 F. D. Vila
F. D. Vila J. J. Kas
J. J. Kas J. J. Rehr
J. J. Rehr K. Kowalski
K. Kowalski B. Peng
B. Peng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 22 September 2021
Sec. Theoretical and Computational Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.734945
This article is part of the Research Topic Many-Body Green’s Functions and the Bethe-Salpeter Equation in Chemistry: From Single Molecules to Complex Systems View all 10 articles
Green’s function methods provide a robust, general framework within many-body theory for treating electron correlation in both excited states and x-ray spectra. Conventional methods using the Dyson equation or the cumulant expansion are typically based on the GW self-energy approximation. In order to extend this approximation in molecular systems, a non-perturbative real-time coupled-cluster cumulant Green’s function approach has been introduced, where the cumulant is obtained as the solution to a system of coupled first order, non-linear differential equations. This approach naturally includes non-linear corrections to conventional cumulant Green’s function techniques where the cumulant is linear in the GW self-energy. The method yields the spectral function for the core Green’s function, which is directly related to the x-ray photoemission spectra (XPS) of molecular systems. The approach also yields very good results for binding energies and satellite excitations. The x-ray absorption spectrum (XAS) is then calculated using a convolution of the core spectral function and an effective, one-body XAS. Here this approach is extended to include the full coupled-cluster-singles (CCS) core Green’s function by including the complete form of the non-linear contributions to the cumulant as well as all single, double, and triple cluster excitations in the CC amplitude equations. This approach naturally builds in orthogonality and shake-up effects analogous to those in the Mahan-Noizeres-de Dominicis edge singularity corrections that enhance the XAS near the edge. The method is illustrated for the XPS and XAS of NH3.
The core-level x-ray absorption spectra (XAS) μ(ω) is typically described formally by Fermi’s golden rule. However, this formulation requires calculations of and summations over eigenstates of the many-body Hamiltonian H and is computationally intractable. Simplifications such as the determinantal ΔSCF approach, in terms of Slater determinants (Liang et al., 2017; Liang and Prendergast, 2019; Nozieres and Combescot, 1971) have similar limitations. Although still computationally demanding, Green’s function methods provide an attractive alternative since summation over final states is implicit (Lee et al., 2012; Bertsch and Lee, 2014; Rehr et al., 2009). Real-time approaches can also be advantageous as they avoid explicit calculations of eigenstates. Our treatment here exploits real-time approaches, following several recent developments: 1) Equation of motion coupled-cluster (EOM-CC) approaches for molecular systems have been formulated for the Green’s function in energy-space (Peng and Kowalski, 2016; Peng and Kowalski, 2018a), 2) An approach has also been developed (Rehr et al., 2020) for calculations of many-body XAS μ(ω) in terms of the convolution of a one-body XAS μ1(ω) and the spectral function of the core-hole Ac(ω)
This result originates from the time-correlation approach (Nozieres and de Dominicis, 1969) that was used to solve the x-ray edge-singularity problem. In this approach the time-domain XAS transition amplitude μ(t) is given by the factorization
Here L(t) is an effective one-body transition amplitude and Ac(ω) = −(1/π)Im Gc(t). 3) A real-time EOM-CC approach for the cumulant core Green’s function Gc has been developed including excitations up to CC-singles (CCS). Intrinsic losses induced by the sudden creation of the core hole lead to shake-up effects, characterized by satellites in Ac(ω), as observed in x-ray photoemission spectra (XPS). The effective one-body XAS μ1(ω) also builds in orthogonality corrections leading to edge enhancements, as predicted by Mahan, Nozieres and de Dominicis (Mahan, 1967). Our goal here is to review these developments and to combine the one-particle absorption spectrum with a more accurate treatment of the core Green’s function, including the complete form of the CCS cumulant, as well as the full single, doubles and triple cluster excitations in the cluster amplitude equations.
In the rest of this review, Section 2 describes the theoretical approaches used, in particular a brief introduction to the cumulant approach (Section 2.1), the real-time equation of motion, coupled cluster (RT-EOM-CC) approach (Section 2.2), the frequency space implementation of the Green’s Function coupled clusters (GFCC) approach (Section 2.3), and the application to XAS (Section 2.4). Section 3 presents results for the core binding energies of small molecules, and a comparison of the theory to XPS and XAS experimental results for NH3. Finally, Section 4 presents a summary and discusses future developments.
Within the cumulant approximation, the core-level Green’s function is defined by an exponential expression
where
As a consequence, the cumulant Green’s function is naturally normalized, with an occupation Gc(t = 0) = 1. One can also analyze A(ω) = −(1/π)Im Gc(ω), which is the natural quantity to compare to experimental x-ray photoemission spectra, and to assess the quality of many-body correlation approximations since satellites appearing in spectral quantities such as XPS are directly related to those seen in the spectral function. The above form of the cumulant also permits a natural separation into quasiparticle and satellite contributions. Separating the terms in the expression for the cumulant above, we have
where a = ∫dωβ(ω)/ω2 is the net satellite strength, Δ = ∫dωβ(ω)/ω is the quasiparticle shift, or core-level “relaxation energy”, and
where Zc = e−a is the quasiparticle renormalization, Ec = ϵc −Δ is the quasiparticle energy, and Asat(ω) = β(ω)/ω2 is the satellite spectral function (Aryasetiawan et al., 1996).
