 Timon Geib
Timon Geib Ghazaleh Moghaddam
Ghazaleh Moghaddam Aimee Supinski
Aimee Supinski Lekha Sleno
Lekha Sleno
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 20 August 2021
Sec. Analytical Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.736788
Acetaminophen (APAP) is a mild analgesic and antipyretic used commonly worldwide. Although considered a safe and effective over-the-counter medication, it is also the leading cause of drug-induced acute liver failure. Its hepatotoxicity has been linked to the covalent binding of its reactive metabolite, N-acetyl p-benzoquinone imine (NAPQI), to proteins. The aim of this study was to identify APAP-protein targets in both rat and mouse liver, and to compare the results from both species, using bottom-up proteomics with data-dependent high resolution mass spectrometry and targeted multiple reaction monitoring (MRM) experiments. Livers from rats and mice, treated with APAP, were homogenized and digested by trypsin. Digests were then fractionated by mixed-mode solid-phase extraction prior to liquid chromatography-tandem mass spectrometry (LC-MS/MS). Targeted LC-MRM assays were optimized based on high-resolution MS/MS data from information-dependent acquisition (IDA) using control liver homogenates treated with a custom alkylating reagent yielding an isomeric modification to APAP on cysteine residues, to build a modified peptide database. A list of putative in vivo targets of APAP were screened from data-dependent high-resolution MS/MS analyses of liver digests, previous in vitro studies, as well as selected proteins from the target protein database (TPDB), an online resource compiling previous reports of APAP targets. Multiple protein targets in each species were found, while confirming modification sites. Several proteins were modified in both species, including ATP-citrate synthase, betaine-homocysteine S-methyltransferase 1, cytochrome P450 2C6/29, mitochondrial glutamine amidotransferase-like protein/ES1 protein homolog, glutamine synthetase, microsomal glutathione S-transferase 1, mitochondrial-processing peptidase, methanethiol oxidase, protein/nucleic acid deglycase DJ-1, triosephosphate isomerase and thioredoxin. The targeted method afforded better reproducibility for analysing these low-abundant modified peptides in highly complex samples compared to traditional data-dependent experiments.
Drugs are generally metabolized by the liver into biologically inactive forms and eliminated from the body through bile and urine. However during these processes, they can also be bioactivated by hepatic enzymes into reactive electrophilic intermediates and subsequently react with nucleophilic sites of proteins to form covalent adducts (Xu et al., 2005; Park et al., 2011). Reactive metabolites can in turn cause oxidative stress, depleting intracellular glutathione, and affect energy metabolism by binding to mitochondrial protein complexes (Pessayre, 1995; Srivastava et al., 2010).
Acetaminophen (N-acetyl p-aminophenol, APAP) is one of the most commonly used analgesic and antipyretic, and is known to be the number one cause of acute liver failure in North America (Bateman, 2016). APAP is deemed safe as long as recommended doses are not exceeded, but very high doses can cause severe hepatotoxicity, liver injury and even death in both children and adults (Tittarelli et al., 2017). APAP metabolism primarily occurs in the liver, where major biotransformations include sulfate and glucuronide conjugation forming readily excretable phase II metabolites. A smaller proportion, between 10 and 15%, is converted to the reactive electrophile N-acetyl p-benzoquinone imine (NAPQI), by multiple cytochrome P450 enzymes (Laine et al., 2009). NAPQI can generally be detoxified via glutathione conjugation and further excreted via the mercapturic acid pathway. However, excess NAPQI can bind to nucleophilic sulfhydryl groups in cysteines of cellular proteins, leading to possible protein dysfunction due to conformational changes, potentially leading to acute liver failure (James et al., 2003; Graham and Scott, 2005). Mitochondrial protein adducts have been shown to directly correlate to the initiation of hepatotoxicity through mitochondrial dysfunction and DNA fragmentation (Tirmenstein and Nelson, 1989; McGill et al., 2012; McGill et al., 2014; Xie et al., 2015; Ramachandran and Jaeschke, 2020). Identifying and monitoring modified proteins from in vivo samples could help better understand the metabolic processes involved in APAP-related hepatotoxicity and potentially lead to the detection of important biomarkers (Cohen et al., 1997; Bissell et al., 2001).
In this study, liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) was employed to identify in vivo protein targets of APAP, via the formation of NAPQI, in mouse and rat. Liver homogenates were digested by trypsin and fractionated by mixed-mode solid-phase extraction (SPE) prior to LC-MS/MS analysis. A scheduled multiple reaction monitoring (MRM) was developed based on data-dependent LC-MS/MS results as well as previous reports of APAP-modified proteins, while using a custom alkylation reagent to prepare isomerically-modified peptides from liver homogenates. This comparative analysis in rat and mouse could potentially provide novel mechanistic insight into APAP-related hepatotoxicity.
Urea was purchased from BioRad (Mississauga, ON, Canada). APAP, trypsin (TPCK-treated, from bovine pancreas), sodium n-dodecyl sulfate (SDS), dithiothreitol (DTT), iodoacetamide (IAM), thiourea, ammonium bicarbonate, ammonium acetate, ammonium hydroxide, formic acid, acetic acid, acetonitrile (ACN), methanol (MeOH) and all other reagents were from Sigma-Aldrich (St. Louis, MO, United States). Labeling agent N-(4-hydroxyphenyl)-2-iodoacetamide (HP-IAM) was synthesized in-house as previously described (LeBlanc et al., 2014). Ultra-pure water was produced using a Millipore Synergy UV system (Billerica, MA, United States).
Four Sprague-Dawley male rats (450−550 g) were dosed with 600 mg/kg APAP (IP; solubilized in 60% PEG 200) or two animals were treated with vehicle for control samples. Rat livers were collected after 24 h post dosing. Male C57BL/6 mice (27−35 g) were treated (IP, in saline) with 150 mg/kg (2 and 6 h) and 300 mg/kg (2 and 6 h), as well as with vehicle for control samples. Two mice were treated at each dose and timepoint. All experiments were performed at INRS Centre National de Biologie Expérimentale (Laval, QC, Canada). The protocol was approved by the Ethics Committee of the INRS Centre National de Biologie Expérimentale under the ethical practices of the Canadian Council on Animal Care (project UQLK.14.02).
Frozen liver samples, stored at −80°C, were homogenized in 100 mM ammonium bicarbonate (ABC buffer, pH 8–8.5) at 5 ml/g tissue weight using a hand-held homogenizer (Tissuemiser; Thermo Fisher Scientific, Waltham, MA, United States), and 100 μl aliquots were combined with 25 μl of 0.1% SDS, and heated for 5 min at 95°C. After cooling samples, 50 μl of a solution of 7 M urea and 2 M thiourea in water was added, and a probe sonicator was used ((XL-2000; Qsonica, Newtown, CT, United States) for three cycles of 10 s, followed by another 15 min in an ultrasonic bath (Branson 2510; Branson Ultrasonics, Brookfield, CT, United States). The resulting protein extract was then diluted with 500 μl of 100 mM ABC buffer. Reductive alkylation was performed by adding DTT (30 μl, 100 mM; 37°C for 20 min), followed by IAM (45 μl, 100 mM; 37°C for 30 min in the dark). Similarly, control samples, from vehicle-treated animals, were alkylated using HP-IAM (by simply substituting the IAM mentioned above). Samples were then digested by trypsin (30 μl, 1 mg/ml; 37°C for 18 h), followed by the addition of 300 μl of 2% formic acid prior to fractionation by solid phase extraction (SPE) on OASIS MCX cartridges (1 cc, 30 mg; Waters, Milford, MA, United States). Loaded cartridges were washed in three steps with 2% formic acid in water, 100% MeOH and 50% MeOH (1 ml each). Eight fractions were collected by eluting with 15, 20, 25, 35, 50, and 200 mM ammonium acetate in 50% MeOH, then 0.1 and 3% ammonium hydroxide in 50% MeOH (1 ml each). Fractions were dried under vacuum and stored at −30°C until analysis.
SPE fractions were reconstituted in 100 µl 10% acetonitrile and injected (20 μl) onto an Aeris PEPTIDE XB-C18 column (100 × 2.1 mm, 1.7 μm) (Phenomenex, Torrance, CA, United States) using a Nexera UHPLC system (Shimadzu, Columbia, MD, United States) with water (A) and ACN (B), both containing 0.1% formic acid, at a flow rate of 300 μl/min (at 40°C). The elution gradient started at 5% B for 2.5 min, and was linearly increased to 30% B in 47.5 min, to 50% B in 5 min, then to 90% B in 1 min, held for another 4 min at 90% B.
