 Norma Flores-Holguín
Norma Flores-Holguín Juan Frau
Juan Frau Daniel Glossman-Mitnik
Daniel Glossman-Mitnik- 1Laboratorio Virtual NANOCOSMOS, Departamento de Medio Ambiente y Energía, Centro de Investigación en Materiales Avanzados, Chihuahua, Mexico
- 2Departament de Química, Universitat de les Illes Balears, Palma de Mallorca, Spain
This research presents the outcomes of a computational determination of the chemical reactivity and bioactivity properties of two plant cyclopeptides isolated from Rosaceae through the consideration of Computational Peptidology (CP), a protocol employed previously in the research of similar molecular systems. CP allows the prediction of the global and local descriptors that are the integral foundations of Conceptual Density Functional Theory (CDFT) and which could help in getting in the understanding of the chemical reactivity properties of the two plant cyclopeptides under study, hoping that they could be related to their bioactivity. The methodology based on the Koopmans in DFT (KID) approach and the MN12SX/Def2TZVP/H2O model chemistry has been successfully validated. Various Chemoinformatics tools have been used to improve the process of virtual screening, thus identifying some additional properties of these two plant cyclopeptides connected to their ability to behave as potentially useful drugs. With the further objective of analyzing their bioactivity, the CP protocol is complemented with the estimation of some useful parameters related to pharmacokinetics, their predicted biological targets, and the Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) parameters related to the bioavailability of the two plant cyclopeptides under study are also reported.
1 Introduction
Plant-based bioactive compounds have drawn attention of all communities around the world due to their unique biochemical activities and health benefits. Research studies have confirmed the safeguarding effects of certain plant-based diets on cardiovascular diseases, obesity, cancer, diabetes, etc. (Guha et al., 2021). Plant-based drugs from secondary metabolites constitute more than 25% of approved new drugs during last 30 years. Also, 50% of the commercially successful medicinal components were developed based on knowledge from plant secondary metabolites and their structures (Chaudhari and Chakraborti, 2021).
Bioactive peptides are organic substances formed by amino acids joined by covalent bonds known as amide or peptide bonds. Although peptides can exist free in terrestrial plants and marine sources, the vast majority of known bioactive peptides are enclosed within the structure of the proteins and can be released using enzymatic processes. Bioactive peptides play a significant role in human health by affecting the digestive, endocrine, cardiovascular, immune, and nervous systems. The increasing interest in bioactive peptides has incentivized the scientific community in the exploration and development of new therapeutic drugs based on these peptides (Sánchez and Vázquez, 2017).
Cyclic peptides can be considered as an alternative scaffold. The smaller size and several functional groups of peptides help to make the contact area large enough to provide good selectivity. Their ability to form several hydrogen bonds make easier to obtain strong binding affinity. Moreover, the cyclization of peptides helps in the generation of structural and functional features that are considered to be critical for their use as pharmaceutical drugs, including resistance to degradation by blood proteases. Also, the cyclization of the peptides facilitates the passage through the cell membrane. Because of such favorable features, many cyclic peptides from terrestrial plant and marine sources and their derivatives have been considered for drug design and development (Gang et al., 2018). Besides these biological features, cyclopeptides have smaller sizes than proteins and reduced flexibilities, exhibiting lower conformations than their linear counterparts, thus making easier and affordable the DFT calculations of their structures and properties.
By considering that the knowledge of the chemical reactivity properties of a given molecule is essential for the development of new therapeutic drugs, we are currently researching on new families of cyclopeptides obtained from terrestrial plants and marine sources hoping that the obtained information could be of help for the design of pharmaceutical based on these peptides (Kim, 2013). The objective of the present work is to report the global and local chemical reactivity descriptors of two plant cyclopeptides, Pashinintides A and B, that have been isolated from Rosacea (Cai et al., 2014) by making use of the Conceptual DFT (CDFT) methodology (Parr and Yang, 1989; Chermette, 1999; Geerlings et al., 2003; Toro-Labbé, 2007; Chattaraj, 2009; Geerlings et al., 2020; Chakraborty and Chattaraj, 2021). A recent review has highlighted the basic electronic structure principles and various reactivity descriptors defined within the premise of CDFT (Chakraborty and Chattaraj, 2021). The study is complemented by considering the report of some additional properties of potential application in Structure Activity Relationships (SAR) research for the development of therapeutic drugs, and also with the bioactivity radars related to the drug-like behavior of the studied peptides, their predicted biochemical targets and the values associated with Pharmacokinetics and ADMET properties (Daina et al., 2017; Pires et al., 2015; Daina et al., 2019) through standard Chemoinformatics procedures (Begam and Kumar, 2012; González-Medina et al., 2017). By considering this integrative strategy, called Conceptual DFT-based Computational Peptidology as a branch of Computational Chemistry dedicated to the study of peptides and cyclopeptides, the current research represents an extension of our recent studies on the properties of some families of therapeutic peptides of marine origin (Frau et al., 2018; Flores-Holguín et al., 2019a; Frau et al., 2019; Flores-Holguín et al., 2019c; Flores-Holguín et al., 2020; Flores-Holguín et al. 2020a; Flores-Holguín et al., 2020b; Flores-Holguín et al., 2021).
2 Materials and Methods
2.1 In Silico Pharmacokinetics Analysis and Absorption, Distribution, Metabolism, Excretion and Toxicity Study
The starting molecular structures of the two plant cyclopeptides to be studied, shown in Figure 1, lwere obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov), which is an open chemistry database.
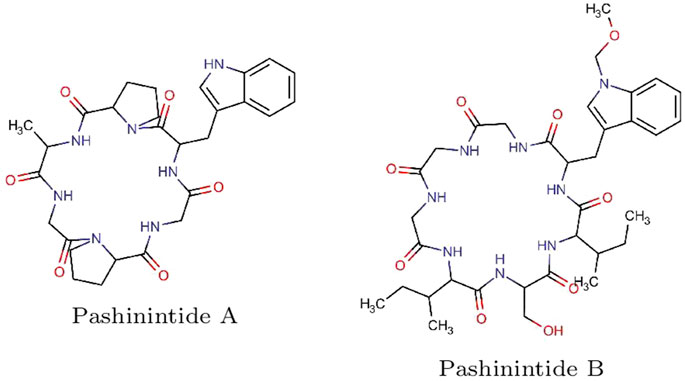
FIGURE 1. Graphical sketches of the molecular structures of the Pashinintides A and B plant cyclopeptides.
As a first step, the SMILES (Simplified Molecular Input Line Entry Specification) notation of every studied compound, which was obtained by accessing ChemDoodle 11.3.0 software, was fed into the online program Chemicalize, a software developed by ChemAxon (http://www.chemaxon.com), which was used for naming, molecular finger prints, structure generation and the prediction of several properties related to Chemoinformatics (http://chemicalize.com/) (accessed March 2021).
The similarity searches in the chemical space of compounds with molecular structures similar to those that are being studied was accomplished using the online available Molinspiration software from Molinspiration Cheminoformatics (https://www.molinspiration.com/) (accessed, March 2021) which was used for the prediction of the bioactivity scores for different drug targets.
