in Band Theory. Phys. Rev. B 12, 3060–3083. doi:10.1103/physrevb.12.3060
Aryasetiawan, F., and Gunnarsson, O. (1994). Product-basis Method for Calculating Dielectric Matrices. Phys. Rev. B 49, 16214–16222. doi:10.1103/physrevb.49.16214
Baer, R., Livshits, E., and Salzner, U. (2010). Tuned Range-Separated Hybrids in Density Functional Theory. Annu. Rev. Phys. Chem. 61, 85–109. doi:10.1146/annurev.physchem.012809.103321
Barawi, M., Ferrer, I. J., Flores, E., Yoda, S., Ares, J. R., and Sánchez, C. (2016). Hydrogen Photoassisted Generation by Visible Light and an Earth Abundant Photocatalyst: Pyrite (FeS2). J. Phys. Chem. C 120, 9547–9552. doi:10.1021/acs.jpcc.5b11482
Baroni, S., de Gironcoli, S., Dal Corso, A., and Giannozzi, P. (2001). Phonons and Related crystal Properties from Density-Functional Perturbation Theory. Rev. Mod. Phys. 73, 515–562. doi:10.1103/revmodphys.73.515
Bechstedt, F., Del Sole, R., Cappellini, G., and Reining, L. (1992). An Efficient Method for Calculating Quasiparticle Energies in Semiconductors. Solid State. Commun. 84, 765–770. doi:10.1016/0038-1098(92)90476-p
Becke, A. D. (1993a). A New Mixing of Hartree-Fock and Local Density‐functional Theories. J. Chem. Phys. 98, 1372–1377. doi:10.1063/1.464304
Becke, A. D. (1993b). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Betzinger, M., Friedrich, C., Blügel, S., and Görling, A. (2011). Local Exact Exchange Potentials within the All-Electron Flapw Method with Pseudopotential Results and a Comparison. Phys. Rev. B 83, 045105. doi:10.1103/physrevb.83.045105
Betzinger, M., Friedrich, C., Görling, A., and Blügel, S. (2015). Precise All-Electron Dynamical Response Functions: Application to Cohsex and the Rpa Correlation Energy. Phys. Rev. B 92, 245101. doi:10.1103/physrevb.92.245101
Betzinger, M., Friedrich, C., Görling, A., and Blügel, S. (2012). Precise Response Functions in All-Electron Methods: Application to the Optimized-Effective-Potential Approach. Phys. Rev. B 85, 245124. doi:10.1103/physrevb.85.245124
Birkholz, M., Fiechter, S., Hartmann, A., and Tributsch, H. (1991). Sulfur Deficiency in Iron Pyrite (FeS2−x) and its Consequences for Band-Structure Models. Phys. Rev. B 43, 11926–11936. doi:10.1103/physrevb.43.11926
Blaha, P., Schwarz, K., Madsen, G. K. H., Kvasnicka, D., and Luitz, J. (2001). WIEN2K, an Augmented Plane Wave + Local Orbitals Program For Calculating Crystal Properties (Karlheinz Schwarz, Techn. Universität Wien, Austria).
