 Aleša Bricelj
Aleša Bricelj Christian Steinebach2
Christian Steinebach2 Michael Gütschow
Michael Gütschow Izidor Sosič
Izidor Sosič- 1Faculty of Pharmacy, University of Ljubljana, Ljubljana, Slovenia
- 2Pharmaceutical Institute, University of Bonn, Bonn, Germany
Proteolysis-targeting chimeras (PROTACs) have received tremendous attention as a new and exciting class of therapeutic agents that promise to significantly impact drug discovery. These bifunctional molecules consist of a target binding unit, a linker, and an E3 ligase binding moiety. The chemically-induced formation of ternary complexes leads to ubiquitination and proteasomal degradation of target proteins. Among the plethora of E3 ligases, only a few have been utilized for the novel PROTAC technology. However, extensive knowledge on the preparation of E3 ligands and their utilization for PROTACs has already been acquired. This review provides an in-depth analysis of synthetic entries to functionalized ligands for the most relevant E3 ligase ligands, i.e. CRBN, VHL, IAP, and MDM2. Less commonly used E3 ligase and their ligands are also presented. We compare different preparative routes to E3 ligands with respect to feasibility and productivity. A particular focus was set on the chemistry of the linker attachment by discussing the synthetic opportunities to connect the E3 ligand at an appropriate exit vector with a linker to assemble the final PROTAC. This comprehensive review includes many facets involved in the synthesis of such complex molecules and is expected to serve as a compendium to support future synthetic attempts towards PROTACs.
Introduction
The ubiquitin-proteasome system (UPS) plays a cardinal role in maintaining intracellular protein homeostasis by eliminating misfolded, damaged, and worn-out proteins (Amm et al., 2014). This process consists of a cascade of distinct steps, starting with ubiquitin activation by enzyme E1. Ubiquitin is then passed to the E2 or ubiquitin-conjugating enzyme by trans-thioesterification. Subsequently, E3 ubiquitin ligase promotes the transfer of ubiquitin onto a lysine of the substrate protein. Ubiquitin’s own internal lysine residues allow binding of additional ubiquitins, resulting in polyubiquitin tags, which serve as a signal for protein degradation via the 26S proteasome (Kleiger and Mayor, 2014).
Hijacking the UPS and utilizing its functions to degrade the selected protein of interest (POI) has been made possible by proteolysis-targeting chimeras (PROTACs) (Burslem and Crews, 2020) (Figure 1). These hetero-bifunctional molecules are composed of a POI ligand connected to an E3 ubiquitin ligase ligand by a linker (Figure 1) (Pettersson and Crews, 2019). A functional PROTAC instigates the formation of a ternary complex POI-PROTAC-E3 ligase, which results in the ubiquitination of the POI, followed by proteasomal degradation (Scheepstra et al., 2019). This new modality began accumulating recognition and significance in medicinal chemistry since 2001 when the first proof-of-concept experiments were published (Sakamoto et al., 2001; Burslem and Crews, 2020).
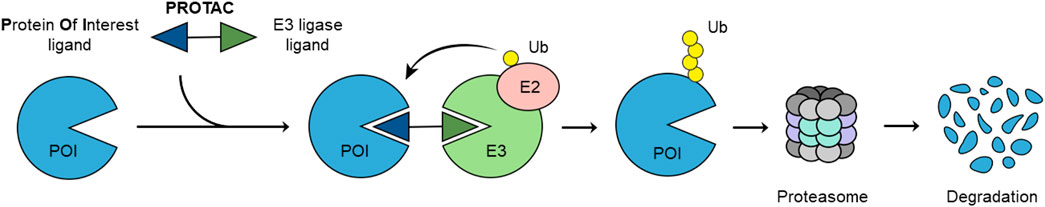
FIGURE 1. PROTAC-induced degradation of target proteins.
E3 Ligases
The human genome includes two members of the E1 enzyme family, roughly 40 E2s, and more than 600 E3 ubiquitin ligases (Kleiger and Mayor, 2014). E3 ligases represent a crucial element in protein ubiquitination due to their role in substrate selection and modulation of the cascade's efficiency (Buetow and Huang, 2016; Zheng and Shabek, 2017). They are categorized into three classes, based on their mechanism of ubiquitin transfer. The first and the most abundant class includes approximately 600 RING (Really Interesting New Gene) E3 ligases, which catalyze the direct transfer of ubiquitin from E2 to a substrate. In contrast, the less represented E3 classes HECT (Homologous to E6AP C-terminus) and RBR (RING-between-RING) form a thioester intermediate with ubiquitin via a catalytic cysteine before the transfer to the substrate protein (Buetow and Huang, 2016). Although our understanding of substrate recognition and regulation of ubiquitination is incomplete, the genome’s selection of roughly 600 E3 ligases is capable of ubiquitinating a much larger number of protein substrates in a controlled manner with ample specificity (Fisher and Phillips, 2018).
Despite the vast selection of known E3 ligases, only a handful have been successfully utilized in PROTAC compounds (Burslem and Crews, 2020). Following the first utilization of a poorly permeable phosphopeptide moiety to hijack Skp1–Cullin–F box complex (SCFβ-TRCP) to degrade methionine aminopeptidase-2 (Sakamoto et al., 2001), and targeting the von Hippel-Lindau (VHL) tumor suppressor protein with a seven amino acid long sequence ALAPYIP (Schneekloth, et al., 2004), the field has evolved tremendously, resulting in numerous small-molecule E3 ligands, that allow for the development of cell-permeable and biologically active PROTACs (Sun X. et al., 2019). The first of its kind was a PROTAC targeting the androgen receptor, using nutlin (Figure 8) to recruit the mouse double minute 2 homologue (MDM2) E3 ligase (Schneekloth et al., 2008). Following that, the number of successfully degraded targets using various E3 ligases, such as cellular inhibitor of apoptosis (cIAP) (Itoh et al., 2010), VHL, and cereblon (CRBN) (Sun X. et al., 2019), steeply increased. More recently, additional E3s were explored and used successfully in degraders, i.e., RING-type zinc-finger protein 114 (RNF114) (Spradlin et al., 2019), damage-specific DNA binding protein 1 (DDB1)-CUL4 associated factor 16 (DCAF16) (Zhang et al., 2019), and Kelch-like ECH-associated protein 1 (KEAP1) (Tong et al., 2020a). However, the majority of recently reported PROTACs still utilize either VHL or CRBN as E3 ligases (Burslem and Crews, 2020); a fact that is corroborated by a high number of different synthetic approaches to obtain these PROTAC building blocks.
Various aspects of degraders have been extensively reviewed in the scientific literature in recent years (Toure and Crews, 2016; Lai and Crews, 2017; An and Fu, 2018; Sun X. et al., 2019; Pettersson and Crews, 2019; Schapira et al., 2019). However, a thorough overview of synthetic efforts leading to the most commonly used ligands for E3 ligases has not been done. Therefore, in this review, we focus on E3 ligase ligands utilized in successful PROTACs. More precisely, we overview the synthetic routes to obtain the E3 ligands and illustrate the possible linker attachment points and types of bonds used to connect the ligands with linkers. The preparation of specific building blocks was reported, as expected, in many publications. However, if no yields were reported or the authors only referred to the original or previously described work, these publications were not referenced in this paper. In addition, for the most commonly used ligases, a statistical overview of the prevalence of E3 ligase ligands and linker attachment options utilized in PROTACs is provided, along with highlighting the contributions of these building blocks to the physicochemical properties of final PROTAC molecules. This review provides the reader with a concise picture of the current state and enables all newcomers to the field a quick go-to-guide in terms of synthetic access to PROTAC building blocks. We hope that this thorough overview will aid in future successful contributions in the protein degradation field.
Cereblon
CRBN is a 442-amino acid protein that forms a Cullin-4-RING E3 ubiquitin ligase (CRL4) complex and interacts with the adaptor protein damaged DNA–binding protein 1 (DDB1) (Ito et al., 2011; Chamberlain et al., 2014). Within the CRL4 complex, CRBN acts as a substrate-specificity receptor (Chamberlain et al., 2014). Known ligands for CRBN include thalidomide and other derived immunomodulatory imide drugs (IMiDs). Upon binding of IMiDs to CRBN, the E3 ubiquitin ligase activity of CRBN is re-modulated (Zhu et al., 2011; Chamberlain et al., 2014; Krönke et al., 2014; Krönke et al., 2015). As a result, an increase in the recruitment of the transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) occurs, which leads to their subsequent ubiquitination and proteasomal degradation. This interaction and its outcome are responsible for the antiproliferative effects of thalidomide, lenalidomide, and pomalidomide in multiple myeloma (Chamberlain et al., 2014; Krönke et al., 2014; Krönke et al., 2015).
To date, CRBN has been successfully utilized as the E3 ligase in PROTAC targeting more than 30 different proteins, ranging from those involved in various cancers (Sun X. et al., 2019) and immune disorders (Bassi et al., 2018), to neurodegenerative disease-associated protein Tau (Silva et al., 2019), and even hepatitis C virus protein NS3 (de Wispelaere et al., 2019). The collection of CRBN ligands with different linker attachment options are presented in Figure 2. The majority of CRBN-targeting PROTACs employ derivatives of pomalidomide (Figure 2, A1, A2), 4-hydroxythalidomide (Figure 2, B1, B2), alkyl-connected thalidomide derivatives (Figure 2, C1), or lenalidomide (Figure 2, D1–D3). However, alternatives are possible, and these include examples with substitution at position 5 of the phthalimide fragment. For organisational purposes and clarity of this section, the complexes between CRBN ligands and linkers are categorized based on the structure of the ligase ligand and further based on the bond type for linker attachment.
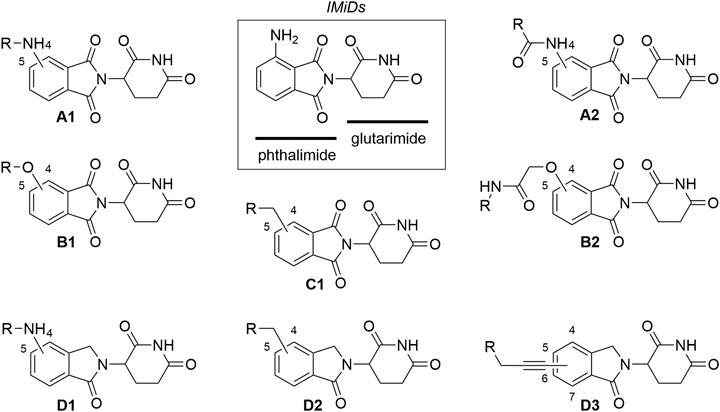
FIGURE 2. Commonly utilized thalidomide-derived CRBN ligands and possible linker attachment styles. (A1–A2) pomalidomide derivatives; (B1–B2) 4-hydroxythalidomide derivatives; (C1) alkyl type attachment to thalidomide; (D1–D3) lenalidomide derivatives.
Pomalidomide-Based Ligands
We categorized the possible synthetic routes based on the common phthalic anhydride precursor, as most syntheses start from either 3-fluorophthalic anhydride, which is then subjected to condensation with the glutarimide ring and subsequent nucleophilic substitution by a linker with a primary amine, or 3-nitrophtalic anhydride, which is subsequently reduced to pomalidomide.
3-Fluorophthalic Anhydride as a Precursor for Pomalidomide-Based Derivatives
Several options are available to obtain pomalidomide-based PROTAC precursors when 3-fluorophthalic anhydride (4) is used as the main synthon. The glutarimide subunit can be incorporated into 4-fluorothalidomide (5) by using compound 2, which can be formed easily by converting Boc-Gln-OH (1) into 2via an intramolecular coupling (Scheme 1, steps a-b) (Steinebach et al., 2018). Another option to afford the desired precursor 2 over three steps with a 57% yield was presented where l-glutamine was used as starting material (Varala and Adapa, 2005). Alternatively, 3-aminopiperidine-2,6-dione hydrochloride can be used in place of 2 (Zhou et al., 2018; An et al., 2019). The 3-fluorophthalic anhydride (4) is usually used as a commercially available building block. However, it can be easily prepared in high yield by refluxing 3-fluorophthalic acid (3) in acetic anhydride (Zhou et al., 2018). Following condensation of the glutarimide subunit with 3-fluorophthalic anhydride (4), 4-fluorothalidomide (5) was obtained in a higher yield by using NaOAc in AcOH under reflux conditions (Steinebach et al., 2018; Zhou et al., 2018), rather than by a method using Et3N in THF at 80°C (An et al., 2019) (Scheme 1, step d). An alternative synthesis towards 5 was reported where l-glutamine was reacted with 3-fluorophthalic acid (3) to form 6, followed by a CDI-mediated intramolecular cyclization (Scheme 1, steps e–f). However, the desired product 5 was obtained in an approximately 14% overall yield using this approach (Lu et al., 2015). Compound 5 then allows for simple linker introduction using primary amines and DIPEA in DMF at 90°C, leading to alkylated pomalidomide derivatives 7 (Scheme 1, step g) (Lu et al., 2015; Steinebach et al., 2018; An et al., 2019). It was reported to replace DMF with DMSO for the linker attachment step because of the thermal decomposition of DMF at high temperatures in the presence of a tertiary amine, forming dimethylamine, which can result in the formation of the undesired 4-(dimethylamino)-thalidomide (Steinebach et al., 2018; Steinebach et al., 2019). Recent advances showed that performing the nucleophilic aromatic substitution of compound 5 with primary or secondary amines at elevated temperatures (130°C) generally resulted in a higher yield of desired pomalidomide derivatives (Brownsey et al., 2021).
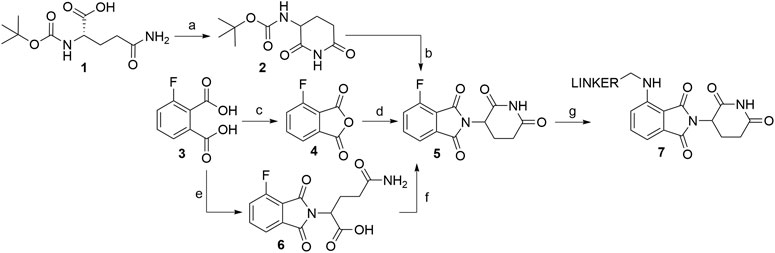
SCHEME 1. Syntheses of compound 7. Reagents and conditions: a) CDI, DMAP, THF, reflux, 4 h, 84% yield (Steinebach et al., 2018); b) 4, NaOAc, AcOH, reflux, 6 h, 89% yield; (Steinebach et al., 2018); c) Ac2O, reflux, 2 h, 92% yield (An et al., 2019); d) 3-aminopiperidine-2,6-dione hydrochloride, Et3N, THF, 80°C, 69% yield (An et al., 2019); d) 3-aminopiperidine-2,6-dione hydrochloride, NaOAc, AcOH, 140°C, 12 h, 88% yield (Zhou et al., 2018); e) l-glutamine, DMF, 90°C, 8 h, 53% yield (Lu et al., 2015); f) CDI, DMAP, MeCN, reflux, 5 h, 26% yield (Lu et al., 2015); g) reagents, conditions, and yields are collected in Table 1.
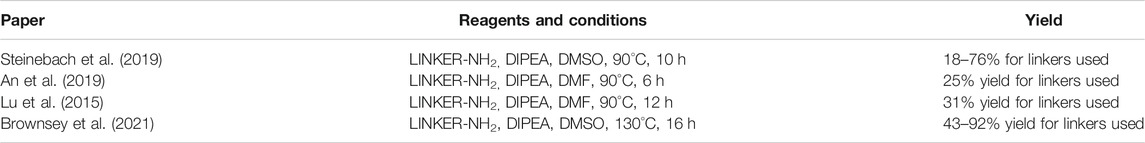
TABLE 1. Reagents, conditions, and yields for converting compound 5 to 7 (Scheme 1, step g).
Numerous studies include thalidomide derivatives with N-alkylated glutarimide ring (e.g., compounds 9 and 10, Scheme 2) as negative controls since they are incapable of binding to CRBN (Buhimschi et al., 2018). Two options are presented for synthesizing such negative controls, the first being the alkylation of glutarimide moiety 8 before conjugation into the final 4-fluorothalidomide (9) (Steinebach et al., 2018). Alternatively, the imide nitrogen of 5 can be alkylated after the condensation of glutarimide and phthalimide parts (An et al., 2019), or a methyl group can be introduced via Mitsunobu reaction (Steinebach et al., 2018).
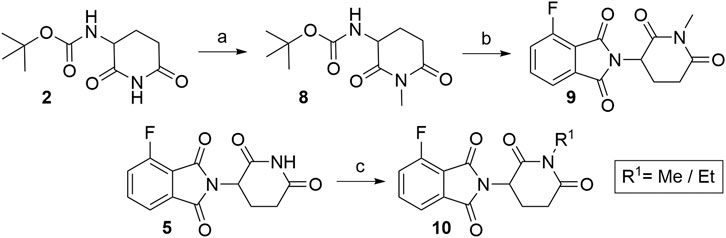
SCHEME 2. Syntheses of compounds 9 and 10, precursors to prepare negative controls. Reagents and conditions: a) K2CO3, MeI, DMF, rt, 2 h, 29% yield; b) 3, NaOAc, AcOH, reflux, 6 h, 82% yield (Steinebach et al., 2018); c) MeOH, PPh3, DIAD, THF, sonication bath, 1 h, 25% yield (Steinebach et al., 2018); c) EtI, K2CO3, acetone, reflux, 3 h, 65% yield (An et al., 2019).
A significant number of reported PROTACs incorporate a triazole fragment (e.g., compound 13, Scheme 3; compound 34, Scheme 7; compound 39, Scheme 8) as a result of utilizing click reactions between azides and alkynes (e.g., compound 12 in Scheme 3), under conditions for a typical copper-catalyzed Huisgen 1,3-dipolar cycloaddition. Deemed the ‘privileged scaffold for PROTACs’, triazoles represent numerous advantages since they are easily accessible in high yields under mild reaction conditions, which are highly compatible with other functional groups (Xia et al., 2019).

SCHEME 3. Preparation of pomalidomide derivative available for azide-alkyne cycloaddition click reaction. Reagents and conditions: a) propargylamine, DIPEA, DMF, 90°C, 12 h, 30% yield; b) linker-N3, CuSO4, Na ascorbate, H2O/t-BuOH, rt, 16 h, 40–83% yield for linkers used (Wu et al., 2019).
3-Nitrophthalic Anhydride as a Precursor for Pomalidomide-Based Derivatives
When 3-nitrophthalic anhydride (15) was used as the starting compound (either commercially available or prepared from 14) (Huang et al., 2016), it was usually immediately condensed with the glutarimide ring into 4-nitrothalidomide (16). Similarly to synthesizing 4-fluorothalidomide (5), this condensation yielded the desired product in a significantly higher yield if the reaction was performed under NaOAc, AcOH/reflux conditions (Steinebach et al., 2018; Rana et al., 2019), instead of treating the mixture with Et3N in THF (Chen et al., 2018). The following reduction to pomalidomide (17) was notably more efficient using Pd/C-catalyzed hydrogenation with reports of near quantitative yield (Steinebach et al., 2018), in contrast to using iron-ammonium chloride (Chen et al., 2018) or Pd/C and ammonium formate (Huang et al., 2016) as reducing agents (Scheme 4, steps a–c). Another option to obtain 17 is through the synthesis of intermediate 18, its reduction to 19, and final cyclization to the desired product with an overall yield of 65% (Scheme 4, steps d–f). Although reported to be efficient, practical and environmentally friendly (Huang et al., 2016), the yield was still inferior to the route via16, which had an overall yield of 94% (Steinebach et al., 2018). Alternatively, Huang et al. reported a method where the 3-nitrophthalimide (20) was first reacted with glutamine via amination to yield 21, which was then reduced to 22, and finally cyclized under CDI-mediated conditions. This resulted in a low pomalidomide (17) yield, mostly due to the poor reduction conversion (Scheme 4, steps g–i) (Huang et al., 2016).
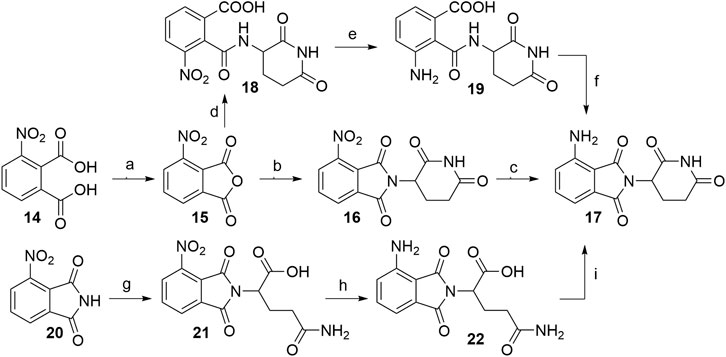
SCHEME 4. Alternative routes for the synthesis of pomalidomide 17. Reagents and conditions: a) Ac2O, reflux, 2 h, 93% yield (Huang et al., 2016); b) tert-butyl (2,6-dioxopiperidin-3-yl)carbamate 2, NaOAc, AcOH, reflux, 6 h, 95% yield (Steinebach et al., 2018); b) 3-aminopiperidine-2,6-dione hydrochloride, NaOAc, AcOH, 130°C, 48 h, 92% yield (Rana et al., 2019); b) 3-aminopiperidine-2,6-dione trifluoroacetate, Et3N, THF, 80°C, 6 h, 69% yield (Chen et al., 2018); b) 3-aminopiperidine-2,6-dione hydrochloride, NaOAc, AcOH, 80°C, 12 h, 73% yield (Huang et al., 2016); c) Pd/C, H2, DMF, rt, 24 h, 99% yield (Steinebach et al., 2018); c) Fe, NH4Cl, EtOH/H2O, rt, overnight, 44% yield (Chen et al., 2018); c) HCOONH4, Pd/C, MeOH, rt, 2 h, 68% yield (Huang et al., 2016); d) 3-aminopiperidine-2,6-dione hydrochloride, Et3N, THF, ≤20°C, 30 min, 91% yield; e) Pd/C, H2, 145 psi, MeOH, rt, 30 min, quant.; f) MeOH, reflux, 2 h, 71% yield (Huang et al., 2016); g) glutamine, DMF, 80–87°C, 8 h, 70% yield (Huang et al., 2016); h) Pd/C, H2, 50 psi, MeOH, 2.5 h, 10% yield; i) CDI, MeCN, reflux, 4.5 h, 88% yield (Huang et al., 2016).
The route towards pomalidomide derivatives with a two-carbon spacer was nicely elaborated (Zhou et al., 2018) (Scheme 5). Treating 3-nitrophthalic anhydride (15) with benzyl alcohol and benzyl bromide, reducing the nitro group with stannous chloride, and alkylating the resulting amine group with tert-butyl bromoacetate yielded compound 25. This intermediate was then condensed with 3-aminopiperidine-2,6-dione hydrochloride into an N-alkylated pomalidomide derivative 26, containing a two-carbon spacer, that allows the attachment of primary amine linkers via the formation of an amide bond (Scheme 5, steps a–e) (Zhou et al., 2018). Alternatively, the synthesis of 26 was described by treating 4-fluorothalidomide (5) with tert-butyl 2-aminoacetate and cleaving the protecting ester with TFA in a 68% overall yield (Scheme 5, step f) (Powell et al., 2018).
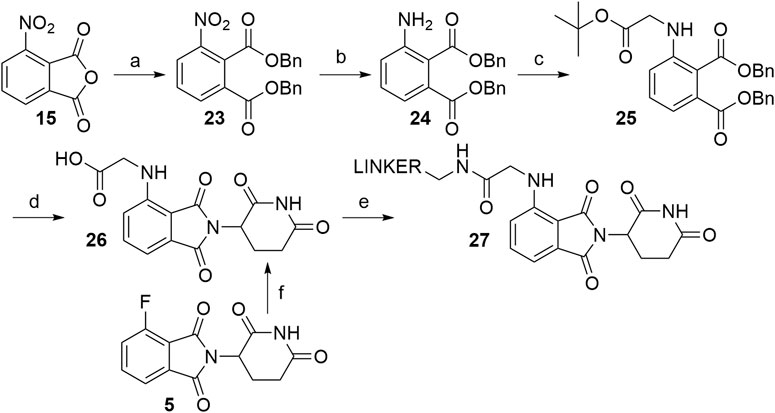
SCHEME 5. Alternative route to pomalidomide derivatives, containing a two carbon spacer. Reagents and conditions: a) i. TsOH × H2O, BnOH, 100°C, 12 h; ii. BnBr, KI, KHCO3, DMF, 100°C, 6 h, 80% yield; b) SnCl2 × 2 H2O, EtOAc, 50°C, 12 h; 90% yield; c) tert-butyl bromoacetate, DIPEA, DMF, 90°C, 12 h, 40% yield; d) i. Pd/C, H2, EtOH, rt; ii. 3-aminopiperidine-2,6-dione hydrochloride, pyridine, 110°C, overnight; iii. TFA, rt, 2 h, 40% yield for 3 steps; e) linker-NH2, HATU, DIPEA; DMF; rt, 2 h, 75% yield for linker used (Zhou et al., 2018); f) i. tert-butyl 2-aminoacetate, DIPEA, DMSO, 90°C, 24 h, 68% yield; ii. TFA, CH2Cl2, rt, overnight, quant (Powell et al., 2018).