The cumulant kernel or excitation spectrum β(ω) can be approximated in a variety of ways. The most common approximation is to expand the Green’s function to low order either in the bare Coulomb interaction, giving β(ω) in terms of the second order self-energy (Vila et al., 2020), or by expanding in terms of the screened Coulomb interaction, which produces an approximation in terms of the GW self-energy (Hedin, 1999; Guzzo et al., 2011; Zhou et al., 2015). Approximate non-linear corrections can be included using real-time TDDFT (Tzavala et al., 2020). Here, as described in the next section, we calculate the cumulant including non-linear corrections within a non-perturbative approach, by expressing the Green’s function in terms of the time-dependent EOM-CC states.
Our treatment of the core-hole Green’s function Gc(t) is based on the CC-ansatz for the time-evolved initial state of the system with a core-hole created at t = 0+: |Ψc (0)⟩ ≡|Ψc⟩ = cc|Ψ⟩ where cc is the core annihilation operator and |Ψ⟩ is the ground state Hartree-Fock Slater determinant:
Then
Calculations of Gc(t) are based on the real-time equation of motion coupled cluster (RT-EOM-CC) ansatz of Schönhammer and Gunnarsson (SG) (Schönhammer and Gunnarsson, 1978), in which the time-evolution of |Ψc(t)⟩ is carried out using an initial value problem and propagation via the Schrödinger equation of motion i ∂|Ψc(t)⟩/∂t = H|Ψc(t)⟩. The time-evolved wave-function |Ψc(t)⟩ can then be expressed using the CC ansatz in Eq. 7. The use of a single-excitations CC ansatz is justified for a single-determinant reference state approximation due to Thouless’ theorem (Thouless, 1961). In Eq. 7Nc(t) is a normalization factor, while the CC operator T(t) is defined in terms of single, double, etc., excitation creation operators
For example, for single excitations n = (i, a) and
where
where
This amplitude accounts for the transfer of oscillator strength from the main peak in XPS to the satellite excitations at frequencies ω. The cumulant initial conditions C (0) = C′(0) = 0 guarantee the normalization of the spectral function. In addition, they ensure that its centroid remains invariant at the Koopmans’ energy −ϵc. After some straightforward diagrammatic analysis to compute the matrix elements in Eqs. 10, 12, we obtain a compact expression for the time-derivative of the cumulant
where
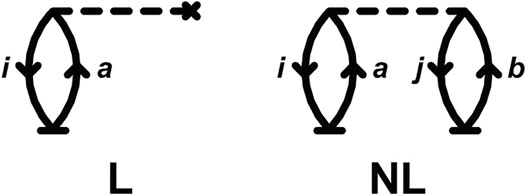
FIGURE 1. Linear (L) and non-linear (NL) CC diagrams (Crawford and Schaefer, 2000; Brandow, 1967) for the time-derivative of the cumulant in Eq. 14. Unlike in traditional EOM-CC diagrams, the base vertices (t) are time-dependent.
Remarkably, these diagrams are completely analogous to the time-independent diagrams for the CCSD energy if only single excitations (T1) are included. It is interesting to note that only one more diagram is needed to obtain the complete CC cumulant for the core-hole Green's function, namely that from double excitations similar to the NL diagram in Figure 1, but with a cluster line joining the base vertices representing the T2 operator.
The EOM for the matrix elements of the CC amplitudes in Eq. 11 can be calculated using similar diagrammatic analysis which yield a set of coupled first-order differential equations.
where ϵp are the bare single-particle energies. The low order terms are identical to those in our original paper (Rehr et al., 2020). However, now the complete CCS T1 approximation is used, including terms up to third order in
1) Lowest order approximation, with only the leading terms, i.e., Eq. 15a: At this level the EOM is exactly solvable giving
2) Core-valence approximation: Eq. 15a together with the dominant, first four sums in Eq. 15b. This approximation includes the linear core- and valence-valence contributions and a non-linear term corresponding to excitations linked to the core-hole.
3) Full second order approximation: Keeping all terms of level 2, and the non-linear valence-valence excitations from the two sums in Eq. 15c. This gives corrections that close the gap between the QP peak and the satellites.
4) Full third order T1 level: This approximation retains all the terms in the T1 EOM in Eq. 15, including the cubic term in Eq. 15d.
Each of the approximation levels to Eq. 15 can be combined with either the linear (L), or both the linear and non-linear (NL) components of C(t) defined in Eq. 14. As we have demonstrated previously (Vila et al., 2020), the NL component is key for obtaining accurate core binding energies. Consequently it is useful to focus on the 2NL, 3NL and 4NL results only. For comparison results are also shown for the solution of the Dyson equation (DSE2) (Linderberg and Öhrn, 2004; Szabo and Ostlund, 1996)
where Σ(2)(ω) is the second order self-energy (Szabo and Ostlund, 1996); and for the frequency space Green’s function GFCCSD and GFCC-i (2, 3) methods (Peng and Kowalski, 2018a; Peng and Kowalski, 2018b).