Data-dependent experiments were performed to collect MS and MS/MS spectra on a high-resolution quadrupole-time-of-flight TripleTOF 5600 mass spectrometer (Sciex, Concord, ON, Canada) equipped with a DuoSpray ion source in positive electrospray mode set at 5 kV source voltage, 500°C source temperature and 50 psi GS1/GS2 gas flows, with a declustering potential of 80 V. The instrument performed a survey TOF-MS acquisition from m/z 140–1250 (250 ms accumulation time), followed by MS/MS on the 15 most intense precursor ions with a minimum intensity of 250 cps from m/z 300–1250. Precursor ion were excluded for 20 s after two occurrences in data-dependent acquisition (DDA) mode with dynamic background subtraction. Each MS/MS acquisition (m/z 80–1500, high sensitivity mode) had an accumulation time of 50 ms and collision energy of 30 ± 10 V, with a total cycle time was 1.05 s. The MS instrument was calibrated at every four injections using an in-house calibration mix. Analyst TF software (1.7.1; Sciex) was used for data acquisition and raw data was visualized with PeakView 2.2 with MasterView 1.1 (Sciex). The mass spectrometry proteomics data from high-resolution data-dependent expriments have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD027674 and 10.6019/PXD027674.
Targeted assays were developed using scheduled LC-MRM on a Sciex QTRAP 5500 hybrid quadrupole-linear ion trap system with a TurboIonSpray ion source in positive mode, with identical UHPLC conditions as for data-dependent high resolution experiments. Source parameters were as follows: ionspray voltage 5 kV; temperature 550°C; GS1 and GS2 50 psi; and curtain gas 35 psi. Declustering, entrance and collision cell exit potentials were set at 80, 10 and 13 V, respectively. Collision-induced dissociation was performed at a collision energy of 30 V. Scheduled MRM time windows were set at 240 s, with a targeted scan time at 1.25 s. Minimum and maximum dwell times were 10 and 250 ms, respectively. MRM transitions monitored for SPE fractions two to eight can be found in Supplementary Tables S1, S2, for rat and mouse, respectively. Sciex Analyst software 1.7 was used for data acquisition.
Raw data files from quadrupole-time-of-flight experiments were searched against the rat or mouse UniProtKB/Swiss-Prot protein database (release date: 07/18/2018, including common protein contaminants) using Sciex ProteinPilot 5.0. To detect APAP covalent adducts, a custom modification was added to the Paragon algorithm (Shilov et al., 2007) with a probability of 50% for APAP modification on cysteine residues (C8H8NO2 replacing H, Δm = +149.04766 u). The search was performed for +2 to +4 charge states at a MS tolerance of 0.05 u on precursor ions and 0.1 u on fragments. For protein identifications, a target-decoy approach was applied at a 1% false discovery rate (Aggarwal and Yadav, 2016). From the list of identified APAP-modified peptides, those with spectral confidence less than 95% were removed, as were those with modifications other than APAP and cysteine carbamidomethylation (CAM).
LC-MRM peaks were integrated and verified using MultiQuant 3.0.2 (Sciex) to ensure that APAP-modified peptides matched with N-(4-hydroxyphenyl)-2-carbamidomethylated (HP-CAM) peptides from control samples. Confirmation of a modified peptide was based on the following criteria: 1) signal-to-noise (S/N) of all transitions above 10; 2) ≤20% deviation of relative abundance (seeEq. 1) (Geib et al., 2019) for the first transition and ≤30% deviation for second and third transitions) from HP-CAM-modified peptide standard; and 3) within 60 s of the corresponding HP-CAM peptide’s retention time, to correct for small drifts in RT over the acquisition batch.
APAP-modified peptides with multiple cysteines, containing at least one CAM-cysteine, did not meet the second (or third) criteria, due to no standard samples available for comparison (all cysteines were HP-CAM modified in reference samples). Therefore, confirmation for these modified peptides incorporated into the MRM method was achieved by the absence of corresponding signals in HP-CAM reference samples.
The goal of this study was to assess the applicability of a targeted LC-MS/MS method for in vivo biomonitoring of liver protein adducted by reactive metabolites in multiple samples from rat and mouse, and to compare the results obtained via data-dependent high-resolution MS/MS assays. With the aim of identifying low abundant APAP-modified peptides in this highly complex biological matrix, tryptic digests were fractionated by mixed-mode solid-phase extraction. These fractions were subjected to data-dependent high-resolution tandem mass spectrometry (HRMS/MS) analyses as well as targeted MRM assays, and results from these analyses were compared.
Four liver digests from each species were initially subjected to untargeted high-resolution MS/MS to identify potentially novel target proteins. In total, 15 APAP-modified mouse peptides (from 14 target proteins) were identified (with over 95% peptide confidence). These included five peptides with more than one cysteine, one being APAP-bound and the other(s) carbamidomethylated, however the exact location of the APAP modification was found in each case, based on unique y and b-ions in their high-resolution MS/MS spectra. A specifically challenging example was seen for peptides KPIGLC174C175IAPVLAAK and C174C175IAPVLAAK from glutamine amidotransferase-like class 1 domain-containing protein 3A (and its rat ES1 protein homolog) (seeSupplementary Figure S1), where the APAP modification could be pinpointed on Cys175. From the rat liver digests, 17 distinct peptides were identified as APAP-modified from 16 proteins. Interestingly, the modified peptide found from triosephosphate isomerase in rat, which has two cysteine residues (CLGELICTLNAAK), was detected both as singly and doubly APAP-modified. A summary of all APAP-modified peptides identified from data-dependent experiments can be found in Table 1. From these experiments, several protein orthologs were found in both rodent species, modified at the same cysteine site.
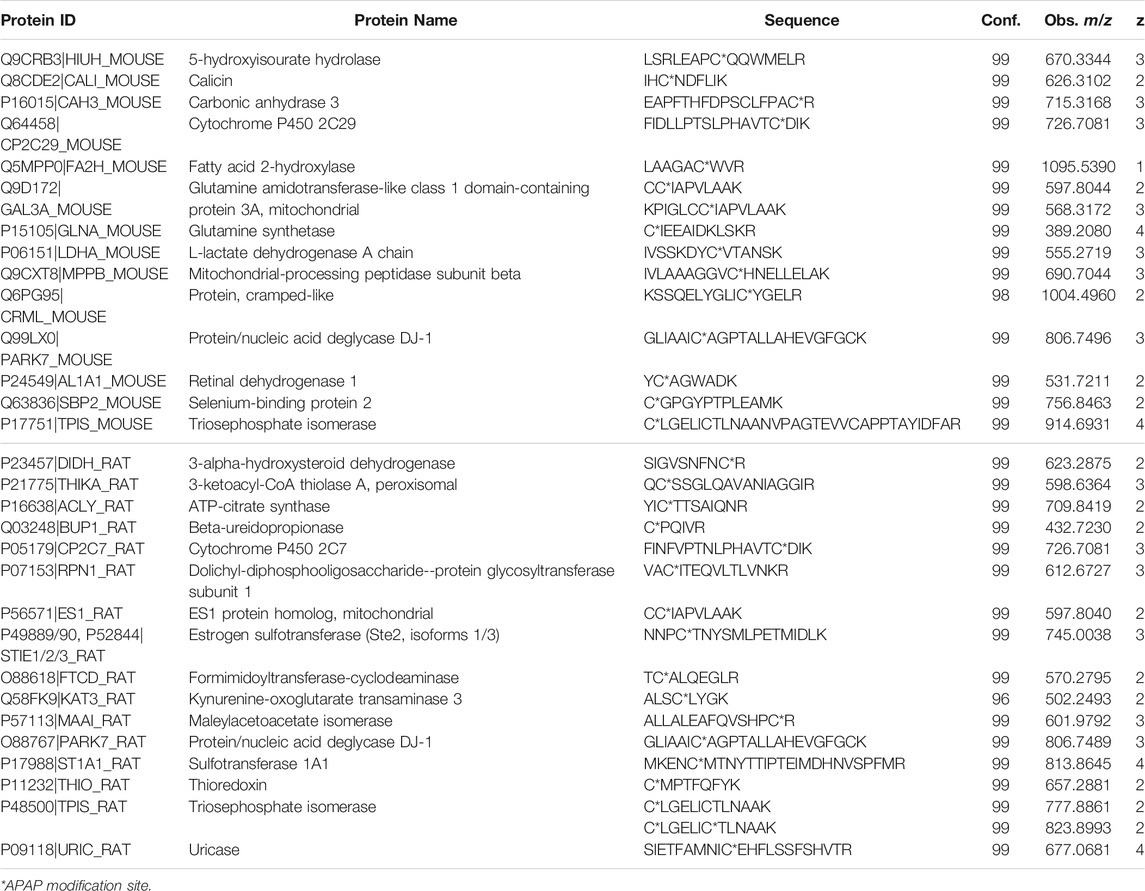
TABLE 1. APAP-modified peptides in rat and mouse liver identified by data-dependent high-resolution MS/MS.