Linker Attachment to Pomalidomide
Coupling pomalidomide (17) with the desired linkers was described by numerous authors, utilizing various acyl chloride-bearing linkers in THF under reflux for a various amount of time (Scheme 6). The exact conditions and reported yields are collected in Table 2 and may provide a better understanding of the achievable yield range (Lai et al., 2016; Buhimschi et al., 2018; Li et al., 2018; Rana et al., 2019, 6). It should be noted that a side reaction can occur, i.e., acylation of the imide nitrogen as described (Man et al., 2003). In contrast, alkylation of pomalidomide with alkyl halides is considered to be an inferior strategy for linker attachment due to the low yield and poor chemoselectivity of the reaction (Brownsey et al., 2021).
SCHEME 6. Linker attachment to pomalidomide (17) through amide bond formation. Reagents and conditions: a) linker-COCl, THF, reflux, various times and yields (Lai et al., 2016; Buhimschi et al., 2018; Li et al., 2018; Rana et al., 2019).

TABLE 2. Reaction times and yields for the conversion of compound 17 to 28 (Scheme 6).
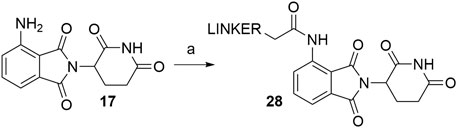
As an alternative approach to N-acylated derivatives, pomalidomide (17) was reacted with bromoacetyl chloride to obtain 29 or chloroacetyl chloride to obtain 30. Compounds 29 and 30 were then refluxed with NaN3 in acetone overnight to form azide 31 in a 84% (Chen et al., 2019) and 76% (Chen et al., 2018) yield over two steps. The azide was then reduced to amine 32, which presents an attachment point for carboxylic acid linkers via amide bond formation (Scheme 7, steps e–f) (Chen et al., 2019). On the other hand, azide 31 was also subjected to click reaction conditions together with a propargyl linker-POI ligand conjugate to form a triazole ring and final PROTAC compounds of type 34 (Scheme 7, step g) (Chen et al., 2018).
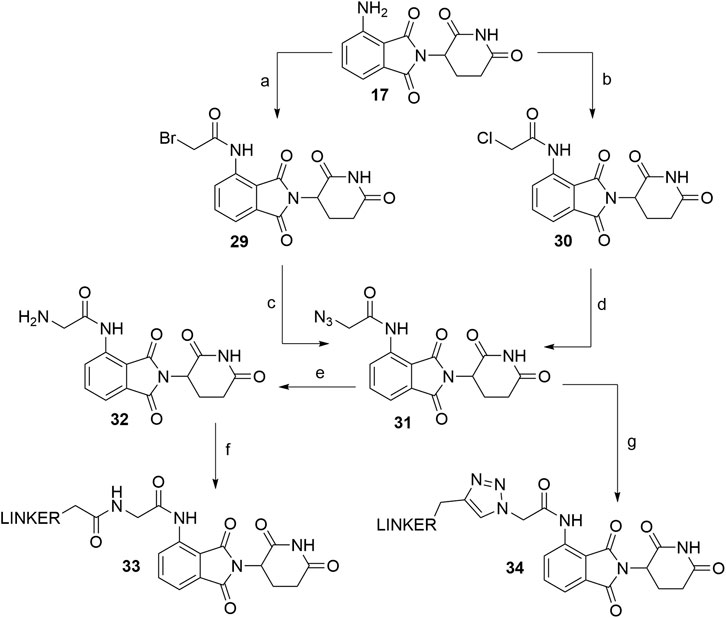
SCHEME 7. Alternate linker attachment to pomalidomide 17 through amide bond formation. Reagents and conditions: a) bromoacetyl chloride, THF; reflux, overnight, 97% yield (Chen et al., 2019); b) chloroacetyl chloride, THF, reflux, overnight, 87% yield (Chen et al., 2018); c) NaN3, acetone, reflux, overnight, 87% yield (Chen et al., 2019); d) NaN3, acetone, reflux, overnight, 76% yield (Chen et al., 2018); e) Pd/C, H2, MeOH, rt, 6 h, 64% yield; f) carboxylic acid linker, TBTU, Et3N, DMF, 50°C, 24 h, 42–59% yield for conjugates used (Chen et al., 2019); g) linker-CH2-C≡CH, CuSO4 × 5 H2O, sodium ascorbate, THF/H2O, rt, overnight, 42–80% yield for linkers used (Chen et al., 2018).
5-Aminothalidomide Derivatives
Derivatives of 5-aminothalidomide are less commonly utilized in PROTACs, despite that this substitution pattern still presents a valid option for targeting CRBN (Sun X. et al., 2019). Reagents, conditions and yields for the synthesis of 5-fluorothalidomide (36) are comparable to those in Scheme 1 for the preparation of the 4-fluoro analog. The introduction of primary amine linkers by heating the mixture of 36 and the chosen linker alongside DIPEA to obtain 5-aminothalidomide derivatives 37 has been reported (Scheme 8, step b) (Ishoey et al., 2018). Interestingly, the yields for this aromatic nucleophilic substitution were notably lower in comparison with reactions on a 4-fluoro analog. Using propargylamine as the nucleophile provided compound 38, which again offered a facile option for attaching an azide linker-POI ligand conjugate to form final PROTACs of type 39 (Wu et al., 2019) (Scheme 8, steps c–d).
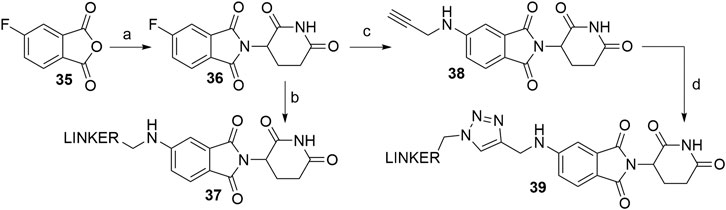
SCHEME 8. Syntheses of 5-aminothalidomide-based conjugates. Reagents and conditions: a) 3-aminopiperidine-2,6-dione hydrochloride, KOAc, AcOH, 90°C, overnight, 88% yield; b) linker-NH2, DIPEA, NMP, 90°C, overnight, 12–23% yield for linkers used (Ishoey et al., 2018); c) propargylamine, DIPEA, DMF, 90°C, 12 h, 17% yield; d) linker-N3, CuSO4 × 5 H2O, Na ascorbate, H2O/t-BuOH, rt, 16 h, 30–49% yield for linkers used (Wu et al., 2019).
An alternative synthesis of 5-aminothalidomide (42) was achieved by condensing 4-nitrophthalic anhydride (40) with 3-aminopiperidine-2,6-dione trifluoroacetate to form 41, followed by reduction to the desired product 42 (Capitosti et al., 2003) (Scheme 9, steps a–b). Interestingly, the reported yield for the condensation step is lower than those reported for the synthesis of pomalidomide precursor 16 (Scheme 4, step b) (Capitosti et al., 2003). Acyl chlorides were employed to attach the desired linker (Buhimschi et al., 2018) (Scheme 9, step c).

SCHEME 9. Synthesis of 5-aminothalidomide 42 and linker attachment through amide bond formation. Reagents and conditions: a) 3-aminopiperidine-2,6-dione trifluoroacetate, AcOH, reflux, 2 h, 58% yield; b) Pd/C, H2, rt, 20 h, 80% yield (Capitosti et al., 2003); c) linker-COCl, THF, reflux, 4 h, 44% yield for linker used (Note: yield includes a following Finkelstein reaction on the linkers) (Buhimschi et al., 2018).
4-Hydroxythalidomide-Based Ligands
4-Hydroxythalidomide with Ether-Bound Linkers
Using 3-hydroxyphthalic anhydride (44) as starting material, the condensation with the glutarimide ring is possible with various reaction conditions (Scheme 10, step a). The highest yield for the desired product 4-hydroxythalidomide (45) was described to be 96% (KOAc, AcOH, reflux) (Robb et al., 2017), whereas yields for other reported procedures span between 83 and 90% (Burslem et al., 2018; Chessum et al., 2018; Zhou et al., 2018; Jiang et al., 2019; Papatzimas et al., 2019), which is comparable to the reactions used for the synthesis of 4-fluoro- and 4-nitrothalidomide (Schemes 1 and 4). Derivatization of 45 (Scheme 10, step b) is possible by attaching iodo (Robb et al., 2017), bromo (Zhou et al., 2018), or tosylate (Rana et al., 2019) groups as linker termini to form ether bond-containing derivatives 46 Linkers with terminal hydroxyl group can be attached to 45via Mitsunobu reaction (Chessum et al., 2018).
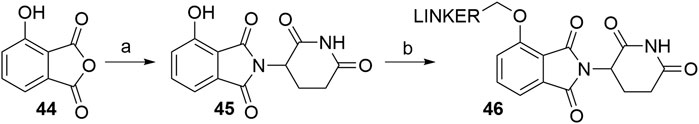
SCHEME 10. Syntheses of 4-hydroxythalidomide 45 and linker attachment. Reagents and conditions: a) 3-aminopiperidine-2,6-dione hydrochloride, KOAc, AcOH, reflux, 24 h, 96% yield (Robb et al., 2017); a) 3-aminopiperidine-2,6-dione hydrochloride, Et3N, toluene, reflux, 12 h, 90% yield (Zhou et al., 2018); a) 3-aminopiperidine-2,6-dione, pyridine, 110°C, 14 h, 88% yield (Jiang et al., 2019); a) i. 3-aminopiperidine-2,6-dione, Et3N, DMF, reflux, 4 h; ii. DCC, reflux, 72 h, 83% yield (Papatzimas et al., 2019); a) i. 3-aminopiperidine-2,6-dione hydrochloride, THF, reflux, 24h; ii. EDC, DMAP, reflux, 24 h, 84% yield (Chessum et al., 2018); a) tert-butyl (2,6-dioxopiperidin-3-yl)carbamate 2, CF3CH2OH, 150°C, 2 h, 86% yield (Burslem et al., 2018); b) reagents, conditions, and yields are collected in Table 3.

TABLE 3. Reagents, conditions, and yields for the conversion of 45 to 46 (Scheme 10, step b).
4-Hydroxythalidomide Used in In-Cell Self-Assembly CLIPTACs
Intracellular formation of PROTAC molecules is possible by the so-called in-cell self-assembly CLIPTACs, an example of which was described for the degradation of bromodomain-containing protein 4 (BRD4) and extracellular signal-regulated kinase 1/2. In this case, 4-hydroxythalidomide was tagged with tetrazine, while the ligands for the POIs were tagged with trans-cyclo-octene. The combination of the two precursors underwent a bio-orthogonal click reaction to form the active chimera intracellularly. Utilizing this concept might overcome the cellular permeability issues of some PROTACs since the two small precursor molecules have a higher ability to pass through cellular membranes than one large compound (Lebraud et al., 2016).
As presented in Scheme 11, methanolysis and subsequent methylation of 3-hydroxyphthalic anhydride (44) yielded dimethyl ester 47, which was then alkylated under Mitsunobu conditions leading to O-alkylated derivative 48. This represents an alternative to most syntheses, in which the linker attachment is performed only after the thalidomide portion of the molecule is fully assembled. Basic reaction conditions resulted in the hydrolysis of the methyl esters of 48, followed by the condensation with the glutarimide ring and tert-butyl ester cleavage under acidic conditions to obtain 4-O-alkylated thalidomide derivative 49. Amide coupling for the attachment of the tetrazine moiety yielded compound 50, which was finally reacted intracellularly with trans-cyclo-octene, bound to the POI ligand (Lebraud et al., 2016).
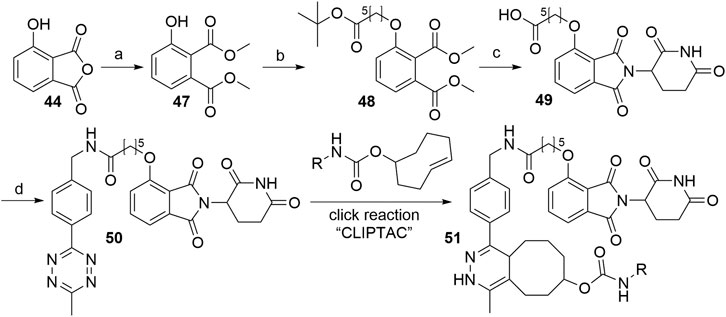
SCHEME 11. Synthesis of CLIPTAC component 50. Reagents and conditions: a) i. MeOH, reflux, 3 h; ii. MeI, NaHCO3, DMF, 55°C, 3h, 94% yield; b) tert-butyl 6-hydroxyhexanoate, PPh3, DIAD, THF, rt, 18 h; c) i. 1 M NaOH, THF/MeOH, rt, 2 h; ii. 3-aminopiperidine-2,6-dione hydrochloride, pyridine, 110°C, 17 h; iii. TFA, rt, 3 h, 10% yield over 4 steps; d) methyltetrazine amine, HATU, DIPEA, DMF, rt, 2 h, 57% yield (Lebraud et al., 2016).
4-Hydroxythalidomide Derivatives With a Two-Carbon Spacer
Alkylating the 4-hydroxyl group of 45 with tert-butyl bromoacetate or benzyl glycolate and subsequent removal of the protecting group produces compound 53, a standard building block, containing a flexible ‘spacer’, which is ready for linker attachment via an amide bond (Scheme 12). The highest yield of over two steps to obtain 53 was reported to be 78% (Remillard et al., 2017). In another study, a yield of only 41% was reached, primarily due to the low conversion rate of Boc-protected derivative 52a to 53 using formic acid (Chessum et al., 2018). A synthesis of 53 was reported by using benzyl glycolate and Mitsunobu conditions, which yielded the desired product 52b in 73% (Lohbeck and Miller, 2016). Coupling reaction yields span between 34 and 85% for a selection of different linkers (Table 4) (Lohbeck and Miller, 2016; Remillard et al., 2017; Chessum et al., 2018; Zhou et al., 2018, Fischer et al., 2014). Importantly, the selective alkylation of the phenolic group was confirmed by means of HMBC spectra (Lohbeck and Miller, 2016). Alternatively, a 2-chloro-N-acetamide-bearing linker was attached onto phenol 47 to obtain an O-alkylated ester 55. This compound was then first converted to 56, followed by condensation with 3-aminopiperidine-2,6-dione to form 54 (Scheme 12, steps d–f). The overall yield of this reaction sequence was approximately 16% (Fischer et al., 2014).
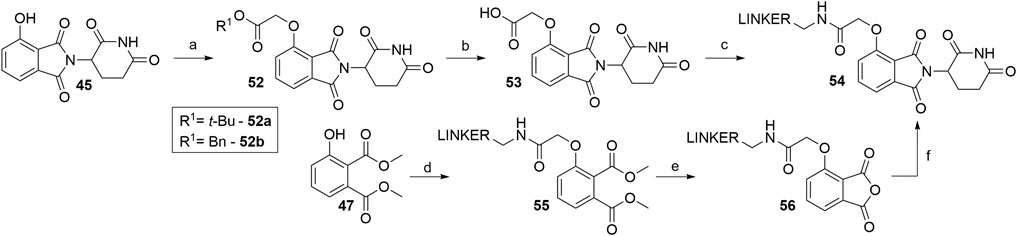
SCHEME 12. Syntheses of 4-hydroxythalidomide derivatives with a CO-CH2 spacer and amide bond-connected linker. Reagents and conditions: a) tert-butyl 2-hydroxyacetate, PPh3, DTBAD, THF, 0°C to rt, overnight, 75% yield (Chessum et al., 2018); a) tert-butyl bromoacetate, KI, KHCO3, DMF, 60°C, 12 h, 80% yield (Zhou et al., 2018); a) tert-butyl bromoacetate, K2CO3, DMF, rt, 2 h, 93% yield (Remillard et al., 2017); a) benzyl glycolate, PPh3, DIAD, THF, 0°C to rt, 18 h, 73% yield (Lohbeck and Miller, 2016); b) HCO2H, CH2Cl2, 40°C, overnight, 54% yield (Chessum et al., 2018); b) TFA, rt, 4 h, 84% yield (Remillard et al., 2017); b) Pd/C, H2, MeOH, rt, 3 h, quant. (Lohbeck and Miller, 2016); c) reagents, conditions, and yields are collected in Table 4; d) linker-CH2-NH-CO-CH2Cl, Cs2CO3, MeCN, 80°C, 12 h, 70% yield for linker used; e) 3 M NaOH, EtOH, 80°C, 2 h; f) 3-aminopiperidine-2,6-dione, pyridine, reflux, 12 h, 23% yield for linker used (Note: yield includes a following Boc deprotection on the linker) (Fischer et al., 2014).

TABLE 4. Reagents, conditions, and yields for the conversion of 53 to 54 (Scheme 12, step c).
Alkyl-Connected Thalidomide Derivatives
PROTACs that utilize alkyl-linked thalidomide derivatives to hijack CRBN include both the alkyne-containing linkers 59, as well as the reduced analogs 60, which provide either a more rigid or a more flexible connection between the linker and the ligase ligand (Scheme 13) (Zhou et al., 2018; Su et al., 2019). By following the standard procedure for condensation (NaOAc, AcOH, reflux) of the glutarimide ring with 3-bromophthalic anhydride (57), 4-bromothalidomide (58) was prepared in a straightforward fashion (Zhou et al., 2018). Propargyl-containing linkers were then attached employing Sonogashira coupling to give derivatives 59 in 72–89% yield (Zhou et al., 2018; Su et al., 2019). The alkyne group was then efficiently reduced using Pd/C-catalyzed hydrogenation (Scheme 13, steps a–c) (Zhou et al., 2018). In place of 4-bromothalidomide (58), the iodo analog 64 could be used, which was synthesized by subjecting 2,3-dimethylaniline (61) to a Sandmeyer-type iodination and potassium permanganate-mediated oxidation to yield 62. This was then treated with acetic anhydride (forming 63) and finally combined with the glutarimide moiety to obtain 64 (Scheme 13, steps d–f) (Stewart et al., 2010; Yeung et al., 2011). Relatively lower yields were noted for the Sonogashira coupling using 4-iodothalidomide (64) (Stewart et al., 2010), in comparison to reports by other authors for reactions with 4-bromothalidomide (58).
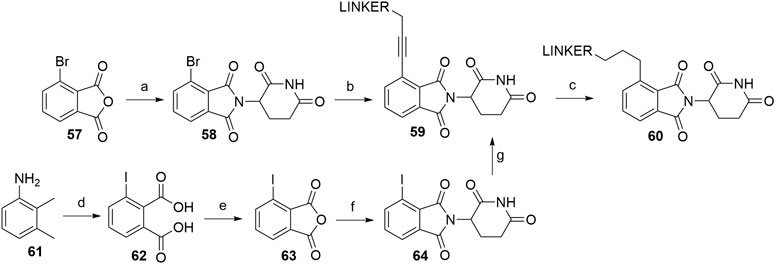
SCHEME 13. Syntheses of thalidomide derivatives with alkyl-connected linkers. Reagents and conditions: a) 3-aminopiperidine-2,6-dione hydrochloride, NaOAc, AcOH, 140°C, 12 h, 80% yield; b) linker-CH2-C≡CH, Pd(PPh3)2Cl2, CuI, Et3N, DMF, 70°C, 3 h, 72% yield for linker used (Zhou et al., 2018); b) linker-CH2-C≡CH, Pd(PPh3)2Cl2, CuI, Et3N, THF, 70°C, 12 h, 87–89% for linkers used (Su et al., 2019); c) Pd/C, H2, EtOH, rt, 12 h, (80%. Note: yield includes a following Boc deprotection on the linker) (Zhou et al., 2018); d) i. HCl, NaNO2, KI, H2O, -15 to 55°C, 5 min, then rt, 16 h, 66% yield; ii. KMnO4, H2O, 80°C, 4 days, 41% yield; e) Ac2O, reflux, 3 h, 68% yield; f) 3-aminopiperidine-2,6-dione trifluoroacetate, Et3N, THF, reflux, 24 h, 56% yield (Stewart et al., 2010); f) 3-aminopiperidine-2,6-dione trifluoroacetate, Et3N, THF, reflux, 88% yield (Yeung et al., 2011); g) linker-CH2-C≡CH, Pd(PPh3)2Cl2, CuI, DIPEA, THF, reflux, 4–22 h, 21–78% yield for linkers used (Stewart et al., 2010).
Lenalidomide-Based Ligands
Utilizing lenalidomide-based ligands poses some advantages over using thalidomide and its derivatives to hijack CRBN, as the absence of one phthalimide carbonyl group results in a decreased TPSA, better physicochemical properties, and a higher metabolic and chemical stability (Hoffmann et al., 2013). Additionally, some lenalidomide-based PROTACs displayed a higher level of induced target degradation than their pomalidomide-based counterparts (Qiu et al., 2019). Compound 66 was obtained by bromination of the starting nitrobenzene derivative 65 with N-bromosuccinimide (NBS) in CCl4 using azobisisobutyronitrile (AIBN) as an initiator of radical bromination, with reported yields of 88% (Balaev et al., 2013) and 49% (Chaulet et al., 2011). An alternative, high-yielding (98%) and green approach for this bromination was presented, where the reaction was carried out in a non-halogenated solvent, i.e., methyl acetate (Ponomaryov et al., 2015). The following condensation with the glutarimide ring was achieved by the addition of a base and heating the solution at 50–55°C, yielding 57% (Et3N in MeCN; Chaulet et al., 2011), 86% (Et3N in DMF; Balaev et al., 2013), and 89% (K2CO3 in NMP, Ponomaryov et al., 2015) of the desired nitro product 67. Optimal conditions for the subsequent reduction to lenalidomide (68), i.e., a Pd/C-catalyzed hydrogenation, were described (Chaulet et al., 2011). Alternatives include using Pd(OH)2 (Balaev et al., 2013) or iron-ammonium chloride (Ponomaryov et al., 2015), but both procedures led to the product in a lower yield. Selective derivatization of the 4-amino position of lenalidomide (68) were performed with of bromo or iodo linkers and DIPEA in NMP at 110°C for 12 h to yield derivatives 69 (Scheme 14) (Qiu et al., 2019). Carboxylic acid linkers were attached via an amide bond to form derivatives 70 (Zhang F. et al., 2020).
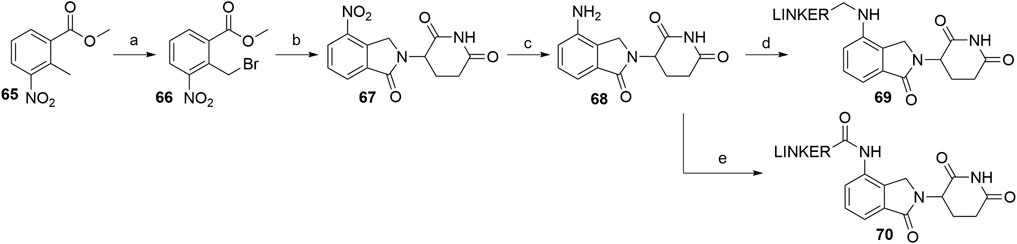
SCHEME 14. Syntheses of lenalidomide (68), N-alkylated lenalidomide derivatives 69, and N-acylated derivatives 70. Reagents and conditions: a) NBS, AIBN, CCl4, reflux, 8 h, 88% yield (Balaev et al., 2013); a) NBS, AIBN, MeOAc, reflux, 18 h, 98% yield (Ponomaryov et al., 2015); a) NBS, AIBN, CCl4, reflux, 24h, 49% yield (Chaulet et al., 2011); b) 3-aminopiperidine-2,6-dione, Et3N, DMF, 50°C, 15 h, 86% yield (Balaev et al., 2013); b) 3-aminopiperidine-2,6-dione hydrochloride, K2CO3, NMP, 35°C for 1 h, then 55 °C for 18 h, 89% yield (Ponomaryov et al., 2015); 3-aminopiperidine-2,6-dione, Et3N, MeCN, 55°C, 18 h, 57% yield (Chaulet et al., 2011); c) Pd(OH)2, H2, dioxane, 50–60°C, 78% yield (Balaev et al., 2013); c) Fe, NH4Cl, H2O, EtOH, 80°C, 4 h, 75% yield (Ponomaryov et al., 2015); c) Pd/C, H2, MeOH, DMF, quant. (Chaulet et al., 2011); d) linker-I or linker-Br, DIPEA, NMP, 110°C, 12 h, 48–84% yield for linkers used (Note: yield includes a following Boc deprotection on the linker) (Qiu et al., 2019); e) linker-CO2H, pyridine, POCl3, MeCN, rt, 3 h, about 40% yield for linkers used (Zhang et al., 2020a).