In this section we briefly review the GFCC formalism introduced by Nooijen et al. (Nooijen and Snijders, 1992; Nooijen and Snijders, 1993; Nooijen and Snijders, 1995, see also Meissner and Bartlett, 1993; Kowalski et al., 2014; Bhaskaran-Nair et al., 2016; Peng and Kowalski, 2016; Peng and Kowalski, 2018a), which draws heavily on the bi-variational CC formalism (Arponen, 1983; Stanton and Bartlett, 1993) where the ground-state bra- ket (〈Ψg|) and (|Ψg〉) states are parametrized in a different way.
where the reference function |Φ⟩ is typically chosen as a Hartree-Fock Slater determinant (for the original papers on the CC ansatz see Coester, 1958; Coester and Kümmel, 1960; Čížek, 1966; Paldus et al., 1972; Purvis and Bartlett, 1982; Paldus and Li, 1999; Bartlett and Musiał, 2007). In the above equations the T and Λ operators refer to the so-called cluster and de-excitation operators, respectively, which can be obtained by solving canonical CC equations for the N-electron system. For simplicity, in this review we will discuss the algebraic form of the retarded part of the frequency dependent CC Green’s function defined by matrix elements
Here H is the electronic Hamiltonian for the N-electron system, E0 the corresponding ground-state energy, η is a broadening factor, and cp (
The similarity transformed operators here
that satisfy equations
Equation 20 can then be re-expressed compactly as
The Xp(ω) operators can be effectively solved using a parallel implementation of the GFCC formalism based on the approximate forms of T, Λ, and Xp(ω) (Peng et al., 2021).
The RT-EOM-CC Green’s function differs from the frequency-space GFCC approaches (Peng and Kowalski, 2016; Peng and Kowalski, 2018b) in several respects. In particular RT-EOM-CC is based on a transformation to an initial value problem with the propagation of the N − 1 particle system carried out after the creation of the core-hole; in contrast the GFCC methods are implemented in frequency-space. In addition, RT-EOM-CC assumes an uncorrelated N-particle single-determinant ground state, while the GFCC approaches calculate this ground state using the CC ansatz (Eq. 17 and Eq. 18). Finally, the RT-EOM-CC cumulant treats the N − 1 particle excited states at the CCS level, while the GFCCSD approximation solves for the excitations of the N − 1 particle system at the approximate CCSD level, keeping only single and double excitations, as discussed near Eq. 14 of Peng and Kowalski, 2018a. Thus high order diagrams are implicitly built in the RT-EOM-CC GF from the exponential form of the cumulant (Gunnarsson et al., 1994; Lange and Berkelbach, 2018). While the RT-EOM-CC utilizes a unique approximation for the time-dependent T(t) operator, the GFCC formalisms permit the use of several levels of approximation for the T, Λ and Xp(ω) operators (for assuring size-extensivity of diagrams defining the
Our treatment here is adapted from that in our original EOM-CC paper (Rehr et al., 2020), but updated here with a more detailed treatment of the EOM discussed above. As outlined in the introduction, the contribution from a deep core level |c⟩ to the XAS is given by the time-correlation function μ(t) = L(t)Gc(t) in Eq. 2, as in Nozieres and de Dominicis, 1969 and Rehr et al., 2020. The core-hole Green’s function Gc can be obtained from the RT-EOM-CC Eq. 8. Calculations of L(t), the one-body time-dependent transition amplitude, can be carried out using coupled equation of motion or equivalent integral equations (Langreth, 1969; Grebennikov et al., 1977; Privalov et al., 2001). Alternatively, propagation based on the overlap integrals uij(t) can also be used, as done by Nozieres and Combescot (NC) (Nozieres and Combescot, 1971). However, it is more convenient to replace the sums over k with the complete set of eigenstates κ of the final state one-particle Hamiltonian
The leading term on the rhs of Eq. 25 can be interpreted as a contribution to Lκ,κ′(t) from the independent particle transition amplitude for the final state when the core-hole is present L0(t) = Σκ|Mcκ|2 exp(iϵκt), consistent with the final-state rule of von Barth and Grossman (von Barth and Grossmann, 1982). The diagonal terms κ = κ′ in Eq. 25 suppress transitions to the occupied states κ < kF by yielding the theta function θ(kF − k). The off-diagonal terms in Lκ,κ′(t) are controlled by states with either κ (or κ′) > kF or κ′ (or κ) < kF. Interestingly the net result can be approximated accurately by the expression
equivalent to the one derived by Friedel (Friedel, 1969), where
Here
As an example of the accuracy of the RT-EOM-CCS method for core ionization energies we show results for CH4, NH3, H2O, HF and Ne, i.e. the ten-electron series, using the experimental geometries (NIST, 2019) for all systems (rCH = 1.087 Å, rNH = 1.012 Å, aHNH = 106.67 degree, rOH = 0.958 Å, aHOH = 104.48 degree, rFH = 0.917 Å), and the aug-cc-pVDZ basis set (Kendall et al., 1992). We also show spectral function and XAS results for NH3 for which experimental values are available in the literature. The ground state single-particle states and molecular orbitals integrals for the RT-EOM-CCS approach and the ΔSCF method were calculated from a Hartree-Fock (HF) reference, while those for the core-excited ΔSCF were derived from a spin-symmetric and occupation-constrained HF reference. The final spectra were broadened to compare with experiment as in Rehr et al., 2020 and Vila et al., 2020 with varying (1–6 eV) broadening to account for the limited basis set for the continuum; similarly XAS used constant broadening consistent with experimental broadening below and 3.5 eV above the binding energy.