A Webtool named SwissTargetPrediction for efficient prediction of protein targets of small molecules has been considered for the determination of the potential bioactivity of the two terrestrial plant cyclopeptides considered in this study (Daina et al., 2019). The associated website allows the estimation of the most probable macromolecular targets of a small molecule, assumed as bioactive. During the process of development of a new therapeutic drug, it is of the most importance to adcquire a knowledge of the fate of the pharmacokinetics, that is, the fate of a compound in the organism. This is usually performed by through individual indices that are called Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) parameters. These parameters are generally obtained using computer models as an alternative to the experimental procedures for their determination. In this research, some ADME parameters were estimated with the aid of Chemicalize and the online available SwissADME software (Daina et al., 2017).Additional information about the Pharmacokinetics parameters and the ADMET properties were obtained by resorting to pkCSM (Pires et al., 2015), a software for the prediction of small-molecule pharmacokinetic properties using SMILES (https://biosig.unimelb.edu.au/pkcsm/) (accessed, March 2021).
2.2 Density Functional Theory Calculations
The goodness of a given density functional can be determined through a comparison of the results that it renders with the experimental values or with the results that can be obtained by means of high-level calculations. However, the lack of experimental results for the molecular systems under study or the large size of the molecules that made computationally impractical the use of some accurate methodologies. Kohn-Sham (KS) methodology includes the determination of the molecular energy, the electronic density and the orbital energies of a given system, related to the frontier orbitals including the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) (Young, 2001; Lewars, 2003; Cramer 2004; Jensen, 2007). This methodology is convenient when thinking of quantitative qualities related with Conceptual DFT descriptors (Parr and Yang, 1989; Chermette, 1999; Geerlings et al., 2003; Toro-Labbé, 2007; Chattaraj, 2009, Geerlings et al., 2020; Chakraborty and Chattaraj, 2021). Range-separated (RS) exchange-correlation density functionals are of extraordinary concern in Kohn-Sham DFT calculations (Iikura et al., 2001; Yanai et al., 2004; Heyd and Scuseria, 2004; Chai and Head-Gordon, 2008; Stein et al., 2009a; Stein et al., 2009b; Stein et al., 2010; Karolewski et al., 2011; Kuritz et al., 2011; Ansbacher et al., 2012; Kronik et al., 2012; Stein et al., 2012). A methodology called KID (Koopmans in DFT) has been established by our research group (Frau et al., 2018; Flores-Holguín et al., 2019a; Frau et al., 2019; Flores-Holguín et al., 2019c; Flores-Holguín et al., 2020; Flores-Holguín et al., 2020a; Flores-Holguín et al., 2020b; Flores-Holguín et al., 2021), t for the validation of a given density functional in terms of its internal coherence. Several descriptors associated with the results of the HOMO and LUMO calculations are related to the results obtained from the estimation of the vertical I and A following the ΔSCF procedure, where SCF refers to the Self-Consistent Field technique. It has been demonstrated that there is a relationship between the KID descriptors and the Koopmans’ theorem or the Ionization Energy theorem, which is its equivalent within the Generalized Kohn-Sham (GKS) version of DFT, by connecting ϵH to -I, ϵL to -A, and their actions by defining the HOMO – LUMO gap JI = |ϵH + Egs(N − 1) − Egs(N)|, JA = |ϵL + Egs(N) − Egs(N + 1)|, and
Taking into account the KID methodology considered in the previous research being integrated into the finite difference approximation (Frau et al., 2018; Flores-Holguín et al., 2019a; Frau et al., 2019; Flores-Holguín et al., 2019c; Flores-Holguín et al., 2020; Flores-Holguín et al., 2020a; Flores-Holguín et al., 2020b; Flores-Holguín et al., 2021), the following definitions can be used for the global descriptors that help in the understanding of the chemical reactivity of the molecular systems (Parr and Yang, 1989; Chermette, 1999; Geerlings et al., 2003; Gázquez et al., 2007; Chattaraj et al., 2009; Chakraborty and Chattaraj, 2021): Electronegativity as
As a complement of these global reactivity descriptors that arise from Conceptual DFT (Parr and Yang, 1989; Chermette, 1999; Geerlings et al., 2003; Gázquez et al., 2007; Chattaraj et al., 2009; Chakraborty and Chattaraj, 2021), Domingo and his collaborators (Domingo et al., 2008; Jaramillo et al., 2008; Domingo and Sáez, 2009; Domingo and Perez, 2011; Domingo et al., 2016) have proposed a Nucleophilicity index N through the consideration of the HOMO energy obtained through the KS scheme with an arbitrary shift of the origin taking the molecule of tetracyanoethylene (TCE) as a reference.
The determination of the conformers of the molecules considered in the current study was performed by resorting to MarvinView 17.15 available from ChemAxon (http://www.chemaxon.com) by doing Molecular Mechanics calculations through the overall MMFF94 force field (Halgren, 1996a, Halgren, 1996b, Halgren, 1999; Halgren and Nachbar, 1996; Halgren, 1996c). This was followed by a geometry optimization and frequency calculation by means of the Density Functional Tight Binding (DFTBA) methodology (Frisch et al., 2016). This last step was required for the verification of the absence of imaginary frequencies as a check for the stability of the optimized structures as being a minimum in the energy landscape. The electronic properties and the chemical reactivity descriptors of the studied molecules involved the use of MN12SX/Def2TZVP/H2O model chemistry (Weigend and Ahlrichs, 2005; Weigend, 2006; Peverati and Truhlar, 2012) on the optimized molecular structures due to is ability in the verification of the “Koopmans in DFT” (KID) protocol (Frau and Glossman-Mitnik, 2018a;; Frau and Glossman-Mitnik, 2018b; Frau and Glossman-Mitnik, 2018c; Frau and Glossman-Mitnik, 2018d; Frau and Glossman-Mitnik, 2018e; Frau and Glossman-Mitnik, 2018f; Flores-Holguín et al., 2019a; Flores-Holguín et al., 2019b; Flores-Holguín et al., 2019d; Frau et al., 2019; Flores-Holguín et al., 2019c, Flores-Holguín et al., 2020; Flores-Holguín et al., 2020a; Flores-Holguín et al., 2020b; Flores-Holguín et al., 2021) using Gaussian 16 (Frisch et al., 2016) and the SMD model for the simulation of the solvent (Marenich et al., 2009). This model chemistry considers the MN12SX screened-exchange density functional (Peverati and Truhlar, 2012) together with the Def2TZVP basis set (Weigend and Ahlrichs, 2005; Weigend, 2006) and in all cases the charge of the molecules is equal to zero while the radical anion and cation have been considered in the doublet spin state.
3 Results and Discussion
3.1 Physicochemical Properties, Bioactivity Scores and Biological Targets
The names, identifiers, molecular fingerprints and basic properties of the two Pashinintides A and B plant cyclopeptides are presented in Table 1, while their geometrical and structural properties are displayed in Table 2.

TABLE 1. Names, identifiers, molecular fingerprints and basic properties of the studied molecular systems.
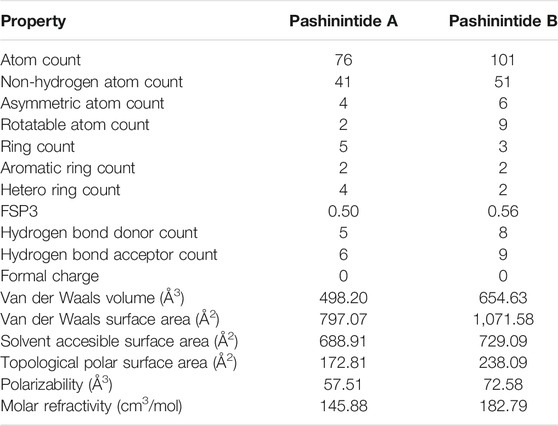
TABLE 2. Geometrical and structural properties of the studied molecular systems.
This information could it be of interest for future SAR studies based on these and other peptides as well as for potential derivatives designed for therapeutical purposes using Peptidomimetics.