Blaha, P., Schwarz, K., Tran, F., Laskowski, R., Madsen, G. K. H., and Marks, L. D. (2020). WIEN2k: An APW+lo Program for Calculating the Properties of Solids. J. Chem. Phys. 152, 074101. doi:10.1063/1.5143061
Blöchl, P. E. (1994). Projector Augmented-Wave Method. Phys. Rev. B 50, 17953–17979. doi:10.1103/physrevb.50.17953
Bronold, M., Pettenkofer, C., and Jaegermann, W. (1994). Surface Photovoltage Measurements on Pyrite (100) Cleavage Planes: Evidence for Electronic Bulk Defects. J. Appl. Phys. 76, 5800–5808. doi:10.1063/1.358393
Brostigen, G., Kjekshus, A., Astrup, E. E., Nordal, V., Lindberg, A. A., and Craig, J. C. (1969). Redetermined crystal Structure of FeS2 (Pyrite). Acta Chem. Scand. 23, 2186–2188. doi:10.3891/acta.chem.scand.23-2186
Brostigen, G., Kjekshus, A., Rømming, C., Gronowitz, S., Koskikallio, J., and Swahn, C.-G. (1973). Compounds with the Marcasite Type crystal Structure. VIII. Redetermination of the Prototype. Acta Chem. Scand. 27, 2791–2796. doi:10.3891/acta.chem.scand.27-2791
Bylander, D. M., and Kleinman, L. (1990). Good Semiconductor Band Gaps with a Modified Local-Density Approximation. Phys. Rev. B 41, 7868–7871. doi:10.1103/physrevb.41.7868
Cabán-Acevedo, M., Kaiser, N. S., English, C. R., Liang, D., Thompson, B. J., Chen, H.-E., et al. (2014). Ionization of High-Density Deep Donor Defect States Explains the Low Photovoltage of Iron Pyrite Single Crystals. J. Am. Chem. Soc. 136, 17163–17179. doi:10.1021/ja509142w
Cao, H., Yu, Z., Lu, P., and Wang, L.-W. (2017). Fully Converged Plane-Wave-Based Self-Consistent GW Calculations of Periodic Solids. Phys. Rev. B 95, 035139. doi:10.1103/physrevb.95.035139
Cappellini, G., Del Sole, R., Reining, L., and Bechstedt, F. (1993). Model Dielectric Function for Semiconductors. Phys. Rev. B 47, 9892–9895. doi:10.1103/physrevb.47.9892
Chattopadhyay, T., and Von Schnering, H. G. (1985). High Pressure X-ray Diffraction Study on P-FeS2, M-FeS2 and MnS2 to 340 Kbar: A Possible High Spin-Low Spin Transition in MnS2. J. Phys. Chem. Sol. 46, 113–116. doi:10.1016/0022-3697(85)90204-5
Chatzitheodorou, G., Fiechter, S., Könenkamp, R., Kunst, M., Jaegermann, W., and Tributsch, H. (1986). Thin Photoactive FeS2 (Pyrite) Films. Mater. Res. Bull. 21, 1481–1487. doi:10.1016/0025-5408(86)90088-7
Chen, W., Miceli, G., Rignanese, G.-M., and Pasquarello, A. (2018). Nonempirical Dielectric-dependent Hybrid Functional with Range Separation for Semiconductors and Insulators. Phys. Rev. Mater. 2, 073803. doi:10.1103/physrevmaterials.2.073803
Choi, S. G., Hu, J., Abdallah, L. S., Limpinsel, M., Zhang, Y. N., Zollner, S., et al. (2012). Pseudodielectric Function and Critical-point Energies of Iron Pyrite. Phys. Rev. B 86, 115207. doi:10.1103/physrevb.86.115207
Cui, Z.-H., Wang, Y.-C., Zhang, M.-Y., Xu, X., and Jiang, H. (2018). Doubly Screened Hybrid Functional: An Accurate First-Principles Approach for Both Narrow- and Wide-gap Semiconductors. J. Phys. Chem. Lett. 9, 2338–2345. doi:10.1021/acs.jpclett.8b00919
Cui, Z.-H., Wu, F., and Jiang, H. (2016). First-principles Study of Relative Stability of Rutile and Anatase Tio2 Using the Random Phase Approximation. Phys. Chem. Chem. Phys. 18, 29914–29922. doi:10.1039/c6cp04973g
Deguchi, D., Sato, K., Kino, H., and Kotani, T. (2016). Accurate Energy Bands Calculated by the Hybrid Quasiparticle Self-consistentGWmethod Implemented in the Ecalj Package. Jpn. J. Appl. Phys. 55, 051201. doi:10.7567/jjap.55.051201
Ennaoui, A., Fiechter, S., Jaegermann, W., and Tributsch, H. (1986). Photoelectrochemistry of Highly Quantum Efficient Single‐Crystalline N ‐ FeS2 (Pyrite). J. Electrochem. Soc. 133, 97–106. doi:10.1149/1.2108553
