 Thillai Govindaraja Senthamaraikannan
Thillai Govindaraja Senthamaraikannan Selvaraj Kaliaperumal2
Selvaraj Kaliaperumal2 Sailaja Krishnamurty
Sailaja Krishnamurty- 1Department of Environmental Engineering, Chungbuk National University, Cheongju, Korea
- 2Nano and Computational Material Lab, Catalysis Division, CSIR-National Chemical Laboratory, Pune, India
- 3Physical Chemistry Division, CSIR-National Chemical Laboratory, Pune, India
Using the periodic density functional theory–based methodology, we propose a potential catalytic system for dinitrogen activation, viz., single metal atoms (Mo, Fe, and V) supported on graphene-based sheets. Graphene-based sheets show an excellent potential toward the anchoring of single atoms on them (Mo, Fe, and V) with adsorption energies ranging between 1.048 and 10.893 eV. Factors such as defects and BN doping are noted to enhance the adsorption energies of single metal atoms on the support. The adsorption of a dinitrogen molecule on metal atom–anchored graphene-based supports is seen to be highly favorable, ranging between 0.620 and 2.278 eV. The adsorption is driven through a direct hybridization between the d orbitals of the metal atom (Mo, Fe, and V) on the support and the p orbital of the molecular nitrogen. Noticeably, BN-doped graphene supporting a single metal atom (Mo, Fe, and V) activates the N2 molecule with a red shift in the N–N stretching frequency (1,597 cm−1 as compared to 2,330 cm−1 in the free N2 molecule). This red shift is corroborated by an increase in the N–N bond length (1.23 Å from 1.09 Å) and charge transfer to an N2 molecule from the catalyst.
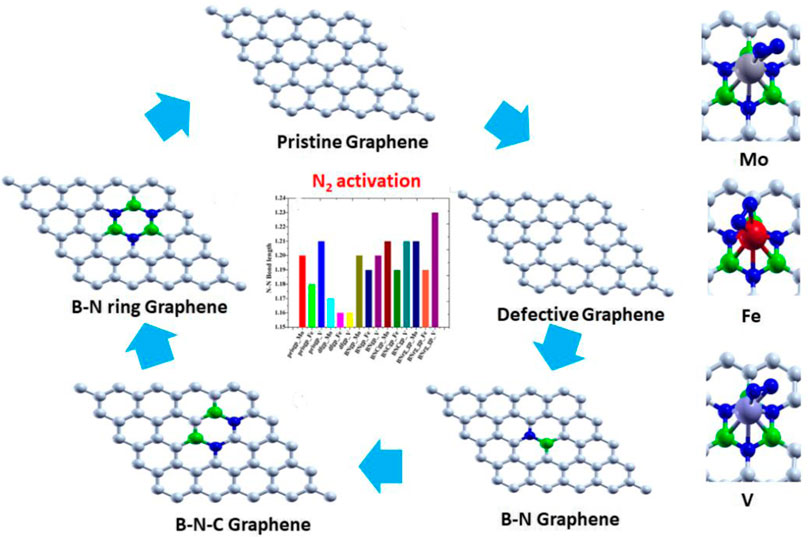
Graphical Abstract. XXX
Introduction
Ammonia is an important chemical substance for the agriculture, pharmaceuticals, and chemical industries. Natural and synthetic N2 fixation is necessary for the existence of all forms of life on Earth. Though the availability of dinitrogen (N2) is abundant in air, it requires high energy for fixation and activation owing to its existence of inert triple bonds between nitrogen atoms. Currently, the well-known Haber–Bosch process invented more than a century ago is used for converting dinitrogen (N2) in the atmosphere into NH3 in the presence of the iron catalyst at an extreme temperature (500°C) and pressure (200 atm) (Fryzuk and Johnson, 2000). The energy- and carbon-intensive Haber–Bosch process consumes 1–2% global energy and, in addition, produces 3% of global CO2 emission (Cherkasov et al., 2015). Nevertheless, N2 fixation can occur readily under mild conditions by nitrogenase mechanism, the enzyme secreted from very few prokaryotic organisms (Kim and Rees, 1992; Sellmann and Sutter, 1997; Einsle et al., 2002). Researchers have demonstrated the occurrence of biological N2 fixation under reasonable or mild conditions in the presence of nitrogenase enzymes, most preferably at the active sites that are rich in Fe and S and also additionally contain Mo or V atoms (Dance, 2008; Stüeken et al., 2015; Tanabe and Nishibayashi, 2016), yet the through kinetics are still disputed. Consequently, exploring an efficient N2 reduction catalyst in ammonia synthesis is the main challenge for the organo-metallic researchers. Naturally, N2 fixation and activation require a potential catalytic active center to promote nitrogen reduction reaction, via electrons overlapping between the σ bond of N2 and the d orbital of the metal center, and the occupied d orbital overlaps with the empty π* bond of N2, resulting in the activation of N2 by a π bond back-donation mechanism.
On accounting for the quantum confinement of electrons, metal clusters are widely explored as catalysts. Using experimental and theoretical strategies, researchers have explored N2 activation on potential inorganic metal clusters (Seh et al., 2017; Liu et al., 2018; Wang et al., 2018). Significantly, Kerpal et al. (2013) have evaluated dinitrogen (N2) activation using infrared multiphoton dissociation (IR-MPD) on neutral Ru clusters. Similarly, Roy et al. (2009) have noticed the red-shifted N–N bond stretching frequency around 810 cm−1 on solid Lin (2 < n < 8) clusters, particularly the Li8 metal cluster showing an exothermic trend in splitting the N–N bond completely. In the midst of metal clusters for evaluating N2 activation reaction, Al clusters play a remarkable role. Previously, Jarrold et al. observed low energy barriers for N2 activation on Al44 and Al100 clusters at high temperatures using concerted experimental and theoretical techniques (Cao et al., 2010). Similarly, in another previous report by this group, N2 activation potential was observed to be dependent on the phase and structure of the metal cluster (Cao et al., 2009). During the course of N2 activation mechanism, conformations with high energy display low energy potential toward the activation of the N2 molecule (Kulkarni et al., 2011). Nevertheless, excited state conformations are meta-stable in nature and are notably present only at some characteristic finite temperatures. Hence, there is an obvious demand for more reliable and stable ground state conformations for N2 activation. Consequently, heteroatoms such as silicon and phosphorus doped on aluminum clusters appear to be a possible alternative and have better activation than their pristine aluminum clusters (Das et al., 2014).