The effect of the geometrical and structural properties on the bioavailability of the Pashinintides A and B presented in Table 2 can be better visualized considering the Bioavailability Radars displayed in Figure 2.
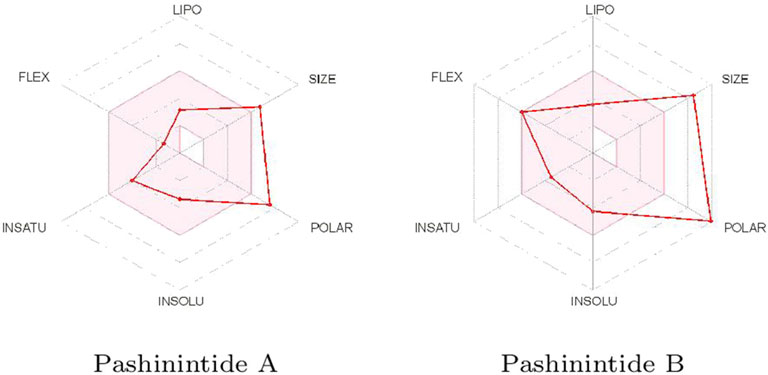
FIGURE 2. Bioavailability radars of the Pashinintides A and B.
It can be appreciated that the two more important properties that could prevent the use of the Pashinintides A and B as therapeutic drugs are their molecular size and their polar character. Although these cyclic peptides violate some of the limits traditionally considered to be important for oral bioavailability of drug-like small molecules, it can be expected that the reduced flexibility could ease oral absorption (Nielsen et al., 2017). However, it must be remarked that these ideal values are based on the Lipinski’s Rule of Five (Lipinski et al., 2001), which is not always applicable to peptides (Zhang and Wilkinson, 2007; Doak et al., 2014; Santos et al., 2016; Nielsen et al., 2017; Sable et al., 2017).
The Bioactivity Scores for the Pashinintides A and B are shown in Table 3.
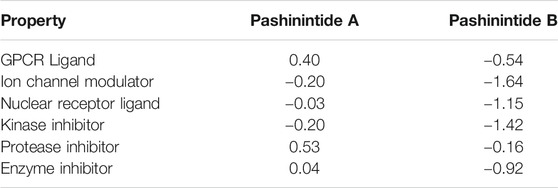
TABLE 3. Bioactivity scores for the Pashinintides A and B.
It can be seen from the results on Table 3 that while the bioactivity of Pashinintide B towards the different targets is considered to be low, for Pashinintide A, its interactions as a GPCR Ligand and a Protease Inhibitors could be of importance for its consideration as a potential therapeutic drug. The same conclusion can be extracted by checking visually the predicted biological targets for these plant cyclopeptides shown in Figure 3.
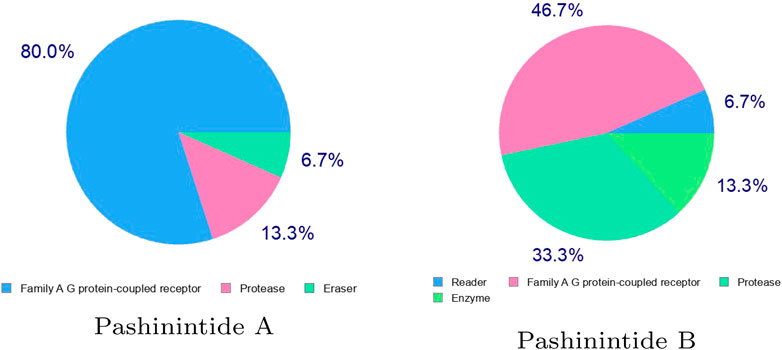
FIGURE 3. Predicted biological targets of the Pashinintides A and B.
3.2 Absorption, Distribution, Metabolism, Excretion and Toxicity Study
An ADMET study is the assessment of pharmacokinetics of a drug which stands for Absorption, Distribution, Metabolism, Excretion and Toxicity. The prediction of the fate of a drug and the effects caused by a drug inside the body, such as how much drug is absorbed if administered orally and how much is absorbed in the gastrointestinal tract, is an indispensable part of drug discovery. In a similar way, if the absorption is poor, its distribution and metabolism would be affected, which can lead to causing neurotoxicity and nephrotoxicity. Ultimately, the study is to understand the disposition of a drug molecule within an organism. Thus, ADMET study is one of the most essential parts of computational drug design.
3.2.1 Absorption
A compound can reach a tissue, if it is taken into the bloodstream. Usually, a drug is administered often through mucous surfaces such as the digestive tract, i.e., intestinal absorption before it is taken up by the target cells. Factors like poor compound solubility, intestinal transit time, gastric emptying time, inability to permeate the intestinal wall and chemical instability in the stomach are responsible for reducing the extent of drug absorption after oral administration. Critically, absorption determines the bioavailability of a compound. Drugs with poor absorption are less desirable for oral administration, such as by inhalation or intravenously (Pires et al., 2015; Jujjavarapu et al., 2019).
The computed absorption properties of the Pashinintides A and B are presented in Table 4.
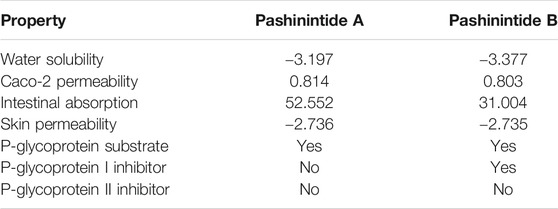
TABLE 4. Absorption properties of the Pashinintides A and B.
The water solubility of a compound (logS) reflects the solubility of the molecule in water at 25°C. The predicted water solubility of a compound is given as the logarithm of the molar concentration (log mol/L) being their values very similar for both cyclopeptides. A compound is considered lo have a high Caco-2 permeability has a Papp >8 × 108 cm/s. Thus, high Caco-2 permeability would translate in predicted values >0.90, presenting the Pashinintides A and B values which are a bit lower than the ideal one. The Intestine is normally the primary site for absorption of a drug from an orally administered solution. A molecule with an Intestinal Absorption of less than 30% is considered lo be poorly absorbed. From Table 4, both plant cyclopeptides will be highly absorbed. The P-glycoprotein has the function of a biological barrier by extruding toxins out of cells. The model predicts whether a given compound is likely to be a substrate of P-glycoprotein or not. The prediction is in the positive direction in both cases. Thus, the study predicts that both cyclopeptides will not act as P-glycoprotein II inhibitors, but Pashinintide A will not be a P-glycoprotein I inhibitor while Pashinintide B is likely to act in that way. Also, it can be predicted whether a given compound is likely to be skin permeable. A compound is considered to have a relatively low skin permeability if it has a log Kp > −2.5. It means that both cyclopeptides could be of interest for the development of transdermal drug delivery (Pires et al. (2015)).
3.2.2 Distribution
The computed distribution properties of the Pashinintides A and B are presented in Table 5.
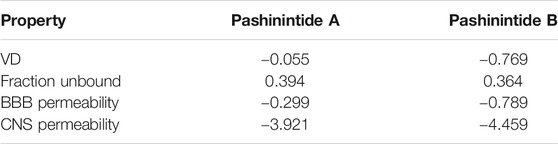
TABLE 5. Distribution properties of the Pashinintides A and B.