Figure 2 shows a comparison of errors vs experiment for the core binding energies of the ten-electron series molecules. The RT-EOM-CCS method shows small errors even at the simplest non-linear level of approximation (2NL), with a mean-absolute error (MAE) across systems of only 0.5 eV, significantly smaller than for the other methods tested. Although better core-optimized basis sets need to be tested to ensure full convergence with basis set size, we find (Vila et al., 2020) similar errors even for smaller basis sets (e.g. DZVP and cc-pVDZ). We emphasize that these accurate results arise from the non-linear terms in
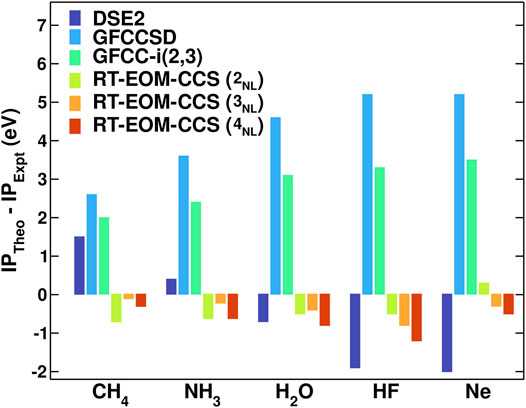
FIGURE 2. Comparison of the theory errors vs experiment (Karlsen et al., 2002; Buttersack et al., 2019; Viñes et al., 2018; Jolly et al., 1984; Williams, 2009) for the core binding energies. The theoretical calculations were performed with the 2-4NL RT-EOM-CCS approximations with the aug-cc-pVDZ basis set. Also shown are results from the second order Dyson equation (DSE2) and the standard GFCCSD and GFCC-i (2, 3) coupled-cluster Green’s function methods (Peng and Kowalski, 2016; Peng and Kowalski, 2018a).
Figure 3 shows results for the spectral function Ac(ω) and the cumulant kernel β(ω). These are shown vs. binding energy to compare more readily to the experimental XPS. β(ω) is dominated by shake-up excitation peaks about 20–30 eV above the quasiparticle peak that correspond precisely with the inelastic losses in Ac(ω). The satellites structure is in reasonable agreement with that observed in XPS (Sankari et al., 2006) once scissors corrections are included, despite the fact that our HF-based Hamiltonian overestimates the excitation energies. From the Landau form in Eq. 4, the strength of the quasi-particle peak is defined by the renormalization constant Z, where for the 4NL approximation to the EOM-CC cumulant Z = exp (−a) = 0.70. The satellite strength is a = ∫dω β(ω)/ω2 = 0.35. This matches the numerical integration over the QP peak that yields Z = 0.70, in good agreement with the ΔSCF value Z = 0.76. The renormalization constant Z is also partly responsible for the amplitude reduction factor
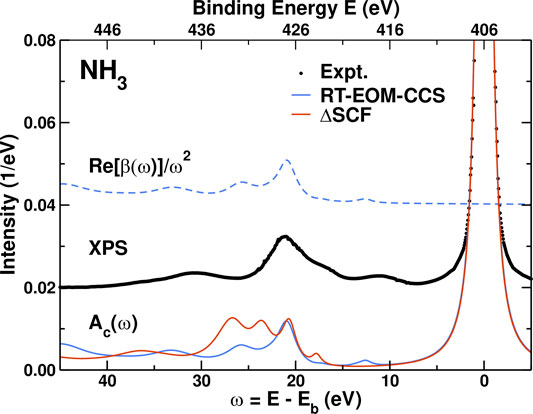
FIGURE 3. Comparison of the 4NL RT-EOM-CCS and ΔSCF core spectral functions Ac(ω) (full lines) and cumulant kernel β(ω) (dashed lines, shown only for the RT-EOM-CC method) for the NH3 molecule, to the experimental XPS (dots) (Sankari et al., 2006). Energies are shows in either absolute binding energy E or versus excitation energy ω = E−Eb with respect to the experimental core binding energy Eb = 405.52 eV (Sankari et al., 2006). All the theoretical results were obtained with the aug-cc-pVDZ basis set. The theoretical Ac(ω) were broadened and include a scissors shift of 3.9 eV.