Lenalidomide-based ligands are also obtainable through derivatization of 4- and 5-bromo substituted analogs. The synthesis of compound 74 was accomplished from starting with benzoic acid derivative 71, which was first converted into a methyl ester 72 and then brominated using NBS and AIBN in MeCN to yield compound 73 (Hansen et al., 2021). The following condensation with the glutarimide ring was achieved by the addition of a base, specifically Et3N (Hansen et al., 2021) or DIPEA (Hayhow et al., 2020), and 5-bromo lenalidomide derivative 74 was obtained. Subsequent derivatization was possible through Buchwald-Hartwig amination, allowing the attachment of sterically hindered linkers containing a piperazine moiety, which are commonly used in numerous latest PROTACs (Hayhow et al., 2020; Hansen et al., 2021, 9000; Crew et al., 2018a; Crew et al., 2018b). Additionally, the Buchwald-Hartwig protocol with various secondary amines gave yields ranging from 21 to 87% (Scheme 15) (Hayhow et al., 2020).
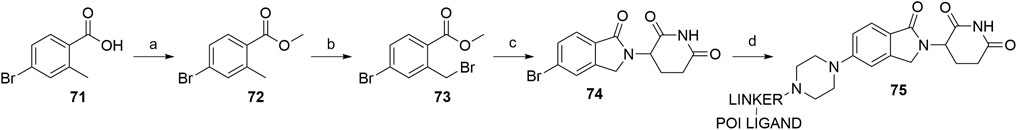
SCHEME 15. Synthesis of 5-amino derivatives with sterically hindered linker attachment points. Reagents and conditions: a) MeOH, H2SO4, 65°C, 18 h, 95% yield; b) NBS, AIBN, MeCN, 85°C, 18 h, 66% yield; c) 3-aminopiperidine-2,6-dione hydrochloride, Et3N, rt, 25 h, 44% yield (Hansen et al., 2021, 9000); c) 3-aminopiperidine-2,6-dione hydrochloride, DIPEA, MeCN, 80°C, 48 h, 85% yield; d) POI ligand-linker-piperazine conjugate, Pd-PEPPSI-IHeptCl, Cs2CO3, dioxane, 100°C, 3.5 h, 27% yield for linker-POI ligand conjugate used (Hayhow et al., 2020).
Alkyl-Connected Lenalidomide Derivatives
Similarly to alkyl-connected thalidomide derivatives (“Alkyl-Connected Thalidomide Derivatives” Section), their lenalidomide analogs are used in PROTACs with both the alkyne-type connection (79) and the reduced linkage (80) (Scheme 16) between the ligase ligand and linker (Su et al., 2019; Wang et al., 2019). The synthesis of these compounds was nicely described recently (Sun Y. et al., 2019). Methyl 3-bromo-2-methylbenzoate (76) was subjected to radical bromination using NBS and AIBN in CHCl3 to yield 77 (Sun Y. et al., 2019). A higher yield of 90% was reported for a similar radical bromination reaction, where benzene was used as the solvent (Zhou et al., 2018). After condensation with the glutarimide ring to yield bromo-lenalidomide (78), linker attachment was achieved through the Sonogashira cross-coupling reaction to afford compounds 79, with yields spanning between 41 and 81%, depending on the linker used (Sun Y. et al., 2019; Li et al., 2019; Wang et al., 2019). The reduction to 80 was carried out through Pd/C-catalyzed hydrogenation (Sun Y. et al., 2019; Li et al., 2019) (Scheme 16).
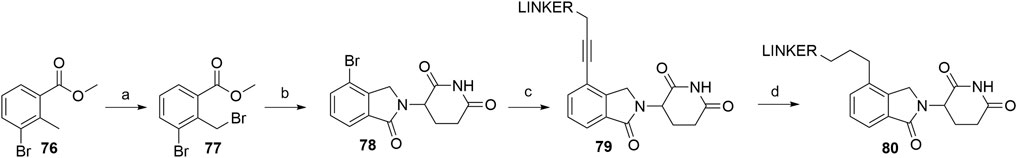
SCHEME 16. Syntheses of lenalidomide derivatives with alkyl-connected linkers. Reagents and conditions: a) NBS, AIBN, CHCl3, reflux, 5 h, 77% yield (Note: the yield was calculated from the following step) (Sun et al., 2019b); a) NBS, dibenzoyl peroxide, benzene, reflux, 6 h, 90% yield (Zhou et al., 2018); b) 3-aminopiperidine-2,6-dione hydrochloride, Et3N, THF, 80°C, 6 h, 69% yield (Sun et al., 2019b); b) 3-aminopiperidine-2,6-dione hydrochloride, NaOAc, AcOH, 140°C, 12 h, 88% yield (Zhou et al., 2018); c) reagents, conditions, and yields are collected in Table 5; d) Pd/C, H2, MeOH/DMF, rt, 12 h, 63% for linker used (Sun et al., 2019b); d) Pd/C, H2, EtOH, rt, 2 h, 85% for linker used (Li et al., 2019).

TABLE 5. Reagents, conditions, and yields for the conversion of 78 to 79 (Scheme 16, step c).
Alkyl-connected lenalidomide analogs can also be synthesized via the Suzuki cross-coupling reaction (Xiao et al., 2020). The amino group of lenalidomide (68) group was converted into an arylboronic ester 81 through a metal-free pinacol borylation reaction under Sandmeyer-type transformation (Scheme 17). Compound 82 was obtained through oxidative hydrolysis and then joined with tert-butyl bromoacetate by using Pd(PPh3)4 as a coupling catalyst for Suzuki cross-coupling. Ester hydrolysis afforded compound 83, which is suitable for amine linker attachment to form compounds 84 (Xiao et al., 2020).
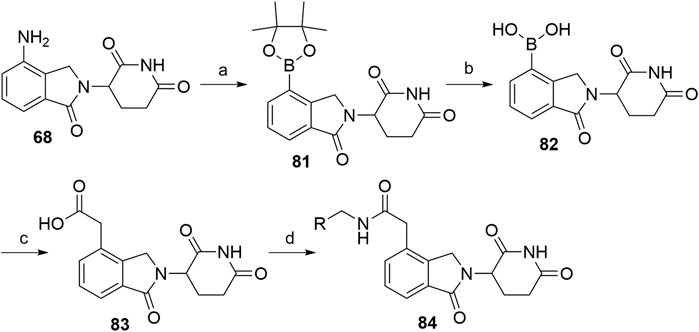
SCHEME 17. Alternative syntheses of lenalidomide derivatives with alkyl-connected linkers. Reagents and conditions: a) t-BuONO, bis(pinacolato)diboron, dibenzoyl peroxide, MeCN, rt, 4 h, 76% yield; b) i. NaIO4, THF, H2O, rt, 2 h; ii. 1 M HCl, rt, 18 h, 67% yield; c) i. tert-butyl bromoacetate, Pd(PPh3)4, CsF, DME, CH2Cl2, reflux, 18 h, 46% yield; ii. TFA, CH2Cl2, rt, 2 h, quant.; d) linker-NH2, HATU, DIPEA, DMF, rt, overnight, 66–89% yield for amines used (Xiao et al., 2020).
It should be mentioned here that synthetic approaches towards hydroxyl analogs of lenalidomide were disclosed recently (Hansen et al., 2020). Although these compounds were not utilized in PROTACs, the syntheses might prove very useful in further research on lenalidomide-derived degraders.
Tricyclic Imide Moiety
The tricyclic imide moiety 86 (Scheme 18) was used in a single PROTAC, which targeted the NS3 protein in virus hepatitis C. Compound 86 had a higher binding affinity for CRBN and did not result in the degradation of neo-substrates, such as IKZF1 and IKZF3 (de Wispelaere et al., 2019). The condensation of 1,8-naphthalic anhydride (85) with the glutarimide ring was performed microwave-assisted (Burslem et al., 2018). 5-Hydroxy derivative 87 was condensed to 88 in a similar way, enabling halogen linker attachment to yield an ether bond-connected linker (Gray et al., 2020).
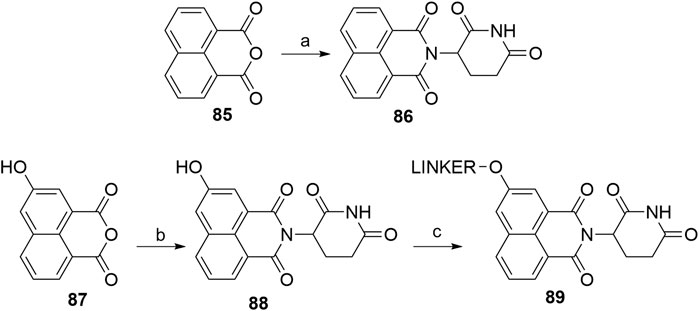
SCHEME 18. Syntheses of tricyclic imide moiety 86 and its 5-hydroxyl derivative 88. Reagents and conditions: a) tert-butyl (2,6-dioxopiperidin-3-yl)carbamate 2, CF3CH2OH, 150°C, 6 h, MW, 80% yield (Burslem et al., 2018); b) 3-aminopiperidine-2,6-dione hydrochloride, THF, 75°C, 1 h (Note: yield not given); c) linker-Br, K2CO3, DMSO, 50°C, overnight, 22% yield for linker used (Gray et al.,. 2020).
Cereblon PHOtochemically TArgeting Chimeras
A recent advance in the field of targeted protein degradation are photoswitchable PROTACs or PHOTACs (PHOtochemically TArgeting Chimeras). In addition to POI and E3 ligase ligands, these compounds possess a photoswitch, which allows them to be reversibly activated with different wavelengths of light, but not display relevant activity in the deactivated conformation. This enables the utilization of PHOTACs as precision therapeutics, capable of avoiding undesired systemic toxicity (Reynders et al., 2020). To date, the strategy has been described twice (Jin et al., 2020; Reynders et al., 2020). Two examples of active PHOTACs (Reynders et al., 2020) are presented in Scheme 19, each incorporating azobenzene photoswitches, with the trans configuration presenting the resting, inactive state. The active cis isomer can be obtained by irradiation with light of specific wavelengths. In one of the cases, the authors incorporated the azobenzene switch directly to lenalidomide (68) to give compound 90. They then derivatized the hydroxyl group to yield 91, which enabled amine linker attachment via an amide bond. Alternatively, compound 53, which contains a flexible two carbon spacer, was coupled with 4,4’-azodianiline to give 93, onto which a POI ligand-linker conjugate with a carboxylic acid was attached to yield 94 (Reynders et al., 2020).
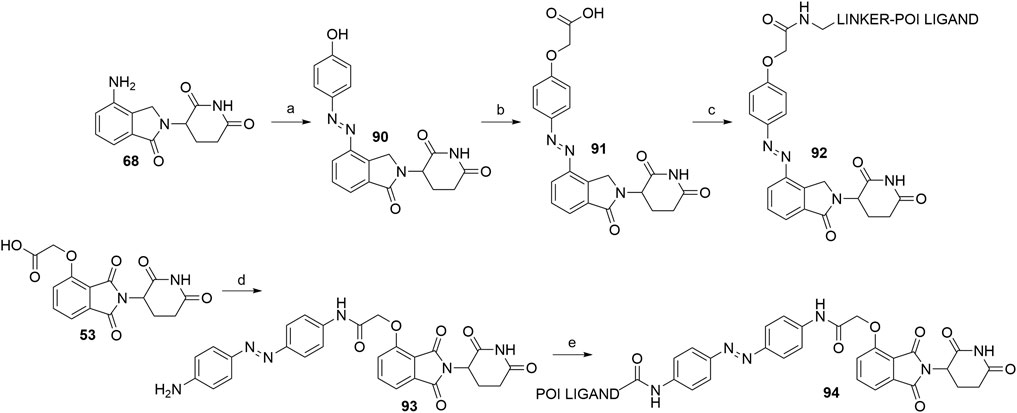
SCHEME 19. Syntheses of CRBN-targeting PHOTACs 92 and 94. Reagents and conditions: a) i. 1 M HCl, HBF4, 2 M NaNO2, 0°C, 1 h; ii. phenol, NaHCO3, Na2CO3, MeOH, H2O, 0°C, 1 h, 86% yield; b) i. tert-butyl bromoacetate, K2CO3, DMF, rt, 2.5 h; ii. TFA, CH2Cl2, rt, 2 h, 56% yield; c) linker-NH2, HATU, DIPEA, DMF, rt, 12 h, 92–99% yield for linkers used; d) 4,4′-azodianiline, HOBt, PyBOP, Et3N, THF, rt, overnight, 79% yield; e) POI ligand-CO2H, HATU, DIPEA, DMF, rt, overnight, 65% yield for the POI ligand used (Reynders et al., 2020).
Caged Cereblon Ligands
Apart from PHOTACs, an alternative option that enables the control of the location and timing of targeted proteolysis is incorporating a photocleavable group into a motif that is essential for binding to the E3 ligase (Xue et al., 2019; Naro et al., 2020). The imide moiety of thalidomide’s glutarimide ring thus presents an ideal position for attaching a photolabile moiety, such as the nitroveratryloxy-carbonyl (Xue et al., 2019) or 6-nitropiperonyloxymethyl (NPOM) group (Naro et al., 2020). In the former study, the imide moiety of starting material 95 was derivatized to form 96 prior to the linker and POI ligand attachment (Xue et al., 2019), while in the latter study, the NPOM group was attached to a conjugate of 4-hydroxythalidomide and a linker (54) to form 98. The POI ligand was then coupled via an amide bond to obtain final PROTAC 99 (Naro et al., 2020) (Scheme 20).
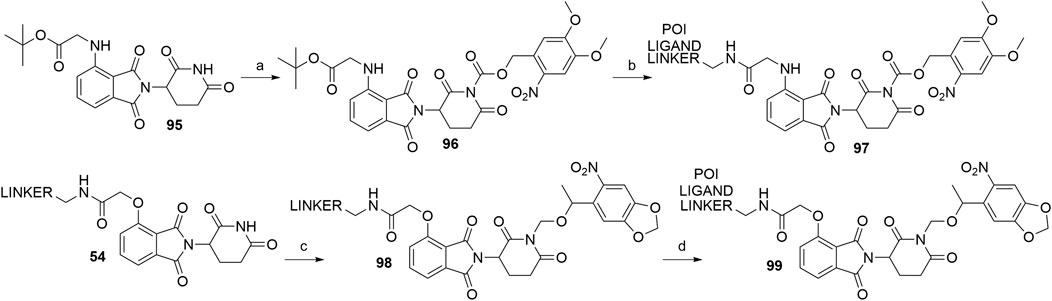
SCHEME 20. Syntheses of caged CRBN degraders 97 and 99. Reagents and conditions: a) i. NaHMDS, CH2Cl2, THF, –80°C to –30°C; ii. 4,5-dimethoxy-2-nitrobenzyl chloroformate, -30°C to rt, 27% yield; b) i. TFA, CH2Cl2, rt, 2 h; ii. POI ligand-linker-NH2, HATU, DIPEA, DMF, rt, overnight, 36% yield for conjugate used (Xue et al., 2019); c) 5-(1-(chloromethoxy)ethyl)-6-nitrobenzo[d][1,3]dioxole, DBU, DMF, 0°C to rt, overnight, 80% yield for conjugate used; d) POI ligand-CO2H, HATU, DIPEA, DMF, 0°C to rt, 16 h, 73% yield for conjugate used (Naro et al., 2020).
Statistical Overview of Utilized Cereblon Ligands
Using data extracted from PROTAC-DB (Weng et al., 2021) (http://cadd.zju.edu.cn/protacdb/, as of the February 26, 2021) a statistical overview was done to determine the frequency of various CRBN ligands and linker attachment options used in PROTAC compounds (Figure 3). An overwhelming majority of PROTACs incorporated an N-alkylated pomalidomide as the E3 ligase ligand, while an acylated pomalidomide was about as commonly represented as 4-hydroxythalidomide derivatives. Interestingly, the 5-amino derivative was utilized in around 5% of PROTACs. Lenalidomide analogs, namely 4-acylated derivatives and alkyl-connected lenalidomide derivatives were similarly frequent at 8 and 7%, respectively.
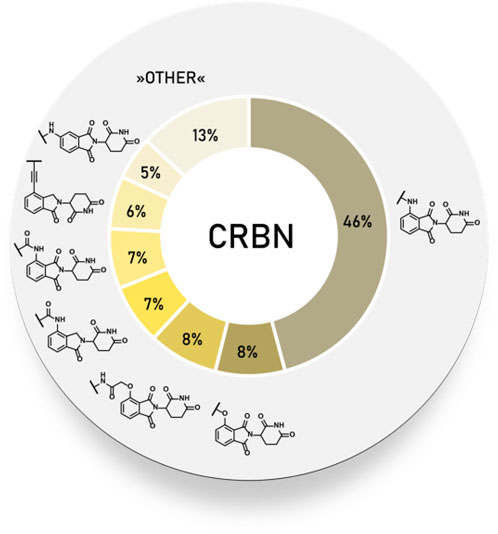
FIGURE 3. Frequency of CRBN ligands used in PROTAC compounds.
Von Hippel–Lindau
The VHL protein is a part of the multiprotein complex, along with elongin B and C, cullin 2 and Rbx-1, which possesses an E3 ubiquitin ligase activity. Within the complex, VHL folds into two domains, one of which is responsible for the binding of specific substrates (Czyzyk-Krzeska and Meller, 2004), most notably the hypoxia-inducible factor (HIF)-1α, leading to its ubiquitination and proteasomal degradation. Early VHL-targeting PROTACs utilized 5-7 amino acid long sequences derived from HIF-1α protein (Schneekloth, et al., 2004; Lee et al., 2007), due to the lack of small-molecule VHL ligands. Peptidomimetic binding moieties with high VHL-binding affinity have been developed in 2012 and widely been used in PROTACs ever since (Buckley et al., 2012a; Buckley et al., 2012b; Van Molle et al., 2012). However, a few more recently reported PROTACs still employ a hydroxylated pentapeptide for hijacking VHL (Wang et al., 2016).
VHL is similarly to CRBN extensively targeted with PROTAC compounds and has been successfully utilized for degrading more than 20 different proteins (Sun X. et al., 2019). The development of VHL ligands with a solid binding affinity included the analysis of co-crystal structures, which helped to locate the solvent-exposed regions. Accordingly, positions that could be derivatized without negatively affecting the critical affinity were identified (Bondeson et al., 2015; Buckley et al., 2015; Zengerle et al., 2015; Maniaci et al., 2017). These include connection via an amide bond after the amino acid tert-leucine (A), phenolic linkage point at the benzene ring (B), link via a thioether at the left-hand side amino acid (C), and via the benzylic methylene group (D) (Figure 4).
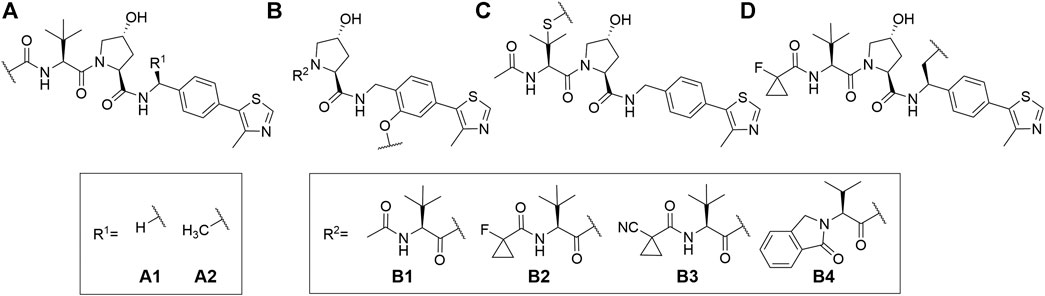
FIGURE 4. VHL ligands found in PROTACs. Linker attachment options are represented with curly bonds and are: (A)via an amide bond after tert-leucine; (B) phenolic linkage point at the benzene ring; (C)via a thioether at the left-hand side amino acid; (D)via the benzylic methylene group.
A: Connection via an Amide Bond after tert-Leucine
von Hippel–Lindau Ligand 1
The key intermediate for the synthesis of VHL ligand 107 (i.e., VHL A1) is compound 105, which can be formed by using a Pd-catalyzed arylation of 4-bromobenzonitrile 103 and subsequent reduction of the nitrile group of 104, for which an array of methods with varying yields has been published (Buckley et al., 2012a; Galdeano et al., 2014; Crew et al., 2018). LiAlH4 was used which resulted in the desired product 105 in 63% yield (Crew et al., 2018), while a NaBH4-CoCl2 combination led to a 29% conversion at 0°C (Galdeano et al., 2014) and 73% at 4°C (Buckley et al., 2012a). Alternatively, a synthetic strategy was reported comprising the conversion of 4-bromobenzylamine (100) into compound 105 in three steps with an overall yield of 18% (Scheme 21, steps a–c) (Buckley et al., 2012a). With compound 105 in hand, the subsequent reaction steps were very straightforward. For example, standard coupling conditions enabled the formation of an amide bond with Boc-l-hydroxyproline, followed by acid-mediated cleavage of the Boc protecting group, which afforded 106. Finally, an amide bond with Boc-l-tert-leucine was formed, and Boc deprotection of the terminal amine yielded compound 107, which allowed derivatization with carboxylic acid linkers to give conjugates 108 (Scheme 21, steps f–h) (Galdeano et al., 2014; Crew et al., 2018; Steinebach et al., 2020a).
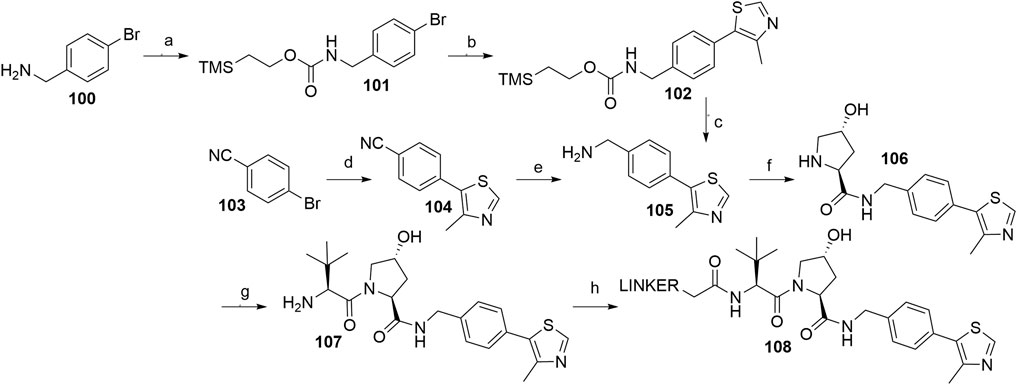
SCHEME 21. Syntheses of VHL ligand 107. Reagents and conditions: a) Teoc-OSu, Et3N, DMF, H2O, rt, 16 h, 84% yield; b) 4-methylthiazole-5-carboxylic acid, Pd(P(tBu)3)2, Bu4NCl × H2O, Cs2CO3, DMF, MW, 170°C, 8 min, 22% yield; c) TBAF, MeCN, rt, 18 h, 96% yield (Buckley et al., 2012a); d) 4-methylthiazole, KOAc, Pd(OAc)2, DMA, 150°C, 5 h, 91% yield (Crew et al., 2018); d) 4-methylthiazole, KOAc, Pd(OAc)2, DMA, 150°C, 5 h, 97% yield (Galdeano et al., 2014); d) 4-methylthiazole, KOAc, Pd(OAc)2, DMA, 150°C, 19 h, 99% yield (Buckley et al., 2012a); e) LiAlH4, THF, reflux, 5 h, 63% yield (Crew et al., 2018); e) NaBH4, CoCl2, MeOH, 0°C, 90 min, 29% yield (Galdeano et al., 2014); e) NaBH4, CoCl2, MeOH, 4°C, 90 min, 73% yield (Buckley et al., 2012a); f) i. Boc-Hyp-OH, HATU, DIPEA, DMF, rt, 2 h; ii. HCl, MeOH, rt, 2 h, 41% yield (Crew et al., 2018); f) i. Boc-Hyp-OH, HATU, DIPEA, DMF, rt, 30 min; ii. TFA, CH2Cl2, rt, 30 min, 93% yield (Galdeano et al., 2014); g) i. Boc-Tle-OH, HATU, DIPEA, DMF, rt, 30 min; ii. TFA, CH2Cl2, rt, 30 min, 96% yield (Galdeano et al., 2014); h) linker-CO2H, HATU, DIPEA, DMF, rt, 2 h, 20% yield for linker used (Crew et al., 2018); h) linker-CO2H, HATU, DIPEA, DMF, rt, 16 h, 59–90% yield for linkers used (Steinebach et al., 2020a).