Ennaoui, A., Fiechter, S., Pettenkofer, C., Alonso-Vante, N., Büker, K., Bronold, M., et al. (1993). Iron Disulfide for Solar Energy Conversion. Solar Energ. Mater. Solar Cell 29, 289–370. doi:10.1016/0927-0248(93)90095-k
Eyert, V., Höck, K.-H., Fiechter, S., and Tributsch, H. (1998). Electronic Structure ofFeS2: The Crucial Role of Electron-Lattice Interaction. Phys. Rev. B 57, 6350–6359. doi:10.1103/physrevb.57.6350
Ferrer, I. J., Nevskaia, D. M., de las Heras, C., and Sánchez, C. (1990). About the Band gap Nature of FeS2 as Determined from Optical and Photoelectrochemical Measurements. Solid State. Commun. 74, 913–916. doi:10.1016/0038-1098(90)90455-k
Folkerts, W., Sawatzky, G. A., Haas, C., Groot, R. A. d., and Hillebrecht, F. U. (1987). Electronic Structure of Some 3d Transition-Metal Pyrites. J. Phys. C: Solid State. Phys. 20, 4135–4144. doi:10.1088/0022-3719/20/26/015
Friedrich, C., Müller, M. C., and Blügel, S. (2011a). Band Convergence and Linearization Error Correction of All-Electron GW Calculations: The Extreme Case of Zinc Oxide. Phys. Rev. B 83, 081101(R). doi:10.1103/physrevb.83.081101
Friedrich, C., Müller, M. C., and Blügel, S. (2011b). Erratum: Band Convergence and Linearization Error Correction of All-Electron GW Calculations: The Extreme Case of Zinc Oxide. Phys. Rev. B 83, 081101(r). 2011Phys. Rev. B 84, 039906. doi:10.1103/physrevb.83.081101
Friedrich, C., Schindlmayr, A., Blügel, S., and Kotani, T. (2006). Elimination of the Linearization Error in GW Calculations Based on the Linearized Augmented-Plane-Wave Method. Phys. Rev. B 74, 045104. doi:10.1103/physrevb.74.045104
Golze, D., Dvorak, M., and Rinke, P. (2019). The GW Compendium: A Practical Guide to Theoretical Photoemission Spectroscopy. Front. Chem. 7, 377. doi:10.3389/fchem.2019.00377
Gong, M., Kirkeminde, A., Kumar, N., Zhao, H., and Ren, S. (2013a). Ionic-passivated FeS2 Photocapacitors for Energy Conversion and Storage. Chem. Commun. 49, 9260–9262. doi:10.1039/c3cc45088k
Gong, M., Kirkeminde, A., Xie, Y., Lu, R., Liu, J., Wu, J. Z., et al. (2013b). Iron Pyrite (FeS2) Broad Spectral and Magnetically Responsive Photodetectors. Adv. Opt. Mater. 1, 78–83. doi:10.1002/adom.201200003
Goodenough, J. B. (1972). Energy Bands in TX2 Compounds with Pyrite, Marcasite, and Arsenopyrite Structures. J. Solid State. Chem. 5, 144–152. doi:10.1016/0022-4596(72)90022-9
Grumet, M., Liu, P., Kaltak, M., Klimeš, J., and Kresse, G. (2018). Beyond the Quasiparticle Approximation: Fully Self-Consistent GW Calculations. Phys. Rev. B 98, 155143. doi:10.1103/physrevb.98.155143
Hedin, L. (1965). New Method for Calculating the One-Particle Green's Function with Application to the Electron-Gas Problem. Phys. Rev. 139, A796–A823. doi:10.1103/physrev.139.a796
Herbert, F. W., Krishnamoorthy, A., Van Vliet, K. J., and Yildiz, B. (2013). Quantification of Electronic Band gap and Surface States on FeS2(100). Surf. Sci. 618, 53–61. doi:10.1016/j.susc.2013.08.014
Heyd, J., Peralta, J. E., Scuseria, G. E., and Martin, R. L. (2005). Energy Band Gaps and Lattice Parameters Evaluated with the Heyd-Scuseria-Ernzerhof Screened Hybrid Functional. J. Chem. Phys. 123, 174101. doi:10.1063/1.2085170
Heyd, J., Scuseria, G. E., and Ernzerhof, M. (2003). Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 118, 8207–8215. doi:10.1063/1.1564060
Heyd, J., Scuseria, G. E., and Ernzerhof, M. (2006). Erratum: “Hybrid Functionals Based on a ScreenedCoulomb Potential” [j. chem. phys.118, 8207 (2003)]. J. Chem. Phys. 124, 219906.