Control liver samples were alkylated with hydroxyphenyl iodoacetamide (HP-IAM) prior to trypsin digestion, SPE fractionation and data-dependent LC-MS/MS analysis (seeFigure 1). HP-CAM modification yields a positional isomer of APAP-modification on all cysteines and modified peptides were identified using the same criteria as APAP-modified peptides from database searching. In total, 1466 HP-CAM-modified proteins with 4554 distinct HP-CAM peptides were identified in the mouse control sample; and 1073 HP-CAM-modified proteins with 3599 distinct HP-CAM-peptides were found in the rat control sample. The resulting list of HP-CAM peptides was further filtered to include peptides of a select list of potential protein targets. These rat and mouse protein targets were based on our data-dependent results, as well as previous APAP in vitro binding studies (Golizeh et al., 2015; Geib et al., 2019), and additional APAP protein targets found in the Target Protein Database (TPDB) (Hanzlik et al., 2020). The resulting filtered HP-CAM-peptide list contained information on which SPE fraction contained the identified peptide, as well as peptide retention time, charge state, and its three most intense fragment ions (preferably with fragment m/z > precursor m/z). These target peptides selected for MRM analysis are in Supplementary Table S3.
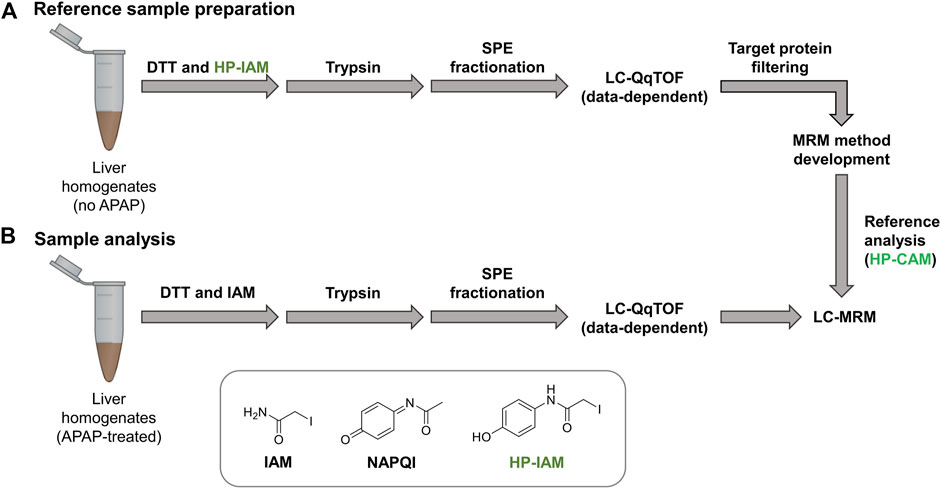
FIGURE 1. Reference samples (A) were prepared using non-treated mouse and rat liver homogenates and HP-IAM as the cysteine alkylation reagent. Modified peptides of interest from potential protein targets were selected for MRM method preparation. Final LC-sMRM methods were used to screen for modified peptides in (B) APAP-treated samples. Data-dependent high-resolution MS/MS experiments were performed on reference samples as well as selected APAP-treated rat and mouse samples for identifying modified peptides via database searching with custom modification (+C8H7NO2 addition onto Cys residues).
Scheduled MRM methods were built, with three transitions per peptide, and separated based on species and SPE fraction. MRM transitions of modified peptides were also monitored in adjacent fractions as those where they were detected in HP-CAM samples, since peptides can often elute in more than one fraction. In the case of two peptides from mouse glutamine amidotransferase and rat ES1 protein homolog, where one Cys was APAP-modified and the other was carbamidomethylated, retention times and fragment ions from data-dependent MS/MS were used to build the MRM method. Unfortunately, several other peptides with multiple cysteines were not monitored by MRM as their signals were not confirmed when developing the targeted method.
APAP-treated and HP-CAM alkylated liver digests were analyzed by scheduled LC-MRM methods specific to each SPE fraction. LC-MRM peaks were integrated, and relative peak areas for each transition (transition/sum of all transitions) as well as retention times were compared with HP-CAM signals for the same peptide. Figure 2 shows a representative example of a confirmed target LC-MRM for a mouse CYP 2C29 peptide, found via data-dependent MS/MS and confirmed by MRM analyses. The analogous peptide in rat CYP2C7 was also confirmed via data-dependent MS/MS and LC-MRM. Figure 2 also shows the example of the tryptic peptide with two cysteines, but only one of them being APAP-modified, from mouse mitochondrial glutamine-amidotransferase-like 1 domain-containing protein 3A and rat mitochondrial ES1 protein homolog. Remarkably, the non-tryptic peptide containing the same modified cysteine was found in our data-dependent analyses in both species, as well as screened for during LC-MRM analyses, with the same modification site being confirmed in all liver samples. These modified peptides were confirmed based on the absence of a corresponding signal in HP-CAM-control samples.
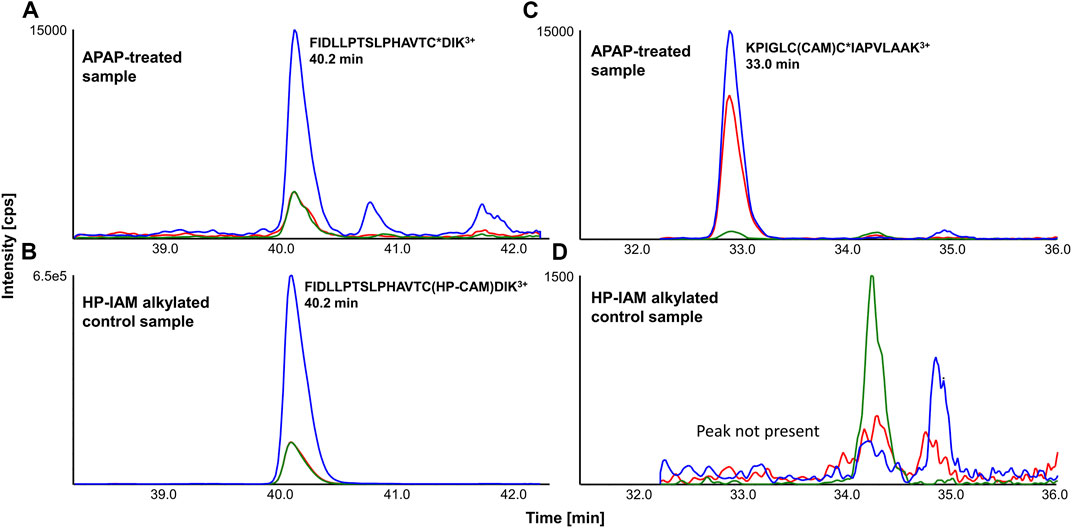
FIGURE 2. Representative LC-MRM chromatograms of (A) FIDLLPTSLPHAVTC*DIK3+ (from cytochrome P450 2C29) from an APAP-treated liver digest and (B) its reference in HP-CAM control sample in mouse (from SPE fraction 3), as well as (C) KPIGLCC*IAPVLAAK3+ (from mitochondrial ES1 protein homolog) from an APAP-treated liver digest and (D) its correspondingly absent signal in the HP-CAM control sample in rat due to the presence of two cysteines in its sequence (from SPE fraction 8). The star (*) in the peptide sequence indicates the confirmed site of APAP modification.
From eight APAP-treated mouse livers (two animal each at 150 and 300 mg/kg, either 2 and 6 h post dose), LC-MRM confirmed 13 distinct APAP-modified sites from 13 different proteins. From Table 2, the distribution of these confirmed targets is shown for each dosing regime, with many of them being confirmed in all samples, and five being detectable only at the 300 mg/kg dosing level. The two dosing levels at 150 and 300 mg/kg in mouse were chosen since this species is known to be more susceptible to APAP-related hepatotoxicity, compared to rat which could tolerate the higher dose at 600 mg/kg. From previous work, it was found that at 600 mg/kg dose in rat, APAP-albumin adducts did not decrease over the 24 h (LeBlanc et al., 2014), however, shorter timepoints (2 and 6 h) were chosen for mouse since they exhibit higher toxicity.