Moreover, an alternative and experimentally supported route is to enhance the activity of metal-based catalysts by anchoring metal centers on 2D material supports such as graphene and BN, which offers a substantial support to the metal centers to adsorb and activate the N2 molecule. Moreover, specific activity per metal atom increases by downsizing the metals from nanoparticles to nanocrystals or hetero-nano framework (Yang et al., 2013; Chen et al., 2014). Single atom catalysts (SACs) have gained more attention in downsizing metals considerably and exhibit the potential of well-dispersed active single atom sites available for atomic utilization (Qiao et al., 2011). Based on these circumstances, SACs exhibiting unique activity with high density of active sites supported on 2D materials can make use of electron sharing for the activation of the inert dinitrogen molecule. A single transition-metal atom or atom clusters supported on N-doped graphene show good nitrogen reduction reaction (NRR) activity (Choi et al., 2015; Li et al., 2016; Fajardo and Peters, 2017; Fei et al., 2018; Yan et al., 2019). Systems such as BiOBr nanosheets, boron anti-sites on BN nanotubes, and Mo-doped boron nitride (BN) have also been reported to have high N2 fixation potential (Li et al., 2015; Kumar and Subramanian, 2017; Zhao and Chen, 2017; Légaré et al., 2018).
In the midst of 2D materials, graphene-based supports attract enormous attention in numerous reactions such as water splitting, Guo et al. (2018), and hydrogen evolution reaction (HER) Ouyang et al. (2018). Few experimental groups reported N2 fixation using a graphene-based catalytic support (Jeon et al., 2013; Lu et al., 2016; Yan et al., 2018). Several computational investigations have also been explored using graphene-based nanomaterials for N2 fixation to compare with the experimental findings. Le et al. reported that the Mo/N-doped graphene-based support dissociates the N2 molecule using the density functional theory (DFT) methodology (Le et al., 2014). In a similar approach, Li et al. observed an N2 molecule activation to nearly 2.5 Å by fixing the FeN3 molecule on a graphene support, in which nitrogen atoms are used as anchoring elements, while iron does the activation job in the FeN3 molecule (Li et al., 2016). Kumar et al. (2016) reported N2 activation using aluminum clusters doped on the BN-doped graphene support. The rare ability of certain transition complexes to bind to N2, which is attributed to their advantageous combination of unoccupied and occupied d-orbitals that have appropriate energy and symmetry to synergistically accept/back-donate electron density from/to N2, can thus be contrived by giving the appropriate environment to a p-block element. In short, activation of N2 is performed by exploiting the electron reservoir property of 2D graphene-based materials. Recently, in our previous investigations, we identified the most active and recyclable SAC/B-graphene composite as the catalyst for NRR activity (Maibam et al., 2019; Maibam and Krishnamurty, 2021). In the present work, using the density functional theory (DFT)-based methodology, we evaluate the possible dinitrogen activation by single metal atoms (Mo, Fe, and V) supported on graphene-based systems such as pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, and BN-ring graphene as support materials.
Computational Details
We use the Vienna Ab Initio Simulation Package (VASP) (Kresse and Furthmller, 1996) with the PBE functional (Perdew et al., 1996) to perform all the first-principles calculations in the present work. The projected augmented wave (PAW) (Blöchl, 1994) method is employed using an energy cutoff of 520 eV to describe the plane wave basis set. The two-dimensional graphene sheets are simulated using periodic boundary conditions. To avoid the interactions between the different nearest neighboring layers, a vacuum space of 20 Å is created along the Z-direction. The 5 × 5 supercell with 50 atoms is used as the graphene surface model, and the optimized C–C bond length in the graphene sheet is 1.42 Å.
Pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, and BN-ring graphene are designed surface supports, and the structures are further optimized. The structural optimization of all geometries is carried out using the conjugate gradient method (Payne et al., 1992). The Brillouin zone is sampled by a (2 × 2×1) K-point grid using the Monkhorst–Pack scheme (Monkhorst and Pack, 1976). For density of states (DOS) calculations, Monkhorst and Pack generated a (9 × 9×1) set of K points.
The ground state geometries of single transition-metal clusters (Mo, Fe, and V) are adsorbed on the above-mentioned supports and the complexes optimized. The adsorption energy of Mo, V, and Fe on these supports is calculated as follows:
where E(M--system) represents the energy of the optimized single transition-metal cluster (Mo, Fe, and V) and the designed surface supports. E(M) and E(system) represent the energy of a single metal and surface support, respectively.
Finally, the N2 molecule is adsorbed on these active metal clusters (Mo, Fe, and V) on graphene-based surface supports. A parallel mode of adsorption (both the nitrogen atoms are exposed to the metal) is used as this mode has been found to be more effective as compared to the vertical mode. In the vertical mode, only one N atom in the N2 molecule interacts with the metal leading to weak activation (Song et al., 2021).
The dissociated adsorption energy of the N2 molecule on the catalytic systems is calculated as follows:
where E(N2----M--system) represents the energy of the dissociated N2 molecule on the catalytic systems. E(N2) and E(M--system) represent the energy of the N2 molecule and metal-adsorbed various surface supports, respectively. Nudged elastic band (NEB) calculations were performed toward prediction of energy barrier of N2 activation on metal-adsorbed BN-doped graphene-based substrates.
Results and Discussion
Anchoring of Single Metal Atom (Mo, Fe, and V) on Various Graphene-Based Supports
Graphene-based 2D materials which act as an electron reservoir are used as the support for adsorbing the single atom cluster (Mo, Fe, and V) which increases the catalytic activity of the metal center. The five graphene-based supports are designed, viz., 1) pristine graphene (50 carbon atoms), 2) defective graphene (49 carbon atoms with a single vacancy at the center), 3) BN-doped graphene (4% heteroatom doping in which boron and nitrogen are substituted instead of carbon in the pristine graphene), 4) BNC-ring graphene (8% heteroatom doping), and 5) BN-ring graphene (12% heteroatom doping). All these graphene-based supports are designed and optimized to the local minima as shown in Figure 1.
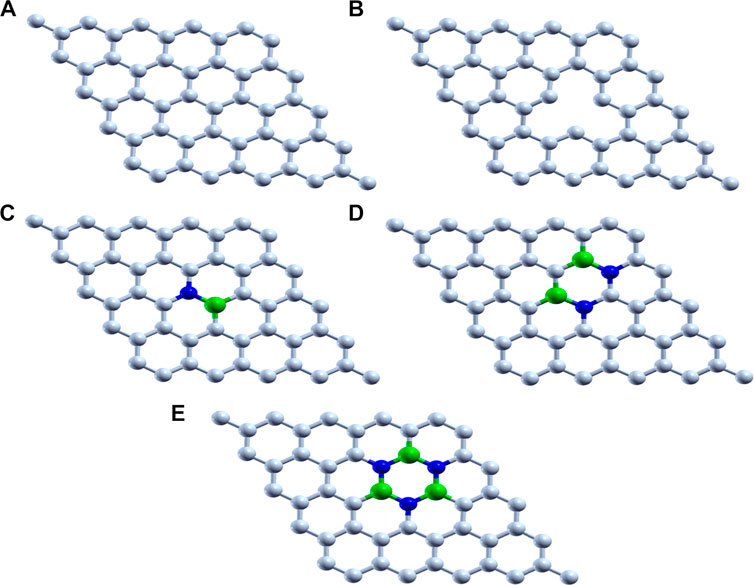
FIGURE 1. Optimized structure of (A) pristine graphene, (B) defective graphene, (C) BN-doped graphene, (D) BNC-ring graphene, and (E) BN-ring graphene (C, white; B, green; and N, blue).