VD is the theoretical volume required by a drug to be uniformly distributed in blood. The higher the VD is, the more of a drug is distributed in tissue rather than plasma. From Table 5 and the usual standards, it can be said that VD for Pashinintide A is low and it is high for Pashinintide B. The Fraction Unbound parameter predicts the fraction that will be unbound in plasma resulting in the values shown in Table 5. The knowledge of the ability of a drug to cross into the brain is an important parameter that may help to reduce side effects and toxicities. A logBBB (for Blood-Brain Barrier) >−0.3 for a given drug is considered to easily cross the BBB while molecules with logBBB >−1 are poorly distributed to the brain, being predicted that both cyclopeptides have a BBB Permeability of the first case. Another measurement is the blood-brain permeability-surface area product or CNS Permeability where compounds with a logPS >−2 will be able to enter the Central Nervous System (CNS), while those with logPS <−3 will be unable to penetrate the CNS. For the current study, both cyclopeptides are predicted to do not penetrate the CNS (Pires et al., 2015).
3.2.3 Metabolism
The computed metabolism properties of the Pashinintides A and B are presented in Table 6.

TABLE 6. Metabolism properties of the Pashinintides A and B.
Cytochrome P450 is an important detoxification enzyme in the body. Many drugs are deactivated by the cytochrome P450 isoforms while some can be activated by it. As can be seen from Table 6, both cyclopeptides are predicted as not being P450 inhibitors for any isoform. It is al important to know if a given compound is likely to be a cytochrome P450 substrate. The predictions indicate that this will be not the case for any of the cyclopeptides (Pires et al., 2015).
3.2.4 Excretion
The computed excretion properties of the Pashinintides A and B are presented in Table 7.
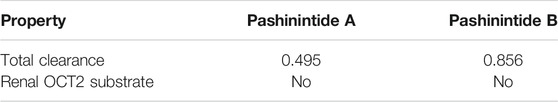
TABLE 7. Excretion properties of the Pashinintides A and B.
Drug clearance occurs as a combination of hepatic clearance and renal clearance (excretion via the kidneys) which is related to bioavailability. The predicted Total Clearance of the Pashinintides A and B are given in log(ml/min/kg) being the value for the former about 55% of the later. OCT2 is a renal uptake transporter that plays an important role in disposition and renal clearance of drugs. In this case, it is predicted that neither of the cyclopeptides will behave as OCT2 substrates (Pires et al., 2015).
3.2.5 Toxicity
The computed excretion properties of the Pashinintides A and B are presented in Table 8.
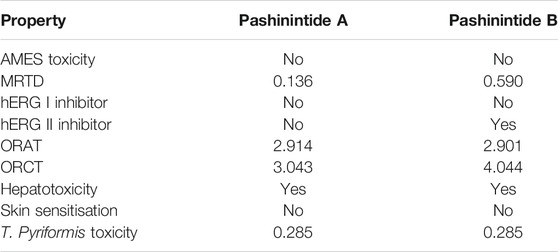
TABLE 8. Toxicity properties of the Pashinintides A and B.
AMES Toxicity s a widely employed methodology considered to check the mutagenic potential of a given drug using bacteria, thus indicating that when the results is positive, the studied compound will be mutagenic and could behave as a carcinogen. From Table 8, the predictions are negative for both cyclopeptides under study. The maximum recommended tolerated dose (MRTD) provides an estimate of the toxic dose threshold of chemicals in humans. A low value for Pashinintide A and high value for Pashinintide B are found from the results in Table 8. Also, the predictions indicate that both cyclopeptides are unlikely to be hERG I inhibitors, but for the case of hERG II, the behavior will be different: Pashinintide A will not be a hERGII inhibitor while Pashinintide B will. The lethal dosage values (LD50) are a standard measurement of acute toxicity and is defined as the amount of a compound that causes the death of 50% of a group of test animals and are measured through the ORAT and ORCT indices where the predicted values are given in mol/kg.Drug-induced liver injury is a major safety concern for drug development. Hepatoxicity is associated with disrupted normal function of the liver and the predicted values for both cyclopeptides are positive. On the other hand, the predicted values for Skin Sensitisation are negative. T. Pyriformis is a protozoa bacteria whose toxicity is often used as a toxicity test. The predicted values for this parameter are the same for both cyclopeptides (Pires et al., 2015).
3.3 Conceptual Density Functional Theory Studies
The optimized molecular structures of the Pashinintides A and B are displayed in Figure 4.
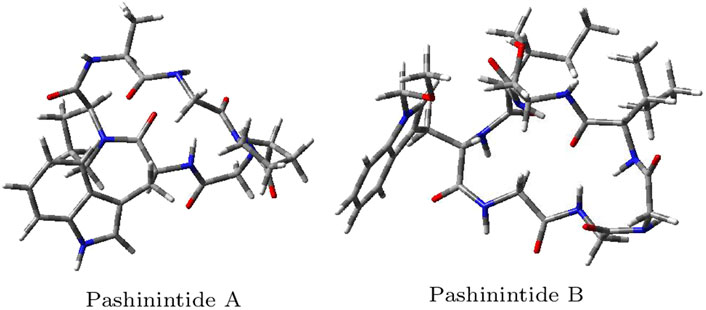
FIGURE 4. Optimized molecular structures of the Pashinintides A and B.
Although the Koopmans-complaint behavior of the MN12SX density functional has been proved previously for the case of marine peptides (Frau et al., 2018; Flores-Holguín et al., 2019a; Frau et al., 2019; Flores-Holguín et al., 2019c; Flores-Holguín et al., 2020; Flores-Holguín et al., 2020a; Flores-Holguín et al., 2020b; Flores-Holguín et al., 2021), we are now performing a further validation for the plant cyclopeptides considered in the present study. This determination has been done by resorting to the in-house developed CDFT software tool and the resulting values are shown in Table 9.

TABLE 9. HOMO, LUMO and SOMO orbital energies, HOMO-LUMO gap and the KID descriptors (all in eV) tested in the verification of the Koopmans-like behavior of the MN12SX/Def2TZVP/H2O model chemistry for the Pashinintides A and B.
As can be seen from the values presented in Table 9, the KID descriptors are all very close to zero meaning that the chosen MN12SX density functional displays a Koopmans-complaint behavior. This in agreement with our previous studies on peptides (Frau et al., 2018; Flores-Holguín et al., 2019a; Frau et al., 2019; Flores-Holguín et al., 2019c; Flores-Holguín et al., 2020; Flores-Holguín et al., 2020a; Flores-Holguín et al., 2020b; Flores-Holguín et al., 2021), thus justifying the adequacy of the MN12SX/Def2TZVP/H2O model chemistry for the purpose of this research.
The results for the global reactivity indices were estimated by making use of the mentioned CDFT tool and are presented in Table 10.

TABLE 10. Global reactivity descriptors (in eV) for the Pashinintides A and B.
As the global hardness η can be regarded as a direct measure of the deformation of the electron density and of the chemical reactivity being related to the HOMO-LUMO gap, it can be seen that Pashininitide A will be slightly more reactive than the other cyclopeptide. The electrodonating ability ω− is more important that its electroaccepting power ω+ for both cyclopeptides because of their molecular structures. However, after a comparison of the values of ω− and ω+ for each molecule, it can be concluded that there are not important differences between them. The electrophilicity ω index encompasses the balance between the tendency to acquire an extra amount of electron density by an electrophile and the resistance of a molecule to exchange electron density with the environment Domingo et al. (2016). By studying the electrophilicities of a series of reagents involved in Diels-Alder reactions (Domingo et al., 2002a; Domingo and Sáez, 2009; Pérez et al., 2003), an electrophilicity ω scale for the classification of organic molecules as strong, moderate or marginal electrophiles was proposed being ω > 1.5 eV for the first case, 0.8 < ω < 1.5 eV for the second case and ω < 0.8 eV for the last case (Domingo et al., 2002a; Domingo and Sáez, 2009; Pérez et al., 2003). By inspection of Table 10, it can be said that both peptides considered in this study may be regarded as moderate electrophiles. Notwithstanding, the overall chemical reactivity is about the same for both cyclopeptides. This information could be of interest for future studies on the potential therapeutic ability of these compounds.