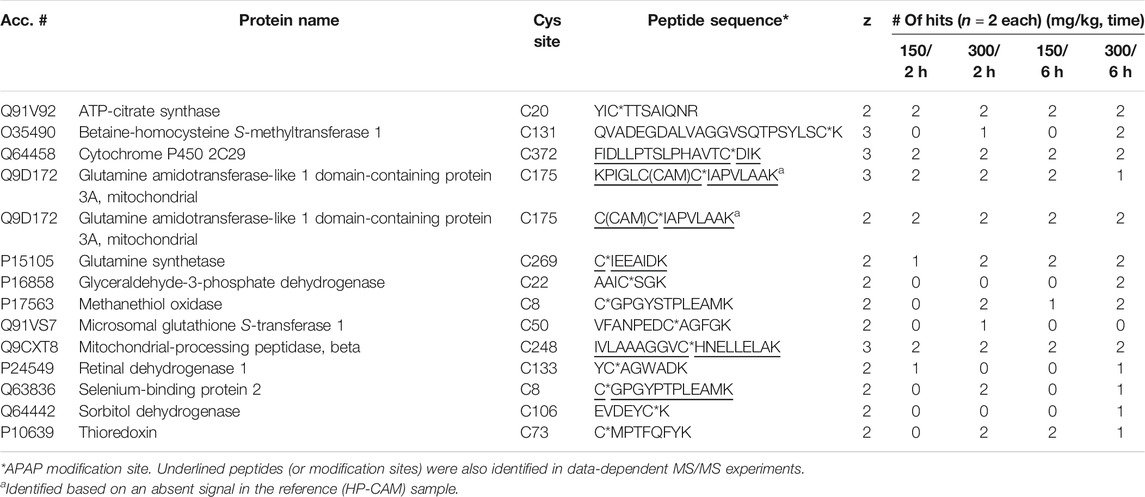
TABLE 2. APAP-modified peptides in mouse liver identified via LC-MRM.
From the analysis of four rat liver samples, 23 distinct APAP-peptides were confirmed, from 21 different proteins (Table 3). Two proteins were close homologs, CYP 2C6 and 2C7, with very similar peptide sequences, though the 2C6 peptide was found via LC-MRM only in one animal, compared to the 2C7 modified peptide being well detected in all rat livers. Two peptides with the same modification site were found for the ES1 protein homolog in all rats. Methanethiol oxidase also had two modified cysteine sites, though one was detectable in only one rat liver, whereas the one modified at Cys8 was detected in all rat livers, as well as in five of eight mice, including all livers at the 300 mg/kg dose. Also in mouse, a very similar protein, selenium-binding protein 2, was modified at the same cysteine site, with a peptide differing only by one amino acid, in three of four mice at the higher dose. Another interesting example is that of triosephosphate isomerase, which was found to have one peptide having two APAP-modified cysteines by data-dependent analyses. LC-MRM results confirmed this doubly modified peptide in all four rat livers. As shown in Table 1, the same peptide with only one cysteine modified was also identified from rat as well as the singly modified mouse peptide, however these peptides were not screened for by LC-MRM. The mouse peptide specifically was a very long peptide with 33 residues and a charge state of 4+, making it quite difficult to screen for via LC-MRM without a corresponding signal in the HP-CAM reference sample. Another example of a target protein found in both species via data-dependent HRMS/MS analyses only is protein/nucleic acid deglycase DJ-1 (also known also Parkinson disease protein 7 homolog), with an identical peptide in mouse and rat incorporating two cysteines, one of which was APAP-modified. This large peptide was not easily amenable to MRM analysis due to the lack of significantly intense fragment ions, as well as no appropriate HP-CAM peptide for method optimisation. LC-MRM data with integrated peak areas and retention times from all samples have been made available in Supplementary Table S4.
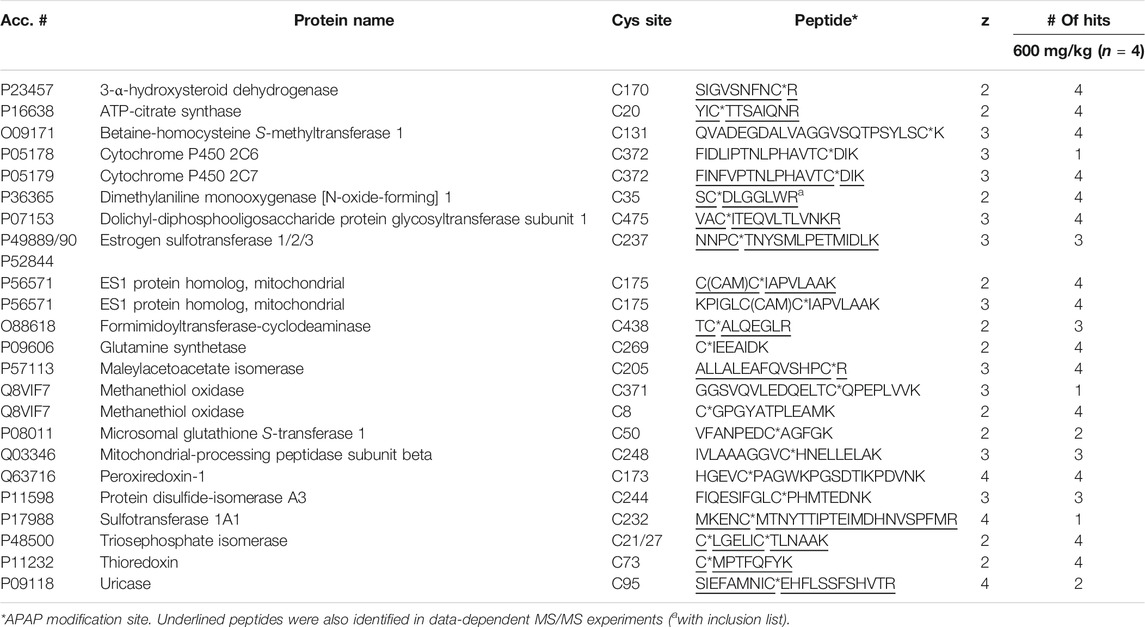
TABLE 3. Identified APAP-modified peptides in rat liver via LC-MRM.
All APAP-modified peptides detected via LC-MRM not detected by data-dependent analyses were screened a second time by incorporating an inclusion list for targeted precursor ions. The same fractions with confirmed LC-MRM hits were analyzed. Only one modified peptide was ultimately detected with sufficient spectral confidence >95% (see high-resolution MS/MS in Supplementary Figure S1) for the modified peptide from rat dimethylaniline (N-oxide forming) monooxygenase, which was confirmed in all four rat livers by LC-MRM. This protein had been added to the LC-MRM method due to its presence as an APAP target in TPDB (Leeming et al., 2017). The same modification site that was previously found was confirmed.
Targeted LC-MS/MS workflows were specifically designed to utilize the high duty cycle and low limit of detection of multiple reaction monitoring (MRM). The detection of modified peptides was optimized using scheduled MRM methods built for each SPE fraction for a given species separately. By using HP-IAM in the reference samples, it was possible to mimic APAP-derived covalent modification on cysteines in the final protein digest. HP-CAM peptides were used to confirm which fractions to monitor for each modified peptide, as well as LC retention time and MS/MS fragmentation behavior, with relatively high signal intensity. Whenever possible, peptide candidates in one species were translated to the closest protein ortholog in the other species. Modified peptides not amenable to MRM transition development were omitted in the final scheduled MRM method, based on giving an appropriate signal for three MRM transitions in reference samples.
In general, targeted LC-MRM analyses showed far superior detectability of APAP-modified peptides in these samples. An important advantage that MRM detection has over data-dependent MS/MS, is that each modified peptide signal is continuously monitored, instead of depending on the automatic selection of a precursor of interest for MS/MS acquisition, which leads unfortunately to much less reproducible data as well as the loss of low-abundant peptides in highly complex samples. For this reason, many modified peptides would not have a chance of being identified with conventional untargeted bottom-up proteomics workflows. An important caveat to the MRM method, however, is that method development is more time-consuming, and in the case of APAP-modified peptides for this study, the custom alkylation HP-CAM peptides were crucial for confirmation. Also, MRM sensitivity depends highly on the fragmentation behavior of the ionized peptide. Certain highly charged or large peptides necessitate more optimization by changing collision energies and selected multiple fragment ions, and without an appropriate standard, this is impossible. High-resolution DDA experiments are more flexible, for example for those with multiple cysteines with only one being modified by APAP. A unique opportunity was afforded to specifically design LC-MRM assays for confirming protein targets by the possibility of having a positional isomer of APAP quantitatively modifying all cysteines in the liver homogenates. Without this standard as a reference, these hits may have been detected but not as easily confirmed.
The aim of this study was to identify protein targets of APAP’s reactive metabolite in mouse and rat liver to help better understand the mechanisms of APAP-induced hepatotoxicity. Using a combination of high-resolution data-dependent MS/MS and scheduled MRM experiments, a multitude of targets have been found for the first time, as well as confirming others previously reported targets of APAP from literature. It was possible to compare protein targets and modification sites in mouse and rat livers. Figure 3 summarizes the proteins confirmed in this study, with those overlapping between both species. All those listed were confirmed by LC-MRM analyses, except protein/nucleic acid deglycase DJ-1, which was found by data-dependent analyses in both species, however the modified peptide was not amenable to MRM analysis. What was quite striking was that for all proteins common to both species, the same modification sites were confirmed. This suggests the selective modification from APAP on these proteins, some of which could be specifically involved in the mechanism of drug-induced liver injury. Of the 24 protein groups listed in Figure 3, 17 were initially identified by DDA experiments and 12 were listed as APAP targets in TPDB. Though not reported as APAP targets, a few were also noted in TPDB as targets of other xenobiotics.