Thus, we have tried to establish the relative reactivity of single atom clusters (Mo, Fe, and V) chemisorbed on the above-mentioned surfaces. The optimized structure of adsorption of Mo (gray), Fe (red), and V (purple) on various surface supports is shown in Figure 2. The adsorption energy of a single metal atom (Mo, Fe, and V) on pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, and BN-ring graphene is 4.653, 2.602, and 3.145 eV; 10.893, 9.329, and 9.744 eV; 3.929, 1.090, and 2.494 eV; 3.864, 1.728, and 2.498 eV; and 3.016, 1.048, and 1.467 eV, respectively. Comparatively, the adsorption energy of Mo on the designed supports is ∼2 eV more due to its bulky nature with respect to other metals (Fe and V). Interestingly, the dangling carbon atoms at the center increase the adsorption energies for a defective graphene support better than the rest, and also the increase in the percentage of heteroatom (B and N) doping decreases the adsorption energies of the single metal atom on supports.
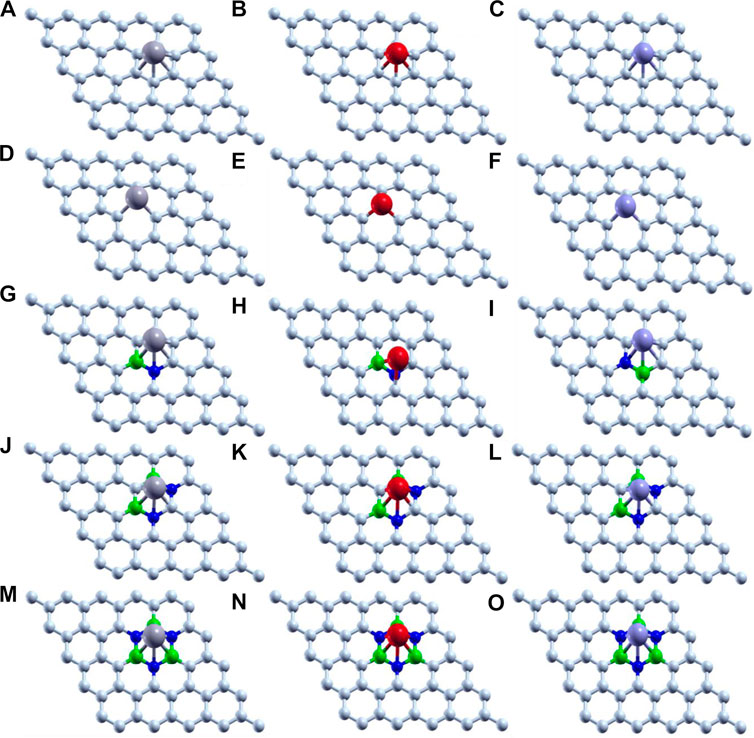
FIGURE 2. Optimized structure of adsorption of Mo (gray), Fe (red), and V (purple) on (A–C) pristine graphene, (D–F) defective graphene, (G–I) BN-doped graphene, (J–L) BNC-ring graphene, and (M–O) BN-ring graphene.
The carbon–metal (C–M) interatomic distance of Mo, Fe, and V on pristine graphene, defective graphene, BN-doped graphene, and BNC-ring graphene is 2.200–2.213, 2.069–2.080, and 2.147–2.170 Å;1.932–1.956, 1.766–1.768, and 1.863–1.873 Å; 2.145–2.261, 2.012–2.473, and 2.079–2.192 Å; and 2.072–2.305, 1.947–2.148, and 2.072–2.306 Å, respectively. The boron–metal (B–M) interatomic distance of Mo, Fe, and V on BN-doped graphene, BNC-ring graphene, and BN-ring graphene is 2.258, 2.303, and 2.215 Å; 2.279–2.28, 2.106–2.218, and 2.279–2.28 Å; and 2.216–2.219, 2.063–2.124, and 2.17–2.227 Å, respectively. The nitrogen–metal (N–M) interatomic distance of Mo, Fe, and V on BN-doped graphene, BNC-ring graphene, and BN-ring graphene is 2.211, 1.861, and 2.172 Å; 2.224–2.226, 2.01–2.225, and 2.224–2.226 Å; and 2.267–2.273, 2.073–2.197, and 2.204–2.226 Å, respectively. The interatomic distances and adsorption energies of Mo, Fe, and V on various graphene-based supports are shown in Table 1. Thus, the significance of the result shows that the adsorption energies of a single metal atom on the surface support provide a stable and potential catalyst for N2 activation. The total density of states and projected density of states of a single metal atom (Mo, Fe, and V) on graphene-based supports are shown in Figure 3. The total density of states (TDOS) and partial density of states (PDOS) reveal that the d-states of a single metal atom (Mo, Fe, and V) strongly hybridize with the p-state of unsaturated carbon atoms and heteroatoms (B and N). The d-state of a single metal atom shows its maximum density of states between −2 and 2 eV. On comparing, the p-state of unsaturated carbon atoms is maximum in pristine and defective supports which reveals that, in the other three supports, the p-state of both boron and nitrogen is hybridized with the d-state of metal.
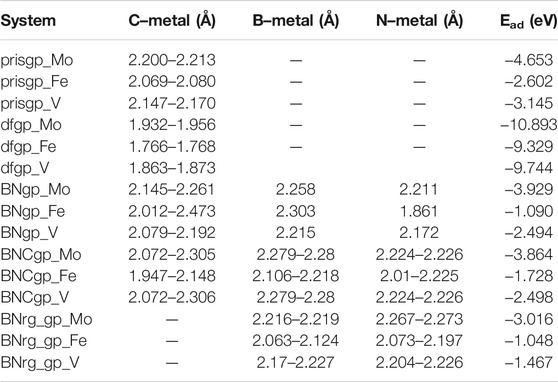
TABLE 1. Interatomic distances and adsorption energies of Mo, Fe, and V on various graphene-based supports (pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, BN-ring graphene, and adsorption energy are abbreviated as prisgp, dfgp, BNgp, BNCgp, BNrg_gp, and Ead).