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous Electron Gas. Phys. Rev. 136, B864–B871. doi:10.1103/physrev.136.b864
Hu, J., Zhang, Y., Law, M., and Wu, R. (2012). First-principles Studies of the Electronic Properties of Native and Substitutional Anionic Defects in Bulk Iron Pyrite. Phys. Rev. B 85, 085203. doi:10.1103/physrevb.85.085203
Hulliger, F., and Mooser, E. (1965b). Semiconductivity in Pyrite, Marcasite and Arsenopyrite Phases. J. Phys. Chem. Sol. 26, 429–433. doi:10.1016/0022-3697(65)90173-3
Hulliger, F., and Mooser, E. (1965a). The Bond Description of Semiconductors: Polycompounds. Prog. Solid State. Chem. 2, 330–377. doi:10.1016/0079-6786(65)90011-7
Husk, D. E., and Seehra, M. S. (1978). Dielectric Constant of Iron Pyrite (FeS2). Solid State. Commun. 27, 1147–1148. doi:10.1016/0038-1098(78)91130-4
Hybertsen, M. S., and Louie, S. G. (1986). Electron Correlation in Semiconductors and Insulators: Band Gaps and Quasiparticle Energies. Phys. Rev. B 34, 5390–5413. doi:10.1103/physrevb.34.5390
Jiang, H., and Blaha, P. (2016). GWwith Linearized Augmented Plane Waves Extended by High-Energy Local Orbitals. Phys. Rev. B 93, 115203. doi:10.1103/physrevb.93.115203
Jiang, H. (2011). Electronic Band Structure from First-Principles Green's Function Approach: Theory and Implementations. Front. Chem. China 6, 253–268. doi:10.1007/s11458-011-0261-6
Jiang, H., Gómez-Abal, R. I., Li, X.-Z., Meisenbichler, C., Ambrosch-Draxl, C., and Scheffler, M. (2013). FHI-gap: A GW Code Based on the All-Electron Augmented Plane Wave Method. Comp. Phys. Commun. 184, 348–366. doi:10.1016/j.cpc.2012.09.018
Jiang, H. (2018). Revisiting the GW Approach to d- and f-electron Oxides. Phys. Rev. B 97, 245132. doi:10.1103/physrevb.97.245132
Khalid, S., Ahmed, E., Khan, Y., Nawaz, S., Ramzan, M., Khalid, N. R., et al. (2018). “Iron Pyrite (FeS2): Sustainable Photovoltaic materialMicro and Nanomanufacturing Volume II,”. Editors M. J. Jackson, and W. Ahmed (Cham: Springer, 281–318. doi:10.1007/978-3-319-67132-1_11
Klimes, J., Kaltak, M., and Kresse, G. (2014). Predictive GW Calculations Using Plane Waves and Pseudopotentials. Phys. Rev. B 90, 075125.
Kolb, B., and Kolpak, A. M. (2013). Ultrafast Band-gap Oscillations in Iron Pyrite. Phys. Rev. B 88, 235208. doi:10.1103/physrevb.88.235208
Koller, D., Blaha, P., and Tran, F. (2013). Hybrid Functionals for Solids with an Optimized Hartree-Fock Mixing Parameter. J. Phys. Condens. Matter 25, 435503. doi:10.1088/0953-8984/25/43/435503
Kotani, T., and van Schilfgaarde, M. (2002). All-electron GW Approximation with the Mixed Basis Expansion Based on the Full-Potential LMTO Method. Solid State. Commun. 121, 461–465. doi:10.1016/s0038-1098(02)00028-5
Kou, W. W., and Seehra, M. S. (1978). Optical Absorption in Iron Pyrite (FeS2). Phys. Rev. B 18, 7062–7068. doi:10.1103/physrevb.18.7062
Krasovskii, E. E. (1997). Accuracy and Convergence Properties of the Extended Linear Augmented-Plane-Wave Method. Phys. Rev. B 56, 12866–12873. doi:10.1103/physrevb.56.12866
Krasovskii, E. E., Yaresko, A. N., and Antonov, V. N. (1994). Theoretical Study of Ultraviolet Photoemission Spectra of noble Metals. J. Electron Spectrosc. Relat. Phenomena 68, 157–166. doi:10.1016/0368-2048(94)02113-9