Results for the XAS, including experiment (Wight and Brion, 1974), are shown in Figure 4. The overall agreement between theory and experiment is quite good for the positions and relative intensities of the first two peaks. The third peak, at 403.5 eV, is almost in the continuum and is more difficult to describe with our limited basis set. For this molecule, the corrections to the independent particle XAS (L0(ϵ)) are clearly visible: First, the edge enhancement factor 1 + χ increases the intensity to L(ϵ). Second, the amplitude reduction factor from the spectral function Ac(ϵ), which has opposite sign and is approximately twice as strong, reduces the intensity to the final μ(ϵ). Since the leading satellites peaks in Ac(ϵ) are 20–30 eV above the QP peak, the corresponding XAS satellite features fall in the continuum and are thus not visible.
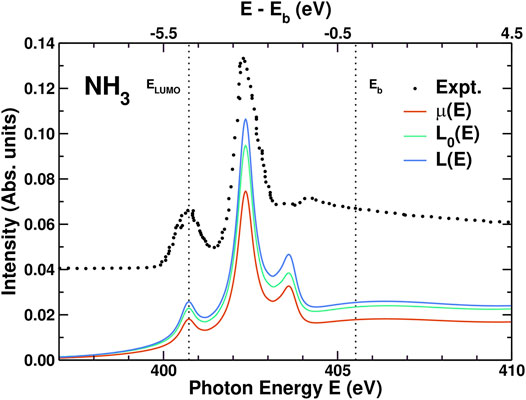
FIGURE 4. Comparison of the experimental XAS μ(E) for NH3 (Wight and Brion, 1974) vs photon energy E to those calculated from the convolution in Eq. 2, the effective one-body XAS L(E) = μ1(E) and the independent particle XAS L0(E) = μ0(E) from Eq. 25. The N x-ray K edge lies just under ELUMO while Eb is the ionization threshold. In order to account for the sparsity of the Gaussian-type orbital basis set in the continuum region above Eb, the comparison to experiment includes variable broadening (see text) that increases to a maximum of 3.5 eV above Eb.
This review describes a combined equation of motion coupled cluster approach in real-time to calculate excitations corresponding to intrinsic losses in XAS and XPS. The approach is based on the cumulant form of the Green’s function representation for the core-hole spectral function that arises naturally from the coupled cluster ansatz. This theoretical connection between the cumulant approach, a powerful tool for computing satellites in solid state physics, to the coupled cluster approach which is the gold standard for accuracy in quantum chemistry brings together two previously mostly unrelated fields, thus opening new areas of research. Unlike our previous treatment of the XAS, where an approximate, effective single-particle Hamiltonian was used, here we use the full two-particle one, yet for simplicity we still limit the representation of the reference wavefunctions functions to single-determinants. We show that the cumulant form aids in both the physical interpretation of many-body effects observed in the spectra as well as the numerical simulations. We find that, for the XAS, a convolution form in terms of an effective single-particle spectrum and the core-hole spectral function is key to accounting for two types of many-body effects: First, inelastic losses caused by shake up excitations, accounted for the spectral function. Second the edge enhancement due to orthogonality. Both effects modulate the XAS amplitude in opposite direction near threshold, despite being non-singular for molecular systems. Interference terms and extrinsic losses from the coupling between the core-hole and the photoelectron are ignored. Nevertheless, these effects tend to cancel due to their opposite signs. The formal behavior of the RT-EOM-CC cumulant Green’s function is similar to that in other approaches, e.g., field-theoretic methods such as the linked-cluster theorem, or the quasi-boson approximation (Nozieres and de Dominicis, 1969; Langreth, 1970; Hedin, 1999). For condensed matter systems, the cumulant kernel function β(ω) is directly connected to the loss function or the screened Coulomb interaction, and represents collective excitations such as density fluctuations arising from the sudden creation of the core-hole (Langreth, 1970; Kas and Rehr, 2017). Other extensions to the approach reviewed here are feasible. For instance, an analogous treatment is possible to study x-ray emission spectra instead of XAS (Nozieres and Combescot, 1971) by changing the unoccupied single-particle states for the occupied ones. Finally, bigger systems computed with a more user-friendly and efficient implementation, including higher excitations, will be presented elsewhere.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
All authors contributed to the derivation of the theory described in this work and to the writing of the manuscript. FV developed the EOM-CC code and performed all the calculations behind the results presented except for the GFCCSD and GFCC-i(2,3) which were performed by BP.
This work was supported by the Computational Chemical Sciences Program of the U.S. Department of Energy, Office of Science, BES, Chemical Sciences, Geosciences and Biosciences Division in the Center for Scalable and Predictive methods for Excitations and Correlated phenomena (SPEC) at the Pacific Northwest National Laboratory. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank N. Hirshberg, X. Li, J. Lisschner, D. Prendergast, K. Schönhammer, and M. Tzavala, for comments. BP also acknowledges the support of Laboratory Directed Research and Development (LDRD) program from PNNL.