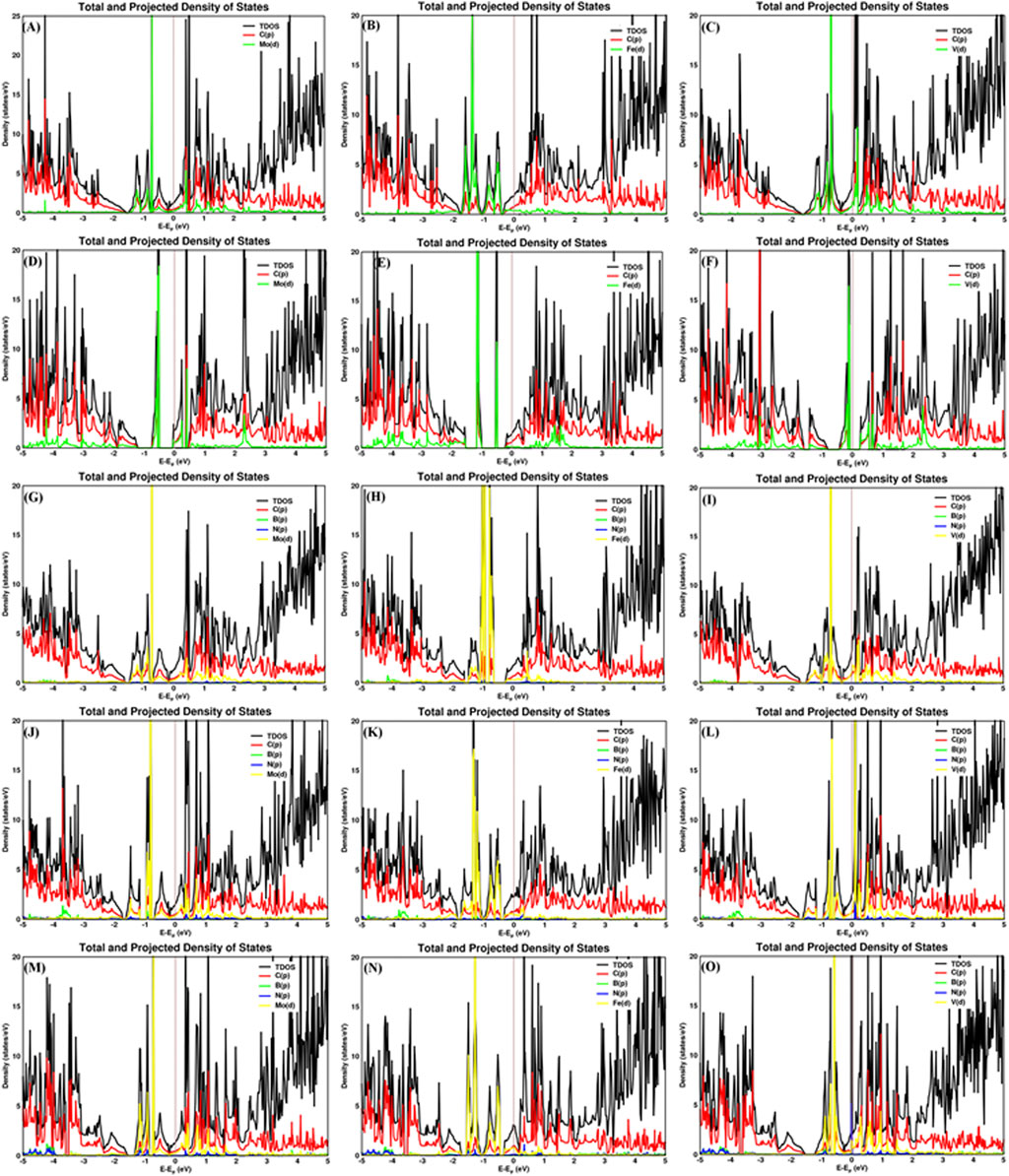
FIGURE 3. Total and projected density of states of Mo, Fe, and V on (A–C) pristine graphene, (D–F) defective graphene, (G–I) BN-doped graphene, (J–L) BNC-ring graphene, and (M–O) BN-ring graphene.
N2 Activation on Single Metal Atom (Mo, Fe, and V) Anchored on Various Graphene-Based Supports
The adsorption energies of N2 on a single metal atom (Mo, Fe, and V) on pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, and BN-ring graphene are 1.739, 1.334, and 1.996 eV; 0.887, 0.620, and 0.628 eV; 1.844, 2.278, and 1.988 eV; 1.870, 1.544, and 2.116 eV; and 1.868, 1.510, and 2.258 eV, respectively. Comparatively, the adsorption energies of N2 on a single metal atom (Mo, Fe, and V) on the defective graphene support are less compared to those on the rest of the support. Moreover, there is an eventual increase in adsorption energies of N2 on V on supports (BN-doped graphene, BNC-ring graphene, and BN-ring graphene) due to more vacant d orbitals (less than half-filled), which is vice versa in Fe (more than half-filled d orbitals) on the same supports. The optimized structure of adsorption of N2 on a single metal atom (Mo, Fe, and V) on various surface supports is shown in Figure 4.
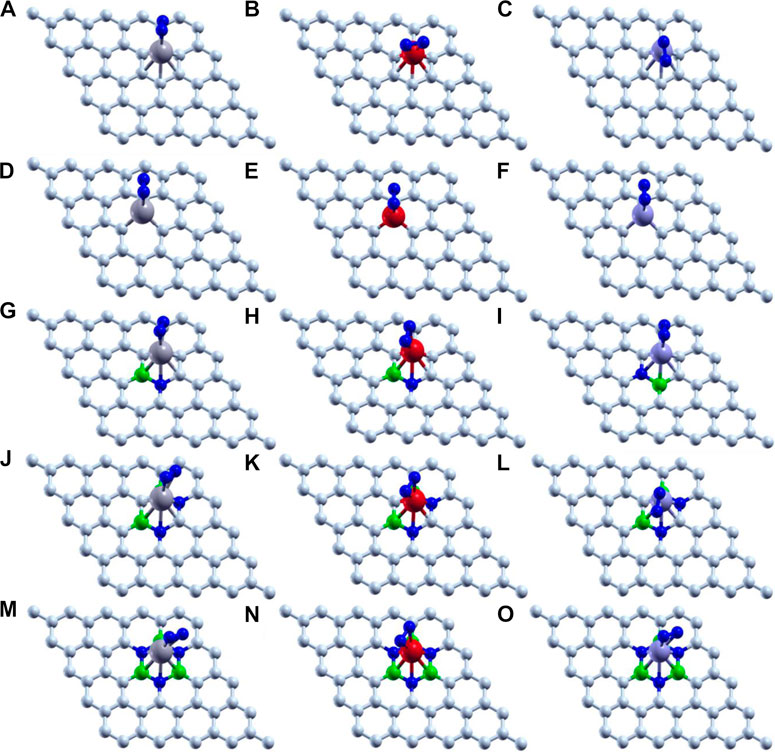
FIGURE 4. Optimized structure of the N2 molecule adsorbed on Mo (gray), Fe (red), and V (purple) on various supports: (A–C) pristine graphene, (D–F) defective graphene, (G–I) BN-doped graphene, (J–L) BNC-ring graphene, and (M–O) BN-ring graphene.
The carbon–metal (C–M) interatomic distance of N2 on a single metal atom (Mo, Fe, and V) on pristine graphene, defective graphene, BN-doped graphene, and BNC-ring graphene is 2.228–2.313, 2.077–2.162, and 2.190–2.264 Å; 1.946–2.013, 1.777–1.847, and 1.871–1.925 Å; 2.194–2.257, 2.096–2.144, and 2.133–2.234 Å; and 2.111–2.319, 2.004–2.172, and 2.048–2.241 Å, respectively. The boron–metal (B–M) interatomic distance of N2 on a single metal atom (Mo, Fe, and V) on BN-doped graphene, BNC-ring graphene, and BN-ring graphene is 2.313, 2.18, and 2.319 Å; 2.316–2.407, 2.219–2.222, and 2.303–2.342 Å; and 2.244–2.41, 2.141–2.216, and 2.256–2.371 Å, respectively. The nitrogen–metal (Ndoped–M) interatomic distance of N2 on a single metal atom (Mo, Fe, and V) on BN-doped graphene, BNC-ring graphene, and BN-ring graphene is 2.32, 2.202, and 2.223 Å; 2.239–2.328, 2.165–2.173, and 2.23–2.242 Å; and 2.274–2.341, 2.083–2.234, and 2.264–2.297 Å, respectively.