Inverting the configuration at the hydroxyproline moiety results in a loss of binding affinity for VHL, and such modified compounds are mostly incorporated into negative control VHL-based PROTACs (Raina et al., 2016). Using N-Boc-cis-4-hydroxy-l-proline (109) in place of Boc-l-hydroxyproline and coupling with 105 yielded the VHL non-binding ligand 111 (Scheme 22) (Crew et al., 2018).
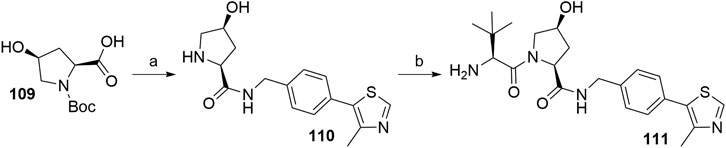
SCHEME 22. Synthesis of VHL non-binding ligand 111. Reagents and conditions: a) i. 105, HATU, DIPEA, DMF, rt, overnight; ii. HCl, MeOH, rt, 2 h, 53% yield; b) i. Boc-Tle-OH, HATU, DIPEA, DMF, rt, 3 h; ii. HCl, dioxane, rt, 3 h, 50% yield (Crew et al., 2018).
The most efficient procedure for the synthesis of VHL ligand 107 started from 4-bromobenzylamine (100) (Scheme 23) which was first Boc-protected to 112 and then underwent the Heck reaction and Boc deprotection to give crucial intermediate 105 (Han et al., 2019). Alternatively, 112 could be prepared through reductive amination of 4-bromobenzaldehyde (Steinebach et al., 2020b). This new synthetic sequence does also allow the introduction of substituents to the central phenylene unit. The final ligand 107 was prepared in a convergent manner by coupling 105 with the dipeptide 116, which was prepared from hydroxyproline methyl ester (115) and N-Boc-l-tert-leucine. Incorporating an element of convergent synthesis helped to increase the overall yield (Han et al., 2019).
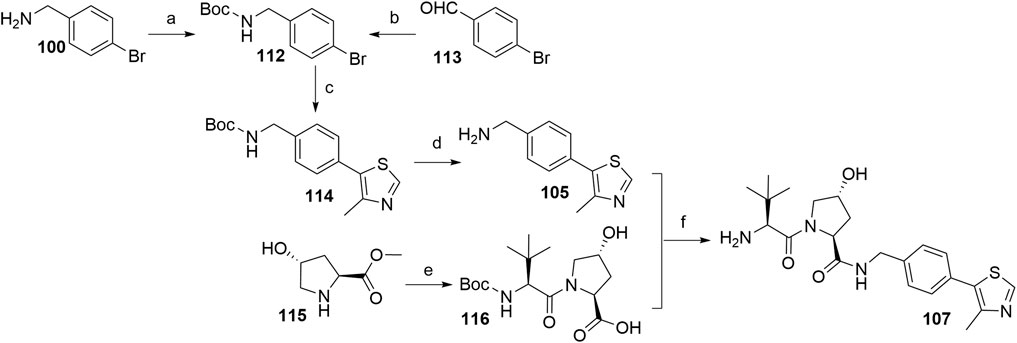
SCHEME 23. Alternative synthesis of VHL ligand 107. Reagents and conditions: a) (Boc)2O, NaHCO3, EtOAc/H2O, rt, 1 h, 95% yield (Han et al., 2019); b) tert-butyl carbamate, Et3SiH, TFA, CH2Cl2, MeCN, rt, overnight, 83% yield (Steinebach et al., 2020b); c) 4-methylthiazole, Pd(OAc)2, KOAc, DMF, 90°C, 2 h, 85% yield; d) TFA, CH2Cl2, rt, 30 min, 95% yield; e) i. Boc-Tle-OH, HATU, DIPEA, DMF, rt, overnight; ii. LiOH, THF/H2O, 85% yield; f) i. HATU, DIPEA, DMF, rt, overnight; ii. TFA, CH2Cl2, rt, 30 min, 88% yield (Han et al., 2019).
von Hippel–Lindau Ligand 2
In the course of design and optimization of VHL ligands, an introduction of an (S)-methyl group on the benzylic carbon atom has improved the binding affinity to VHL. Namely, the potency of the methyl-substituted ligand is three times better than of the non-substituted ligand 107 (Han et al., 2019). Synthesis of the key intermediate 120 is analogous to the synthetic route described in Scheme 23, using (S)-(-)-4-bromo-α-methylbenzylamine (117) as starting material. The left-hand side dipeptide moiety could be assembled either by convergent synthesis (Raina et al., 2016) or linear synthesis (Hu et al., 2019) to yield 122, which was then ready for attaching carboxylic acid linker-POI ligand conjugates by a coupling reaction to give derivatives 123 (Scheme 24) (Raina et al., 2016; Hu et al., 2019).
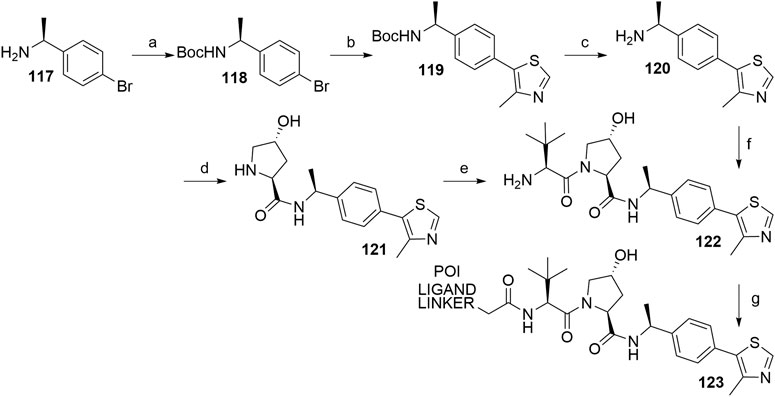
SCHEME 24. Synthesis of VHL ligand 122 with an (S)-methyl group on the benzylic carbon atom of the molecule. Reagents and conditions: a) (Boc)2O, NaHCO3, H2O/EtOAc, rt, 2 h, 99% yield; b) 4-methylthiazole, Pd(OAc)2, KOAc, DMA, 90°C, 18 h, 82% yield; c) 4 M HCl in MeOH, rt, 3 h, 85% yield (Raina et al., 2016); c) 4 M HCl in dioxane, MeOH, rt, 12 h, 100% yield; d) i. Boc-Hyp-OH, HATU, DIPEA, DMF, 0°C to rt, 12 h; ii. 4 M HCl in dioxane, MeOH, rt, 12 h, 80% yield; e) i. Boc-Tle-OH, HATU, DIPEA, DMF, 0°C to rt, 12 h; ii. 4 M HCl in dioxane, MeOH, rt, 12 h, 80% yield (Hu et al., 2019); f) i. 116, HATU, DIPEA, THF, rt, 2 h; ii. 4 M HCl in MeOH, rt, 3 h, 72% yield; g) POI ligand-linker-CO2H, HATU, DIPEA, DMF, 0°C to rt, 20 min, 32% yield for conjugate used (Raina et al., 2016); g) POI ligand-linker-CO2H, HATU, DIPEA, DMF, rt, 1 h, 40% yield for conjugate used (Hu et al., 2019).
B: Linkage via a Phenolic Group at the Phenylene Unit
The recent literature on VHL-recruiting PROTACs confirmed that a phenolic linkage point is well-tolerated (Farnaby et al., 2019), with the left-hand dipeptide part of the molecule permitting the attachment of various substituents. The structure-activity relationship studies resulted in ligands 130 (Buckley et al., 2015), 133 (Maniaci et al., 2017), 134 (Farnaby et al., 2019; Zoppi et al., 2019), and 135 (Maniaci et al., 2017) with high VHL binding affinity (Scheme 25). To synthesize these compounds, the starting 4-bromo-2-hydroxybenzonitrile (126) was transformed into 127 through a Heck reaction, where prolonging the reaction time from 15 h (Buckley et al., 2015) to 20 h (Farnaby et al., 2019) only had a minor effect on the yield. The key intermediate 129 was formed through a reduction to amine 128 using LiAlH4 with low reported yields of 27% (Buckley et al., 2015) and 38% (Farnaby et al., 2019), followed by amide bond formation with Boc-l-hydroxyproline and subsequent Boc deprotection (Scheme 25, steps b–d) (Buckley et al., 2015; Farnaby et al., 2019). Coupling of 129 with 125 formed VHL ligand 130, which allowed for the attachment of linkers with a terminal mesylate group to obtain conjugates 132 in a 37–68% yield for linkers used (Buckley et al., 2015; Steinebach et al., 2020a). Alternatively, 129 was first reacted with Boc-l-tert-leucine and then Boc-deprotected to yield 131, which was then derivatized into VHL ligands 133 using acetylimidazole (Maniaci et al., 2017), 134 using 1-fluorocyclopropanecarboxylic acid (Farnaby et al., 2019; Zoppi et al., 2019), and 135 using 1-cyanocyclopropanecarboxylic acid (Maniaci et al., 2017). Each of those VHL ligands then had mesylate linkers attached to the phenol under standard conditions, i.e. K2CO3, DMF, 70°C (Maniaci et al., 2017; Farnaby et al., 2019; Zoppi et al., 2019). Alternatively, Cs2O3 can be used as a base in place of K2CO3 (Steinebach et al., 2020a).
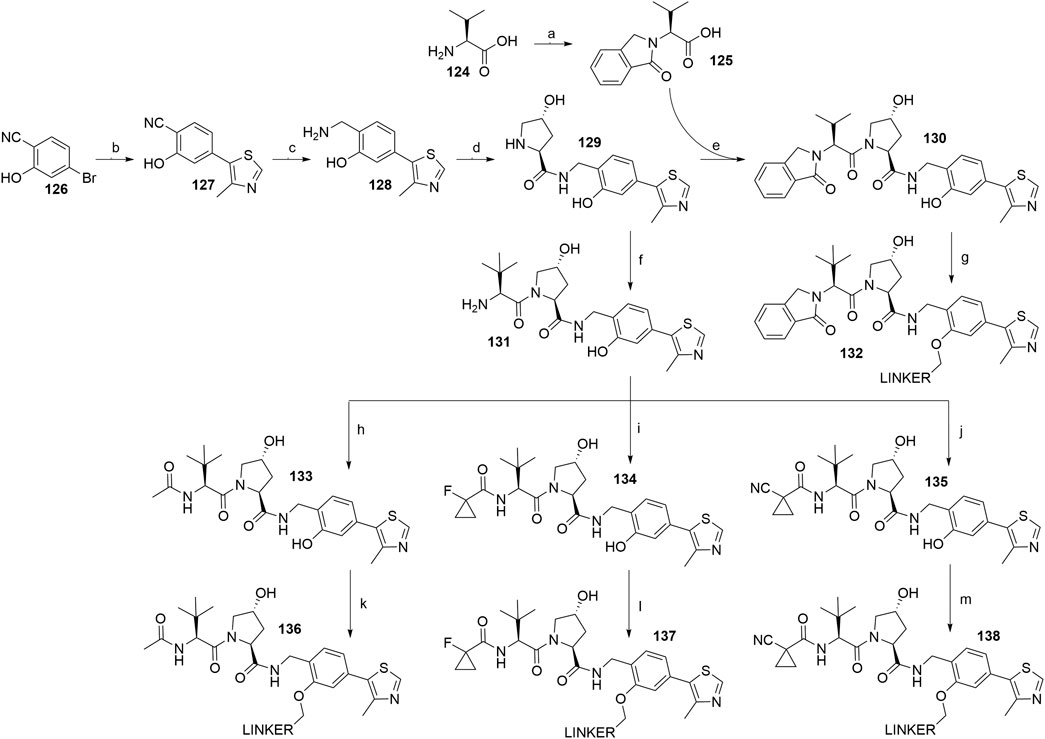
SCHEME 25. Syntheses of VHL ligands 130, 133, 134, and 135. Reagents and conditions: a) phthalaldehyde, MeCN, 90°C, 3.5 h, 83% yield; b) 4-methylthiazole, KOAc, Pd(OAc)2, DMA, 150°C, 15 h, 76% yield (Buckley et al., 2015); b) 4-methylthiazole, KOAc, Pd(OAc)2, DMA, 150°C, 20 h, 77% yield (Farnaby et al., 2019); c) LiAlH4, THF, 50°C, 22 h, 27% yield (Buckley et al., 2015); c) LiAlH4, THF, 0°C, 1 h to rt, overnight, 38% yield (Farnaby et al., 2019); d) i. Boc-Hyp-OH, HATU, HOAt, DIPEA, DMF, rt, 1 h; ii. 4 M HCl in dioxane, CH2Cl2, rt, 2 h, 55% yield (Maniaci et al., 2017); d) i. Boc-Hyp-OH, HATU, DIPEA, DMF, 4°C to rt, 2.5 h; ii. 4 M HCl in dioxane, CH2Cl2, MeOH, rt, 16 h, 52% yield (Buckley et al., 2015); d) i. Boc-Hyp-OH, HATU, DIPEA, DMF, rt, overnight; ii. 4 M HCl in dioxane, CH2Cl2, rt, overnight, 54% yield (Farnaby et al., 2019); e) HATU, DIPEA, DMF, rt, 22 h, 24% yield (Buckley et al., 2015); f) i. Boc-Tle-OH, HATU, HOAt, DIPEA, DMF, rt, 1 h; ii. 4 M HCl in dioxane, CH2Cl2, rt, 2 h, 55% yield (Maniaci et al., 2017); f) i. Boc-Tle-OH, HATU, DIPEA, DMF, rt, overnight; ii. 4 M HCl in dioxane, CH2Cl2, rt, overnight, 55% yield (Farnaby et al., 2019); g) linker-OMs, K2CO3, DMF, 70°C, overnight, 37–60% yield for linkers used (Buckley et al., 2015); g) linker-OMs, Cs2O3, rt, 18 h, then 60°C, 3 h, 49–68% yield for linkers used (Steinebach et al., 2020a); h) acetylimidazole, DIPEA, DMF, rt, 48 h, 78% yield (Maniaci et al., 2017); i) 1-fluorocyclopropanecarboxylic acid, Et3N, DMF, rt, overnight, 76% yield (Farnaby et al., 2019); i) 1-fluorocyclopropanecarboxylic acid, HATU, HOAt, DIPEA, DMF, rt, 2 h, 57% yield (Zoppi et al., 2019); j) 1-cyanocyclopropanecarboxylic acid, HATU, HOAt, DIPEA, DMF, rt, 1 h, 55% yield; k) linker-OMs, K2CO3, DMF, 70°C, overnight, 33% yield for linker used (Maniaci et al., 2017); l) linker-OMs, K2CO3, DMF, 75°C, overnight, 85% yield for linker used (Farnaby et al., 2019); l) linker-OMs, K2CO3, DMF, 70°C, overnight, 35–79% yield for linkers used (Zoppi et al., 2019); l) linker-OMs, Cs2O3, rt, 18 h, then 60°C, 3 h, 52% yield for linker used (Steinebach et al., 2020a); m) linker-OMs, K2CO3, DMF, 70°C, overnight, 33% yield for linker used (Maniaci et al., 2017); m) linker-OMs, Cs2CO3, rt, 18 h, then 60°C, 3 h, 52% yield for linker used (Steinebach et al., 2020a).
An alternative synthetic route used 4-bromo-2-hydroxybenzaldehyde (141) in place of 4-bromo-2-hydroxybenzonitrile (126, Scheme 25) (Steinebach et al., 2020a). Starting material 141 is easily accessible by ortho-formylating 3-bromophenol (139) or by transforming 4-bromosalicyclic acid (140) into a Weinreb amide and its subsequent reduction (Scheme 26). Compound 143 was obtained through reductive amination of 141 with tert-butyl carbamate under mild conditions and Heck coupling in a higher yield compared to the analogous synthesis of compound 128 (Scheme 25, step c). The phenol group of 143 was then protected to prevent the formation of acylated by-products in the following coupling reactions (Scheme 26, steps f–j). The key intermediate 145 was generated from 144 and Boc-l-hydroxyproline and then coupled with 125 and deprotected to give VHL ligand 130. Alternatively, forming an amide bond between 145 and Boc-l-tert-leucine yielded compound 146, which was then Boc-deprotected and derivatized into VHL ligands 134 and 135 (Steinebach et al., 2020b).
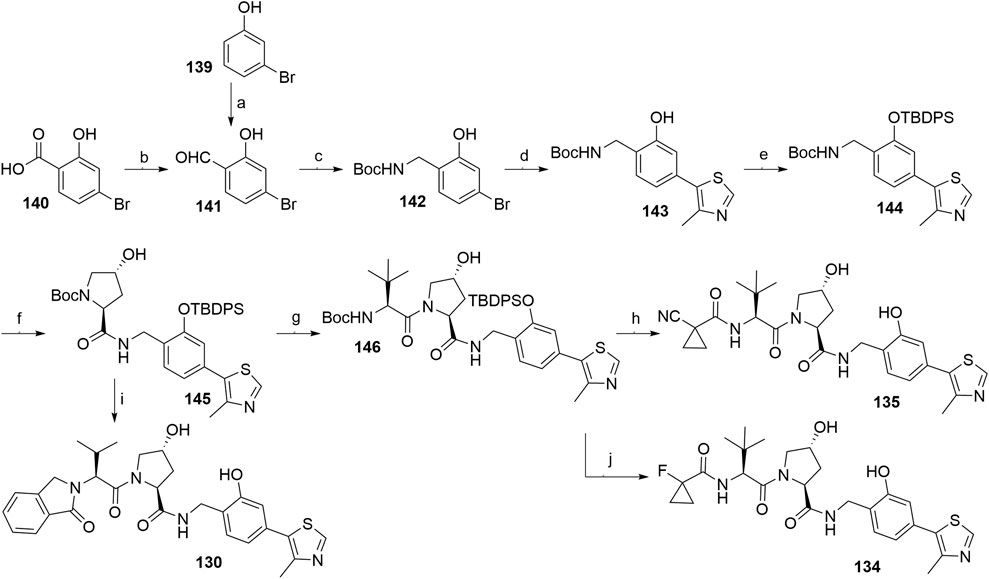
SCHEME 26. Synthesis of VHL ligands 130, 134, and 135. Reagents and conditions: a) paraformaldehyde, Et3N, MgCl2, THF, reflux, 6 h, 32% yield; b) i. N,O-dimethylhydroxylamine, EDC, Et3N, CH2Cl2, rt, 16 h, 78% yield; ii. LiAlH4, THF, 0°C, 30 min, 53% yield; c) tert-butyl carbamate, Et3SiH, TFA, CH2Cl2, MeCN, rt, 18 h, 94% yield; d) 4-methylthiazole, KOAc, Pd(OAc)2, DMA, 130°C, 4 h, 60% yield; e) TBDPSCl, imidazole, DMF, rt, 18 h, 92% yield; f) i. TFA, CH2Cl2, rt, 2 h; ii. Boc-Hyp-OH, HATU, DIPEA, DMF, rt, 18 h, 75% yield over two steps; g) i. TFA, CH2Cl2, rt, 2 h; ii. Boc-Tle-OH, HATU, DIPEA, DMF, rt, 18 h, 60% yield over two steps; h) i. TFA, CH2Cl2, rt, 2 h; ii. 1-cyanocyclopropanecarboxylic acid, HATU, DIPEA, DMF, rt, 18 h, 77% yield over two steps; iii. TBAF, THF, 0°C to rt, 18 h, 98% yield; i) i. TFA, CH2Cl2, rt, 2 h; ii. 125, HATU, DIPEA, DMF, rt, 18 h, 56% yield over two steps; iii. TBAF, THF, 0°C to rt, 18 h; j) i. TFA, CH2Cl2, rt, 2 h; ii. 1-fluorocyclopropanecarboxylic acid, HATU, DIPEA; DMF, rt, 18 h, 69% yield over two steps; iii. TBAF, THF, 0°C to rt, 18 h (Steinebach et al., 2020b).
C: Attachment via a Thioether at the Left-Hand Side Amino Acid
The side chain of tert-leucine group on VHL ligand’s left-hand side represents a possible linker attachment point, so it was replaced with trityl-protected penicillamine to synthesize compound 147. After treating 147 with acetic anhydride to afford 148, and subsequently removing the trityl group, the thiol-containing fragment 149 was obtained, to which mesylate, tosylate or bromo linkers were attached to form thioether conjugates 150 (Scheme 27) (Gadd et al., 2017).
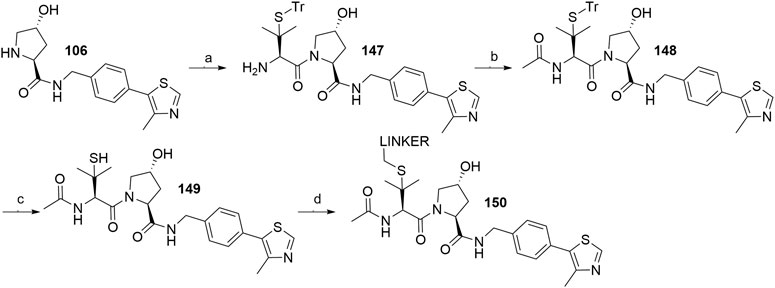
SCHEME 27. Synthesis of derivatives 150 with a thioether bond. Reagents and conditions: a) i. Fmoc-(S)-trityl-l-penicillamine, HATU, HOAt, DIPEA, DMF, rt, 2 h; ii. piperidine, CH2Cl2, rt, 1 h, 75% yield; b) Ac2O, Et3N, CH2Cl2, rt, 2 h, 98% yield; c) TIPS, TFA, CH2Cl2, rt, 2 h, 79% yield; d) linker-OMs/-OTs/Br, DBU, DMF, 0°C to rt, 1–3 h, 70–82% yield for linkers used (Gadd et al., 2017).
D: Connection via the Benzylic Position
Based on analyses of co-crystal structures of VHL ligand 122 (Scheme 24) in the active site of the enzyme, the (S)-methyl group of the VHL ligand was found to be exposed to the solvent and therefore represents a possible liker attachment point for the design of PROTACs. 4-Methylthiazole was coupled with commercially available 151 to yield 152, to which a desired linker-POI ligand conjugate was attached via an amide bond. Boc deprotection afforded compound 153, and the left-hand side dipeptide part was attached to form conjugates 154 (Scheme 28) (Han et al., 2019).
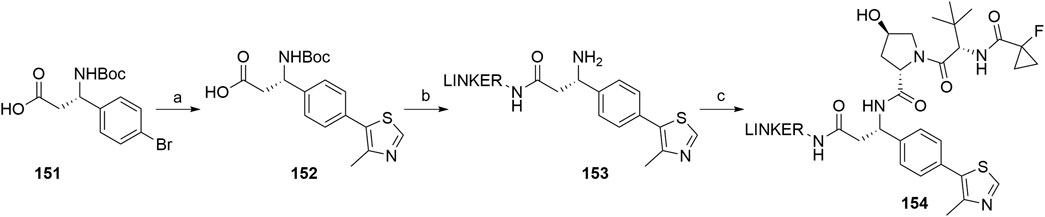
SCHEME 28. Synthesis of conjugates 154 connected with the linker via the benzylic position. Reagents and conditions: a) 4-methylthiazole, Pd(OAc)2, KOAc, Et3N, DMF, 80°C, 4 h, 80% yield; b) i. POI ligand-linker-NH2, HATU, DIPEA, DMF, rt, 30 min; ii. TFA, CH2Cl2, 80% yield for 2 steps; c) (2S,4R)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxylic acid, HATU, DIPEA, DMF, rt, 30 min (Han et al., 2019, 69).
von Hippel–Lindau Photacs
An azobenzene handle was employed in place of a standard linker, which allowed for photoinduced switching between the inactive cis and active trans configuration of the VHL-targeting PHOTAC (Pfaff et al., 2019). Intermediate 159 was generated from 2,6-difluoro-4-iodoaniline (157) (Scheme 29, steps b-c) and then treated with nitrosonium tetrafluoroborate to afford diazonium tetrafluoroborate 160. TBS-protection of (3,5-difluorophenyl)methanol (155) led to compound 156, which was treated with tert-butyllithium and combined with 160, giving 161. Following the TBS-deprotection and oxidation, 162 was coupled with VHL ligand 107, Boc-deprotected and additionally coupled with POI ligand-amine, to generate the finished PHOTAC compound 163 (Pfaff et al., 2019).