Besides global reactivity descriptors, their local counterparts have been developed to get an idea of the differences in chemical reactivity between the atoms within the molecule. Among these local reactivity descriptors are the Fukui functions (Parr and Yang, 1989; Chermette, 1999; Geerlings et al., 2003) and the Dual Descriptor (Toro-Labbé, 2007; Morell et al., 2005, Morell et al., 2006; Martínez-Araya, 2012a; Martínez-Araya, 2012b; Martínez-Araya, 2015), which have been defined as: Nucleophilic Fukui Function (NFF) = f+(r) = ρN+1(r) − ρN(r), Electrophilic Fukui Function (EFF) = f−(r) = ρN(r) − ρN−1(r), and Dual Descriptor (DD) = Δf(r) =
The NFF, f+(r), is associated with the sites within a molecular system which are prone to nucleophilic attacks while the EFF, f+(r), describes those sites that are more susceptible to electrophilic attacks. Although the NFF and the EFF have been used successfully for the identification of reactive sites, the Dual Descriptor Δf(r) or DD, can describe unambiguously nucleophilic and electrophilic sites within a molecule (Martínez-Araya (2015)). A graphical representation of the DD for the Pashinintides A and B cyclopeptides is displayed in Figure 5 showing the zones where DD > 0 and DD < 0.
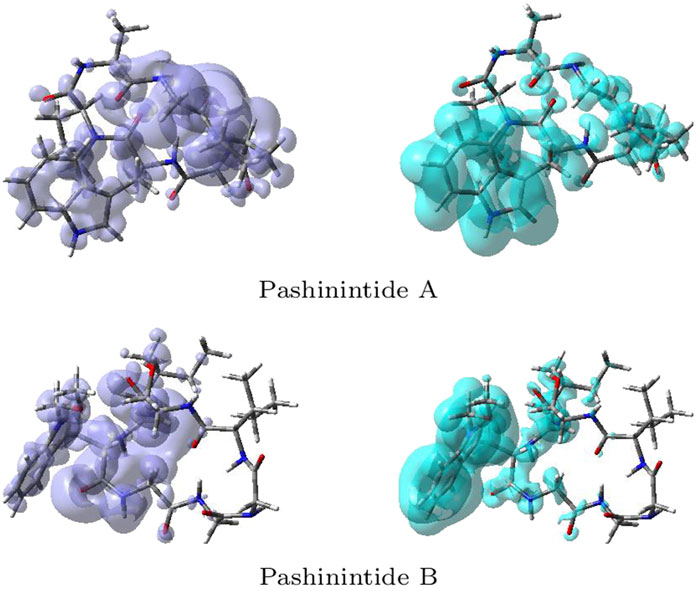
FIGURE 5. Graphical representations of the dual descriptor DD of the Pashinintides A and B. Left: DD > 0, right: DD < 0.
Although these graphical representations allowed to distinguish the regions within the molecules where the Dual Descriptor will be greater or smaller than zero, it can appreciated that there is some overlap between them. Thus, for a better estimation of these reactivity areas it is worth to determine the values of the Condensed Dual Descriptor (Δfk) (Morell et al., 2008; Frau and Glossman-Mitnik, 2018d) over the all the atoms (excluding H) in comparison with the condensed versions of the Electrophilicity, that is, the Condensed Electrophilicity (ωk) (Domingo et al., 2002b), and of the Nucleophilicity, being the Condensed Nucleophilicity Nk (Pérez et al., 2009). The resulting values are displayed in Tables 11 and 12 for the Pashinintides A and B, respectively.
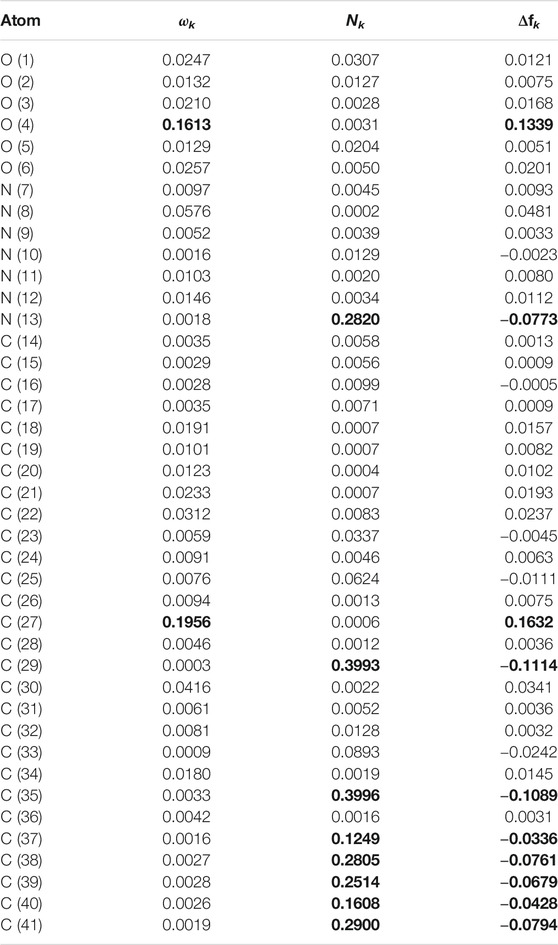
TABLE 11. Comparison of several reactivity descriptors: condensed electrophilicity ωk, condensed nucleophilicity Nk and condensed dual descriptor Δfk, over the atoms of Pashinintide A. H atoms are not shown.
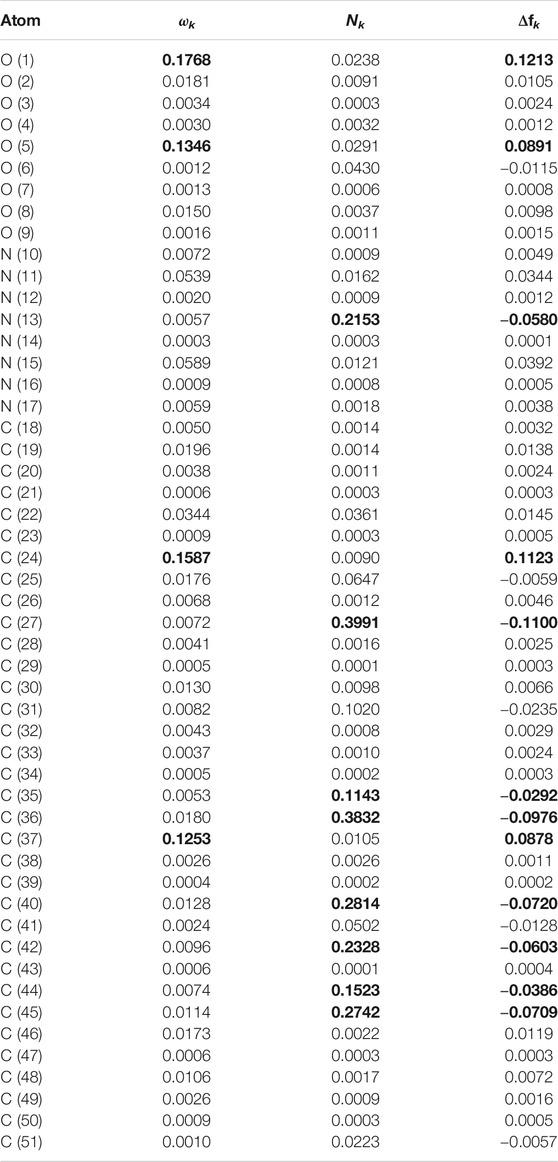
TABLE 12. Comparison of several reactivity descriptors: condensed electrophilicity ωk, condensed nucleophilicity Nk and condensed dual descriptor Δfk, over the atoms of Pashinintide B. H atoms are not shown.