The nitrogen–metal (Nad–M) interatomic distance of N2 on a single metal atom (Mo, Fe, and V) on pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, and BN-ring graphene is 2.039–2.117, 1.92–1.923, and 1.911–1.994 Å; 2.22–2.221, 1.964–2.078, and 2.161–2.218 Å; 2.027–2.091, 1.902–1.907, and 1.918–2 Å; 2–2.057, 1.889–1.89, and 1.908–1.979 Å; and 2–2.068, 1.9, and 1.869–1.928 Å, respectively. The interatomic distances and adsorption energy of N2 on a single metal atom (Mo, Fe, and V) on various substrate systems are shown in Table 2. The total density of states and projected density of states of N2 on a single metal atom (Mo, Fe, and V) on the graphene-based support are shown in Figure 5. The total density of states (TDOS) and partial density of states (PDOS) reveal that the d-states of a single metal atom (Mo, Fe, and V) hybridize with the p-state of adsorbed nitrogen as well as carbon, boron, and nitrogen atoms doped on the support. Thus, the d-state of a single metal atom shares its vacant orbital with the p-state of hybridizing atoms.
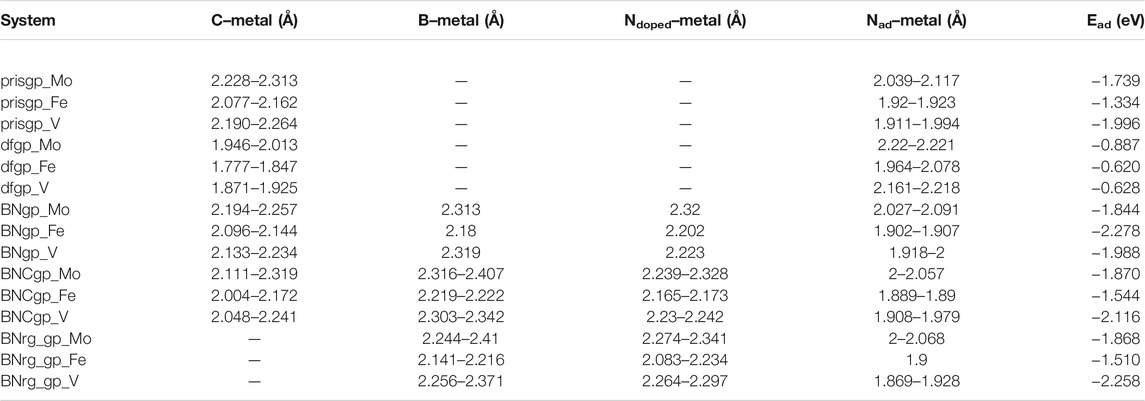
TABLE 2. Interatomic distances and adsorption energies of N2 on Mo, V, and Fe on various graphene-based supports (pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, BN-ring graphene, and adsorption energy are abbreviated as prisgp, dfgp, BNgp, BNCgp, BNrg_gp, and Ead).
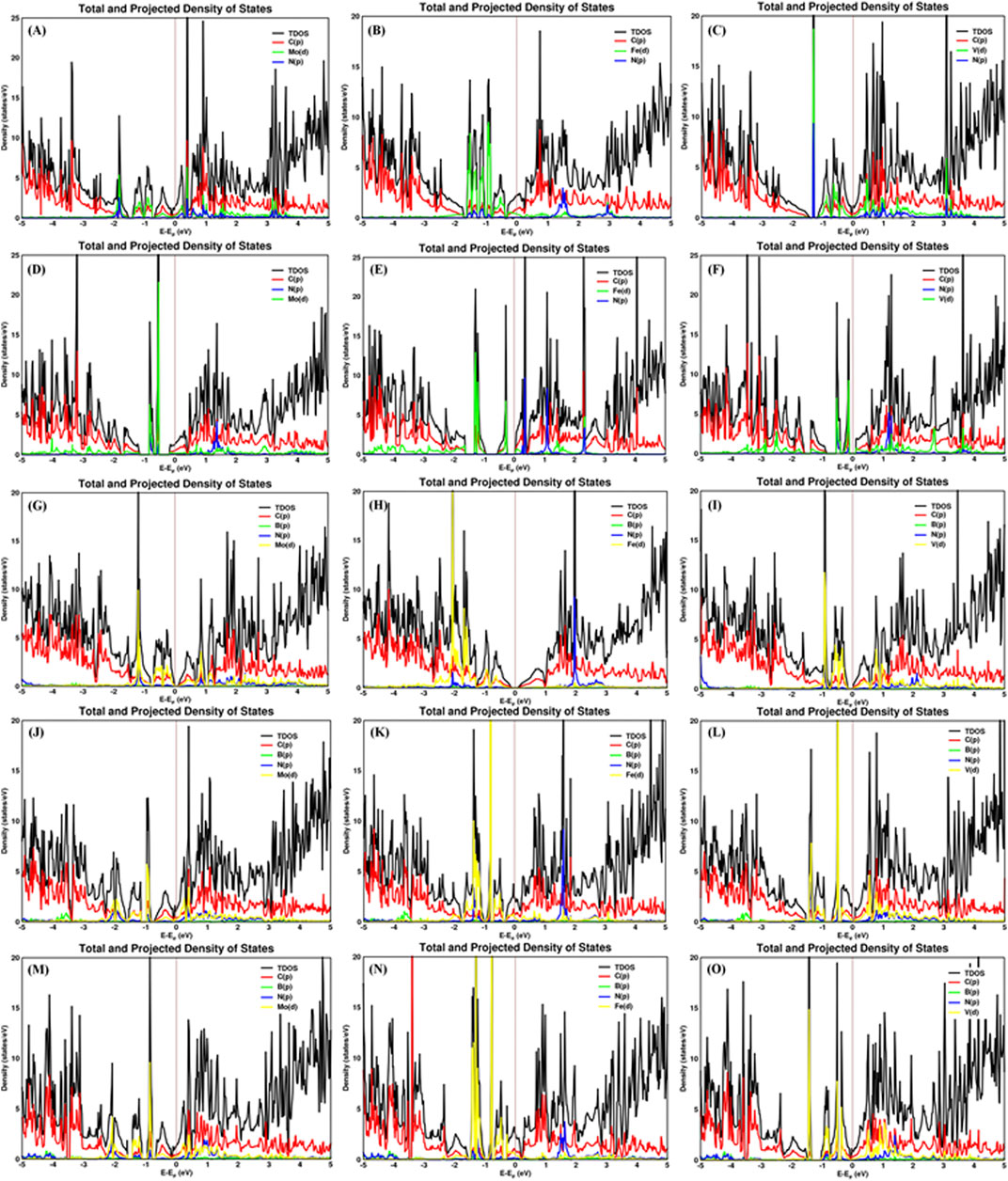
FIGURE 5. Total and projected density of states of the N2 molecule adsorbed on Mo, Fe, and V on various supports: (A–C) pristine graphene, (D–F) defective graphene, (G–I) BN-doped graphene, (J–L) BNC-ring graphene, and (M–O) BN-ring graphene.