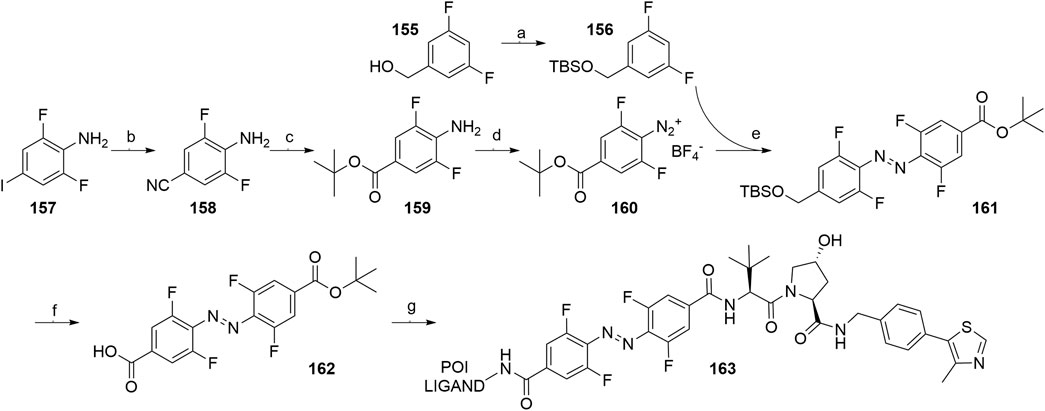
SCHEME 29. Synthesis of VHL-targeting PHOTAC 163. Reagents and conditions: a) imidazole, TBSCl, CH2Cl2, rt, 16 h, 98% yield; b) CuCN, NMP, 180°C, 7 h, 91% yield; c) i. 1 M NaOH (aq), reflux, 1 h, 89% yield; ii. Oxalyl chloride, DMF, rt, 30 min, then t-BuOK, THF, 0°C; iii. N, N′-dimethylethane-1,2-diamine, EtOH, 110°C, 12 h, 50% yield over two steps; d) NOBF4, EtOAc, 0°C, 1 h, 72% yield; e) t-BuLi, THF, –78°C to –50°C, 1 h, then 160, –78°C to rt, 1 h, 76% yield; f) i. TBAF, THF, 0°C, 15 min, 76% yield; ii. TEMPO, NaClO, NaClO2, MeCN/pH 6.8 phosphate buffer, rt, 3 h, quant.; g) i. 107, HATU, DIPEA, DMF, rt, 2 h, 88% yield; ii. TFA, CH2Cl2, rt, 1 h, quant.; iii. POI ligand-NH2, HATU, DUPEA, DMF, rt, 2 h, 51% yield for POI ligand used (Pfaff et al., 2019).
Caged von Hippel–Lindau Ligands
The concept of caged E3 ligase ligands was used for the controlled degradation of BRD4 by incorporating a photocleavable 4,5-dimethoxy-2-nitro-benzyl group (DMNB), bound to the hydroxyproline core of the VHL ligand, connected via a linker to pan-bromodomain inhibitor JQ1. Following irradiation with a wavelength of 365 nm, the PROTAC could be uncaged, which triggered the degradation of BRD4. To prepare a caged PROTAC, the VHL ligand 122 was first N-Boc protected, followed by the functionalization of hydroxyl group by forming an ether bond with the DMNB group using phase transfer catalysis to yield 164. After Boc deprotection, a carboxylic acid linker was introduced via an amide bond to form 165 (Scheme 30) (Kounde et al., 2020). Additionally, the concept was also utilized for the degradation of estrogen related receptor α, where a diethylamino coumarin (DEACM) group was installed at the hydroxyl group of the VHL ligand via a carbonate linkage. Irradiation with a wavelength of 360 nm causes the photolysis and subsequent decaging of the VHL ligand, thus activating the degrader. Compound 167 was obtained from starting material 166 over two steps and then converted to a chloroformate before being attached to a POI ligand-linker-VHL ligand conjugate, forming the final caged PROTAC 168 (Naro et al., 2020).
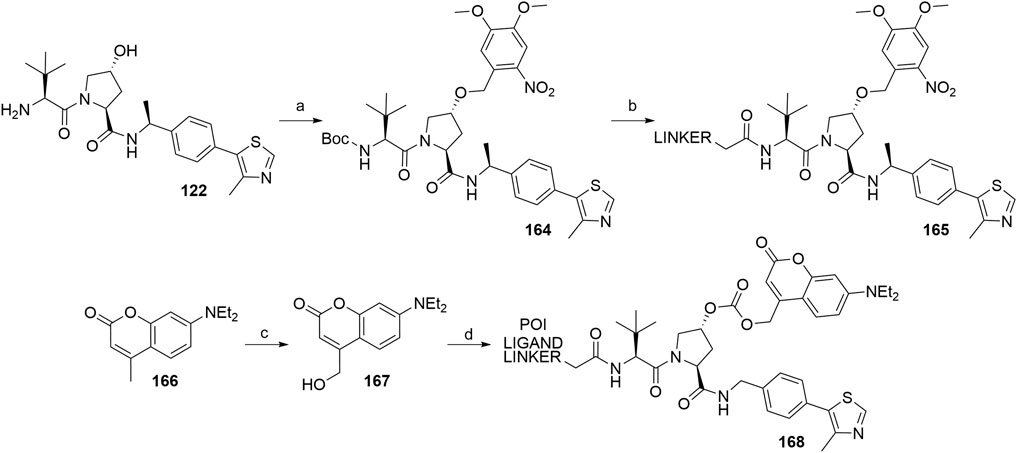
SCHEME 30. Syntheses of caged VHL degraders 165 and 168. Reagents and conditions: a) i. (Boc)2O, Et3N, CH2Cl2, rt, 16 h, 77% yield; ii. 4,5-dimethoxy-2-nitrobenzyl bromide, TBAI, 50% NaOH (aq), CH2Cl2, rt, 2 h, 53% yield; b) i. HCl in dioxane, rt, 4 h, quant.; ii. linker-CO2H, HATU, DIPEA, CH2Cl2, rt, 16 h, 41% yield for linker used (Kounde et al., 2020); c) i. SeO2, dioxane, reflux, 16 h; ii. NaBH4, EtOH, 0°C to rt, 4 h; d) i. diphosgene, CH2Cl2, 0°C to rt, 24 h; ii. POI ligand-linker-VHL ligand A1, DMAP, DIPEA, CH2Cl2, 0°C to rt, 16 h, 59% yield for conjugate used (Naro et al., 2020).
Statistical Overview of Utilized von Hippel–Lindau Ligands
Using data extracted from PROTAC-DB (Weng et al., 2021) (http://cadd.zju.edu.cn/protacdb/, as of the February 26, 2021), a statistical overview was done to determine the frequency of various VHL ligands and linker attachment options used in PROTAC compounds. The vast majority of PROTACs incorporated VHL ligand 1, while the (S)-methyl group-containing ligand occurred in about a third of degraders. Linkage via a phenolic group at benzene ring was less commonly utilized, at about 4%. In comparison, attachment via a thioether at the left-hand side amino acid could be found in only around 1% of PROTACs (Figure 5).
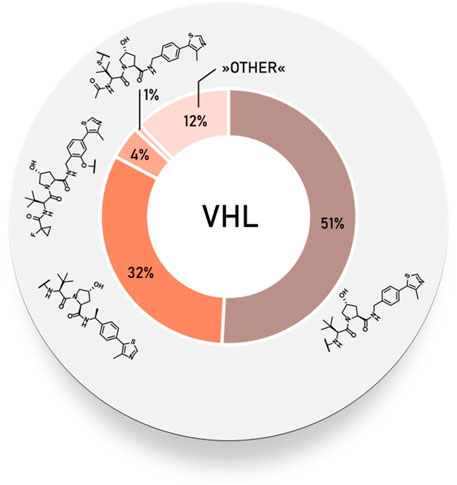
FIGURE 5. Frequency of VHL ligands used in PROTAC compounds.
Inhibitor of Apoptosis Proteins
The family of inhibitor of apoptosis proteins (IAP) includes antiapoptotic proteins, which are commonly overexpressed in some cancer cells and promote their survival, as well as the survival of neuronal cells (Fulda and Vucic, 2012; Ohoka et al., 2017b). All IAP proteins contain one to three baculoviral IAP repeat (BIR) domains that interact with their binding proteins, while some of them [cellular IAP1(c-IAP1), c-IAP2, X chromosome-linked IAP (XIAP), and melanoma IAP (ML-IAP)] also contain a RING finger domain, which provides an E3 ubiquitin ligase activity (Cohen and Tcherpakov, 2010; Itoh et al., 2010; Fulda and Vucic, 2012; Ohoka et al., 2017b). Because of their involvement in multiple malignancies, inhibitors of these proteins represent an attractive strategy for tumor therapy, and many potent peptidomimetic antagonists have been developed based on the endogenous inhibitory IAP protein second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO) (Ohoka et al., 2017b). Interaction between IAP antagonists and their targets results in the autoubiquitylation and proteasomal degradation of cIAP1 (Varfolomeev et al., 2007; Vince et al., 2007).
First hybrid molecules that utilized c-IAP1 for its E3 ubiquitin ligase activity have been described in 2010 and those compounds induced the degradation of cellular retinoic acid-binding proteins (Itoh et al., 2010). Alternatively to PROTACs, for IAP-recruiting degraders, a different terminology is also used, i.e., specific and nongenetic IAP-dependent protein erasers (SNIPERs) (Sun X. et al., 2019). Methyl bestatin derivative (Figure 6, A) was used as the c-IAP1-binding ligand for the early reported degraders recruiting this E3 ligase (Itoh et al., 2010; Okuhira et al., 2011; Okuhira et al., 2013; Okuhira et al., 2016; Demizu et al., 2012; Ohoka et al., 2014). Future development of high-affinity IAP ligands and their incorporation into bifunctional molecules improved the efficiency of SNIPERs in comparison with early bestatin-based compounds (Ohoka et al., 2017b; Okuhira et al., 2018; Naito et al., 2019). Structures of IAP ligands utilized in chimeric molecules are presented in Figure 6.
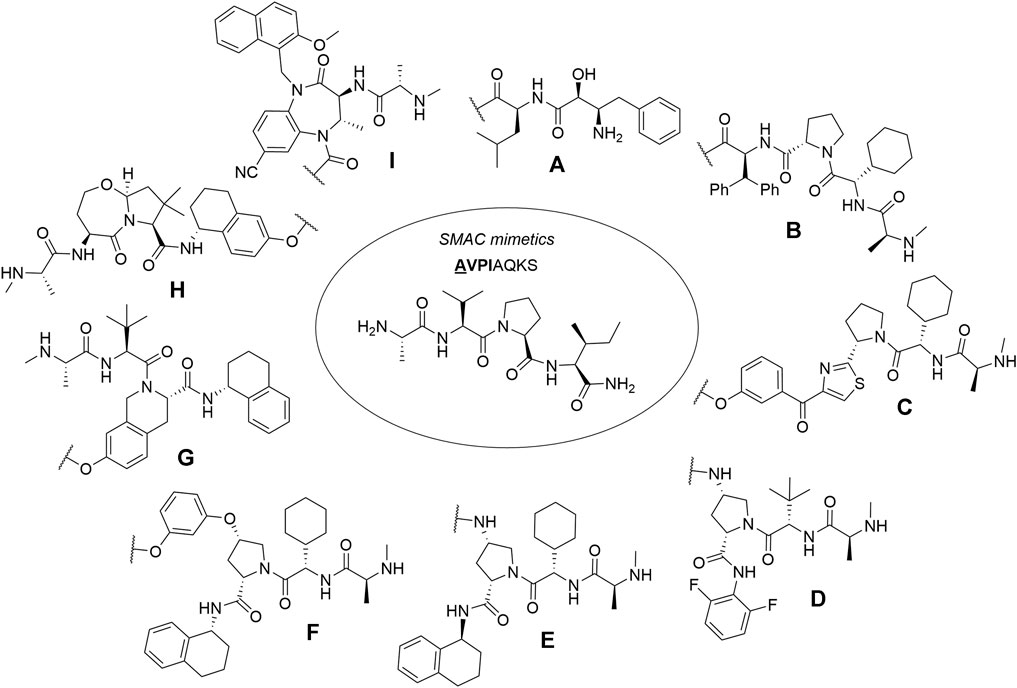
FIGURE 6. IAP ligands. IAP ligand (A) (bestatin), IAP ligand (B) (MV1 derivative), IAP ligand (C) (LCL-161 derivative), IAP ligand (D) (Cmpd37 derivative), IAP ligand (E) (A410099 derivative), IAP ligand (F), IAP ligand (G), IAP ligand (H) (SBP-0636457 derivative), IAP ligand (I).
Inhibitor of Apoptosis Proteins Ligand A: Bestatin
Aromatic α-Aminoaldehydes as a Starting Material for Bestatin Synthesis
Several authors proposed different synthetic routes for the synthesis of bestatin, utilizing aromatic α-aminoaldehydes as starting compounds (Scheme 31). For example, compound 169 was treated with nitromethane to afford a diastereomeric mixture of nitroaldols 170, which were then converted into a mixture of dimethyl oxazolidines, out of which the desired compound 171 was separated by silica gel column chromatography in a 54% yield. Compound 172 was obtained by a Nef reaction and then coupled with l-leucine tert-butyl ester to yield 173. Finally, Boc cleavage using TFA afforded bestatin (179) in an overall yield of 24% (Scheme 30, steps a–e) (Shang et al., 2018). An alternative route started from aldehyde 174, which was converted to syn-aminoalcohol 175 in a 96% yield and a 9.5:1 syn/anti stereochemic ratio. The hydroxyl group was then protected with a Bn group to obtain 176, followed by terminal alkyne oxidation to carboxylic acid 177. Coupling reaction with l-leucine methyl ester afforded compound 178, and removal of the protecting groups led to the desired product 179 with an overall yield of 59% (Scheme 31, steps f–j) (Lee et al., 2003). Furthermore, a one-pot method was described, in which starting materials 180, 181, and 182 were joined into 183 with 63% yield. Following the deprotection, bestatin (179) was obtained in an overall yield of 60% (Scheme 31, steps k–l) (Nemoto et al., 2000).
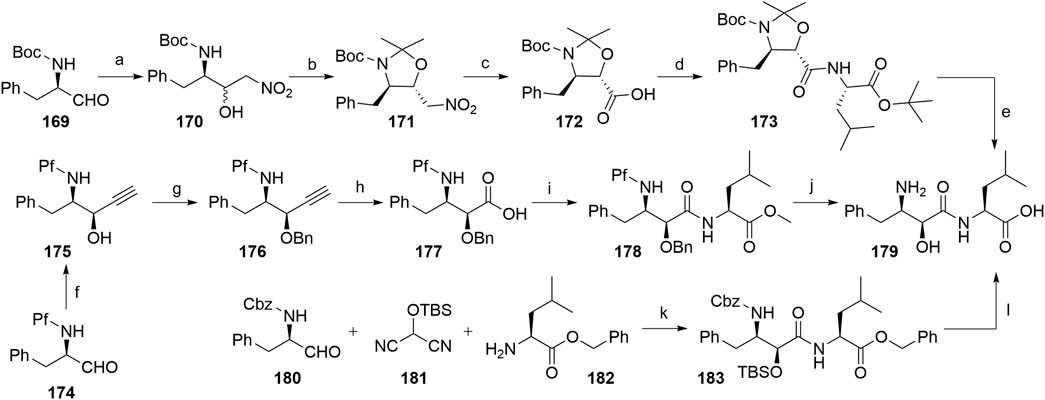
SCHEME 31. Syntheses of bestatin (179). Reagents and conditions: a) nitromethane, NaH, 15-crown-5, EtO2, hexane, 0°C to rt, 22 h, 64% yield; b) 2,2-dimethoxypropane, BF3×OEt2, 0°C, 1 h, then rt, overnight, 54% yield; c) KOH, KMnO4, Na2HPO4, MeOH, H2O, 0°C, 2 h, quant.; d) isobutyl chloroformate, NMP, THF, 0°C to –12°C, 30 min, then l-leucine tert-butyl ester, NMP, DMF, –12°C, 90 min, 69% yield; e) TFA, H2O, 0°C, 2.5 h, quant. (Shang et al., 2018); f) ethynylmagnesium bromide, THF, –40°C, 10 min, 96% yield; g) BnBr, NaH, Bu4NI, THF, 0°C, 97% yield; h) KMnO4, AcOH, H2O, pentane, 87% yield; i) l-leucine methyl ester, DCC, HOBt, TsOH, Et3N, THF, 0°C, 91% yield; j) i. LiOH, THF, H2O, 0°C, 95% yield; ii. Pd/C, H2, MeOH, 50°C, 93% yield (Lee et al., 2003); k) 4-pyrrolidinopyridine, Et2O, 0°C, 5 h, diastereomeric mixture in 80% yield, 79:21 ratio; l) i. Bu4NF, THF, 0°C, 20 min, 96% yield; ii. Pd/C, H2, MeOH, rt, 2 h, quant (Nemoto et al., 2000).
Alternative Routes for the Synthesis of Bestatin
One route included the treatment of (2-nitroethyl)benzene (184) with ethyl glyoxalate in Shibasaki’s asymmetric Henry reaction, which was catalyzed by an optically active lanthanum-(R)-binaphthol complex. Compound 185 was then O-acetylated before reducing the nitro group to yield 186. Following N-Boc protection, 187 was coupled with l-leucine benzyl ester, followed by immediate deprotection of the terminal carboxylic moiety. Both protecting groups of 188 were removed to give bestatin (179) in an overall yield of 26% (Scheme 32) (Gogoi et al., 2005).
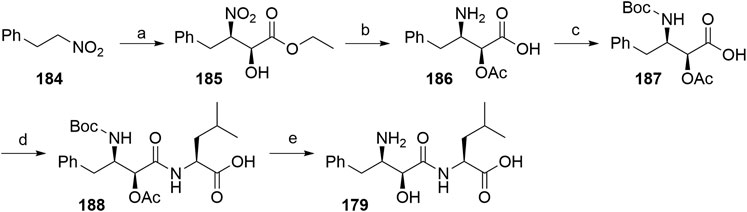
SCHEME 32. The synthesis of bestatin (179) (Gogoi et al., 2005). Reagents and conditions: a) ethyl glyoxalate, La-(R)-BINOL, THF, –50°C, 81% yield, 93% ee; b) i. acetylation, 94% yield (no detailed reagents given); ii. Pd/C, NaBH4, H2, MeOH, 60%, 93% ee; c) (Boc)2O, NaHCO3, H2O, EtOAc, 92% yield; d) i. l-leucine benzyl ester, N-ethylmorpholine, isobutyl chloroformate, THF,–10°C; ii. Pd/C, H 2, MeOH, 77% yield over two steps; e) i. K2CO3, MeOH; ii. TFA, 73% yield over two steps (Gogoi et al., 2005).
A procedure partly derived from the patent literature started with the treatment of the Meldrum’s acid 189 with phenylacetyl chloride to yield 190, which was then chlorinated using sulfuryl chloride to form 191. The following asymmetric hydrogenation using a ruthenium-phosphine complex afforded compound 192, which was then subjected to epoxidation to obtain 193 (Sayo et al., 1996). Compound 194 was synthesized through an MgBr2-mediated ring opening of 193. Treatment with NaN3 afforded the azide derivative 195, which was then hydrogenated and Boc-protected to give compound 196. Hydrolysis of the methyl ester allowed coupling of 197 with l-leucine, and deprotection of 198 yielded bestatin (179) (Scheme 33) (Righi et al., 2003).
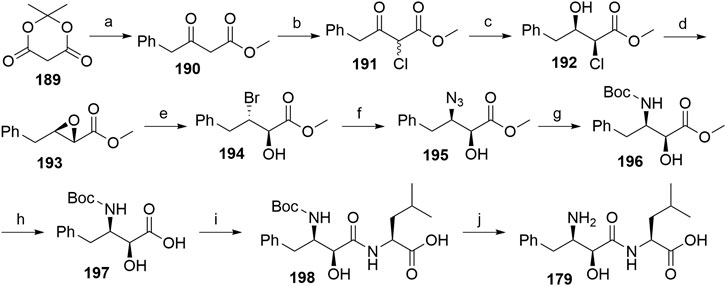
SCHEME 33. Alternative route for the syntheses of bestatin (179). Reagents and conditions: a) phenylacetyl chloride, pyridine, CH2Cl2, 0°C, 92% yield; b) SO2Cl2, 0°C, overnight, 88% yield; c) Ru2Cl4[R-BINAP]2(NEt3)H2, MeOH, rt, 20 h, 95% yield, 63:37 syn/anti; d) MeONa, MeOH, 0°C to rt, 2 h, 75% yield (Sayo et al., 1996). e) MgBr2, Et2O, rt, 2 h, 92% yield; f) NaN3, DMSO, 40°C, 6 h, 73% yield; g) i. Pd/C, H2, EtOAc; ii. (Boc)2O, EtOAc, rt, 5 h, 95% yield; h) Na2CO3, MeOH, H2O, rt, 12 h, 79% yield; i) i. l-leucine benzyl ester tosylate, EDC, HOBt, DIPEA, DMF, CH2Cl2, rt, 12 h, 87% yield; ii. Pd/C, H2, MeOH, rt, 95% yield; j) TFA, CH2Cl2, rt, 12 h, 85% yield (Righi et al., 2003).
Linker Attachment to Bestatin
Bestatin (179) was incorporated into chimeric molecules either via an amide or ester bond, the latter being less frequently utilized. Prior to coupling with the selected POI ligand-amine linker conjugates, the amino group of bestatin was protected (Boc = 198 or Fmoc = 199) (Itoh et al., 2010; Ohoka et al., 2014). A combination of EDC and HOBt for amide coupling yielded bifunctional compounds 200 in a yield spanning from 81 to 89% for various conjugates used (Itoh et al., 2011; Ohoka et al., 2014; Okuhira et al., 2017a). Alternatively, the ester bond was formed under similar conditions, using a POI ligand-linker hydroxyl conjugate. However, the reported yield was much lower (36%) (Itoh et al., 2010). Using the appropriate deprotection procedure, final SNIPER compounds 201 and 202 were synthesized (Scheme 34) (Itoh et al., 2010; Okuhira et al., 2016).
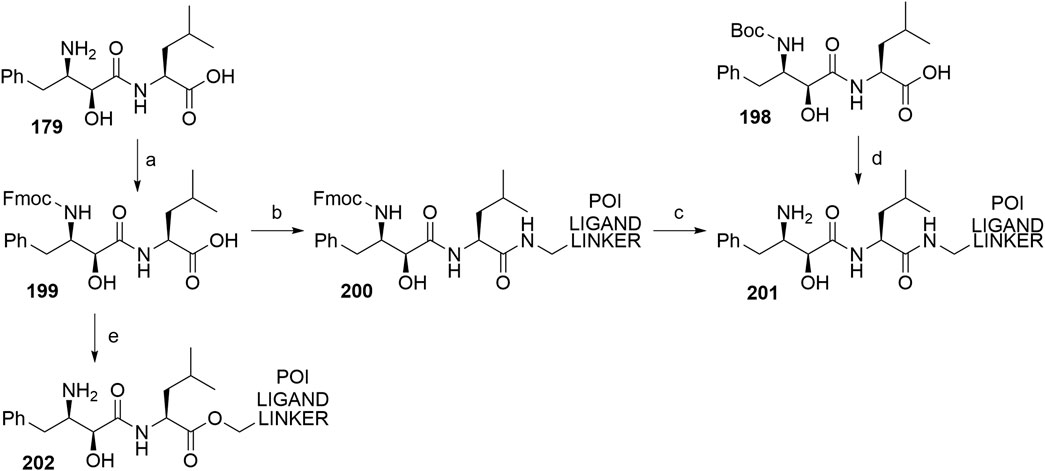
SCHEME 34. Linker attachment via an amide or an ester bond. Reagents and conditions: a) Fmoc-Cl, K2CO3, THF, H2O, 0°C to rt, 24 h, 97% yield (Itoh et al., 2010); a) Fmoc-Cl, K2CO3, THF, H2O, rt, 81% yield (Ohoka et al., 2014); b) reagents, conditions, and yields are collected in Table 6; c) 2 M Me2NH in MeOH, rt, 84–89% yield for conjugates used (Ohoka et al., 2014); 2 M Me2NH in MeOH, rt, 6 h, 37% yield for conjugate used (Ohoka et al., 2017a); d) i. POI ligand-linker-NH2, EDC, HOBt, DIPEA, CH2Cl2, rt, 12 h, 77–86% yield for conjugates used; ii. 6M HCl (aq), THF, rt, 6 h, quant. (Okuhira et al., 2016); e) i. POI ligand-linker-OH, EDC, HOBt, DIPEA, CH2Cl2, 0°C to rt, 17 h, 36% yield for conjugate used; ii. DBU, dodecyl mercaptan, CH2Cl2, rt, 1 h (Itoh et al., 2010).