Arponen, J. (1983). Variational Principles and Linked-Cluster Exp S Expansions for Static and Dynamic many-body Problems. Ann. Phys. 151, 311–382. doi:10.1016/0003-4916(83)90284-1
Aryasetiawan, F., Hedin, L., and Karlsson, K. (1996). Multiple Plasmon Satellites in Na and Al Spectral Functions from Ab Initio Cumulant Expansion. Phys. Rev. Lett. 77, 2268–2271. doi:10.1103/physrevlett.77.2268
Bartlett, R. J., and Musiał, M. (2007). Coupled-cluster Theory in Quantum Chemistry. Rev. Mod. Phys. 79, 291–352. doi:10.1103/revmodphys.79.291
Bertsch, G. F., and Lee, A. J. (2014). Time-dependent Mean-Field Theory for X-ray Near-Edge Spectroscopy. Phys. Rev. B 89, 075135. doi:10.1103/physrevb.89.075135
Bhaskaran-Nair, K., Kowalski, K., and Shelton, W. A. (2016). Coupled Cluster Green Function: Model Involving Single and Double Excitations. J. Chem. Phys. 144, 144101. doi:10.1063/1.4944960
Brandow, B. H. (1967). Linked-cluster Expansions for the Nuclear many-body Problem. Rev. Mod. Phys. 39, 771–828. doi:10.1103/revmodphys.39.771
Buttersack, T., Mason, P. E., McMullen, R. S., Martinek, T., Brezina, K., Hein, D., et al. (2019). Valence and Core-Level X-ray Photoelectron Spectroscopy of a Liquid Ammonia Microjet. J. Am. Chem. Soc. 141, 1838–1841. doi:10.1021/jacs.8b10942
Čížek, J. (1966). On the Correlation Problem in Atomic and Molecular Systems. Calculation of Wavefunction Components in Ursell-type Expansion Using Quantum-Field Theoretical Methods. J. Chem. Phys. 45, 4256–4266.
Coester, F. (1958). Bound States of a many-particle System. Nucl. Phys. 7, 421–424. doi:10.1016/0029-5582(58)90280-3
Coester, F., and Kümmel, H. (1960). Short-range Correlations in Nuclear Wave Functions. Nucl. Phys. 17, 477–485. doi:10.1016/0029-5582(60)90140-1
Crawford, T. D., and Schaefer, H. F. (2000). An Introduction to Coupled Cluster Theory for Computational Chemists. Rev. Comp. Ch. 14, 33–136.
Friedel, J. (1969). X-ray Absorption and Emission Edges in Metals. Comments Solid State. Phys. 2, 21.
Grebennikov, V., Babanov, Y., and Sokolov, O. (1977). Extra-atomic Relaxation and X-ray Spectra of Narrow-Band Metals. I. Formalism. Phys. Stat. Sol. (B) 79, 423. doi:10.1002/pssb.2220790204
Gunnarsson, O., Meden, V., and Schönhammer, K. (1994). Corrections to Migdal’s Theorem for Spectral Functions: A Cumulant Treatment of the Time-dependent Green’s Function. Phys. Rev. B 50, 10462–10473. doi:10.1103/physrevb.50.10462
Guzzo, M., Lani, G., Sottile, F., Romaniello, P., Gatti, M., Kas, J. J., et al. (2011). Valence Electron Photoemission Spectrum of Semiconductors: Ab Initio Description of Multiple Satellites. Phys. Rev. Lett. 107, 166401. doi:10.1103/physrevlett.107.166401
Hedin, L. (1999). On Correlation Effects in Electron Spectroscopies and the GW Approximation. J. Phys. Condens. Matter 11, R489. doi:10.1088/0953-8984/11/42/201
Jolly, W., Bomben, K., and Eyermann, C. (1984). Core-electron Binding Energies for Gaseous Atoms and Molecules. Atom. Data Nucl. Data 31, 433–493. doi:10.1016/0092-640x(84)90011-1
Karlsen, T., Børve, K. J., Sæthre, L. J., Wiesner, K., Bässler, M., and Svensson, S. (2002). Toward the Spectrum of Free Polyethylene: Linear Alkanes Studied by Carbon 1s Photoelectron Spectroscopy and Theory. J. Am. Chem. Soc. 124, 7866–7873. doi:10.1021/ja010649j
Kas, J. J., and Rehr, J. J. (2017). Finite Temperature Green’s Function Approach for Excited State and Thermodynamic Properties of Cool to Warm Dense Matter. Phys. Rev. Lett. 119, 176403. doi:10.1103/physrevlett.119.176403
Keller, L., Blum, V., Rinke, P., and Golze, D. (2020). Relativistic Correction Scheme for Core-Level Binding Energies from GW. J. Chem. Phys. 153, 114110. doi:10.1063/5.0018231
Kendall, R. A., Dunning, T. H., and Harrison, R. J. (1992). Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 96, 6796–6806. doi:10.1063/1.462569
Kowalski, K., Bhaskaran-Nair, K., and Shelton, W. A. (2014). Coupled-cluster Representation of Green Function Employing Modified Spectral Resolutions of Similarity Transformed Hamiltonians. J. Chem. Phys. 141, 094102. doi:10.1063/1.4893527
Lange, M. F., and Berkelbach, T. C. (2018). On the Relation between Equation-Of-Motion Coupled-Cluster Theory and the GW Approximation. J. Chem. Theor. Comput. 14, 4224–4236. doi:10.1021/acs.jctc.8b00455
Langreth, D. C. (1969). Singularities in the X-ray Absorption and Emission of Metals. Phys. Rev. 182, 973–974. doi:10.1103/physrev.182.973
Langreth, D. C. (1970). Singularities in the X-ray Spectra of Metals. Phys. Rev. B 1, 471–477. doi:10.1103/physrevb.1.471
Lee, A. J., Vila, F. D., and Rehr, J. J. (2012). Local Time-Correlation Approach for Calculations of X-ray Spectra. Phys. Rev. B 86, 115107. doi:10.1103/physrevb.86.115107
Liang, Y., and Prendergast, D. (2019). Taming Convergence in the Determinant Approach for X-ray Excitation Spectra. ArXiv:1905.00542v1.