N–N Bond Stretching Frequency Analysis on Designed Catalytic Systems
To probe the stretching frequency of the adsorbed N2 molecule on a single metal atom (Mo, Fe, and V) on the graphene-based support, we investigated the spectral range of 1,300–2,300 cm−1, which covers the typical frequencies of the different N2 species known to exist on transition-metal surfaces. The stretching frequency of the unbound N2 molecule is attributed to 2,330 cm−1, and the N–N bond length is 1.09 Å (Shi and Jacobi, 1992). The N–N bond length and IR stretching frequency ν(N–N) of N2 on a single metal atom (Mo, Fe, and V) on pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, and BN-ring graphene are 1.20 Å (1735 cm−1), 1.18 Å (1823 cm−1), and 1.21 Å (1,692 cm−1); 1.17 Å (1907 cm−1), 1.16 Å (2009 cm−1), and 1.16 Å (1997 cm−1); 1.20 Å (1701 cm−1), 1.19 Å (1802 cm−1), and 1.20 Å (1711 cm−1); 1.21 Å (1,636 cm−1), 1.19 Å (1777 cm−1), and 1.21 Å (1,688 cm−1); and 1.21 Å (1,666 cm−1), 1.19 Å (1796 cm−1), and 1.23 Å (1,597 cm−1), respectively.
The Bader charge analysis (Bader, 1991; Tang et al., 2009) clearly demonstrates the charge redistribution between the activated nitrogen atoms and the active metal centered on support-based catalysts. The structural, electronic, and vibrational properties of various catalytic systems for N2 activation are listed in Table 3. The N–N stretching frequency, N–N bond length, and charge on nitrogen of the N2 molecule adsorbed on Mo, Fe, and V on various graphene supports are shown in Supplementary Figure S1.
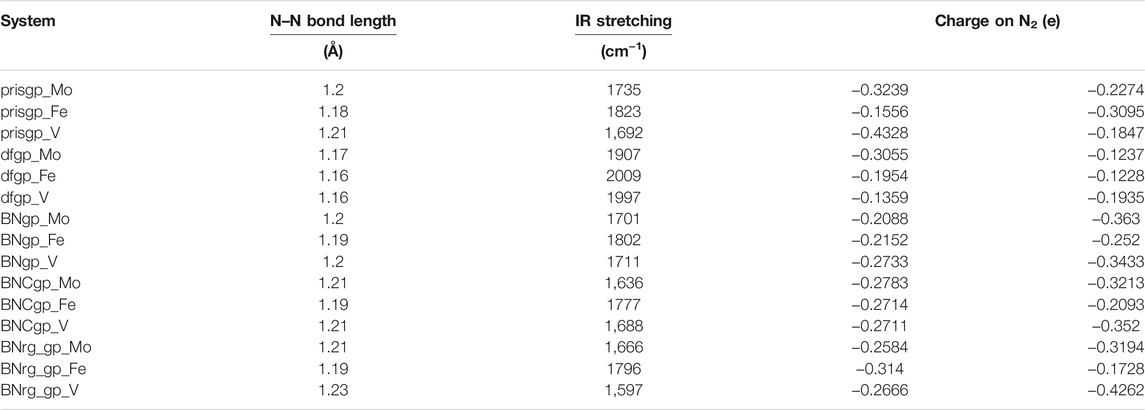
TABLE 3. Structural, electronic, and vibrational properties of various catalytic systems for N2 activation (pristine graphene, defective graphene, BN-doped graphene, BNC-ring graphene, and BN-ring graphene are abbreviated as prisgp, dfgp, BNgp, BNCgp, and BNrg_gp).
As a case study, the potential of the Mo-adsorbed BN-doped graphene catalyst for the activation of N2 is discussed in Supplementary Figure S2. NEB calculation is performed in between these reactants and products to confirm the N2 activation energy barrier. Mo-adsorbed BN-doped graphene and gaseous nitrogen are considered reactants. Thus, the Mo-adsorbed BN-doped graphene catalyst shows more feasible N2 activation with an effective energy barrier of 3.21 eV. The activation barrier plot of the N2 molecule adsorbed on Mo on the BN-doped graphene support is shown in Supplementary Figure S2 (Liu et al., 2021).
Conclusion
In this work, we explore the potential of various graphene-based 2D materials, viz., pristine, defective, BN-doped graphene, etc., as a support for a single atom cluster (Mo, Fe, and V). These graphene-based supports show excellent potential toward the anchoring of a single atom cluster (Mo, Fe, and V) with adsorption energies ranging between 1.048 and 10.893 eV. Thus, the adsorption energies vary substantially with respect to the graphene-based supports, viz., pristine, defective, BN doped, etc. This is attributed to the size and nature of hybridization between the d orbitals of the interacting single metal atom (Mo, Fe, and V) and the sp2 orbitals of unsaturated carbon atoms of various designed graphene-based supports. The catalytic performance of a single metal atom (Mo, Fe, and V) on graphene-supported catalysts is explored for the activation of molecular nitrogen. The adsorption energies of the nitrogen molecule on a graphene-supported single atom cluster (Mo, Fe, and V) range between 0.620 and 2.278 eV, which is attributed to the interacting environment of the active metal centered on the support and the p orbital of adsorbed molecular nitrogen. Bader charge and density of states analyses corroborate an enhanced hybridization between the d states of the single metal atoms (Mo, Fe, and V) and adsorbed molecular nitrogen for activation. The N–N stretching frequencies are found which are considerably red-shifted ranging from 2009 cm−1 (1.16 Å) to 1,597 cm−1 (1.23 Å) compared to that of the unbound N2 molecule (2,330 cm−1 (1.09 Å)). Thus, from the results, we understood that even a single metal atom (Mo, Fe, and V) with functionalized (BN-doped) graphene supports can highlight the excellent potential for nitrogen activation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, and further inquiries can be directed to the corresponding authors.
Author Contributions
SaK conceptualized the research idea. TS investigated the data and wrote the original draft. TS, SeK, and SaK were involved in formal analysis. TS and SaK validated the results and reviewed and edited the paper. SeK and SaK supervised the work. SeK was involved in funding acquisition and project administration.