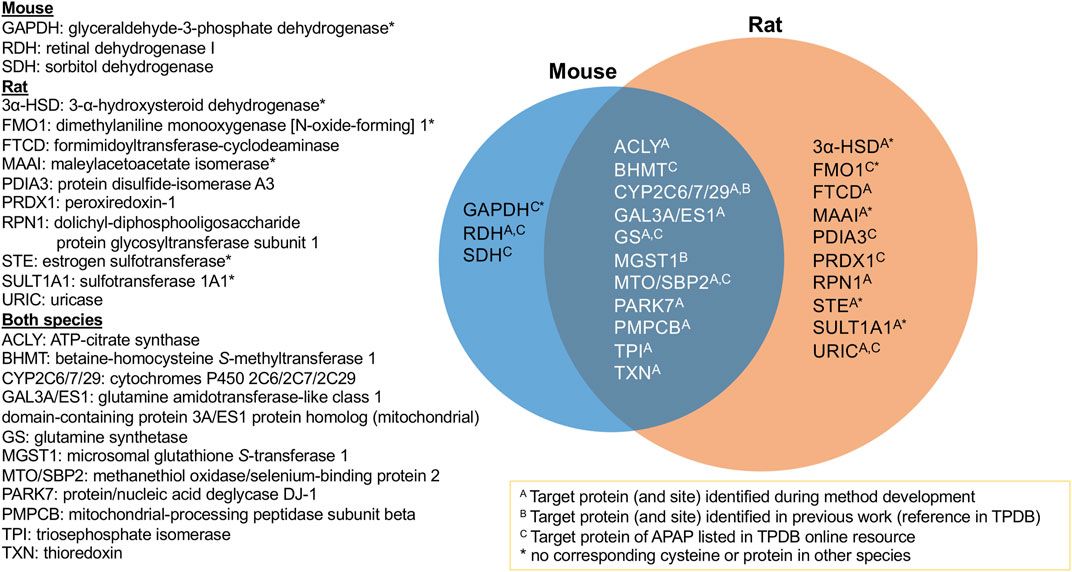
FIGURE 3. Summary of APAP protein targets confirmed in rat and mouse liver samples via targeted LC-MRM analyses.
From the 10 protein targets confirmed in rat only, four of these did not have corresponding cysteine residues in the mouse ortholog, namely two sulfotransferases, dimethylaniline monooxygenase and maleylacetoacetate isomerase. One other protein, 3-alpha hydroxysteroid dehydrogenase (3α-HSD), does not have a corresponding ortholog in mouse at all. In the case of the three targets found uniquely in mouse, one protein has no corresponding cysteine in the rat protein (GAPDH). Retinal dehydrogenase, though the same peptide was monitored for both species, was only found in two of the eight mouse samples. As for sorbitol dehydrogenase, the corresponding ortholog peptide was not incorporated into the MRM method, since it was not found in the rat HP-CAM reference sample and thus could not be optimized in the targeted method.
One example of an APAP target found in rat, with no corresponding cysteine in the mouse ortholog, was estrogen sulfotransferase (EST), a cytosolic enzyme that inhibits estrogen activity by conjugating a sulfonate group to it (Strott, 1996). The APAP-modified peptide NNPC237TNYSMLPETMIDLK, which is common to rat estrogen sulfotransferase isoforms 1, two or 3, was identified by data-dependent experiments, as well as confirmed by LC-MRM. Similarly, modified rat sulfotransferase 1A1 was found from data-dependant experiments, and was also confirmed by LC-MRM.
3α-HSD catalyzes NAD(P)+-dependent oxidoreduction of steroids and dihydrodiols. Multiple forms of 3α-HSD have been identified in rat liver with a role in xenobiotic metabolism and intracellular transport of bile acids (Turley and Dietschy, 1978). In rat liver, 3α-HSD can metabolize polycyclic aromatic hydrocarbon carcinogens and also affects net bile acid transport across hepatocytes (Dufort et al., 2001). Although not previously found to be a target of APAP binding, 3α-HSD has been identified as a target of bromobenzene (Koen et al., 2007), thiobenzamide (Ikehata et al., 2008) and furan (Moro et al., 2012), without site specificity noted. From this study, Cys170 was confirmed as the APAP modification site.
All 11 protein groups confirmed in both species (Figure 3) had the same cysteine modification sites. ATP-citrate synthase (ACLY) was found to be a target of APAP in both species by LC-MRM, following initial identification of the modified rat peptide via data-dependent MS/MS. The modified residue Cys20 is in its ATP-binding domain, which catalyses the conversion of citrate to acetyl-CoA in the TCA cycle. Glutamine synthetase (GS) is a cytosolic enzyme, also associated to mitochondria and ER. GS was previously identified as an APAP target by immunochemical detection in mice (Bulera et al., 1995). Its enzymatic activity was also significantly decreased by APAP in cultured hepatocytes. After identifying the Cys269 modification site by data-dependent MS/MS in mouse, the APAP peptide was confirmed in both species by LC-MRM. Triosephosphate isomerase (TPI) is a glycolytic enzyme necessary for energy production. Cytoplasmic TPI has been reported as a covalent target of bromobenzene (Koen et al., 2007), mycophenolic acid (Asif et al., 2007), diaminochlorotriazine (Dooley et al., 2008), 4-bromophenol (Koen et al., 2012), tienilic acid (Methogo et al., 2007) and thioacetamide (Koen et al., 2013). Although TPI was only confirmed by LC-MRM for the doubly APAP-modified peptide in rat, it was also found to be singly modified at the same site in both species via data-dependent experiments. It, therefore, was classified as a target of APAP in both species.
Rat ES1 protein homolog and mouse glutamine amidotransferase-like class 1 domain-containing protein 3A are orthologous mitochondrial proteins confirmed as modified in both species from these results. ES1 was found to be elevated in Down syndrome, potentially as an antioxidant response to increased mitochondrial ROS production (Shin et al., 2004). Other mitochondrial protein targets confirmed in both species were mitochondrial-processing peptidase subunit beta (PMPCB), responsible for the proteolytic processing of mitochondrially encoded precursor polypeptides, and protein/nucleic acid deglycase DJ-1, also known as Parkinson disease protein 7 homolog, PARK7. PARK7 catalyzes the deglycation of Maillard adducts between proteins or DNA and reactive carbonyls (Richarme et al., 2015; Richarme et al., 2017) and protects cells from H2O2-related death (Sekito et al., 2006). It is required for correct mitochondrial morphology and function. The same modification site (Cys106) of PARK7 was recently found following APAP treatment of liver microtissues using data-independent MS/MS acquisition (Bruderer et al., 2015). Mitochondrial protein targets are of specific interest, as they have been correlated to the initiation of APAP’s hepatotoxicity (Tirmenstein and Nelson, 1989; Ramachandran and Jaeschke, 2019). They have been proposed as a source of APAP-induced mitochondrial dysfunction (Hu et al., 2016), and are believed to be linked to the inhibition of electron transport chain (Lee et al., 2015), reactive oxygen species formation (Jiang et al., 2015) and mitogen-activated protein kinase and c-Jun N terminal kinase activation (Nguyen et al., 2021).
Two protein targets found previously by our group in vitro in rat liver microsomes, CYP2C6 and MGST1, were also confirmed here. Therefore, further in vitro work could be of value for finding biologically relevant targets in other cellular compartments, such as the cytosol and mitochondria. MGST1 had a common APAP-modified peptide in both species confirmed by LC-MRM. Orthologs CYP2C6 and 2C7 in rat, and 2C29 in mouse, were all found to be modified at Cys372 by APAP from data-dependent and MRM experiments. The latter two were consistently found as positive hits by LC-MRM in all animals of their respective species.
Other protein targets common to both species included betaine-homocysteine S-methyltransferase 1 (BHMT), methanethiol oxidase (MTO)/selenium binding protein 2 (SBP2) and thioredoxin (TXN). BHMT catalyzes betaine conversion to homocysteine in the biosynthesis of dimethylglycine and methionine. BHMT deficiency has been linked to hepatocellular carcinoma and fatty liver disease (Teng et al., 2011). BHMT was monitored as a potential target due to its presence in the TPDB list of APAP protein targets (Leeming et al., 2017), and confirmed the previously elucidated modification site. MTO and SBP2 are selenium-binding proteins, previously reported as major APAP targets in the liver (Bartolone et al., 1992; Pumford et al., 1992; Qiu et al., 1998) as well as other xenobiotics (Shipkova et al., 2004; Koen et al., 2007; Meier et al., 2007; Koen et al., 2012). Involved in organo-sulfur degradation, these proteins may be involved in the sensing of reactive xenobiotics in the cytoplasm (Pol et al., 2018).