Even if every atom within the peptides cannot be graphically individualized due to the large size of the molecules, it is clear from the results in Tables 11 and 12 about which are the sites prone to electrophilic and nucleophilic attacks based on the agreement between the values for the Condensed Electrophilicity ωk with the positive values of the Condensed Dual Descriptor Δfk, for one side, and with the values of the Condensed Nucleophilicity Nk and the negative results for the Condensed Dual Descriptor Δfk.
From Table 11 it can be seen that for Pashinintide A the maximum values for the Condensed Electrophilicity ωk (shown in bold) are located over the C (27) and O (4) which correlate well with the maximum positive results for the Condensed Dual Descriptor Δfk over those atoms. The same situation is found for the case of the Condensed Nucleophilicity Nk, whose maximum values over the C (29) and C(35) correlate with the maximum negative values Condensed Dual Descriptor Δfk localized on those atoms. The closeness between C(27) and C (29) explains the overlap between the two regions within the graphical representation of the Dual Descriptor.
For the case of Pashinintide B, it can be appreciated from Table 12 that the maximum values are located over the O (1), C (24), O (5) and C (37) (in that order) correlating in agreement with the greatest positive results for the Condensed Dual Descriptor Δfk, while for the Condensed Nucleophilicity Nk the order of reactivity will be C (27) > C (36) > C (40) > C (45) > N (13) being the same as for those derived from the Condensed Dual Descriptor Δfk. As for the case of the other peptide, the partial overlapping between the different reactive areas could be attributed to nearness between C (24) and C (27).
4 Conclusion
Two cyclic peptides, Pashinintides A and B, isolated from a terrestrial plant have been studied by resorting to some techniques of common use in the process of drug discovery and development through our proposed Computational Peptidology methodology showing that these kind of molecules can be regarded as potential therapeutic drugs.
With the further objective of analyzing their bioactivity, some useful parameters for future Structure Activity Relationships (SAR) research for the development of therapeutic drugs, their predicted biological targets, and the ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) parameters related to the bioavailability and pharmacokinetics of the two plant cyclopeptides under study were predicted and analyzed.
The chemical reactivities of the studied cyclopeptides have been exhaustively analyzed through the optimization of their structures using the DFTBA methodology and the estimation of their electronic properties making use of the MN12SX/Def2TZVP/H2O model chemistry already considered in previous research for the study of peptides, thus verifying its usefulness for this kind of calculations and supplemented with the calculation the Conceptual DFT-derived global and local reactivity descriptors, allowing to identify the preferred reactivity atoms within the molecules.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
NF-H and JF: Research and data analysis; DG-M: Research, data analysis and writing of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
NFH and DGM are researchers of CIMAV and CONACYT and want to thank both institutions for partial support.
References
Ansbacher, T., Srivastava, H. K., Stein, T., Baer, R., Merkx, M., and Shurki, A. (2012). Calculation of Transition Dipole Moment in Fluorescent Proteins-Towards Efficient Energy Transfer. Phys. Chem. Chem. Phys. 14, 4109–4117. doi:10.1039/c2cp23351g
Begam, B. F., and Kumar, J. S. (2012). A Study on Cheminformatics and its Applications on Modern Drug Discovery. Proced. Eng. 38, 1264–1275. doi:10.1016/j.proeng.2012.06.156
Cai, L., Zhao, M., Liu, S., Yin, T., Zhou, H., Dong, J., et al. (2014). Pashinintide A, the First Plant Cyclopeptide from Rosaceae, Included a Sucrose, Suggests a New Natural Receptor for Saccharide. Tetrahedron Lett. 55, 6231–6235. doi:10.1016/j.tetlet.2014.09.063
Chai, J.-D., and Head-Gordon, M. (2008). Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. doi:10.1039/b810189b
Chakraborty, D., and Chattaraj, P. K. (2021). Conceptual Density Functional Theory Based Electronic Structure Principles. Chem. Sci. 12, 6264–6279. doi:10.1039/d0sc07017c
Chattaraj, P. K., Chakraborty, A., and Giri, S. (2009). Net Electrophilicity. J. Phys. Chem. A. 113, 10068–10074. doi:10.1021/jp904674x
P. K. Chattaraj (Editors) (2009). Chemical Reactivity Theory - A Density Functional View (Boca Raton, FL: CRC Press. Taylor & Francis Group).
Chaudhari, R., and Chakraborti, D. (2021). “Bioactive Compounds from In-Vitro Culture of Swertia Chirayita (Roxb. Ex Flem.) Karsten: Identification and Quantification,” in Plant-based Functional Foods and Phytochemicals: From Traditional Knowledge to Present Innovation. Editors M. R. Goyal, A. Nath, H. Suleria, and F. L. Palm Bay (USA Burlington, ON, Canada Boca Raton, FL, USA Abingdon, Oxon, UK: Apple Academic Press Inc. CRC Press), 201–239.
Chermette, H. (1999). Chemical Reactivity Indexes in Density Functional Theory. J. Comput. Chem. 20, 129–154. doi:10.1002/(sici)1096-987x(19990115)20:1<129::aid-jcc13>3.0.co;2-a
Cramer, C. (2004). Essentials of Computational Chemistry - Theories and Models. 2nd edn. Chichester, England: John Wiley & Sons.
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 7, 42717. doi:10.1038/srep42717
Daina, A., Michielin, O., and Zoete, V. (2019). SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 47, W357–W364. doi:10.1093/nar/gkz382
Doak, B. C., Over, B., Giordanetto, F., and Kihlberg, J. (2014). Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 21, 1115–1142. doi:10.1016/j.chembiol.2014.08.013
Domingo, L. R., and Pérez, P. (2011). The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 9, 7168–7175. doi:10.1039/c1ob05856h
Domingo, L. R., and Sáez, J. A. (2009). Understanding the Mechanism of Polar Diels-Alder Reactions. Org. Biomol. Chem. 7, 3576–3583. doi:10.1039/b909611f
Domingo, L. R., Aurell, M. J., Pérez, P., and Contreras, R. (2002a). Quantitative Characterization of the Global Electrophilicity Power of Common diene/Dienophile Pairs in Diels-Alder Reactions. Tetrahedron 58, 4417–4423. doi:10.1016/s0040-4020(02)00410-6
Domingo, L. R., Aurell, M. J., Pérez, P., and Contreras, R. (2002b). Quantitative Characterization of the Local Electrophilicity of Organic Molecules. Understanding the Regioselectivity on Diels−Alder Reactions. J. Phys. Chem. A. 106, 6871–6875. doi:10.1021/jp020715j
Domingo, L. R., Chamorro, E., and Pérez, P. (2008). Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 73, 4615–4624. doi:10.1021/jo800572a
Domingo, L., Ríos-Gutiérrez, M., and Pérez, P. (2016). Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 21, 748. doi:10.3390/molecules21060748
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2019a). Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A-F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint. Computation 7, 52. doi:10.3390/computation7030052
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2019b). Computational Peptidology Assisted by Conceptual Density Functional Theory for the Study of Five New Antifungal Tripeptides. ACS Omega 4, 12555–12560. doi:10.1021/acsomega.9b01463
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2019c). Computational Prediction of Bioactivity Scores and Chemical Reactivity Properties of the Parasin I Therapeutic Peptide of Marine Origin Through the Calculation of Global and Local Conceptual DFT Descriptors. Theor. Chem. Acc. 138, 78. doi:10.1007/s00214-019-2469-3
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2019d). “Conceptual DFT as a Helpful Chemoinformatics Tool for the Study of the Clavanin Family of Antimicrobial Marine Peptides,” in Density Functional Theory Calculations (Rijetia: IntechOpen), 1–11.