Funding
DST-SERB funded the N-PDF (National Post-Doctoral Fellowship) (File Number: PDF/2016/002785).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
TS acknowledges DST-SERB for funding the N-PDF (National Post-Doctoral Fellowship) and Pragnya for supporting with activation barrier calculations. SK and SK both acknowledge the High-Performance Computing facility provided by CSIR-NCL, Pune, and CSIR-4PI, Bangalore. The authors dedicate this article to Sourav Pal for his landmark contributions in the area of computational chemistry in catalysis. He is an excellent teacher and a wonderful guide who has inspired many generations of students.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.733422/full#supplementary-material
References
Bader, R. F. W. (1991). A Quantum Theory of Molecular Structure and its Applications. Chem. Rev. 91, 893–928. doi:10.1021/cr00005a013
Blöchl, P. E. (1994). Projector Augmented-Wave Method. Phys. Rev. B 50, 17953–17979. doi:10.1103/physrevb.50.17953
Cao, B., Starace, A. K., Judd, O. H., Bhattacharyya, I., Jarrold, M. F., López, J. M., et al. (2010). Activation of Dinitrogen by Solid and Liquid Aluminum Nanoclusters: a Combined Experimental and Theoretical Study. J. Am. Chem. Soc. 132, 12906–12918. doi:10.1021/ja103356r
Cao, B., Starace, A. K., Judd, O. H., Bhattacharyya, I., and Jarrold, M. F. (2009). Metal Clusters with Hidden Ground States: Melting and Structural Transitions in Al115+, Al116+, and Al117+. J. Chem. Phys. 131, 124305. doi:10.1063/1.3224124
Chen, C., Kang, Y., Huo, Z., Zhu, Z., Huang, W., Xin, H. L., et al. (2014). Highly Crystalline Multimetallic Nanoframes with Three-Dimensional Electrocatalytic Surfaces. Science 343, 1339–1343. doi:10.1126/science.1249061
Cherkasov, N., Ibhadon, A. O., and Fitzpatrick, P. (2015). A Review of the Existing and Alternative Methods for Greener Nitrogen Fixation. Chem. Eng. Process. Process Intensification 90, 24–33. doi:10.1016/j.cep.2015.02.004
Choi, W. I., Wood, B. C., Schwegler, E., and Ogitsu, T. (2015). Combinatorial Search for High-Activity Hydrogen Catalysts Based on Transition-Metal-Embedded Graphitic Carbons. Adv. Energ. Mater. 5, 1501423. doi:10.1002/aenm.201501423
Dance, I. (2008). The Chemical Mechanism of Nitrogenase: Calculated Details of the Intramolecular Mechanism for Hydrogenation of η2-N2 on FeMo-Co to NH3. Dalton Trans. 43, 5977–5991. doi:10.1039/b806100a
Das, S., Pal, S., and Krishnamurty, S. (2014). Dinitrogen Activation by Silicon and Phosphorus Doped Aluminum Clusters. J. Phys. Chem. C 118, 19869–19878. doi:10.1021/jp505700a
Einsle, O., Tezcan, F. A., Andrade, S. L., Schmid, B., Yoshida, M., Howard, J. B., et al. (2002). Nitrogenase MoFe-Protein at 1.16 A Resolution: A Central Ligand in the FeMo-Cofactor. Science 297, 1696–1700. doi:10.1126/science.1073877
Fajardo, J., and Peters, J. C. (2017). Catalytic Nitrogen-To-Ammonia Conversion by Osmium and Ruthenium Complexes. J. Am. Chem. Soc. 139, 16105–16108. doi:10.1021/jacs.7b10204
Fei, H., Dong, J., Feng, Y., Allen, C. S., Wan, C., Volosskiy, B., et al. (2018). General Synthesis and Definitive Structural Identification of MN4C4 Single-Atom Catalysts with Tunable Electrocatalytic Activities. Nat. Catal. 1, 63–72. doi:10.1038/s41929-017-0008-y
Fryzuk, M. D., and Johnson, S. A. (2000). The Continuing story of Dinitrogen Activation. Coord. Chem. Rev. 200-202, 379–409. doi:10.1016/s0010-8545(00)00264-2
Guo, X., Liu, S., and Huang, S. (2018). Single Ru Atom Supported on Defective Graphene for Water Splitting: DFT and Microkinetic Investigation. Int. J. Hydrogen Energ. 43, 4880–4892. doi:10.1016/j.ijhydene.2018.01.122
Jeon, I. Y., Choi, H. J., Ju, M. J., Choi, I. T., Lim, K., Ko, J., et al. (2013). Direct Nitrogen Fixation at the Edges of Graphene Nanoplatelets as Efficient Electrocatalysts for Energy Conversion. Sci. Rep. 3, 2260–2267. doi:10.1038/srep02260
Kerpal, C., Harding, D. J., Lyon, J. T., Meijer, G., and Fielicke, A. (2013). N2 Activation by Neutral Ruthenium Clusters. J. Phys. Chem. C 117, 12153–12158. doi:10.1021/jp401876b
Kim, J., and Rees, D. (1992). Structural Models for the Metal Centers in the Nitrogenase Molybdenum-Iron Protein. Science 257, 1677–1682. doi:10.1126/science.1529354
Kresse, G., and Furthmüller, J. (1996). Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 6, 15–50. doi:10.1016/0927-0256(96)00008-0
Kulkarni, B. S., Krishnamurty, S., and Pal, S. (2011). Size- and Shape-Sensitive Reactivity Behavior of Aln (N = 2-5, 13, 30, and 100) Clusters toward the N2 Molecule: A First-Principles Investigation. J. Phys. Chem. C 115, 14615–14623. doi:10.1021/jp203452a
Kumar, C. V. S., and Subramanian, V. (2017). Can boron Antisites of BNNTs Be an Efficient Metal-free Catalyst for Nitrogen Fixation? - A DFT Investigation. Phys. Chem. Chem. Phys. 19, 15377–15387. doi:10.1039/c7cp02220d
Kumar, D., Pal, S., and Krishnamurty, S. (2016). N2 Activation on Al Metal Clusters: Catalyzing Role of BN-Doped Graphene Support. Phys. Chem. Chem. Phys. 18, 27721–27727. doi:10.1039/c6cp03342c
Le, Y.-Q., Gu, J., and Tian, W. Q. (2014). Nitrogen-fixation Catalyst Based on Graphene: Every Part Counts. Chem. Commun. 50, 13319–13322. doi:10.1039/c4cc01950d
Légaré, M.-A., Bélanger-Chabot, G., Dewhurst, R. D., Welz, E., Krummenacher, I., Engels, B., et al. (2018). Nitrogen Fixation and Reduction at boron. Science 359, 896–900. doi:10.1126/science.aaq1684
Li, H., Shang, J., Ai, Z., and Zhang, L. (2015). Efficient Visible Light Nitrogen Fixation with BiOBr Nanosheets of Oxygen Vacancies on the Exposed {001} Facets. J. Am. Chem. Soc. 137, 6393–6399. doi:10.1021/jacs.5b03105