Thioredoxin is involved in redox signalling through oxidation of its thiols. The APAP modification site was confirmed on Cys73, the only available free thiol in fully oxidized THX, which is also known to serve as a donor for nitrosylation of proteins under NO stress (Mitchell and Marletta, 2005). APAP overdose-linked oxidation of TXN2 has been previously observed (Ramachandran et al., 2015). Mitochondrial TXN2 and cytosolic TXN1 have been shown to be irreversibly modified by APAP (Jan et al., 2014). APAP-induced oxidation of cytosolic TXN1 results in the dissociation of TXN1 and its binding partner, apoptosis signal-regulating kinase 1 (Nakagawa et al., 2008). A protective effect of administration of albumin-fused recombinant TXN1, 4 h after APAP administration, has also been reported (Tanaka et al., 2014). Interestingly, this is the first report of APAP covalent binding to TXN, though it has been found as a target of other reactive metabolites (Koen et al., 2007; Ikehata et al., 2008; Moro et al., 2012; Koen et al., 2013).
Reactive metabolites can covalently bind to proteins causing downstream immune reactions and/or cell damage (Poprac et al., 2017). APAP protein adduction has long been studied and serves as a model of drug-induced hepatotoxicity, however, its mechanism of action is not yet fully understood (Heard et al., 2016). Results from this study confirmed many previously reported protein targets, as well as their specific modification sites, and adding several new in vivo protein targets in two rodent models. These findings can be further investigated by studying protein and/or site-specific effects and mechanisms related to drug-induced toxicity. Mitochondrial protein targets of APAP would be of specific interest for subsequent research since there is already much evidence of mitochondrial signalling being involved in APAP hepatotoxicity.
An analytical workflow was developed and applied to rat and mouse liver homogenates to investigate protein covalent binding following APAP administration. Using a combination of high-resolution MS/MS and scheduled MRM assays for proteomic analyses and a custom alkylation reagent, many in vivo protein targets and their modification sites in both species have been confirmed or newly identified. The protein targets identified could serve to better understand the reactivity of NAPQI in vivo and could point towards specific targets of interest linked to acetaminophen-related hepatotoxicity. The performance of targeted scheduled LC-MRM has been demonstrated, based on a curated list of potential candidates, as a powerful tool in LC-MS/MS proteomics of low-abundant modified peptides in highly complex biological samples.
MRM transitions and retention time settings used for LC-sMRM analyses of individual SPE fractions (two to eight) from rat and mouse digests (Supplementary Tables S1, S2, respectively). Overview of modified peptides screened for in scheduled MRM experiments (Supplementary Table S3). Results from scheduled MRM experiments, including peak areas for each transition monitored, relative areas, and retention times and calculated deviations to the control HP-CAM peptides (Supplementary Table S4). High-resolution MS/MS spectra for APAP-modified peptides identified using data-dependent acquisition (Supplementary Figure S1).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: ProteomeXchange PRIDE repository, accession no: PXD027674.
The animal study was reviewed and approved by INRS Centre National de Biologie Expérimentale under the ethical practices of the Canadian Council on Animal Care (project UQLK.14.02).
TG, MG, and LS conceived the research. TG, MG, and GM carried out sample preparation, and analyses. All authors contributed to data processing. TG, AS, and LS were involved in the preparation of tables and figures for the manuscript. TG and LS were the main contributors to the writing of the final manuscript. All authors made substantial, direct and intellectual contribution to the work, and revised the manuscript.
Financial support for this study was provided by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant # RGPIN 2016-06034) and a UQAM Institutional Research Chair in Bioanalytical Chemistry for LS. We would like to recognize funding for infrastructure from the Canadian Foundation for Innovation for LC-MS/MS instrumentation used in this study. We also acknowledge support to our mass spectrometry platform from CERMO-FC (Centre d’excellence de recherche sur les maladies orphelines – Fondation Courtois) via the UQAM Foundation.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.736788/full#supplementary-material
ACN, acetonitrile; APAP, acetaminophen; BHMT, betaine—homocysteine S-methyltransferase 1; CA3, carbonic anhydrase 3; CAM, carbamidomethylation; CPS1, mitochondrial carbamoyl-phosphate synthase [ammonia]; DTT, dithiothreitol; ER, endoplasmic reticulum; EST, estrogen sulfotransferase; GS, glutamine synthetase; HDAg, hepatitis delta antigen; HP-CAM, N-(4-hydroxyphenyl)-2-carbamidomethylation; HP-IAM, N-(4-hydroxyphenyl)-2-iodoacetamide; 3α-HSD, 3-alpha-hydroxysteroid dehydrogenase; IAM, iodoacetamide; IDA, information-dependent acquisition; IP, intraperitoneal; LC-MS/MS, liquid chromatography-tandem mass spectrometry; MeOH, methanol; MGST1, microsomal glutathione S-transferase 1; MRM, multiple reaction monitoring; MTO, methanethiol oxidase; NAPQI, N-acetyl p-benzoquinone imine; PMPCB, mitochondrial-processing peptidase subunit beta; SDS, n-dodecyl sulfate; S/N, signal-to-noise; SPE, solid-phase extraction; TPDB, Target Protein Database; TPI, triosephosphate isomerase; TXN, thioredoxin.
Aggarwal, S., and Yadav, A. K. (2016). False Discovery Rate Estimation in Proteomics. Methods Mol. Biol. 1362, 119–128. doi:10.1007/978-1-4939-3106-4_7
Asif, A. R., Armstrong, V. W., Voland, A., Wieland, E., Oellerich, M., and Shipkova, M. (2007). Proteins Identified as Targets of the Acyl Glucuronide Metabolite of Mycophenolic Acid in Kidney Tissue from Mycophenolate Mofetil Treated Rats. Biochimie 89, 393–402. doi:10.1016/j.biochi.2006.09.016
Bartolone, J. B., Birge, R. B., Bulera, S. J., Bruno, M. K., Nishanian, E. V., Cohen, S. D., et al. (1992). Purification, Antibody Production, and Partial Amino Acid Sequence of the 58-kDa Acetaminophen-Binding Liver Proteins. Toxicol. Appl. Pharmacol. 113, 19–29. doi:10.1016/0041-008x(92)90004-c
Bateman, D. N. (2016). “Acetaminophen (Paracetamol),” in Critical Care Toxicology - Diagnosis And Management Of the Critically Poisoned Patient. Editors J. Brent, K. Burkhart, P. Dargan, B. Hatten, B. Megarbane, R. Palmeret al. (Cham, Switzerland: Springer), 1–25. doi:10.1007/978-3-319-20790-2_108-2
Bissell, D., Gores, G. J., Laskin, D. L., and Hoofnagle, J. H. (2001). Drug-induced Liver Injury: Mechanisms and Test Systems. Hepatology 33, 1009–1013. doi:10.1053/jhep.2001.23505
Bruderer, R., Bernhardt, O. M., Gandhi, T., Miladinović, S. M., Cheng, L.-Y., Messner, S., et al. (2015). Extending the Limits of Quantitative Proteome Profiling with Data-independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell Proteomics 14, 1400–1410. doi:10.1074/mcp.M114.044305
Bulera, S. J., Birge, R. B., Cohen, S. D., and Khairallah, E. A. (1995). Identification of the Mouse Liver 44-kDa Acetaminophen-Binding Protein as a Subunit of Glutamine Synthetase. Toxicol. Appl. Pharmacol. 134, 313–320. doi:10.1006/taap.1995.1197
Cohen, S. D., Pumford, N. R., Khairallah, E. A., Boekelheide, K., Pohl, L. R., Amouzadeh, H. R., et al. (1997). Selective Protein Covalent Binding and Target Organ Toxicity. Toxicol. Appl. Pharmacol. 143, 1–12. doi:10.1006/taap.1996.8074
Dooley, G. P., Reardon, K. F., Prenni, J. E., Tjalkens, R. B., Legare, M. E., Foradori, C. D., et al. (2008). Proteomic Analysis of Diaminochlorotriazine Adducts in Wister Rat Pituitary Glands and LβT2 Rat Pituitary Cells. Chem. Res. Toxicol. 21, 844–851. doi:10.1021/tx700386f
Dufort, I., Labrie, F., and Luu-The, V. (2001). Human Types 1 and 3 3α-Hydroxysteroid Dehydrogenases: Differential Lability and Tissue Distribution1. J. Clin. Endocrinol. Metab. 86, 841–846. doi:10.1210/jcem.86.2.7216
Geib, T., Lento, C., Wilson, D. J., and Sleno, L. (2019). Liquid Chromatography-Tandem Mass Spectrometry Analysis of Acetaminophen Covalent Binding to Glutathione S-Transferases. Front. Chem. 7, 558. doi:10.3389/fchem.2019.00558
Golizeh, M., LeBlanc, A., and Sleno, L. (2015). Identification of Acetaminophen Adducts of Rat Liver Microsomal Proteins Using 2D-LC-MS/MS. Chem. Res. Toxicol. 28, 2142–2150. doi:10.1021/acs.chemrestox.5b00317
Graham, G. G., and Scott, K. F. (2005). Mechanism of Action of Paracetamol. Am. J. Ther. 12, 46–55. doi:10.1097/00045391-200501000-00008
Hanzlik, R. P., Koen, Y. M., and Garrett, M. J. (2020). Reactive Metabolite Target Protein Database (TPDB). Available at: https://kuscholarworks.ku.edu/handle/1808/30592 (Accessed November 23, 2020).