TABLE 6. Reagents, conditions, and yields for the conversion of 199 to 200 (Scheme 34, step b).
Inhibitor of Apoptosis Proteins Ligand B: MV1 Derivative
A similar stepwise peptide synthesis of IAP ligand B using starting biphenyl 203 was described (Itoh et al., 2012; Shibata et al., 2017b). Coupling with N-Boc-l-proline yielded compound 204, the following coupling with Boc-l-cyclohexylglycine gave 205, and finally adding Boc-N-methyl-l-alanine afforded 206. Catalytic reduction cleaved the O-benzyl group and allowed for coupling of the resulting 207 (Boc-protected MV1 derivative) with POI ligand-linker amine conjugates, giving bifunctional derivatives 208, which were Boc-deprotected to obtain final SNIPER compounds 209 (Scheme 35) (Itoh et al., 2012; Ohoka et al., 2017b; Shibata et al., 2017b).
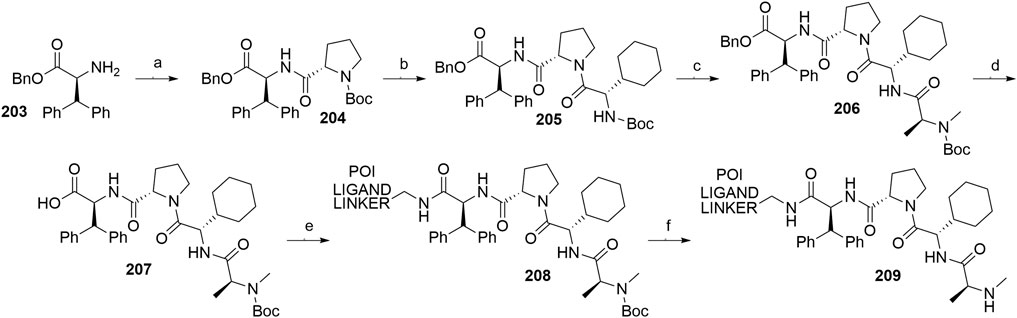
SCHEME 35. Synthesis of IAP ligand B. Reagents and conditions: a) N-Boc-l-proline, EDC, HOBt, DIPEA, DMF, rt, 22 h, 84% yield (Itoh et al., 2012); a) N-Boc-l-proline, EDC, HOBt, DIPEA, DMF, rt, 3 h, 95% yield (Shibata et al., 2017b); b) i. HCl, dioxane, rt, 4.5 h; ii. Boc-l-cyclohexylglycine, EDC, HOBt, DIPEA, DMF, rt, 13 h, 86% yield (Itoh et al., 2012); b) i. 4 M HCl, CPME, rt, 3 h; ii. Boc-l-cyclohexylglycine, EDC, HOBt, DIPEA, DMF, overnight, 98% yield (Shibata et al., 2017b); c) i. HCl, dioxane, rt, 4.5 h; ii. Boc-N-methyl-l-alanine, EDC, HOBt, DIPEA, DMF, rt, 15 h, 80% yield (Itoh et al., 2012); c) i. 4 M HCl, CPME, rt, 3 h; ii. Boc-N-methyl-l-alanine, EDC, HOBt, DIPEA, DMF, rt, overnight, 94% yield (Shibata et al., 2017b); d) Pd/C, H2, dioxane, rt, 6.5 h, quant. (Itoh et al., 2012); d) Pd/C, H2, EtOH, rt, overnight, quant. (Shibata et al., 2017b); e) and f) reagents, conditions, and yields are collected in Table 7.

TABLE 7. Reagents, conditions, and yields for the conversion of 207 to 209 (Scheme 35, steps e–f).
A solid-phase peptide synthesis for an IAP ligand B derivative on a 2-chlorotrityl chloride resin was reported. The stepwise procedure was performed using HCTU, HOBt and DIPEA for coupling, followed by the addition of 20% piperidine in DMF to remove the Fmoc group after each step (Scheme 36, steps a–d). Finally, 214 was treated with 1% TFA in CH2Cl2 to remove the resin and to obtain 207 (Boc-protected IAP ligand B) (Ohoka et al., 2017b).
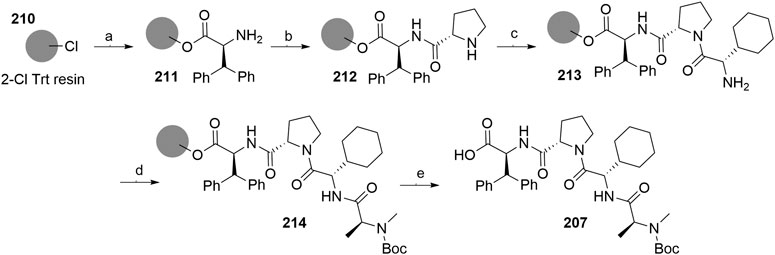
SCHEME 36. The synthesis of Boc-protected IAP ligand B (207) using 2-chlorotrityl chloride resin. Reagents and conditions: a) i. Fmoc-l-3,3-diphenylalanine, DIPEA, CH2Cl2, rt, 2 days; ii. 20% piperidine in DMF, rt, 10 min; b) i. Fmoc-l-Pro-OH, HCTU, HOBt, DIPEA, DMF, rt, 1.5 h; ii. 20% piperidine in DMF, rt, 10 min; c) Fmoc-l-cyclohexylglycin, HCTU, HOBt, DIPEA, DMF, rt, 1.5 h; ii. 20% piperidine in DMF, rt, 10 min; d) i. Fmoc-l-Ala-OH, HCTU, HOBt, DIPEA, DMF, rt, 1.5 h; ii. 20% piperidine in DMF, rt, 10 min; e) 1% TFA in CH2Cl2, rt, 5 min, 70% yield (Ohoka et al., 2017b).
Inhibitor of Apoptosis Proteins Ligand C: LCL-161 Derivative
The procedure for the synthesis of IAP ligand C (Ohoka et al., 2017b; Shibata et al., 2017b) is shown in Scheme 37. The starting (tert-butoxycarbonyl)-l-proline (215) was converted into 216 over two steps before building the thiazole fragment to give 217. Following the attachment of the 3-hydroxyphenyl building block to obtain 218, the hydroxyl group was deprotected, and the right-hand side of the molecule was built by coupling with Boc-l-cyclohexylglycine to yield 219, and Boc-N-methyl-l-alanine to produce the Boc-protected IAP ligand C (compound 220) (Ohoka et al., 2017b; Shibata et al., 2017b). Tosylate-containing POI ligand-linker conjugates were attached to the phenol of 220 by heating in DMF or DMSO using K2CO3 as a base, with yields spanning between 62 and 81%, depending on the conjugate used (Ohoka et al., 2017b; Shibata et al., 2017b; Shibata et al., 2017a; Shimokawa et al., 2017). Alternatively, POI ligand-linker conjugates with a terminal hydroxyl group were attached under Mitsunobu reaction conditions (Shibata et al., 2017b). Final SNIPER molecules of type 222 were obtained by Boc-deprotection of bifunctional conjugates 221 (Scheme 37) (Ohoka et al., 2017b; Shibata et al., 2017b; Shimokawa et al., 2017).
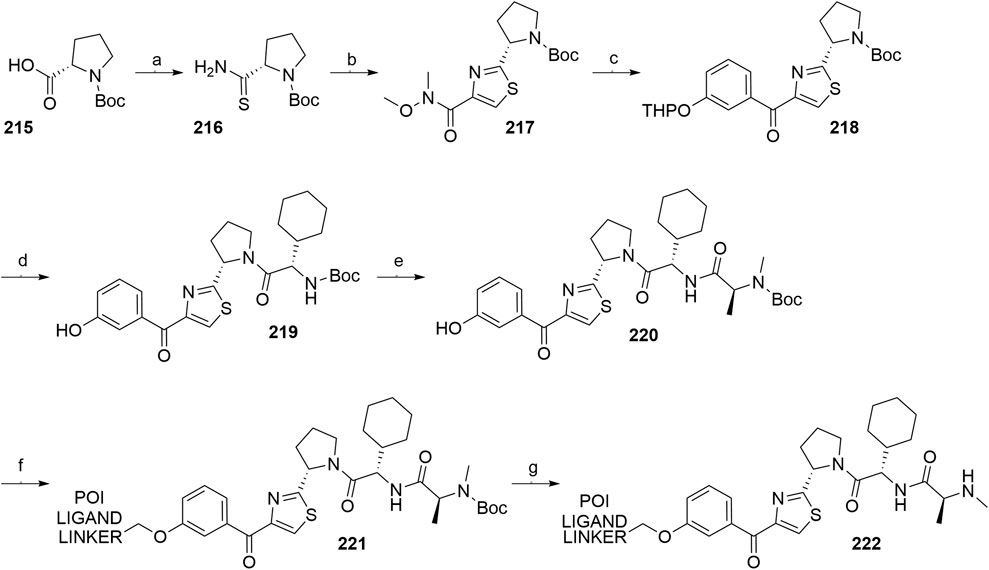
SCHEME 37. Synthesis of Boc-protected IAP ligand C (compound 220) and derived degraders 222. Reagents and conditions: a) i. HOBt·NH3, EDC, DMF, THF, 0°C to rt, 3 h, 96% yield; ii. Lawesson’s reagent, THF, 60°C, 2 h, 82% yield; b) i. ethyl bromopyruvate, EtOH, 60°C, 3 h; ii. (Boc)2O, NaHCO3, THF, H2O, rt, 3 h, 89% yield over two steps; iii. 1 M NaOH in MeOH, rt, overnight, 93%; iv. N,O-dimethylhydroxylamine hydrochloride, EDC, HOBt, DIPEA, DMF, rt, overnight, 97% yield (Ohoka et al., 2017b); c) i. 0.5 M 3-(2-tetrahydro-2H-pyranoxy)phenylmagnesium bromide in THF, –55 °C to –10 °C over 50 min, 99% yield; d) i. 2 M HCl in MeOH, rt, overnight; ii. Boc-l-cyclohexylglycine, EDC, HOBt, THF, 0°C, 1 h; iii. K2CO3, MeOH, 0°C, 2 h, 99% yield over three steps; e) i. 4 M HCl in CPME, MeOH, THF, rt, 4 h; ii. Boc-N-methyl-l-alanine, EDC, HOBt, DIPEA, DMF, rt, 2 h; iii. K2CO3, MeOH, H2O, 0 °C, 2 h, 42% yield over three steps; (Ohoka et al., 2017b; Shibata et al., 2017b) f) reagents, conditions, and yields are collected in Table 8; g) 4 M HCl in CPME, THF, rt, 4 h, 32–61% yield for conjugates used (Ohoka et al., 2017b); g) TFA, rt, 10 min, 70–90% yield for conjugates used (Shibata et al., 2017b); g) TFA, rt, 30 min, 50–68% yield for conjugates used (Shimokawa et al., 2017).
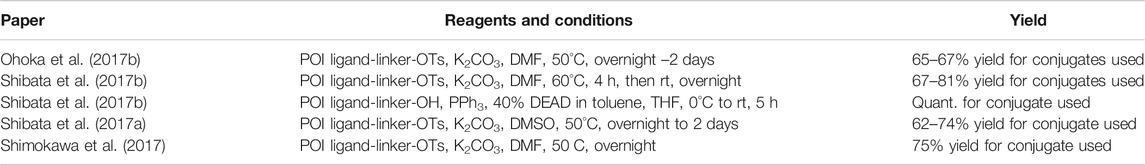
TABLE 8. Reagents, conditions, and yields for the conversion of 220 to 221 (Scheme 37, step f).
An N-methylated analog of LCL-161 loses the ability to bind to IAP, which allows for the use of such derivatives as negative controls (Ohoka et al., 2017b). The methylation can be achieved before linker attachment by first protecting the phenol group of 220 to obtain benzyl protected derivative 223, followed by N-methylation using NaH and MeI to form 224 (Scheme 38, steps a–b) (Ohoka et al., 2017b). Alternatively, the N-methylation of the POI ligand-linker-LCL-161 conjugates 221 was reported (Shimokawa et al., 2017). In the latter case, however, other possible liable atoms for methylation should be identified before performing the reaction.
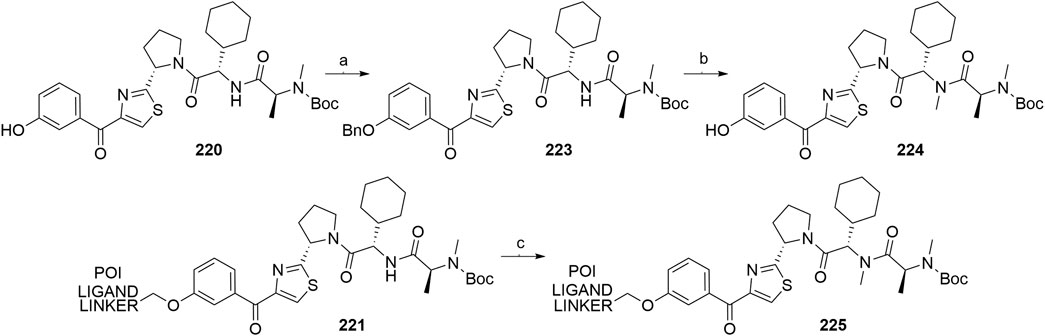
SCHEME 38. Syntheses of negative control degraders. Reagents and conditions: a) benzyl bromide, K2CO3, DMF, rt, 4 h, 87% yield; b) i. NaH, DMF, 0°C, 10 min, then MeI, 0°C, 1 h, 81% yield; ii. TFA, rt, 12 h; iii. (Boc)2O, THF, rt, 12 h, 34% yield over two steps (Ohoka et al., 2017b); c) MeI, 60% NaH in mineral oil, DMF, rt, 1 h, 68% yield for conjugate used (Shimokawa et al., 2017).
Inhibitor of Apoptosis Proteins Ligand D
Building block 227 was synthesized from N-(tert-butoxycarbonyl)-N-methyl-l-alanine (226) as starting compound using EDC/HOBt-mediated coupling and subsequent hydrolysis of the methyl ester (Casillas et al., 2016; Mares et al., 2020). Compound 227 was then coupled with 230, which was synthesized by subsequent protection of the hydroxyl group and deprotection of the carboxylic acid of 228 to form (2S,4R)-1-(tert-butoxycarbonyl)-4-(tosyloxy)pyrrolidine-2-carboxylic acid (229), followed by the coupling of 2,6-difluoroaniline. The tosylate group of 231 was transformed into an azide and then reduced to an amine yielding 232, which was then coupled with a carboxylic acid-containing POI ligand-linker conjugate and Boc-deprotected to yield final SNIPER compounds 233 (Scheme 39) (Anderson et al., 2020, 6; Mares et al., 2020).
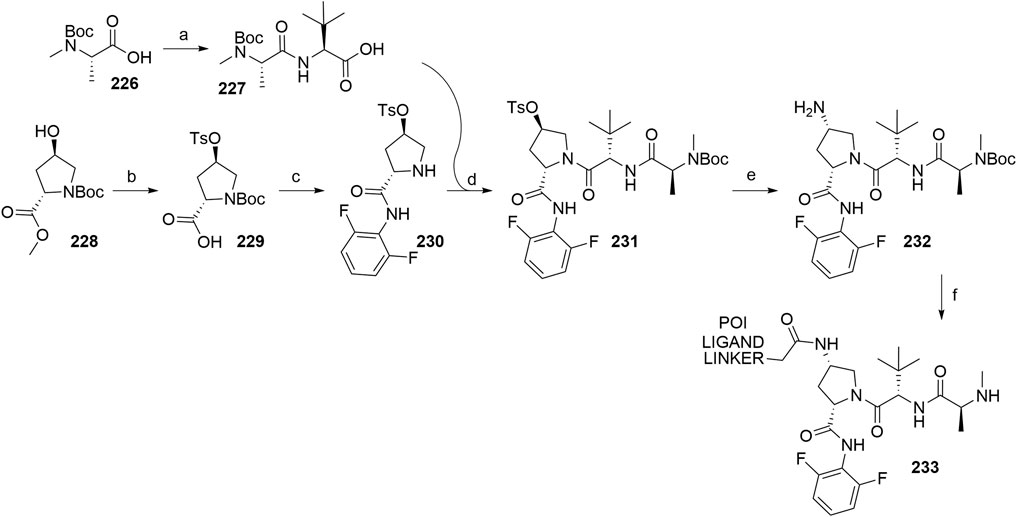
SCHEME 39. Synthesis of Boc-protected IAP ligand D (232) and derived SNIPERs 233. Reagents and conditions: a) i. l-tert-Leucine methyl ester, EDC, HOBt, THF, rt, 18 h, 90% yield; ii. LiOH, H2O, MeOH, MeOH, THF, rt, 18 h, 84% yield (Casillas et al., 2016); a) i. l-tert-Leucine methyl ester, EDC, HOBt, 4-methylmorpholine, CH2Cl2, 0°C to rt, overnight; ii. LiOH, H2O, MeOH, MeOH, THF, rt, 18 h, 99% yield over two steps (Mares et al., 2020); b) i. TsCl, DIPEA, DMAP, CH2Cl2, rt, overnight; ii. LiOH, MeOH, H2O, rt, overnight, 82% yield over two steps; c) i. 2,6-difluoroaniline, DCC, CH2Cl2, rt, overnight; ii. TFA, CH2Cl2, rt, 3 h, 66% yield over two steps; d) EDC, HOBt, CH2Cl2, –20°C, 1 h, then rt, overnight, 54% yield; e) i. NaN3, DMF, 80°C, overnight; ii. Pd/C, H2, MeOH, rt, 3 h, 10% yield; f) i. POI ligand-linker-CO2H, HATU, DIPEA, DMF, rt, 3 h; ii. 4 M HCl in dioxane, rt, 3 h, 8% yield for conjugate used (Anderson et al., 2020; Mares et al., 2020).
Inhibitor of Apoptosis Proteins Ligand E: A410099 Derivative
The synhesis of IAP ligand E is described in patents and consists of a stepwise coupling procedure (Scheme 40). The orthogonally protected 234 was first coupled with (S)-1,2,3,4-tertahydronaphthalen-1-amine to form 235, which was then Boc-deprotected and coupled with Boc-l-cyclohexylglycine. The resulting 236 was Boc-deprotected and coupled with Boc-N-methyl-l-alanine to yield 237. The Fmoc protection of 4-amino group on the proline fragment allowed for the selective deprotection to obtain 238 (Borzilleri et al., 2014; Mischke, 2014). Carboxylic acid-containing POI ligand-linker conjugates were attached and the N-terminal amino group was Boc-deprotected to give final SNIPER compounds 239 (Shah et al., 2020). As an alternative, 238 was coupled with a carboxylic acid-containing linker, the product was Boc-deprotected and the POI ligand was attached to obtain the desired SNIPER compounds (Nunes et al., 2019).
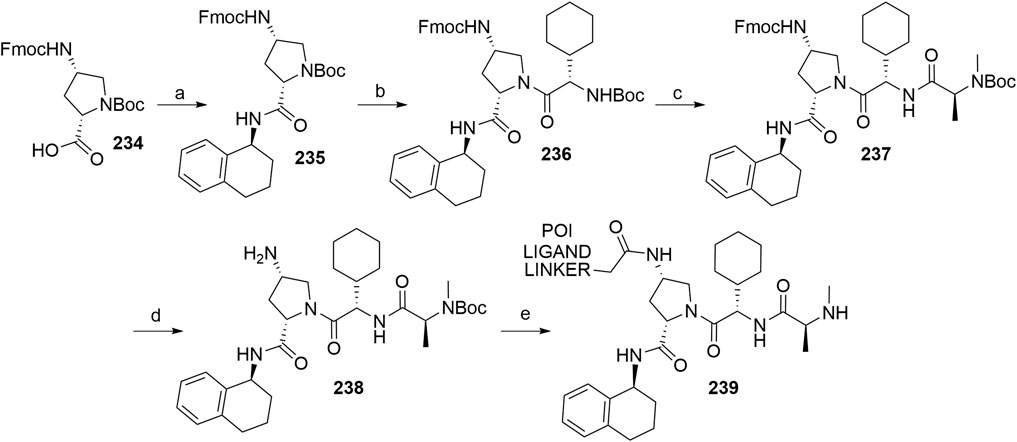
SCHEME 40. Synthesis of Boc protected IAP ligand E (compound 238) and derived degraders 239. Reagents and conditions: a) (S)-1,2,3,4-tertahydronaphthalen-1-amine, EDC, HOAt, NMP; b) i. TFA; ii. Boc-l-cyclohexylglycine, EDC, HOAt, NMP; c) i. TFA; ii. Boc-N-methyl-l-alanine, EDC, HOAt, NMP; d) piperidine (Note: no yields, solvents, temperatures, or reaction times given) (Borzilleri et al., 2014); e) i. POI ligand-linker-CO2H, HATU, Et3N, DMF, rt, 15 h; ii. TFA, rt, 16 h, 30–54% yield for conjugates used over two steps (Shah et al., 2020); e) i. linker-CO2H, HATU, DIPEA, DMF, rt, 16 h, 79–91% yield for conjugates used; ii. 4 M HCl in dioxane, rt, 2 h, 91–96% yield for conjugates used; iii. POI ligand attachment (Nunes et al., 2019).
Inhibitor of Apoptosis Proteins Ligand F
A procedure towards IAP ligand F used appropriately di-protected 4-hydroxyproline 240 as a starting compound, which was subjected to Mitsunobu conditions to attach 3-(benzyloxy)phenol, giving compound 241 (Ohoka et al., 2018). 4-(Benzyloxy)phenol was also used in place of 3-(benzyloxy)phenol to yield a para-hydroxy substituted IAP ligand. The right-hand side of the molecule was then assembled through coupling reactions with Boc-l-cyclohexylglycine to yield 242 and Boc-N-methyl-l-alanine to produce 243. Pd/C-catalyzed hydrogenation cleaved both benzyl protecting groups, resulting in compound 244, which was then coupled with (R)-1,2,3,4-tetrahydronaphthalen-1-amine into 245. A tosylate-containing POI ligand-linker conjugate was then attached, and the compounds were Boc-deprotected to give final products 246 (Scheme 41) (Ohoka et al., 2018). Alternatively, a mesylate-containing linker was incorporated, followed by the POI linker attachment and Boc-deprotection (Steinebach et al., 2020a).
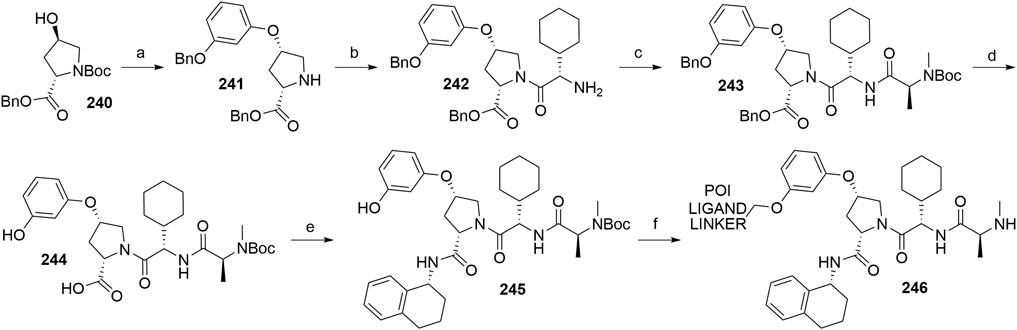
SCHEME 41. Syntheses of Boc-protected IAP ligand F (compound 245) and derived SNIPERs 246. Reagents and conditions: a) i. 3-(benzyloxy)phenol, PPh3, DEAD, THF, –10°C to rt, 16 h, 32% yield; ii. 4 M HCl in EtOAc, 0°C to rt, 1 h, 76% yield; b) i. Boc-l-cyclohexylglycine, EDC, HOBt, DIPEA, CH2Cl2, 0°C to rt, 16 h, 99% yield; ii. 4 M HCl in EtOAc, rt, 16 h, 99% yield; c) Boc-N-methyl-l-alanine, EDC, HOBt, DIPEA, CH2Cl2, rt, 16 h, 77% yield; d) Pd/C, H2, MeOH, rt, 16 h, 95% yield; e) (R)-1,2,3,4-tetrahydronaphthalen-1-amine, EDC, HOBt, DIPEA, CH2Cl2, rt, 16 h, 36% yield; f) i. POI ligand-linker-OTs, K2CO3, DMF, 60°C, 6 days, 91% yield for conjugate used; ii. 6 M HCl, THF, rt, 4 h, 46% yield (Ohoka et al., 2018); f). linker-OMs, K2CO3, DMF, 60°C, 24 h; ii. POI ligand attachment; iii. 1 M HCl in EtOAc, rt, 4 h, 79% yield for conjugates used (Steinebach et al., 2020a).