Li, X.-F., Li, Q.-K., Cheng, J., Liu, L., Yan, Q., Wu, Y., et al. (2016). Conversion of Dinitrogen to Ammonia by FeN3-Embedded Graphene. J. Am. Chem. Soc. 138, 8706–8709. doi:10.1021/jacs.6b04778
Liu, B., Manavi, N., Deng, H., Huang, C., Shan, N., Chikan, V., et al. (2021). Activation of N2 on Manganese Nitride-Supported Ni3 and Fe3 Clusters and Relevance to Ammonia Formation. J. Phys. Chem. Lett. 12, 6535–6542. doi:10.1021/acs.jpclett.1c01752
Liu, J.-C., Tang, Y., Wang, Y.-G., Zhang, T., and Li, J. (2018). Theoretical Understanding of the Stability of Single-Atom Catalysts. Natl. Sci. Rev. 5, 638–641. doi:10.1093/nsr/nwy094
Lu, Y., Yang, Y., Zhang, T., Ge, Z., Chang, H., Xiao, P., et al. (2016). Photoprompted Hot Electrons from Bulk Cross-Linked Graphene Materials and Their Efficient Catalysis for Atmospheric Ammonia Synthesis. ACS nano 10, 10507–10515. doi:10.1021/acsnano.6b06472
Maibam, A., Govindaraja, T., Selvaraj, K., and Krishnamurty, S. (2019). Dinitrogen Activation on Graphene Anchored Single Atom Catalysts: Local Site Activity or Surface Phenomena. J. Phys. Chem. C 123, 27492–27500. doi:10.1021/acs.jpcc.9b06757
Maibam, A., and Krishnamurty, S. (2021). Nitrogen Activation to Reduction on a Recyclable V-SAC/BN-graphene Heterocatalyst Sifted through Dual and Multiphilic Descriptors. J. Colloid Interf. Sci. 600, 480–491. doi:10.1016/j.jcis.2021.05.027
Monkhorst, H. J., and Pack, J. D. (1976). Special Points for Brillouin-Zone Integrations. Phys. Rev. B 13, 5188–5192. doi:10.1103/physrevb.13.5188
Ouyang, Y., Li, Q., Shi, L., Ling, C., and Wang, J. (2018). Molybdenum Sulfide Clusters Immobilized on Defective Graphene: a Stable Catalyst for the Hydrogen Evolution Reaction. J. Mater. Chem. A. 6, 2289–2294. doi:10.1039/c7ta09828f
Payne, M. C., Teter, M. P., Allan, D. C., Arias, T. A., and Joannopoulos, J. D. (1992). Iterative Minimization Techniques Forab Initiototal-Energy Calculations: Molecular Dynamics and Conjugate Gradients. Rev. Mod. Phys. 64, 1045–1097. doi:10.1103/revmodphys.64.1045
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Qiao, B., Wang, A., Yang, X., Allard, L. F., Jiang, Z., Cui, Y., et al. (2011). Single-atom Catalysis of CO Oxidation Using Pt1/FeOx. Nat. Chem 3, 634–641. doi:10.1038/nchem.1095
Roy, D., Navarro-Vazquez, A., and Schleyer, P. v. R. (2009). Modeling Dinitrogen Activation by Lithium: A Mechanistic Investigation of the Cleavage of N2 by Stepwise Insertion into Small Lithium Clusters. J. Am. Chem. Soc. 131, 13045–13053. doi:10.1021/ja902980j
Seh, Z. W., Kibsgaard, J., Dickens, C. F., Chorkendorff, I., Nørskov, J. K., and Jaramillo, T. F. (2017). Combining Theory and experiment in Electrocatalysis: Insights into Materials Design. Science 355, eaad4998. doi:10.1126/science.aad4998
Sellmann, D., and Sutter, J. (1997). In Quest of Competitive Catalysts for Nitrogenases and Other Metal Sulfur Enzymes. Acc. Chem. Res. 30, 460–469. doi:10.1021/ar960158h
Shi, H., and Jacobi, K. (1992). Evidence for Physisorbed N2 in the Monolayer on Ru(001) at 40 K. Surf. Sci. 278, 281–285. doi:10.1016/0039-6028(92)90664-r
Song, R., Yang, J., Wang, M., Shi, Z., Zhu, X., Zhang, X., et al. (2021). Theoretical Study on P-Coordinated Metal Atoms Embedded in Arsenene for the Conversion of Nitrogen to Ammonia. ACS omega 6, 8662–8671. doi:10.1021/acsomega.1c00581
Stüeken, E. E., Buick, R., Guy, B. M., and Koehler, M. C. (2015). Isotopic Evidence for Biological Nitrogen Fixation by Molybdenum-Nitrogenase from 3.2 Gyr. Nature 520, 666–669. doi:10.1038/nature14180
Tanabe, Y., and Nishibayashi, Y. (2016). Catalytic Dinitrogen Fixation to Form Ammonia at Ambient Reaction Conditions Using Transition Metal-Dinitrogen Complexes. Chem. Rec. 16, 1549–1577. doi:10.1002/tcr.201600025
Tang, W., Sanville, E., and Henkelman, G. (2009). A Grid-Based Bader Analysis Algorithm without Lattice Bias. J. Phys. Condens. Matter 21, 084204. doi:10.1088/0953-8984/21/8/084204
Wang, A., Li, J., and Zhang, T. (2018). Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2, 65–81. doi:10.1038/s41570-018-0010-1
Yan, H., Zhao, X., Guo, N., Lyu, Z., Du, Y., Xi, S., et al. (2018). Atomic Engineering of High-Density Isolated Co Atoms on Graphene with Proximal-Atom Controlled Reaction Selectivity. Nat. Commun. 9, 3197–3199. doi:10.1038/s41467-018-05754-9
Yan, X., Liu, D., Cao, H., Hou, F., Liang, J., and Dou, S. X. (2019). Nitrogen Reduction to Ammonia on Atomic‐Scale Active Sites under Mild Conditions. Small Methods 3, 1800501. doi:10.1002/smtd.201800501
Yang, X.-F., Wang, A., Qiao, B., Li, J., Liu, J., and Zhang, T. (2013). Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 46, 1740–1748. doi:10.1021/ar300361m
Keywords: N2 activation, single metal atom, pristine graphene, defective graphene, BN-functionalized graphene
Citation: Senthamaraikannan TG, Kaliaperumal S and Krishnamurty S (2021) Role of Chemical Structure of Support in Enhancing the Catalytic Activity of a Single Atom Catalyst Toward NRR: A Computational Study. Front. Chem. 9:733422. doi: 10.3389/fchem.2021.733422
Received: 30 June 2021; Accepted: 12 August 2021;
Published: 08 September 2021.
Edited by:
Lalith Perera, National Institute of Environmental Health Sciences (NIEHS), United StatesReviewed by:
Michael Springborg, Saarland University, GermanyDebdutta Chakraborty, KU Leuven, Belgium
Copyright © 2021 Senthamaraikannan, Kaliaperumal and Krishnamurty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sailaja Krishnamurty, ay5zYWlsYWphQG5jbC5yZXMuaW4=; Thillai Govindaraja Senthamaraikannan, dGhpbGxhaW5jbEBnbWFpbC5jb20=