Kresse, G., and Furthmüller, J. (1996). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186. doi:10.1103/physrevb.54.11169
Kronik, L., Stein, T., Refaely-Abramson, S., and Baer, R. (2012). Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theor. Comput. 8, 1515–1531. doi:10.1021/ct2009363
Kümmel, S., and Kronik, L. (2008). Orbital-dependent Density Functionals: Theory and Applications. Rev. Mod. Phys. 80, 3–60. doi:10.1103/revmodphys.80.3
Laskowski, R., and Blaha, P. (2014). Calculating NMR Chemical Shifts Using the Augmented Plane-Wave Method. Phys. Rev. B 89, 014402. doi:10.1103/physrevb.89.014402
Laskowski, R., and Blaha, P. (2012). Calculations of NMR Chemical Shifts with Apw-Based Methods. Phys. Rev. B 85, 035132. doi:10.1103/physrevb.85.035132
Lazić, P., Armiento, R., Herbert, F. W., Chakraborty, R., Sun, R., Chan, M. K., et al. (2013). Low Intensity Conduction States in FeS2: Implications for Absorption, Open-Circuit Voltage and Surface Recombination. J. Phys. Condens Matter 25, 465801. doi:10.1088/0953-8984/25/46/465801
Lehner, S. W., Newman, N., van Schilfgaarde, M., Bandyopadhyay, S., Savage, K., and Buseck, P. R. (2012). Defect Energy Levels and Electronic Behavior of Ni-, Co-, and As-Doped Synthetic Pyrite (FeS2). J. Appl. Phys. 111, 083717. doi:10.1063/1.4706558
Li, B., Huang, L., Zhong, M., Wei, Z., and Li, J. (2015). Electrical and Magnetic Properties of FeS2 and CuFeS2 Nanoplates. RSC Adv. 5, 91103–91107. doi:10.1039/c5ra16918f
Li, E. K., Johnson, K. H., Eastman, D. E., and Freeouf, J. L. (1974). Localized and Bandlike Valence-Electron States in FeS2and NiS2. Phys. Rev. Lett. 32, 470–472. doi:10.1103/physrevlett.32.470
Li, Y., Chen, J., Chen, Y., Zhao, C., Lee, M.-H., and Lin, T.-H. (2018). DFT+U Study on the Electronic Structures and Optical Properties of Pyrite and Marcasite. Comput. Mater. Sci. 150, 346–352. doi:10.1016/j.commatsci.2018.04.009
Limpinsel, M., Farhi, N., Berry, N., Lindemuth, J., Perkins, C. L., Lin, Q., et al. (20141974–1989). An Inversion Layer at the Surface of N-type Iron Pyrite. Energy Environ. Sci. 7. doi:10.1039/c3ee43169j
Liu, J., Xu, A., Meng, Y., He, Y., Ren, P., Guo, W.-P., et al. (2019). From Predicting to Correlating the Bonding Properties of Iron Sulfide Phases. Comput. Mater. Sci. 164, 99–107. doi:10.1016/j.commatsci.2019.04.001
Liu, P., Franchini, C., Marsman, M., and Kresse, G. (2020). Assessing Model-dielectric-dependent Hybrid Functionals on the Antiferromagnetic Transition-Metal Monoxides MnO, FeO, CoO, and NiO. J. Phys. Condens. Matter 32, 015502. doi:10.1088/1361-648x/ab4150
Madsen, G. K. H., Blaha, P., Schwarz, K., Sjöstedt, E., and Nordström, L. (2001). Efficient Linearization of the Augmented Plane-Wave Method. Phys. Rev. B 64, 195134. doi:10.1103/physrevb.64.195134
Maier, T. M., Arbuznikov, A. V., and Kaupp, M. (2019). Local Hybrid Functionals: Theory, Implementation, and Performance of an Emerging New Tool in Quantum Chemistry and beyond. WIREs Comput. Mol. Sci. 9, e1378. doi:10.1002/wcms.1378