Inhibitor of Apoptosis Proteins Ligand G
Compound 250 was synthesized by first combining 1,2,3,4-tetrahydroisoquinoline 247 with (R)-1,2,3,4-tetrahydronaphthalen-1-amine and Boc-deprotecting the product to give 248. Coupling with Boc-l-tert-leucine and subsequent Boc-deprotection yielded 249, which was further converted with Boc-N-methyl-l-alanine to give 250. A POI ligand-linker chloro conjugate was then connected via the phenol group, giving compound 251, which was Boc-deprotected to afford final SNIPER molecules 252 (Scheme 42) (Casillas et al., 2016; Mares et al., 2020).
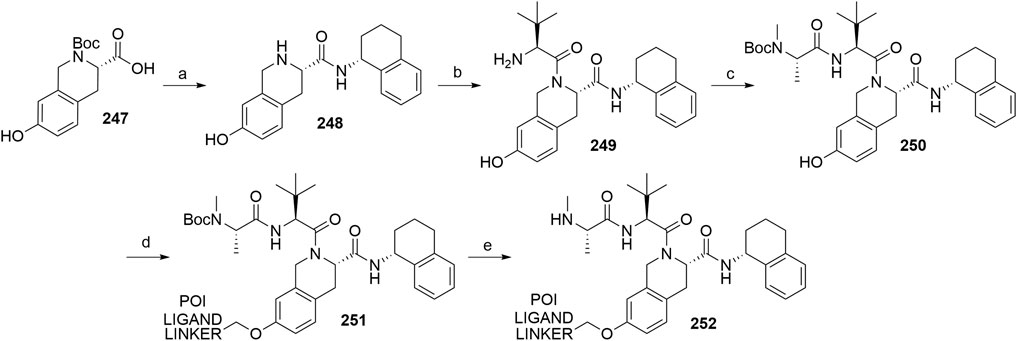
SCHEME 42. Synthesis of Boc-protected IAP ligand G (250) and derived degraders 246. Reagents and conditions: a) i. (R)-1,2,3,4-tetrahydronaphthalen-1-amine, HATU, DIPEA, DMF, rt, 30 min, 82% yield; ii. 4 M HCl in dioxane, THF, rt, overnight, 95% yield; b) i. Boc-Tle-OH, HATU, DIPEA; DMF, rt, 1 h, 66% yield; ii. 4 M HCl in dioxane, THF, rt, overnight, 97% yield; c) Boc-N-methyl-l-alanine, HATU, DIPEA, DMF, rt, overnight, 77% yield; d) POI ligand-linker-Cl, K2CO3, DMF, 80°C, overnight, 63% yield for conjugate used; e) TFA, CH2Cl2, rt, 3 h, 54% yield for conjugate used (Casillas et al., 2016; Mares et al., 2020).
Inhibitor of Apoptosis Proteins Ligand H
The synthesis of IAP ligand H (Zhang et al., 2020b) started with the treatment of compound 253 with ethyl formate to form formamide 254. Isocyanide 255 was obtained by dehydration in the presence of POCl3 and triethylamine. The 7,5-heterobicycle 256 was obtained as a mixture of diastereomers through an Ugi four-component reaction and subsequent treatment with trifluoroacetic acid. Coupling with Boc-N-methyl-l-alanine yielded a mixture of diastereomers, and flash column separation gave the desired compound 257 in a 38% yield. Hydrogenolysis cleaved the benzyl protective group and gave product 258, to which tosylate- or bromo-containing linkers were attached to form conjugates 259 (Scheme 43) (Zhang et al., 2020b).
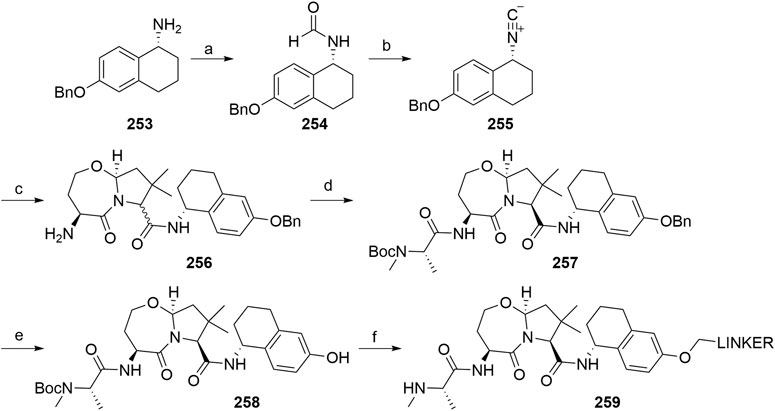
SCHEME 43. Synthesis of IAP ligand H. Reagents and conditions: a) ethyl formate, 80°C, 16 h, 84% yield; b) POCl3, Et3N, CH2Cl2, 0 °C to rt, 1 h, 89% yield; c) i. N-Boc-l-homoserine, 4,4-dimethoxy-2,2-dimethylbutanal, 7 M NH3 in MeOH, rt, 16 h; ii. TFA, CH2Cl2, rt, 16 h, 74% yield over two steps; d) Boc-N-methyl-l-alanine, HATU, DIPEA, CH2Cl2, rt, 1 h, 38% yield; e) Pd(OH)2, H2, MeOH, 40°C, 2 h, quant.; f) i. linker-OTs or -Br, K2CO3, NaI, DMF, 70°C, overnight; ii. TFA, CH2Cl2, rt, 16 h (Note: yields not given) (Zhang et al., 2020b).
Inhibitor of Apoptosis Proteins Ligand I
IAP ligand I was utilized in PROTACs (Dragovich et al., 2020) and could be synthesized based on the following procedure (Kester et al., 2013). By coupling compound 254 with methyl 4-(chlorocarbonyl)benzoate and the subsequent methyl ester hydrolysis, intermediate 255 was formed. POI ligand-linker-amine conjugates were then coupled, and the obtained products were Boc-deprotected to give the final PROTAC compound 256 (Scheme 44) (Dragovich et al., 2020).
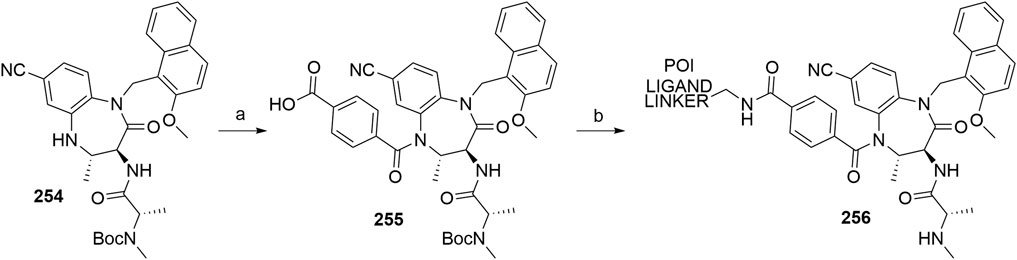
SCHEME 44. Synthesis of IAP ligand I-based PROTAC 256. Reagents and conditions: a) i. methyl 4-(chlorocarbonyl)benzoate, Et3N, 1,2-dichloroethane, 80°C, 24 h, 59% yield; ii. LiOH × H2O, THF/H2O, rt, 1 h; b) i. POI ligand-linker-NH2, HATU, DIPEA, DMF, rt, 1 h, 68% yield for conjugate used; ii. 4 M HCl in dioxane, CH2Cl2, rt, 1 h, 41% yield for conjugate used (Dragovich et al., 2020).
Statistical Overview of Utilized Inhibitor of Apoptosis Proteins Ligands
Using data extracted from PROTAC-DB (Weng et al., 2021) (http://cadd.zju.edu.cn/protacdb/, as of the February 26, 2021) a statistical overview was done to determine the frequency of various IAP ligands and linker attachment options used in PROTAC compounds (Figure 7). LCL-161 derivatives were most commonly utilized, with around 30% of PROTACs incorporating its structure. Following closely was bestatine, with MV1 derivatives and IAP ligand E having a lower presence at 10 and 9%, respectively. Other IAP ligands are less common, with fewer than 3% representation.
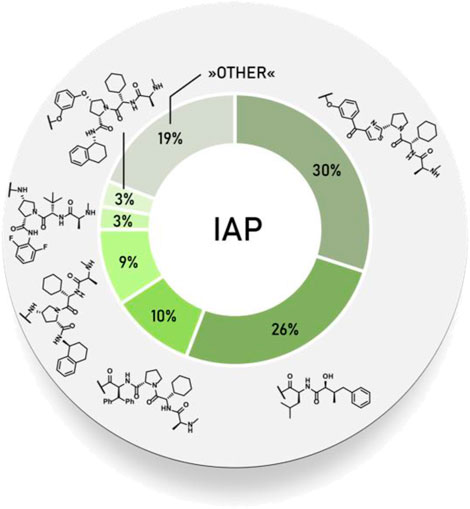
FIGURE 7. Frequency of IAP ligands used in PROTAC compounds.
MDM2
The p53 protein is a product of the tumor-suppressor gene and acts as a transcription factor that gets activated when cell stress occurs, especially upon the occurrence of DNA damage. Activation of the p53 network results in the inhibition of the cell cycle and can lead to apoptosis of damaged cells to prevent their unhindered growth, thus acting as an important tumor suppressor (Levine, 1997; Vogelstein et al., 2000). The effects of p53 are controlled in an autoregulatory negative feedback loop involving MDM2, which belongs to the family of RING finger ubiquitin ligases (Michael and Oren, 2003). p53 induces the expression of MDM2, which in turn leads to the repression of p53 activity through binding of MDM2 to p53 and blocking its function, as well as through MDM2-mediated ubiquitination and subsequent degradation of p53 by the proteasome (Michael and Oren, 2003; Vassilev et al., 2004). Excessive activity of MDM2 has been observed in numerous malignancies, which makes it a promising target for the treatment of tumors due to its dual-mode mechanism of interaction with p53 (Momand et al., 1998).
The E3 ligase activity of MDM2 was utilized in the first small-molecule PROTAC by incorporating a molecule belonging to a class of imidazoline derivatives called nutlins, which bind to MDM2 in a nano-to micromolar range (Vassilev et al., 2004; Schneekloth et al., 2008). Additional proteins to be successfully degraded through MDM2-mediated ubiquitination include BTK (Sun et al., 2018), PARP1 (Zhao et al., 2019), and TrkC (Zhao and Burgess, 2019), among others. Utilized ligands are collected in Figure 8.
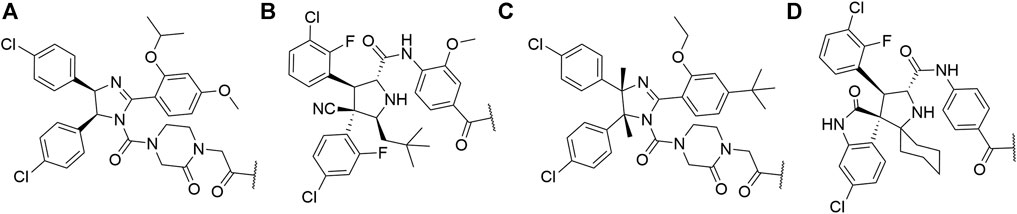
FIGURE 8. MDM2 ligands. (A) Nutlin-3a derivative, (B) Idasanutlin derivative, (C) RG7112-derivative, (D) MI-1061 derivative.
Nutlin-3-Based Ligands and Derivatives
The first nutlin-derived PROTAC (Schneekloth et al., 2008) has been prepared as follows. Starting material 257 was first reacted with meso-1,2-bis(4-chlorophenyl)-1,2-ethylenediamine to form imidazoline 258via a radical pathway induced by NBS. Triphosgene was used to create an activated formic acid ester which was then reacted with tert-butyl 2-(2-oxopiperazin-1-yl)acetate and deprotected by TFA to give 259 in an 88% yield over three steps(Schneekloth et al., 2008). HATU-facilitated coupling was then employed to attach POI ligand-linker-NH2 conjugates to form 260 (Scheme 45) (Schneekloth et al., 2008; Zhao et al., 2019).
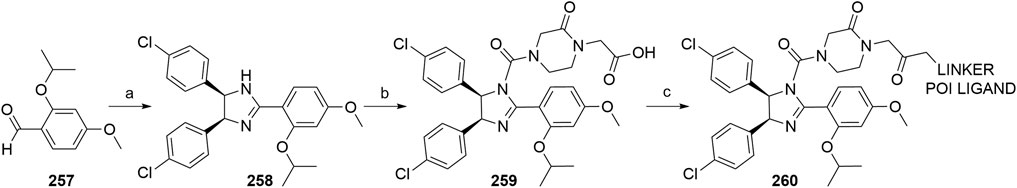
SCHEME 45. Synthesis of Nutlin-3a derivative 259 and degraders 260. Reagents and conditions: a) 1,2-bis(4-chlorophenyl)-1,2-ethanediamine, CH2Cl2, 0°C, 2 h, then NBS, 0°C to rt, 16 h, 88% yield; b) i. triphosgene, Et3N, THF, 0°C, 2.5 h, 96% yield; ii. tert-butyl 2-(2-oxopiperazin-1-yl)acetate, CH2Cl2, 0°C, 1.5 h, 96% yield; iii. TFA, CH2Cl2, 96% yield; c) POI ligand-linker-NH2, HATU, DIPEA, DMF, rt, 16.5 h, 61% yield for conjugate used (Schneekloth et al., 2008); c) linker-NH2, HATU, Et3N, DMF, CH2Cl2, 5 h, 63% yield for linker used (Zhao et al., 2019).
Close Nutlin-3a derivatives were also incorporated into PROTACs (Wang et al., 2019). Starting material 261 was coupled with 4-(tert-butoxy)-2-ethoxybenzoic acid to yield 262, which was subsequently Boc-deprotected and underwent a CDI-mediated urea formation to obtain 263. Cyclization to 264 was done using TPPO and triflic anhydride. Linker attachment was obtained by alkylation using bromo linkers alongside K2CO3 under reflux or by coupling with carboxylic acid linkers (Scheme 46) (Wang et al., 2019).
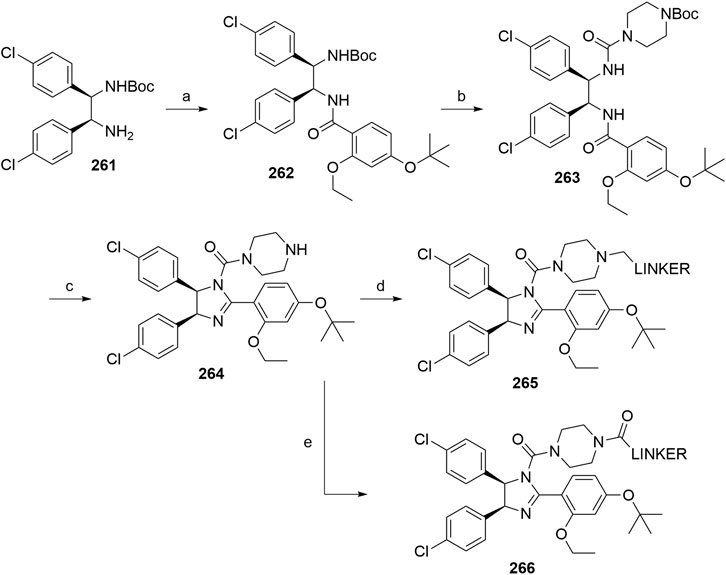
SCHEME 46. Synthesis of Nutlin-3 derivative 264 and linker conjugates 265 and 266. Reagents and conditions: a) 4-(tert-butoxy)-2-ethoxybenzoic acid, EDC, DMAP CH2Cl2, rt, 16 h, 96% yield; b) i. TFA, CH2Cl2, rt, 30 min, 92%; ii. CDI, CH2Cl2, N-Boc-piperazine, rt, overnight, 84% yield; c) TPPO, triflic anhydride, CH2Cl2, 0°C, 1.5 h, 97% yield; d) Br-linker, K2CO3, THF, reflux, overnight, 61–72% for linkers used; e) linker-CO2H, HATU, DIPEA, DMF, rt, 3 h, 83–98% yield for linkers used (Wang et al., 2019).
An alternative synthetic strategy was devised (Scheme 47), by attaching the linker to tert-butyl 3-oxopiperazine-1-carboxylate 268 prior to urea bond formation with 267 to give compound 270. Cyclization to 271 was obtained similarly as depicted in Scheme 45 (Nietzold et al., 2019).
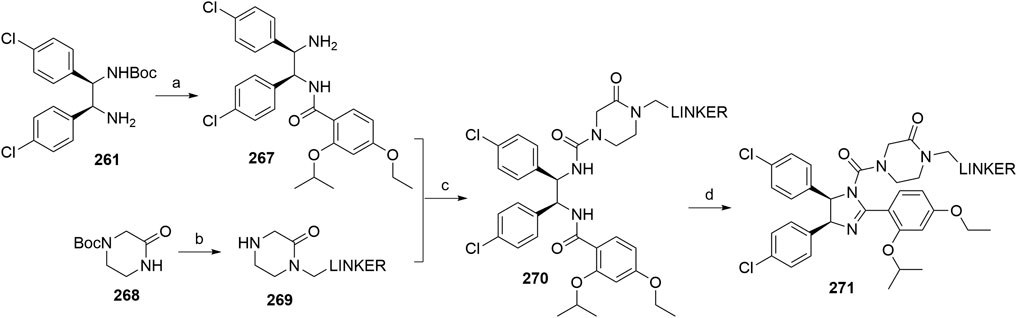
SCHEME 47. Alternative synthesis of Nutlin-3 derivative—linker conjugates 271. Reagents and conditions: a) i. 2-isopropoxy-4-methoxybenzoic acid, EDC, DMAP, CH2Cl2, rt, 17 h, 79% yield; ii. TFA, CH2Cl2, rt, 3 h, 97% yield; b) i. Br-linker, NaH, DMF, 0°C to rt, 15 h, 75% yield for linker used; ii. TFA, CH2Cl2, rt, 5 h, 82% yield for conjugate used; c) CDI, CH2Cl2, rt, 2 h, then 269, rt, 14 h, 86% yield for conjugate used; d) triflic anhydride, TPPO, CH2Cl2, 45°C, 15 h, quant. for conjugate used (Nietzold et al., 2019).
Idasanutlin-Based Ligands
The established synthesis of idasanutlin begins with a Knoevenagel condensation between starting material 272 and 3-chloro-2-fluorobenzaldehyde (Ding et al., 2013; Rimmler et al., 2016). Compound 273 can then be joined with 274 to give methyl ester 275, which is then hydrolyzed and coupled with methyl 4-amino-3-methoxybenzoate and again hydrolyzed to yield idasanutlin 277. The procedure requires a chiral chromatographic phase for separation of the desired product (Rimmler et al., 2016). Alternatively, imine 279 can be formed using 3,3-dimethylbutanal and 278, followed by a Ag(I) or Cu(I) catalyzed asymmetric [3 + 2] cycloaddition. Isomerization with NaOH in THF/EtOH yields the desired stereoisomer (Rimmler et al., 2016; Zou et al., 2020). POI ligand-linker-NH2 conjugates were coupled with idasanutlin to yield MDM2-targeting PROTACs 280 (Hines et al., 2019; Zhang et al., 2020c). By starting the PROTAC preparation with racemic idasanutlin, the chiral separation was performed after the coupling reaction (Hines et al., 2019) (Scheme 48).
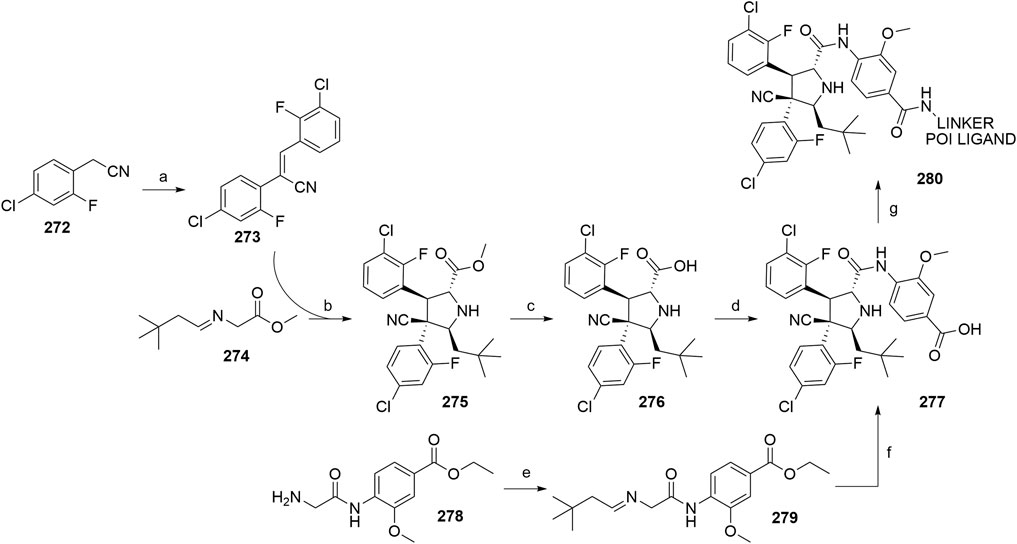
SCHEME 48. Synthesis of idasanutlin 277. Reagents and conditions: a) 3-chloro-2-fluorobenzaldehyde, NaOMe, MeOH, 50°C, 3 h (yield not given) (Ding et al., 2013); a) 3-chloro-2-fluorobenzaldehyde, NaOMe, MeOH, EtOH, H2O, 50°C, 3 h, 91% yield (Rimmler et al., 2016, 2); b) AgF, Et3N, 1,2-dichloroethane, 31% yield; c) i. 2 M NaOH, MeOH, 97% yield; ii. chiral SFC separation, 49% yield; d) i. methyl 4-amino-3-methoxybenzoate, Ph2POCl2, DIPEA, CH2Cl2, 81% yield; ii. NaOH (aq), MeOH, THF, 80% yield (Shu et al., 2016); e) 3,3-dimethylbutanal, Et3N, MTBE, rt, 6 h, 94% yield (Rimmler et al., 2016); f) i. 273, AgOAc, (R)-MeOBIPHEP, 2-Me-THF, 2–4°C, 16 h; ii. LiOH, 2-Me-THF, 65°C, 19 h, 97% yield over 2 steps; iii. LiOH, isopropanol, 63–67°C, 6 h then 15–20°C, 8 h, 45% yield; f) i. 273, (R)-BINAP, CuOAc, Et3N, 2-Me-THF, 5 h; ii. NaOH (aq), THF, EtOH, rt, 18 h, 78% yield over 2 steps; iii. THF, EtOAc, 65°C, 2 h, 79% yield (Rimmler et al., 2016, 2); f) i. 273, Cu(MeCN)4PF6/ent-Phosferrox, Cs2CO3, THF, –40°C, 93% yield; ii. NaOH (aq), THF, EtOH, rt, 18 h; iii. THF, EtOAc, 65°C, 2 h (Zou et al., 2020); g) POI ligand-linker-NH2, HATU, DIPEA, MeCN, rt, 30 min, 42–60% yield for conjugates used (Zhang et al., 2020c); POI ligand-linker-NH2, HATU, DIPEA, DMF, rt, 24 h, 28% yield for conjugate used (Hines et al., 2019).
RG7112-Based Derivatives
Diamine 281 was reacted with methyl 4-(tert-butyl)-2-ethoxybenzoate using trimethylaluminum to give imidazoline 282. Carbamoyl chloride 283 was obtained through treatment with phosgene. Coupling with an appropriate piperazine derivative (exact procedure is not described) afforded compound 284, which was then coupled with amine linkers to yield derivatives 285 (Scheme 49) (Vu et al., 2013; Sun et al., 2018).
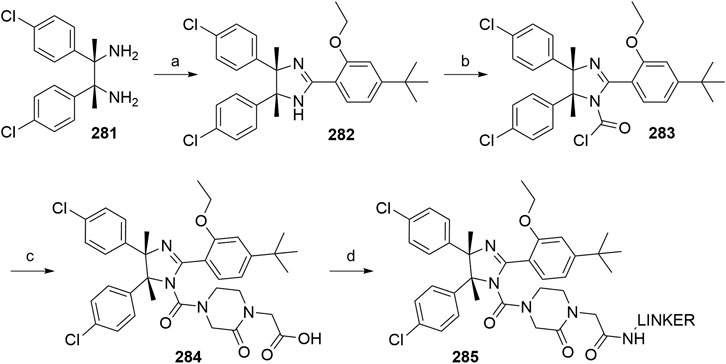
SCHEME 49. Synthesis of RG7112 derivative 284 and linker attachment to form conjugate 285. Reagents and conditions: a) AlMe3, toluene, 0°C to rt, 20 min, then methyl 4-(tert-butyl)-2-ethoxybenzoate, reflux, 2.5 h, 18% yield; b) Et3N, phosgene, toluene, 0°C, 30 min, 87% yield; c) exact procedure is not disclosed; piperazine derivative, Et3N, CH2Cl2, 0°C, 7 h (Vu et al., 2013); d) linker-NH2, HATU, DIPEA, DMF, rt, 2 h, 51% yield for linker used (Sun et al., 2018).