Heard, K., Green, J. L., Anderson, V., Bucher-Bartelson, B., and Dart, R. C. (2016). Paracetamol (Acetaminophen) Protein Adduct Concentrations During Therapeutic Dosing. Br. J. Clin. Pharmacol. 81, 562–568. doi:10.1111/bcp.12831
Hu, J., Kholmukhamedov, A., Lindsey, C. C., Beeson, C. C., Jaeschke, H., and Lemasters, J. J. (2016). Translocation of Iron from Lysosomes to Mitochondria during Acetaminophen-Induced Hepatocellular Injury: protection by Starch-Desferal and Minocycline. Free Radic. Biol. Med. 97, 418–426. doi:10.1016/j.freeradbiomed.2016.06.024
Ikehata, K., Duzhak, T. G., Galeva, N. A., Ji, T., Koen, Y. M., and Hanzlik, R. P. (2008). Protein Targets of Reactive Metabolites of Thiobenzamide in Rat Liver In Vivo. Chem. Res. Toxicol. 21, 1432–1442. doi:10.1021/tx800093k
James, L. P., Mayeux, P. R., and Hinson, J. A. (2003). Acetaminophen-induced Hepatotoxicity. Drug Metab. Dispos. 31, 1499–1506. doi:10.1124/dmd.31.12.1499
Jan, Y.-H., Heck, D. E., Dragomir, A.-C., Gardner, C. R., Laskin, D. L., and Laskin, J. D. (2014). Acetaminophen Reactive Intermediates Target Hepatic Thioredoxin Reductase. Chem. Res. Toxicol. 27, 882–894. doi:10.1021/tx5000443
Jiang, J., Briedé, J. J., Jennen, D. G. J., Van Summeren, A., Saritas-Brauers, K., Schaart, G., et al. (2015). Increased Mitochondrial ROS Formation by Acetaminophen in Human Hepatic Cells Is Associated with Gene Expression Changes Suggesting Disruption of the Mitochondrial Electron Transport Chain. Toxicol. Lett. 234, 139–150. doi:10.1016/j.toxlet.2015.02.012
Koen, Y. M., Gogichaeva, N. V., Alterman, M. A., and Hanzlik, R. P. (2007). A Proteomic Analysis of Bromobenzene Reactive Metabolite Targets in Rat Liver Cytosol In Vivo. Chem. Res. Toxicol. 20, 511–519. doi:10.1021/tx6003166
Koen, Y. M., Hajovsky, H., Liu, K., Williams, T. D., Galeva, N. A., Staudinger, J. L., et al. (2012). Liver Protein Targets of Hepatotoxic 4-bromophenol Metabolites. Chem. Res. Toxicol. 25, 1777–1786. doi:10.1021/tx3002675
Koen, Y. M., Sarma, D., Hajovsky, H., Galeva, N. A., Williams, T. D., Staudinger, J. L., et al. (2013). Protein Targets of Thioacetamide Metabolites in Rat Hepatocytes. Chem. Res. Toxicol. 26, 564–574. doi:10.1021/tx400001x
Laine, J. E., Auriola, S., Pasanen, M., and Juvonen, R. O. (2009). Acetaminophen Bioactivation by Human Cytochrome P450 Enzymes and Animal Microsomes. Xenobiotica 39, 11–21. doi:10.1080/00498250802512830
LeBlanc, A., Shiao, T. C., Roy, R., and Sleno, L. (2014). Absolute Quantitation of NAPQI-Modified Rat Serum Albumin by LC-MS/MS: Monitoring Acetaminophen Covalent Bindingin Vivo. Chem. Res. Toxicol. 27, 1632–1639. doi:10.1021/tx500284g
Lee, K. K., Imaizumi, N., Chamberland, S. R., Alder, N. N., and Boelsterli, U. A. (2015). Targeting Mitochondria with Methylene Blue Protects Mice Against Acetaminophen-Induced Liver Injury. Hepatology 61, 326–336. doi:10.1002/hep.27385
Leeming, M. G., Donald, W. A., and O’Hair, R. A. J. (2017). Nontargeted Identification of Reactive Metabolite Protein Adducts. Anal. Chem. 89, 5748–5756. doi:10.1021/acs.analchem.6b04604
McGill, M. R., Li, F., Sharpe, M. R., Williams, C. D., Curry, S. C., Ma, X., et al. (2014). Circulating Acylcarnitines as Biomarkers of Mitochondrial Dysfunction after Acetaminophen Overdose in Mice and Humans. Arch. Toxicol. 88, 391–401. doi:10.1007/s00204-013-1118-1
McGill, M. R., Williams, C. D., Xie, Y., Ramachandran, A., and Jaeschke, H. (2012). Acetaminophen-induced Liver Injury in Rats and Mice: Comparison of Protein Adducts, Mitochondrial Dysfunction, and Oxidative Stress in the Mechanism of Toxicity. Toxicol. Appl. Pharmacol. 264, 387–394. doi:10.1016/j.taap.2012.08.015
Meier, B. W., Gomez, J. D., Kirichenko, O. V., and Thompson, J. A. (2007). Mechanistic Basis for Inflammation and Tumor Promotion in Lungs of 2,6-Di-Tert-Butyl-4-Methylphenol-Treated Mice: Electrophilic Metabolites Alkylate and Inactivate Antioxidant Enzymes. Chem. Res. Toxicol. 20, 199–207. doi:10.1021/tx060214f
Methogo, R. M., Dansette, P. M., and Klarskov, K. (2007). Identification of Liver Protein Targets Modified by Tienilic Acid Metabolites Using a Two-Dimensional Western Blot-Mass Spectrometry Approach. Int. J. Mass Spectrom. 268, 284–295. doi:10.1016/j.ijms.2007.06.002
Mitchell, D. A., and Marletta, M. A. (2005). Thioredoxin Catalyzes the S-Nitrosation of the Caspase-3 Active Site Cysteine. Nat. Chem. Biol. 1, 154–158. doi:10.1038/nchembio720
Mitterberger, M. C., Kim, G., Rostek, U., Levine, R. L., and Zwerschke, W. (2012). Carbonic Anhydrase III Regulates Peroxisome Proliferator-Activated Receptor-γ2. Exp. Cel Res. 318, 877–886. doi:10.1016/j.yexcr.2012.02.011
Moro, S., Chipman, J. K., Antczak, P., Turan, N., Dekant, W., Falciani, F., et al. (2012). Identification and Pathway Mapping of Furan Target Proteins Reveal Mitochondrial Energy Production and Redox Regulation as Critical Targets of Furan Toxicity. Toxicol. Sci. 126, 336–352. doi:10.1093/toxsci/kfs005
Nakagawa, H., Maeda, S., Hikiba, Y., Ohmae, T., Shibata, W., Yanai, A., et al. (2008). Deletion of Apoptosis Signal-Regulating Kinase 1 Attenuates Acetaminophen-Induced Liver Injury by Inhibiting C-Jun N-Terminal Kinase Activation. Gastroenterology 135, 1311–1321. doi:10.1053/j.gastro.2008.07.006
Nerland, D. E., Cai, J., and Benz, F. W. (2003). Selective Covalent Binding of Acrylonitrile to Cys 186 in Rat Liver Carbonic Anhydrase III In Vivo. Chem. Res. Toxicol. 16, 583–589. doi:10.1021/tx0256883
Nguyen, N. T., Du, K., Akakpo, J. Y., Umbaugh, D. S., Jaeschke, H., and Ramachandran, A. (2021). Mitochondrial Protein Adduct and Superoxide Generation Are Prerequisites for Early Activation of C-Jun N-Terminal Kinase within the Cytosol after an Acetaminophen Overdose in Mice. Toxicol. Lett. 338, 21–31. doi:10.1016/j.toxlet.2020.12.005
Park, B. K., Boobis, A., Clarke, S., Goldring, C. E. P., Jones, D., Kenna, J. G., et al. (2011). Managing the Challenge of Chemically Reactive Metabolites in Drug Development. Nat. Rev. Drug Discov. 10, 292–306. doi:10.1038/nrd3408
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 47 (D1), D442–D450. doi:10.1093/nar/gky1106
Pessayre, D. (1995). Role of Reactive Metabolites in Drug-Induced Hepatitis. J. Hepatol. 23 (Suppl. 1), 16–24. doi:10.1016/s0168-8278(21)80002-1
Pol, A., Renkema, G. H., Tangerman, A., Winkel, E. G., Engelke, U. F., de Brouwer, A. P. M., et al. (2018). Mutations in SELENBP1, Encoding a Novel Human Methanethiol Oxidase, Cause Extraoral Halitosis. Nat. Genet. 50, 120–129. doi:10.1038/s41588-017-0006-7