Mamiya, K., Mizokawa, T., Fujimori, A., Takahashi, H., Môri, N., Miyadai, T., et al. (1997). Photoemission Study of Pyrite-type Transition-Metal Chalcogenides MS2−xSex (M+Fe, Co, Ni). Physica B: Condensed Matter 237-238, 390–391. doi:10.1016/s0921-4526(97)00243-3
Marques, M. A. L., Vidal, J., Oliveira, M. J. T., Reining, L., and Botti, S. (2011). Density-based Mixing Parameter for Hybrid Functionals. Phys. Rev. B 83, 035119. doi:10.1103/physrevb.83.035119
Marsman, M., Paier, J., Stroppa, A., and Kresse, G. (2008). Hybrid Functionals Applied to Extended Systems. J. Phys. Condens. Matter 20, 064201. doi:10.1088/0953-8984/20/6/064201
Michalicek, G., Betzinger, M., Friedrich, C., and Blügel, S. (2013). Elimination of the Linearization Error and Improved Basis-Set Convergence within the Flapw Method. Comp. Phys. Commun. 184, 2670–2679. doi:10.1016/j.cpc.2013.07.002
Muscat, J., Hung, A., Russo, S., and Yarovsky, I. (2002). First-principles Studies of the Structural and Electronic Properties of Pyrite FeS2. Phys. Rev. B 65, 054107. doi:10.1103/physrevb.65.054107
Nabok, D., Gulans, A., and Draxl, C. (2016). Accurate All-Electron G0W0 Quasiparticle Energies Employing the Full-Potential Augmented Plane-Wave Method. Phys. Rev. B 94, 035118. doi:10.1103/physrevb.94.035118
Nesbitt, H. W., Uhlig, I., Bancroft, G. M., and Szargan, R. (2003). Resonant XPS Study of the Pyrite Valence Band with Implications for Molecular Orbital Contributions. Am. Mineral. 88, 1279–1286. doi:10.2138/am-2003-8-910
Ohsawa, A., Yamamoto, H., and Watanabe, H. (1974). X-ray Photoelectron Spectra of Valence Electrons in FeS2, CoS2 and NiS2. J. Phys. Soc. Jpn. 37, 568. doi:10.1143/jpsj.37.568
Ollonqvist, T., Perälä, R., and Väyrynen, J. (1997). Unoccupied Electronic States of the FeS2(100) Surface Studied by Inverse Photoemission. Surf. Sci. 377-379, 201–205. doi:10.1016/s0039-6028(96)01351-9
Ouarab, N., and Boumaour, M. (2017). First-principles Calculations of Electronic and Optical Properties of Fe 1−x Zn x S 2 and Zn 1−x Mg x O Alloys. Curr. Appl. Phys. 17, 1169–1180. doi:10.1016/j.cap.2017.05.008
Paier, J., Marsman, M., Hummer, K., Kresse, G., Gerber, I. C., and Ángyán, J. G. (2006a). Erratum: "Screened Hybrid Density Functionals Applied to Solids" [J. Chem. Phys. 124, 154709 (2006)]. J. Chem. Phys. 125, 249901. J. Chem. Phys. 124, 154709 (2006). doi:10.1063/1.2403866”
Paier, J., Marsman, M., Hummer, K., Kresse, G., Gerber, I. C., and Ángyán, J. G. (2006b). Screened Hybrid Density Functionals Applied to Solids. J. Chem. Phys. 124, 154709. doi:10.1063/1.2187006
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996a). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Perdew, J. P., Ernzerhof, M., and Burke, K. (1996b). Rationale for Mixing Exact Exchange with Density Functional Approximations. J. Chem. Phys. 105, 9982–9985. doi:10.1063/1.472933
Perdew, J. P., Parr, R. G., Levy, M., and Balduz, J. L. (1982). Density-functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy. Phys. Rev. Lett. 49, 1691–1694. doi:10.1103/physrevlett.49.1691
Perdew, J. P., and Schmidt, K. (2001). Jacob’s Ladder of Density Functional Approximations for the Exchange-Correlation Energy. AIP Conf. Proc. 577, 1. doi:10.1063/1.1390175