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2020). A Fast and Simple Evaluation of the Chemical Reactivity Properties of the Pristinamycin Family of Antimicrobial Peptides. Chem. Phys. Lett. 739, 137021. doi:10.1016/j.cplett.2019.137021
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2020a). Conceptual DFT-Based Computational Peptidology of Marine Natural Compounds: Discodermins A-H. Molecules 25, 4158. doi:10.3390/molecules25184158
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2020b). Virtual Screening of Marine Natural Compounds by Means of Chemoinformatics and CDFT-Based Computational Peptidology. Mar. Drugs 18, 478. doi:10.3390/md18090478
Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D. (2021). “Conceptual DFT as a Helpful Chemoinformatics Tool for the Study of the Clavanin Family of Antimicrobial Marine Peptides,” in Density Functional Theory. Editors S. R. De Lazaro, L. H. Da Silveira Lacerda, and R. A. Pontes Ribeiro (London, UK: IntechOpen), 57–67. chap. 3. doi:10.5772/intechopen.88657
Frau, J., and Glossman-Mitnik, D. (2018a). Blue M2: An Intermediate Melanoidin Studied via Conceptual DFT. J. Mol. Model. 24, 1–13. doi:10.1007/s00894-018-3673-0
Frau, J., and Glossman-Mitnik, D. (2018b). Chemical Reactivity Theory Applied to the Calculation of the Local Reactivity Descriptors of a Colored Maillard Reaction Product. Chem. Sci. Int. J. 22, 1–14. doi:10.9734/csji/2018/41452
Frau, J., and Glossman-Mitnik, D. (2018c). Computational Study of the Chemical Reactivity of the Blue-M1 Intermediate Melanoidin. Comput. Theor. Chem. 1134, 22–29. doi:10.1016/j.comptc.2018.04.018
Frau, J., and Glossman-Mitnik, D. (2018d). Local Molecular Reactivity of the Colored Dansylglycine in Water and Dioxane Studied through Conceptual DFT. J. Chem. 2018, 1–7. doi:10.1155/2018/3172412
Frau, J., and Glossman-Mitnik, D. (2018e). Conceptual DFT Study of the Local Chemical Reactivity of the Dilysyldipyrrolones A and B Intermediate Melanoidins. Theor. Chem. Acc. 137, 1210. doi:10.1007/s00214-018-2244-x
Frau, J., and Glossman-Mitnik, D. (2018f). Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 23, 559. doi:10.3390/molecules23030559
Frau, J., Flores-Holguín, N., and Glossman-Mitnik, D. (2018). Chemical Reactivity Properties, pKa Values, AGEs Inhibitor Abilities and Bioactivity Scores of the Mirabamides A-H Peptides of Marine Origin Studied by Means of Conceptual DFT. Mar. Drugs 16, 302–319. doi:10.3390/md16090302
Frau, J., Flores-Holguín, N., and Glossman-Mitnik, D. (2019). Chemical Reactivity Theory and Empirical Bioactivity Scores as Computational Peptidology Alternative Tools for the Study of Two Anticancer Peptides of Marine Origin. Molecules 24, 1115. doi:10.3390/molecules24061115
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 Revision C.01. Wallingford CT: Gaussian Inc. [Dataset].
Gang, D., Kim, D., and Park, H.-S. (2018). Cyclic Peptides: Promising Scaffolds for Biopharmaceuticals. Genes 9, 557. doi:10.3390/genes9110557
Gázquez, J. L., Cedillo, A., and Vela, A. (2007). Electrodonating and Electroaccepting Powers. J. Phys. Chem. A. 111, 1966–1970. doi:10.1021/jp065459f
Geerlings, P., De Proft, F., and Langenaeker, W. (2003). Conceptual Density Functional Theory. Chem. Rev. 103, 1793–1874. doi:10.1021/cr990029p
Geerlings, P., Chamorro, E., Chattaraj, P. K., De Proft, F., Gázquez, J. L., Liu, S., et al. (2020). Conceptual Density Functional Theory: Status, Prospects, Issues. Theor. Chem. Acc. 139, 36. doi:10.1007/s00214-020-2546-7
González-Medina, M., Naveja, J. J., Sánchez-Cruz, N., and Medina-Franco, J. L. (2017). Open Chemoinformatic Resources to Explore the Structure, Properties and Chemical Space of Molecules. RSC Adv. 7, 54153–54163. doi:10.1039/c7ra11831g
Guha, M., Das, N., and Savarimuthu, X. (2021). “Natural Phytobiactives: Let’s Eat Smart!,” in Plant-based Functional Foods and Phytochemicals: From Traditional Knowledge to Present Innovation. Editors M. R. Goyal, A. Nath, H. Suleria, and F. L. Palm Bay (FL, USA Burlington, ON, Canada Boca Raton, FL, USA Abingdon, Oxon, UK: Apple Academic Press Inc. CRC Press), 201–239.
Halgren, T. A., and Nachbar, R. B. (1996). Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 17, 587–615. doi:10.1002/(sici)1096-987x(199604)17:5/6<587::aid-jcc4>3.0.co;2-q
Halgren, T. A. (1996a). Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 17, 490–519. doi:10.1002/(sici)1096-987x(199604)17:5/6<490::aid-jcc1>3.0.co;2-p
Halgren, T. A. (1996b). Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 17, 520–552. doi:10.1002/(sici)1096-987x(199604)17:5/6<520::aid-jcc2>3.0.co;2-w
Halgren, T. A. (1996c). Merck Molecular Force Field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 17, 616–641. doi:10.1002/(sici)1096-987x(199604)17:5/6<616::aid-jcc5>3.0.co;2-x
Halgren, T. A. (1999). MMFF VI. MMFF94s Option for Energy Minimization Studies. J. Comput. Chem. 20, 720–729. doi:10.1002/(sici)1096-987x(199905)20:7<720::aid-jcc7>3.0.co;2-x
Heyd, J., and Scuseria, G. E. (2004). Efficient Hybrid Density Functional Calculations in Solids: Assessment of the Heyd-Scuseria-Ernzerhof Screened Coulomb Hybrid Functional. J. Chem. Phys. 121, 1187–1192. doi:10.1063/1.1760074
Iikura, H., Tsuneda, T., Yanai, T., and Hirao, K. (2001). A Long-Range Correction Scheme for Generalized-Gradient-Approximation Exchange Functionals. J. Chem. Phys. 115, 3540–3544. doi:10.1063/1.1383587
Jaramillo, P., Domingo, L. R., Chamorro, E., and Pérez, P. (2008). A Further Exploration of a Nucleophilicity Index Based on the Gas-Phase Ionization Potentials. J. Mol. Struct. THEOCHEM 865, 68–72. doi:10.1016/j.theochem.2008.06.022
Jensen, F. (2007). Introduction to Computational Chemistry. 2nd edn. Chichester, England: John Wiley & Sons.