Liang, Y., Vinson, J., Pemmaraju, S., Drisdell, W. S., Shirley, E. L., and Prendergast, D. (2017). Accurate X-ray Spectral Predictions: An Advanced Self-Consistent-Field Approach Inspired by many-body Perturbation Theory. Phys. Rev. Lett. 118, 096402. doi:10.1103/PhysRevLett.118.096402
Linderberg, J., and Öhrn, Y. (2004). Propagators in Quantum Chemistry. Wiley. Theoretical chemistry.
Mahan, G. D. (1967). Excitons in Metals: Infinite Hole Mass. Phys. Rev. 163, 612. doi:10.1103/physrev.163.612
Meissner, L., and Bartlett, R. J. (1993). Electron Propagator Theory with the Ground State Correlated by the Coupled-Cluster Method. Int. J. Quan. Chem. 48, 67–80. doi:10.1002/qua.560480810
Nist, (2019). in NIST Computational Chemistry Comparison and Benchmark Database. Editor R. D. Johnson III (NIST). vol. Release 20 of NIST Standard Reference Database Number 101. doi:10.18434/T47C7Z
Nooijen, M., and Snijders, J. G. (1992). Coupled Cluster Approach to the Single-Particle Green’s Function. Int. J. Quan. Chem. 44, 55–83. doi:10.1002/qua.560440808
Nooijen, M., and Snijders, J. G. (1993). Coupled Cluster Green’s Function Method: Working Equations and Applications. Int. J. Quan. Chem. 48, 15–48. doi:10.1002/qua.560480103
Nooijen, M., and Snijders, J. G. (1995). Second Order many-body Perturbation Approximations to the Coupled Cluster Green’s Function. J. Chem. Phys. 102, 1681–1688. doi:10.1063/1.468900
Nozieres, P., and Combescot, M. (1971). Infrared Catastrophy and Excitons in the X-ray Spectra of Metals. J. de Physique 32, 11.
Nozieres, P., and de Dominicis, C. (1969). Singularities in the X-ray Absorption and Emission of Metals. III. One-Body Theory Exact Solution. Phys. Rev. 178, 1097. doi:10.1103/physrev.178.1097
Paldus, J., Čížek, J., and Shavitt, I. (1972). Correlation Problems in Atomic and Molecular Systems. IV. Extended Coupled-Pair many-electron Theory and its Application to the BH3 Molecule. Phys. Rev. A. 5, 50–67. doi:10.1103/physreva.5.50
Paldus, J., and Li, X. (1999). A Critical Assessment of Coupled Cluster Method in Quantum Chemistry. Adv. Chem. Phys. 110, 1–175.