Perdew, J. P., Yang, W., Burke, K., Yang, Z., Gross, E. K. U., Scheffler, M., et al. (2017). Understanding Band Gaps of Solids in Generalized Kohn-Sham Theory. Proc. Natl. Acad. Sci. USA 114, 2801–2806. doi:10.1073/pnas.1621352114
Pickett, W. E., Krakauer, H., and Allen, P. B. (1988). Smooth Fourier Interpolation of Periodic Functions. Phys. Rev. B 38, 2721–2726. doi:10.1103/physrevb.38.2721
Rahman, M., Boschloo, G., Hagfeldt, A., and Edvinsson, T. (2020). On the Mechanistic Understanding of Photovoltage Loss in Iron Pyrite Solar Cells. Adv. Mater. 32, 1905653. doi:10.1002/adma.201905653
Ren, X., Merz, F., Jiang, H., Yao, Y., Rampp, M., Lederer, H., et al. (2021). All-electron Periodic G0W0 Implementation with Numerical Atomic Orbital Basis Functions: Algorithm and Benchmarks. Phys. Rev. Mater. 5, 013807. doi:10.1103/physrevmaterials.5.013807
Ren, X., Rinke, P., Blum, V., Wieferink, J., Tkatchenko, A., Sanfilippo, A., et al. (2012). Resolution-of-identity Approach to Hartree-Fock, Hybrid Density Functionals, RPA, MP2 andGWwith Numeric Atom-Centered Orbital Basis Functions. New J. Phys. 14, 053020. doi:10.1088/1367-2630/14/5/053020
Rojas, H. N., Godby, R. W., and Needs, R. J. (1995). Space-Time Method forAb InitioCalculations of Self-Energies and Dielectric Response Functions of Solids. Phys. Rev. Lett. 74, 1827–1830. doi:10.1103/physrevlett.74.1827
Sánchez, C., Flores, E., Barawi, M., Clamagirand, J. M., Ares, J. R., and Ferrer, I. J. (2016). Marcasite Revisited: Optical Absorption gap at Room Temperature. Solid State. Commun. 230, 20–24. doi:10.1016/j.ssc.2016.01.004
Schena, T., Bihlmayer, G., and Blügel, S. (2013). First-Principles Studies of FeS2 Using Many-Body Perturbation Theory in the G0W0 Approximation. Phys. Rev. B 88, 235203. doi:10.1103/physrevb.88.235203
Schlegel, A., and Wachter, P. (1976). Optical Properties, Phonons and Electronic Structure of Iron Pyrite (FeS2). J. Phys. C: Solid State. Phys. 9, 3363–3369. doi:10.1088/0022-3719/9/17/027
Seidl, A., Görling, A., Vogl, P., Majewski, J. A., and Levy, M. (1996). Generalized Kohn-Sham Schemes and the Band-gap Problem. Phys. Rev. B 53, 3764–3774. doi:10.1103/physrevb.53.3764
Shen, T., Zhang, X.-W., Shang, H., Zhang, M.-Y., Wang, X., Wang, E.-G., et al. (2020). Influence of High-Energy Local Orbitals and Electron-Phonon Interactions on the Band Gaps and Optical Absorption Spectra of Hexagonal boron Nitride. Phys. Rev. B 102, 045117. doi:10.1103/physrevb.102.045117
Shimazaki, T., and Asai, Y. (2008). Band Structure Calculations Based on Screened Fock Exchange Method. Chem. Phys. Lett. 466, 91–94. doi:10.1016/j.cplett.2008.10.012
Shishkin, M., and Kresse, G. (2007). Self-consistentGWcalculations for Semiconductors and Insulators. Phys. Rev. B 75, 235102. doi:10.1103/physrevb.75.235102
Shukla, S., Xing, G., Ge, H., Prabhakar, R. R., Mathew, S., Su, Z., et al. (2016). Origin of Photocarrier Losses in Iron Pyrite (FeS2) Nanocubes. ACS Nano 10, 4431–4440. doi:10.1021/acsnano.6b00065
Singh, D. (1991). Ground-state Properties of Lanthanum: Treatment of Extended-Core States. Phys. Rev. B 43, 6388–6392. doi:10.1103/physrevb.43.6388
Singh, D. J., and Nordström, L. (2006). Planewaves, Pseudopotentials and the LAPW Method. 2nd ed. edn. New York: Springer.