Jujjavarapu, S. E., Dhagat, S., and Yadav, M. (2019). Computer-Aided Design of Antimicrobial Lipopeptides as Prospective Drug Candidates. Boca Raton: CRC Press LLC.
Karolewski, A., Stein, T., Baer, R., and Kümmel, S. (2011). Communication: Tailoring the Optical Gap in Light-Harvesting Molecules. J. Chem. Phys. 134, 151101–151105. doi:10.1063/1.3581788
Kim, S.-K. (2013). Marine Proteins and Peptides - Biological Activities and Applications. Chichester, UK: Wiley-Blackwell.
Kronik, L., Stein, T., Refaely-Abramson, S., and Baer, R. (2012). Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theor. Comput. 8, 1515–1531. doi:10.1021/ct2009363
Kuritz, N., Stein, T., Baer, R., and Kronik, L. (2011). Charge-Transfer-Like π→π* Excitations in Time-Dependent Density Functional Theory: A Conundrum and its Solution. J. Chem. Theor. Comput. 7, 2408–2415. doi:10.1021/ct2002804
Lewars, E. (2003). Computational Chemistry - Introduction to the Theory and Applications of Molecular and Quantum Mechanics. Dordrecht: Kluwer Academic Publishers.
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (2001). Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings 1PII of Original Article: S0169-409X(96)00423-1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997) 3-25. 1. Adv. Drug Deliv. Rev. 46, 3–26. doi:10.1016/s0169-409x(00)00129-0
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 113, 6378–6396. doi:10.1021/jp810292n
Martínez-Araya, J. I. (2015). Why Is the Dual Descriptor a More Accurate Local Reactivity Descriptor Than Fukui Functions?. J. Math. Chem. 53, 451–465. doi:10.1007/s10910-014-0437-7
Martínez-Araya, J. I. (2012a). Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. J. Mol. Model. 19, 2715–2722. doi:10.1007/s00894-012-1520-2
Martínez-Araya, J. I. (2012b). Revisiting Caffeate's Capabilities as a Complexation Agent to Silver Cation in Mining Processes by Means of the Dual Descriptor-A Conceptual DFT Approach. J. Mol. Model. 18, 4299–4307. doi:10.1007/s00894-012-1405-4
Morell, C., Grand, A., and Toro-Labbé, A. (2005). New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A. 109, 205–212. doi:10.1021/jp046577a
Morell, C., Grand, A., and Toro-Labbé, A. (2006). Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 425, 342–346. doi:10.1016/j.cplett.2006.05.003
Morell, C., Hocquet, A., Grand, A., and Jamart-Grégoire, B. (2008). A Conceptual DFT Study of Hydrazino Peptides: Assessment of the Nucleophilicity of the Nitrogen Atoms by Means of the Dual Descriptor Δf(r). J. Mol. Struct. THEOCHEM 849, 46–51. doi:10.1016/j.theochem.2007.10.014
Nielsen, D. S., Shepherd, N. E., Xu, W., Lucke, A. J., Stoermer, M. J., and Fairlie, D. P. (2017). Orally Absorbed Cyclic Peptides. Chem. Rev. 117, 8094–8128. doi:10.1021/acs.chemrev.6b00838
Parr, R., and Yang, W. (1989). Density-Functional Theory of Atoms and Molecules. New York: Oxford University Press.
Pérez, P., Domingo, L. R., José Aurell, M., and Contreras, R. (2003). Quantitative Characterization of the Global Electrophilicity Pattern of Some Reagents Involved in 1,3-Dipolar Cycloaddition Reactions. Tetrahedron 59, 3117–3125. doi:10.1016/s0040-4020(03)00374-0
Pérez, P., Domingo, L. R., Duque-Noreña, M., and Chamorro, E. (2009). A Condensed-To-Atom Nucleophilicity Index. An Application to the Director Effects on the Electrophilic Aromatic Substitutions. J. Mol. Struct. THEOCHEM 895, 86–91. doi:10.1016/j.theochem.2008.10.014
Peverati, R., and Truhlar, D. G. (2012). Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 14, 16187–16191. doi:10.1039/c2cp42576a
Pires, D. E. V., Blundell, T. L., and Ascher, D. B. (2015). pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 58, 4066–4072. doi:10.1021/acs.jmedchem.5b00104
Sable, R., Parajuli, P., and Jois, S. (2017). Peptides, Peptidomimetics, and Polypeptides from Marine Sources: A Wealth of Natural Sources for Pharmaceutical Applications. Mar. Drugs 15, 124. doi:10.3390/md15040124
Sánchez, A., and Vázquez, A. (2017). Bioactive Peptides: A Review. Food Qual. Saf. 1, 29–46. doi:10.1093/fqs/fyx006
Santos, G. B., Ganesan, A., and Emery, F. S. (2016). Oral Administration of Peptide-Based Drugs: Beyond Lipinski's Rule. ChemMedChem 11, 2245–2251. doi:10.1002/cmdc.201600288
Stein, T., Kronik, L., and Baer, R. (2009a). Prediction of Charge-Transfer Excitations in Coumarin-Based Dyes Using a Range-Separated Functional Tuned from First Principles. J. Chem. Phys. 131, 244119. doi:10.1063/1.3269029
Stein, T., Kronik, L., and Baer, R. (2009b). Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 131, 2818–2820. doi:10.1021/ja8087482
Stein, T., Eisenberg, H., Kronik, L., and Baer, R. (2010). Fundamental Gaps in Finite Systems from Eigenvalues of a Generalized Kohn-Sham Method. Phys. Rev. Lett. 105, 266802–266804. doi:10.1103/physrevlett.105.266802
Stein, T., Autschbach, J., Govind, N., Kronik, L., and Baer, R. (2012). Curvature and Frontier Orbital Energies in Density Functional Theory. J. Phys. Chem. Lett. 3, 3740–3744. doi:10.1021/jz3015937
A. Toro-Labbé (Editors) (2007). Theoretical Aspects of Chemical Reactivity (Amsterdam: Elsevier Science).
Weigend, F., and Ahlrichs, R. (2005). Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/b508541a
Weigend, F. (2006). Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057–1065. doi:10.1039/b515623h
Yanai, T., Tew, D. P., and Handy, N. C. (2004). A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3lyp). Chem. Phys. Lett. 393, 51–57. doi:10.1016/j.cplett.2004.06.011
Young, D. (2001). Computational Chemistry - A Practical Guide for Applying Techniques to Real-World Problems. New York: John Wiley & Sons.
Keywords: plant cyclopeptides, pharmacokinetics, ADMET, conceptual density functional theory, Koopmans in density functional theory
Citation: Flores-Holguín N, Frau J and Glossman-Mitnik D (2021) In Silico Pharmacokinetics, ADMET Study and Conceptual DFT Analysis of Two Plant Cyclopeptides Isolated From Rosaceae as a Computational Peptidology Approach. Front. Chem. 9:708364. doi: 10.3389/fchem.2021.708364
Received: 11 May 2021; Accepted: 08 July 2021;
Published: 12 August 2021.
Edited by:
Jorge M. del Campo, Universidad Nacional Autónoma de México, MexicoReviewed by:
Ramon Miranda Quintana, University of Florida, United StatesTanmoy Chakraborty, Sharda University, India
Copyright © 2021 Flores-Holguín, Frau and Glossman-Mitnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Glossman-Mitnik, ZGFuaWVsLmdsb3NzbWFuQGNpbWF2LmVkdS5teA==