Poprac, P., Jomova, K., Simunkova, M., Kollar, V., Rhodes, C. J., and Valko, M. (2017). Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 38, 592–607. doi:10.1016/j.tips.2017.04.005
Pumford, N. R., Martin, B. M., and Hinson, J. A. (1992). A Metabolite of Acetaminophen Covalently Binds to the 56 kDa Selenium Binding Protein. Biochem. Biophysical Res. Commun. 182, 1348–1355. doi:10.1016/0006-291x(92)91881-p
Qiu, Y., Benet, L. Z., and Burlingame, A. L. (1998). Identification of the Hepatic Protein Targets of Reactive Metabolites of Acetaminophen in Vivoin Mice Using Two-Dimensional Gel Electrophoresis and Mass Spectrometry. J. Biol. Chem. 273, 17940–17953. doi:10.1074/jbc.273.28.17940
Ramachandran, A., and Jaeschke, H. (2020). A Mitochondrial Journey through Acetaminophen Hepatotoxicity. Food Chem. Toxicol. 140, 111282. doi:10.1016/j.fct.2020.111282
Ramachandran, A., and Jaeschke, H. (2019). Acetaminophen Hepatotoxicity: A Mitochondrial Perspective. Adv. Pharmacol. 85, 195–219. doi:10.1016/bs.apha.2019.01.007
Ramachandran, A., Lebofsky, M., Yan, H.-M., Weinman, S. A., and Jaeschke, H. (2015). Hepatitis C Virus Structural Proteins Can Exacerbate or Ameliorate Acetaminophen-Induced Liver Injury in Mice. Arch. Toxicol. 89, 773–783. doi:10.1007/s00204-015-1498-5
Renner, S. W., Walker, L. M., Forsberg, L. J., Sexton, J. Z., and Brenman, J. E. (2017). Carbonic Anhydrase III (Car3) Is Not Required for Fatty Acid Synthesis and Does Not Protect against High-Fat Diet Induced Obesity in Mice. PLoS One 12, e0176502. doi:10.1371/journal.pone.0176502
Richarme, G., Liu, C., Mihoub, M., Abdallah, J., Leger, T., Joly, N., et al. (2017). Guanine Glycation Repair by DJ-1/Park7 and its Bacterial Homologs. Science 357, 208–211. doi:10.1126/science.aag1095
Richarme, G., Mihoub, M., Dairou, J., Bui, L. C., Leger, T., and Lamouri, A. (2015). Parkinsonism-associated Protein DJ-1/Park7 Is a Major Protein Deglycase that Repairs Methylglyoxal- and Glyoxal-Glycated Cysteine, Arginine, and Lysine Residues. J. Biol. Chem. 290, 1885–1897. doi:10.1074/jbc.M114.597815
Sekito, A., Koide-Yoshida, S., Niki, T., Taira, T., Iguchi-Ariga, S. M. M., and Ariga, H. (2006). DJ-1 Interacts with HIPK1 and Affects H2O2-Induced Cell Death. Free Radic. Res. 40, 155–165. doi:10.1080/10715760500456847
Shilov, I. V., Seymour, S. L., Patel, A. A., Loboda, A., Tang, W. H., Keating, S. P., et al. (2007). The Paragon Algorithm, A Next Generation Search Engine that Uses Sequence Temperature Values and Feature Probabilities to Identify Peptides from Tandem Mass Spectra. Mol. Cell Proteomics 6, 1638–1655. doi:10.1074/mcp.T600050-MCP200
Shin, J., Weitzdoerfer, R., Fountoulakis, M., and Lubec, G. (2004). Expression of Cystathionine β-synthase, Pyridoxal Kinase, and ES1 Protein Homolog (Mitochondrial Precursor) in Fetal Down Syndrome Brain. Neurochem. Int. 45, 73–79. doi:10.1016/j.neuint.2003.12.004
Shipkova, M., Beck, H., Voland, A., Armstrong, V. W., Gröne, H.-J., Oellerich, M., et al. (2004). Identification of Protein Targets for Mycophenolic Acid Acyl Glucuronide in Rat Liver and colon Tissue. Proteomics 4, 2728–2738. doi:10.1002/pmic.200300836
Srivastava, A., Maggs, J. L., Antoine, D. J., Williams, D. P., Smith, D. A., and Park, B. K. (2010). “Role of Reactive Metabolites in Drug-Induced Hepatotoxicity,” in Adverse Drug Reactions. Editor J. Uetrcht (Berlin, Heidelberg, Germany: Springer), 165–194. doi:10.1007/978-3-642-00663-0_7
Strott, C. A. (1996). Steroid Sulfotransferases. Endocr. Rev. 17, 670–697. doi:10.1210/edrv-17-6-670
Tanaka, R., Ishima, Y., Maeda, H., Kodama, A., Nagao, S., Watanabe, H., et al. (2014). Albumin Fusion Prolongs the Antioxidant and Anti-inflammatory Activities of Thioredoxin in Mice with Acetaminophen-Induced Hepatitis. Mol. Pharmaceutics 11, 1228–1238. doi:10.1021/mp400690v
Teng, Y.-W., Mehedint, M. G., Garrow, T. A., and Zeisel, S. H. (2011). Deletion of Betaine-Homocysteine S-Methyltransferase in Mice Perturbs Choline and 1-carbon Metabolism, Resulting in Fatty Liver and Hepatocellular Carcinomas. J. Biol. Chem. 286, 36258–36267. doi:10.1074/jbc.M111.265348
Tirmenstein, M. A., and Nelson, S. D. (1989). Subcellular Binding and Effects on Calcium Homeostasis Produced by Acetaminophen and a Nonhepatotoxic Regioisomer, 3′-hydroxyacetanilide, in Mouse Liver. J. Biol. Chem. 264, 9814–9819. doi:10.1016/s0021-9258(18)81731-8
Tittarelli, R., Pellegrini, M., Scarpellini, M. G., Marinelli, E., Bruti, V., Di Luca, N. M., et al. (2017). Hepatotoxicity of Paracetamol and Related Fatalities. Eur. Rev. Med. Pharmacol. Sci. 21(N.1 Suppl), 95–101. Available at: https://www.europeanreview.org/article/12436.
Tuin, A. W., Mol, M. A. E., van den Berg, R. M., Fidder, A., van der Marel, G. A., Overkleeft, H. S., et al. (2009). Activity-based Protein Profiling Reveals Broad Reactivity of the Nerve Agent Sarin. Chem. Res. Toxicol. 22, 683–689. doi:10.1021/tx8004218
Turley, S. D., and Dietschy, J. M. (1978). Re-evaluation of the 3 Alpha-Hydroxysteroid Dehydrogenase Assay for Total Bile Acids in Bile. J. Lipid Res. 19, 924–928. doi:10.1016/s0022-2275(20)40709-6
Xie, Y., Ramachandran, A., Breckenridge, D. G., Liles, J. T., Lebofsky, M., Farhood, A., et al. (2015). Inhibitor of Apoptosis Signal-Regulating Kinase 1 Protects against Acetaminophen-Induced Liver Injury. Toxicol. Appl. Pharmacol. 286, 1–9. doi:10.1016/j.taap.2015.03.019
Keywords: acetaminophen, rodent model, reactive metabolites, liver proteins, LC-MS/MS, LC-MRM, protein modification, NAPQI, N-acetyl-p-benzoquinone, mouse, rat, covalent binding
Citation: Geib T, Moghaddam G, Supinski A, Golizeh M and Sleno L (2021) Protein Targets of Acetaminophen Covalent Binding in Rat and Mouse Liver Studied by LC-MS/MS. Front. Chem. 9:736788. doi: 10.3389/fchem.2021.736788
Received: 05 July 2021; Accepted: 04 August 2021;
Published: 20 August 2021.
Edited by:
Marcus S Cooke, University of South Florida, United StatesReviewed by:
Hartmut Jaeschke, University of Kansas Medical Center Research Institute, United StatesCopyright © 2021 Geib, Moghaddam, Supinski, Golizeh and Sleno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lekha Sleno, c2xlbm8ubGVraGFAdXFhbS5jYQ==
†Present address: Department of Mathematical and Physical Sciences, Concordia University of Edmonton, Edmonton, AB, Canada
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.