Skone, J. H., Govoni, M., and Galli, G. (2014). Self-consistent Hybrid Functional for Condensed Systems. Phys. Rev. B 89, 195112. doi:10.1103/physrevb.89.195112
Spagnoli, D., Refson, K., Wright, K., and Gale, J. D. (2010). Density Functional Theory Study of the Relative Stability of the Iron Disulfide Polymorphs Pyrite and Marcasite. Phys. Rev. B 81, 094106. doi:10.1103/physrevb.81.094106
Stankovski, M., Antonius, G., Waroquiers, D., Miglio, A., Dixit, H., Sankaran, K., et al. (2011). G0W0band gap of ZnO: Effects of Plasmon-Pole Models. Phys. Rev. B 84, 241201. (R). doi:10.1103/physrevb.84.241201
Sun, R., Chan, M. K. Y., and Ceder, G. (2011). First-principles Electronic Structure and Relative Stability of Pyrite and Marcasite: Implications for Photovoltaic Performance. Phys. Rev. B 83, 235311. doi:10.1103/physrevb.83.235311
Tian, A., Xu, Q., Shi, X., Yang, H., Xue, X., You, J., et al. (2015). Pyrite Nanotube Array Films as an Efficient Photocatalyst for Degradation of Methylene Blue and Phenol. RSC Adv. 5, 62724–62731. doi:10.1039/c5ra07434g
van der Heide, H., Hemmel, R., van Bruggen, C. F., and Haas, C. (1980). X-ray Photoelectron Spectra of 3d Transition Metal Pyrites. J. Solid State. Chem. 33, 17–25. doi:10.1016/0022-4596(80)90543-5
van Setten, M. J., Giantomassi, M., Bousquet, E., Verstraete, M. J., Hamann, D. R., Gonze, X., et al. (2018). The PseudoDojo: Training and Grading a 85 Element Optimized Norm-Conserving Pseudopotential Table. Comp. Phys. Commun. 226, 39–54. doi:10.1016/j.cpc.2018.01.012
van Setten, M. J., Giantomassi, M., Gonze, X., Rignanese, G.-M., and Hautier, G. (2017). Automation Methodologies and Large-Scale Validation for GW : Towards High-Throughput GW Calculations. Phys. Rev. B 96, 155207. doi:10.1103/physrevb.96.155207
Wadia, C., Alivisatos, A. P., and Kammen, D. M. (2009). Materials Availability Expands the Opportunity for Large-Scale Photovoltaics Deployment. Environ. Sci. Technol. 43, 2072–2077. doi:10.1021/es8019534
Walter, J., Zhang, X., Voigt, B., Hool, R., Manno, M., Mork, F., et al. (2017). Surface Conduction in N-type Pyrite FeS2 Single Crystals. Phys. Rev. Mater. 1, 065403. doi:10.1103/physrevmaterials.1.065403
Wang, D.-Y., Jiang, Y.-T., Lin, C.-C., Li, S.-S., Wang, Y.-T., Chen, C.-C., et al. (2012). Solution-processable Pyrite FeS2 Nanocrystals for the Fabrication of Heterojunction Photodiodes with Visible to NIR Photodetection. Adv. Mater. 24, 3415–3420. doi:10.1002/adma.201200753
Wilson, J. A. (1972). Systematics of the Breakdown of Mott Insulation in Binary Transition Metal Compounds. Adv. Phys. 21, 143–198. doi:10.1080/00018737200101278
Wu, L., Dzade, N. Y., Gao, L., Scanlon, D. O., Öztürk, Z., Hollingsworth, N., et al. (2016). Enhanced Photoresponse of FeS2Films: The Role of Marcasite-Pyrite Phase Junctions. Adv. Mater. 28, 9602–9607. doi:10.1002/adma.201602222
Zhang, M.-Y., Cui, Z.-H., and Jiang, H. (2018). Relative Stability of FeS2 Polymorphs with the Random Phase Approximation Approach. J. Mater. Chem. A. 6, 6606–6616. doi:10.1039/c8ta00759d
Zhang, M.-Y., and Jiang, H. (2019). Electronic Band Structure of Cuprous and Silver Halides: An All-Electron GW Study. Phys. Rev. B 100, 205123. doi:10.1103/physrevb.100.205123
Zhang, M. Y., Cui, Z. H., Wang, Y. C., and Jiang, H. (2020). Hybrid Functionals with System‐dependent Parameters: Conceptual Foundations and Methodological Developments. WIREs Comput. Mol. Scimol. Sci. 10, 1476. doi:10.1002/wcms.1476
Keywords: iron disulfide, band structure, GW approximation, self-energy, linearized augmented plane waves, hybrid functionals
Citation: Zhang M-Y and Jiang H (2021) Accurate Prediction of Band Structure of FeS2: A Hard Quest of Advanced First-Principles Approaches. Front. Chem. 9:747972. doi: 10.3389/fchem.2021.747972
Received: 27 July 2021; Accepted: 14 September 2021;
Published: 28 September 2021.
Edited by:
Patrick Rinke, Aalto University, FinlandReviewed by:
Walter Lambrecht, Case Western Reserve University, United StatesCopyright © 2021 Zhang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Jiang, amlhbmdoY2hlbUBwa3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.