Peng, B., and Kowalski, K. (2016). Coupled-cluster Green’s Function: Analysis of Properties Originating in the Exponential Parametrization of the Ground-State Wave Function. Phys. Rev. A. 94, 062512. doi:10.1103/physreva.94.062512
Peng, B., and Kowalski, K. (2018b). Green’s Function Coupled Cluster Formulations Utilizing Extended Inner Excitations. J. Chem. Phys. 149, 214102. doi:10.1063/1.5046529
Peng, B., and Kowalski, K. (2018a). Green’s Function Coupled-Cluster Approach: Simulating Photoelectron Spectra for Realistic Molecular Systems. J. Chem. Theor. Comput. 14, 4335–4352. doi:10.1021/acs.jctc.8b00313
Peng, B., Panyala, A., Kowalski, K., and Krishnamoorthy, S. (2021). GFCCLib: Scalable and Efficient Coupled-Cluster Green’s Function Library for Accurately Tackling many-body Electronic Structure Problems. Computphys. Commun. 265, 108000. doi:10.1016/j.cpc.2021.108000
Peng, B., Van Beeumen, R., Williams-Young, D. B., Kowalski, K., and Yang, C. (2019). Approximate Green’s Function Coupled Cluster Method Employing Effective Dimension Reduction. J. Chem. Theor. Comput. 15, 3185–3196. doi:10.1021/acs.jctc.9b00172
Privalov, T., Gel’mukhanov, F., and Ågren, H. (2001). Role of Relaxation and Time-dependent Formation of X-ray Spectra. Phys. Rev. B 64, 165115. doi:10.1103/physrevb.64.165115
Pueyo Bellafont, N., Viñes, F., and Illas, F. (2016). Performance of the Tpss Functional on Predicting Core Level Binding Energies of Main Group Elements Containing Molecules: A Good Choice for Molecules Adsorbed on Metal Surfaces. J. Chem. Theor. Comput. 12, 324–331. doi:10.1021/acs.jctc.5b00998
Purvis, G., and Bartlett, R. (1982). A Full Coupled-Cluster Singles and Doubles Model: The Inclusion of Disconnected Triples. J. Chem. Phys. 76, 1910–1918. doi:10.1063/1.443164
Rehr, J. J., Kas, J. J., Prange, M. P., Sorini, A. P., Takimoto, Y., and Vila, F. (2009). Ab Initio theory and Calculations of X-ray Spectra. CR Phys. 10, 548–559. doi:10.1016/j.crhy.2008.08.004
Rehr, J. J., Stern, E. A., Martin, R. L., and Davidson, E. R. (1978). Extended x-ray-absorption fine-structure Amplitudes—Wave-Function Relaxation and Chemical Effects. Phys. Rev. B 17, 560–565. doi:10.1103/physrevb.17.560
Rehr, J. J., Vila, F. D., Kas, J. J., Hirshberg, N., Kowalski, K., and Peng, B. (2020). Equation of Motion Coupled-Cluster Cumulant Approach for Intrinsic Losses in X-ray Spectra. J. Chem. Phys. 152, 174113. doi:10.1063/5.0004865
Sankari, R., Ehara, M., Nakatsuji, H., Fanis, A. D., Aksela, H., Sorensen, S., et al. (2006). High Resolution O 1s Photoelectron Shake-Up Satellite Spectrum of H2O. Chem. Phys. Lett. 422, 51–57. doi:10.1016/j.cplett.2006.02.018
Schönhammer, K., and Gunnarsson, O. (1978). Time-dependent Approach to the Calculation of Spectral Functions. Phys. Rev. B 18, 6606–6614. doi:10.1103/physrevb.18.6606
Stanton, J. F., and Bartlett, R. J. (1993). The Equation of Motion Coupled-Cluster Method. A Systematic Biorthogonal Approach to Molecular Excitation Energies, Transition Probabilities, and Excited State Properties. J. Chem. Phys. 98, 7029–7039. doi:10.1063/1.464746
Szabo, A., and Ostlund, N. (1996). Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory. Dover Publications). Dover Books on Chemistry.
Tzavala, M., Kas, J. J., Reining, L., and Rehr, J. J. (2020). Nonlinear Response in the Cumulant Expansion for Core-Level Photoemission. Phys. Rev. Res. 2, 033147. doi:10.1103/PhysRevResearch.2.033147
Vila, F. D., Rehr, J. J., Kas, J. J., Kowalski, K., and Peng, B. (2020). Real-time Coupled-Cluster Approach for the Cumulant Green’s Function. J. Chem. Theor. Comput. 16, 6983–6992. doi:10.1021/acs.jctc.0c00639
Viñes, F., Sousa, C., and Illas, F. (2018). On the Prediction of Core Level Binding Energies in Molecules, Surfaces and Solids. Phys. Chem. Chem. Phys. 20, 8403–8410. doi:10.1039/c7cp08503f
von Barth, U., and Grossmann, G. (1982). Dynamical Effects in X-ray Spectra and the Final-State Rule. Phys. Rev. B 25, 5150–5179. doi:10.1103/physrevb.25.5150
Wight, G., and Brion, C. (1974). K-shell Excitation of CH4, NH3, H2O, CH3OH, CH3OCH3 and CH3NH2 by 2.5 keV Electron Impact. J. Electron. Spectrosc. Relat. Phenom. 4, 25–42. doi:10.1016/0368-2048(74)80040-x
Williams, G. P. (2009). X-Ray Data Booklet: Section 1.1 ELECTRON BINDING ENERGIES. Berkeley): Lawrence Berkeley National Laboratory.
Keywords: coupled cluster, cumulant, green’s function, excited states, x-ray
Citation: Vila FD, Kas JJ, Rehr JJ, Kowalski K and Peng B (2021) Equation-of-Motion Coupled-Cluster Cumulant Green’s Function for Excited States and X-Ray Spectra. Front. Chem. 9:734945. doi: 10.3389/fchem.2021.734945
Received: 01 July 2021; Accepted: 06 September 2021;
Published: 22 September 2021.
Edited by:
Bjoern Baumeier, Eindhoven University of Technology, NetherlandsReviewed by:
Juhan Matthias Kahk, University of Tartu, EstoniaCopyright © 2021 Vila, Kas, Rehr, Kowalski and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. J. Rehr, ampyQHV3LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.