MI-1061-Based Derivatives
Further development of MDM2 inhibitors yielded the potent and efficacious compound MI-1061 (289, Scheme 50). Using starting material 286, an asymmetric 1,3-dipolar cycloaddition reaction was employed to create the spiro-oxindole scaffold 287. Ring-opening and oxidative removal of the chiral auxiliary (1,2-diphenylethanol) on the pyrrolidine nitrogen yielded the free carboxylic acid 288. Following CDI coupling with methyl 4-aminobenzoate and ester hydrolysis yielded inhibitor 289 (Aguilar et al., 2014). Attachment of POI ligand-linker conjugates to 288 (Yang et al., 2019) or incorporation of the full MI-1061 structure (289) (Li et al., 2019) provided corresponding PROTACs.
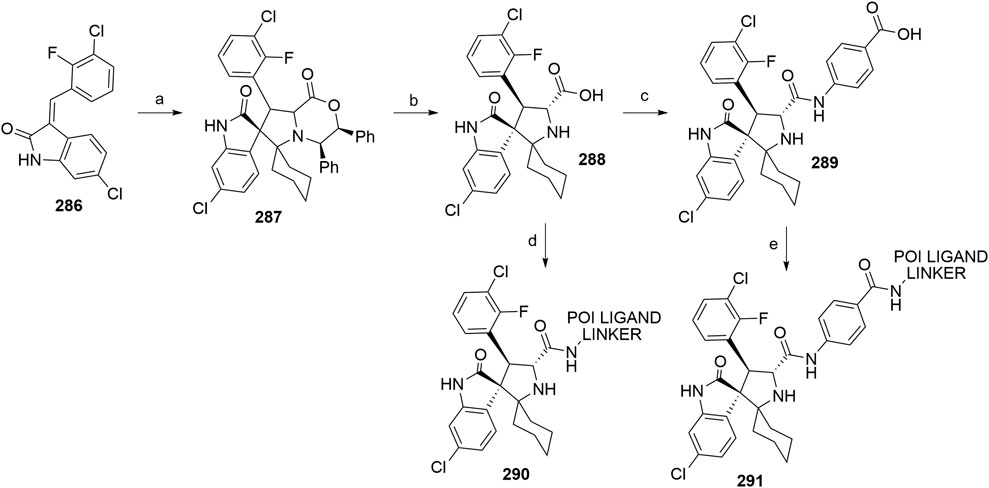
SCHEME 50. Synthesis of MI-1061 based derivative 289. Reagents and conditions: a) (5R,6S)-5,6-diphenyl-2-morpholinone, cyclohexanone, toluene, 140°C, 2 h, 64% yield; b) i. conc. H2SO4, MeOH, 50°C, 57% yield; ii. MeCN, H2O, CAN, rt, 30 min, 92% yield; c) i. CDI, DIPEA, DMAP, 1,2-dichloroethane, 40°C, 30 min; ii. methyl 4-aminobenzoate, reflux, overnight, 41% yield; ii. LiOH × H2O, NaOH, THF, MeOH, rt, 2 h; ii. TFA, rt, briefly, 31% (Aguilar et al., 2014); d) POI ligand-linker-NH2, HATU, DIPEA, DMF, rt, 10 min (Yang et al., 2019); e) POI ligand-linker-NH2, HATU, DIPEA, DMF, DMSO rt, 30 min, 17–75% yield for conjugates used (Li et al., 2019).
RNF114
Gene transcription is regulated in a crucial way by the zinc-finger gene family. One of the members is ZNF313, also known as RNF114 or ZNF228, which contains both C2H2 and RING-finger structure. Along with the N-terminal RING domain, it also has a C-terminal ubiquitin-interacting motif (UIM), both of which are responsible for RNF114s E3 ligase activity (Bijlmakers et al., 2011; Han et al., 2013). Nimbolide, a limonoid natural product derived from the Neem tree (Azadirachta indica), was recently found to primarily target RNF114 by covalently modifying its cysteine-8, thus leading to impaired E3 ligase activity of RNF114. As a result, substrate recognition is blocked, which leads to the stabilization of tumor suppressors p21 and p57, explaining nimbolide’s antiproliferative effects. By incorporating nimbolide into a BRD4-targeting PROTAC, RNF114 was successfully established as a viable E3 ligase option for targeted protein degradation (Spradlin et al., 2019).
Nimbolide
A procedure for extracting nimbolide from commercial neem leaf powder has been reported (Tong et al., 2020b). After a maceration of 450 g neem leave powder in anhydrous MeOH, the crude extract was filtered through celite and concentrated under vacuum. The residue was partitioned between EtOAc and H2O. The organic phase was then washed with an aqueous solution of NaHCO3 and Na2S2O3, dried over Na2SO4 and concentrated in vacuo. Two subsequent column chromatographies on silica gel with hexanes/EtOAc and CH2Cl2/acetone were performed. Finally, trituration yielded around 1.2 g of nimbolide (Tong et al., 2020b). Derivatization is possible as described (Spradlin et al., 2019). Hence, treatment of nimbolide with NBS led to a selectively brominated product, which was then reacted with 4-formylphenylboronic acid following a Suzuki coupling procedure to yield 293 (Spradlin et al., 2019). Performing a Pinnick oxidation gave carboxylic acid 294, which was then available for attaching POI ligand-amine linker conjugates through HATU-mediated coupling (Scheme 51) (Spradlin et al., 2019; Tong et al., 2020b).
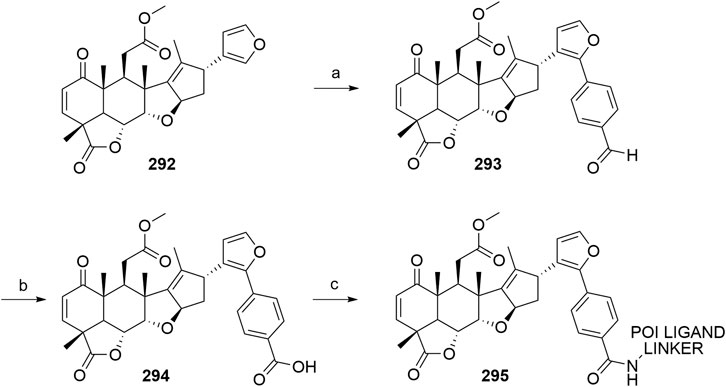
SCHEME 51. Derivatization of nimbolide (292) for use in PROTACs. Reagents and conditions: a) i. NBS, DMF, 0°C, 1 h, 95% yield; ii. 4-formylphenylboronic acid, Pd2(dba)3, SPhos, K3PO4, toluene, 60°C, 48 h, 92% yield; b) 2-methyl-2-butene, NaClO2, NaH2PO4, t-BuOH, H2O, rt, 6 h, yield not given; c) POI ligand-linker-NH2, HATU, DIPEA, DMF, 0–4°C, 16 h, 42–45% yield for conjugates used (Spradlin et al., 2019); c) POI ligand-linker-NH2, HATU, DIPEA; CH2Cl2, rt, 10 h, 26–54% yield for conjugates used (Tong et al., 2020b).
Alternative RNF114 Ligand
A small molecule, that accessed the same cysteine targeted by nimbolide was incorporated into a BRD4-targeting PROTAC (Luo et al., 2021). Starting material 296 was first protected with a tetrahydropyranyl ether to form 297 and then reacted with 4-bromoacetophenone. Following a deprotection, 298 was obtained and the OH alkylated with a bromo linker, yielding 299. Finally, compound 300 can be achieved over 2 steps (Scheme 52) (Luo et al., 2021).
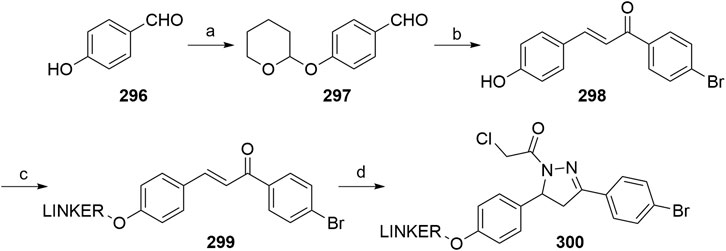
SCHEME 52. Synthesis of a fully synthetic small molecule as a covalent recruiter of RNF114 and its derivatization into PROTACs. Reagents and conditions: a) 3,4-dihydro-2H-pyran, pyridinium p-toluenesulfonate, CH2Cl2, rt, overnight, 78% yield; b) i. 4-bromoacetophenone, EtOH, 0°C, 15 min, then 10% NaOH, 0°C, 20 min, then rt, 3 h, 83% yield; ii. pyridinium p-toluenesulfonate, MeOH, 55°C, 3 h, 72% yield; c) linker-Br, K 2CO3, DMF, 60°C, 5 h, 87% yield for linker used; d) i. Hydrazine monohydrate, EtOH, 80°C, 5 h; ii. chloroacetyl chloride, Et3N, CH2Cl2, 0°C, 30 min, then rt, overnight, 58% yield (Luo et al., 2021).
DCAF16
DCAF16 is a member of the damage-specific DNA binding protein 1 (DDB1)-CUL4 associated factor (DCAF) protein group, which act as substrate-recognition receptors within the UPS (Liang et al., 2017). It consists of 216 amino acids and contains eight cysteine residues, four of which are clustered together between amino acids 173 and 179. By using broadly reactive, cysteine-directed electrophilic fragments, a successful covalent modification of DCAF16 was achieved to induce the degradation of BRD4 and FKBP12 (Zhang et al., 2019). The authors suggested that utilizing such electrophilic PROTACs may provide certain advantages in the field of targeted protein degradation, as DCAF16 seems to exclusively promote the degradation of nuclear proteins, sparing cytosolic ones. The covalent interaction between DCAF16 and the chimeric molecule allows for protein degradation at low fractional engagement and could prolong the degradation effect, as it should correlate with DCAF16 turnover, as opposed to the clearance of the PROTAC. This strategy may additionally have a minimal effect on DCAF16’s endogenous substrates. However, the broadly reactive fragments, utilized in DCAF16-hijacking PROTACs, still modified numerous other cellular proteins and required further optimization to enable a future for electrophilic PROTACs (Zhang et al., 2019).
Electrophilic DCAF16 Binder 1
First of the reactive fragments used was compound 302, which was synthesized by treating 6-hydroxy-1,2,3,4-tetrahydroquinoline (301) with chloroacetyl chloride. tert-Butyl bromoacetate was then attached to the hydroxyl group and subsequently Boc-deprotected to yield 303, which allowed for the coupling with primary amine linkers to form conjugates 304 (Scheme 53) (Zhang et al., 2019).
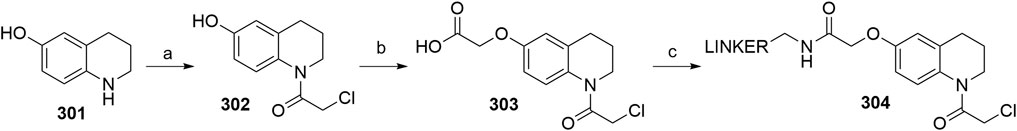
SCHEME 53. Synthesis of DCAF-hijacking degraders 304. Reagents and conditions: a) chloroacetyl chloride, NaOH, dioxane, H2O, rt, 4 h; b) i. tert-butyl bromoacetate, Cs2CO3, DMF, rt, 3 h; ii. TFA, CH2Cl2, H2O, rt, 2 h, 76% yield over three steps; c) linker-NH2, COMU, NMP, DMF, 1 h, rt, 18–22% yield for linkers used (Zhang et al., 2019).
Electrophilic DCAF16 Binder 2
To synthesize conjugates 308, β-alanine tert-butyl ester was coupled with starting aniline derivative 305 to form 306. The aromatic amine group underwent a reaction with chloroacetyl chloride, giving compound 307, which was then treated with TFA to remove the Boc-protecting group and enable coupling with primary amine linkers (Scheme 54) (Zhang et al., 2019).
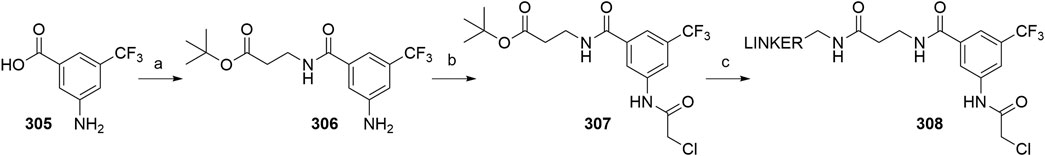
SCHEME 54. Synthesis of DCAF-hijacking degraders 308. Reagents and conditions: a) β-alanine tert-butyl ester, COMU, NMP, DMF, rt, 2 h, 72% yield; b) chloroacetyl chloride, DIPEA, CH2Cl2, 0°C to rt, 2 h, 78% yield; c) i. TFA, CH2Cl2, H2O, rt, 2 h; ii. linker-NH2, COMU, NMP, DMF, rt, 1 h, 13% yield for linker used (Zhang et al., 2019).
Electrophilic DCAF16 Binder 3
For the third structural type of DCAF binders (compounds 312), the synthesis started with the methyl ester 309, which was hydrolyzed to form 310 and then coupled with primary amine linkers. Compound 311 was then treated with acryloyl chloride to incorporate a cysteine-targeting moiety into conjugates 312 (Scheme 55) (Zhang et al., 2019).
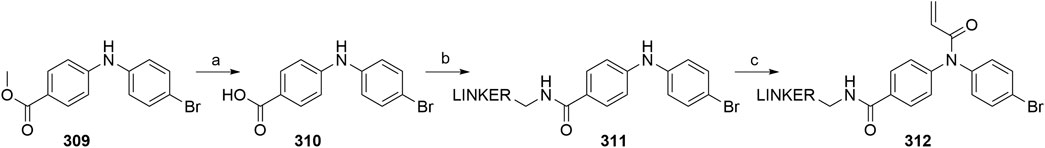
SCHEME 55. Syntheses of DCAF-hijacking degraders 312. Reagents and conditions: a) KOH, EtOH, H2O, 100°C, 4 h; b) LINKER-NH2, COMU, NMP, DMF; rt, 2 h, 57% yield for linker used over 2 steps; c) acryloyl chloride, DIPEA, DMAP, CH2Cl2, 0°C to rt, 6 h (Note: the compound was set in the next step (POI ligand attachment) without purification) (Zhang et al., 2019).
DCAF15
DDB1-and-Cul4-associated factor 15 (DCAF15) is a substrate adaptor of the E3 ligase Rbx-Cul4-DDA1-DDB1-DCAF15. Through studying aryl sulfonamides with known anticancer activity, it has been found that indisulam stabilizes the interaction between DCAF15 and an essential splicing factor RBM39, which leads to RBM39 polyubiquitination and proteasomal degradation, thus inhibiting cell growth (Han et al., 2017; Bussiere et al., 2020).
Building on the known pharmacological activity of indisulam, its structure was modified to enable linker attachment and incorporation of the E3 ligase ligand into PROTACs (Zoppi et al., 2019). Starting material 313 was reacted with 4-formylbenzenesulfonyl chloride to obtain sulfonamide 314. Reductive amination lead to amine 315, which was then coupled with POI ligand-linker-NH2 conjugates to give PROTACs 316, thus successfully expanding the E3 ligase options for targeted protein degradation (Scheme 56). However, activity of such PROTACs was only moderate (Zoppi et al., 2019).
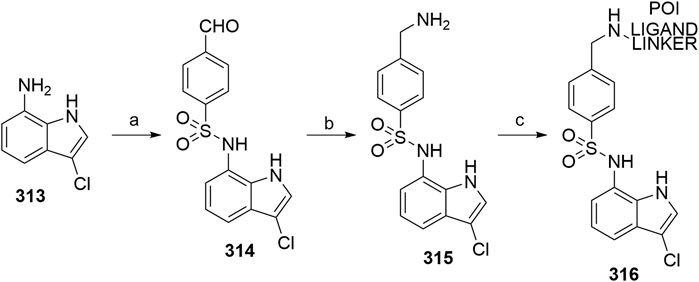
SCHEME 56. Synthesis of indisulam derivative 315 and its incorporation into degraders 316. Reagents and conditions: a) 4-formylbenzenesulfonyl chloride, pyridine, EtOAc, rt, 3 h, 62% yield; b) sat. NH4OAc in EtOAc, NaBH3CN, 100°C, 15 min, 35% yield; c) POI ligand-linker-CO2H, HATU, HOAt, DIPEA, DMF, rt, 2 h, 34–37% yield for conjugates used (Zoppi et al., 2019).
KEAP1
Kelch-like ECH-associated protein 1 (KEAP1) plays a key role in regulating the nuclear factor erythroid 2–related factor 2 (NRF2), which is involved in the cellular cytoprotective response to electrophiles and reactive oxygen species. Being a part of a homodimeric KEAP1/Cul3 complex that possesses E3 ubiquitin ligase activity, KEAP1 works as a substrate receptor and is responsible for selectively recognizing NRF2 and linking it to Cul3 for its ubiquitination (Davies et al., 2016; Jiang et al., 2016). Through forming reversible covalent interactions with cysteines of KEAP1, BRD4 degradation was achieved by recruiting KEAP1/Cul3 E3 ligase activity using the highly electron-deficient cyanoenone moiety-containing triterpene derivative bardoxolone (Tong et al., 2020a).
Bardoxolone Derivatives
With oleanolic acid 317 as starting material, an efficient, five-step synthesis of bardoxolone methyl (321) was accomplished (Fu and Gribble, 2013) (Scheme 57). After forming a methyl ester 318, the compound was oxidized to give 319, which was then transformed into 320 over two steps. The final step consisted of substituting the bromo group with a cyano, to form bardoxolone methyl with an overall yield of 50% (Fu and Gribble, 2013). Both bardoxolone methyl and bardoxolone-CO2H are commercially available.
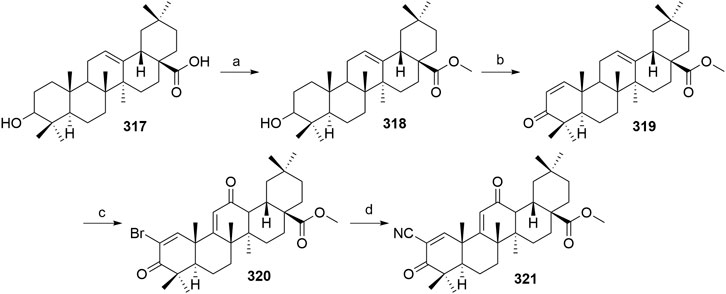
SCHEME 57. Syntheses of bardoxolone methyl (321) from oleanolic acid 317. Reagents and conditions: a) K2CO3, MeI, DMF, 0°C to rt, 24 h, 99% yield; b) iodobenzoic acid, fluorobenzene, DMSO, 85°C, 24 h, 87% yield; c) i. mCPBA, CH2Cl2, 0°C to rt, 24 h; ii. HBr, Br2, acetic acid, rt to 35°C, 24 h, 82% yield; d) CuCN, KI, DMF, 120°C, 24 h, 73% yield (Fu and Gribble, 2013).
Final PROTACs were assembled by using bardoxolone (322) and coupling it with POI ligand-linker-NH2 conjugates under standard conditions using HATU and DIPEA. Alternatively, bardoxolone was subjected to Pd/C-catalyzed hydrogenation to give compound 324, which was incorporated into negative-control compounds 325 with the same coupling procedure (Scheme 58) (Tong et al., 2020a).
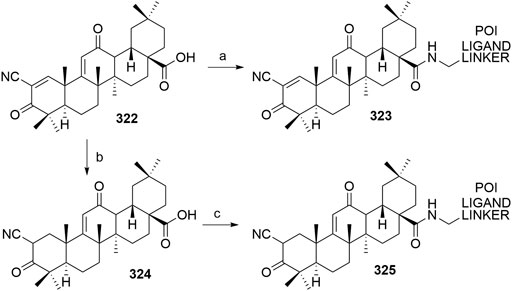
SCHEME 58. Synthesis of final KEAP1-hijacking PROTAC 323 and negative control compound 325. Reagents and conditions: a) HATU, DIPEA, CH2Cl2, rt, 12 h, 48% yield for conjugate used; b) Pd/C, H2, EtOAc, rt, 25 min, 96% yield; c) HATU, DIPEA, CH2Cl2, rt, 12 h, 45% yield for conjugate used (Tong et al., 2020a).
FEM1B
FEM1B was recently discovered to play a role in regulating the cellular response to reductive stress, which can lead to various diseases, such as diabetes, cardiomyopathy, or cancer. During a depletion of reactive oxygen species (ROS), FEM1B recognizes reduced cysteines on its target Folliculin-interacting protein 1 (FNIP1) and induces its ubiquitination and subsequent degradation, which restores mitochondrial activity and redox homeostasis of the cell (Manford et al., 2020). The key cysteine residue C186 was noted to present a possible target for developing a FEM1B recruiter useful in the field of targeted protein degradation. Through screening a library of cysteine-reactive covalent ligands, compound EN106 was identified and its structure modified to be incorporated into BRD4-targeting PROTACs (Henning et al., 2021).
An acetate spacer was attached to starting material 326 to give methyl ester 327. After the nitro group was reduced, amine 328 was alkylated with acrylonitrile, followed by acylation with chloroacetyl chloride to yield 329. After hydrolyzing the methyl ester, POI ligand-linker-NH2 conjugate was attached through coupling to provide PROTAC 330 (Scheme 59) (Henning et al., 2021).
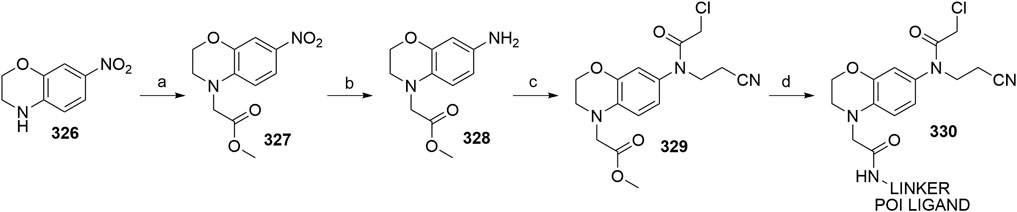
SCHEME 59. Synthesis of FEM1B-recruiting PROTACs 330. Reagents and conditions: a) methyl bromoacetate, NaH, DMF, 0°C to rt, 94% yield; b) Fe, NH4Cl, EtOH, H2O, 80°C; c) i. acrylonitrile, alumina, reflux; ii. chloroacetyl chloride, Et3N, CH2Cl2, 0°C, 65% yield over 3 steps; d) i. LiOH (aq), MeOH, rt; ii. POI ligand-linker-NH2, HATU, DIPEA, DMF, rt, 50% yield over 2 steps for conjugate used (Henning et al., 2021).
Arylhydrocarbon Receptor
The arylhydrocarbon receptor (AhR), a member of the basic helix-loop-helix (bHLH)/Per- Arnt-Sim (PAS) family of proteins, functions as a ligand-activated transcription factor. In addition to playing a role in response to xenobiotic-induced toxicity and carcinogenesis, it also acts as an adaptor protein for substrate recognition as part of CUL4AhR E3 ligase complex (Ohtake et al., 2007; Luecke-Johansson et al., 2017). Studies of AhR agonists, such as 3-methylcholanthrene, α- and β-naphthoflavone (α- and β-NF) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), have shown that these hydrophobic compounds activate AhR, leading to the degradation of the sex steroid receptors ERα and AR, along with the self-ubiquitination of AhR (Ma and Baldwin, 2000; Ohtake et al., 2007; Busbee et al., 2013; Luecke-Johansson et al., 2017).
β-Naphthoflavone Derivatives
Building on established AhR agonists, β-NF and 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), chimeric molecules have been developed that induce the degradation of cellular retinoic acid binding proteins (CRABPs) and BRD by hijacking AhR for its E3 ligase activity. Naphthoflavone derivative 332 was synthesized from 2-hydroxy-1′-acetonaphthone (331) over three steps. The product was then subjected to radical bromination using NBS and AIBN to yield 333, which was then alkylated with a hydroxy-containing linker to give 334 (Scheme 60) (Ohoka et al., 2019).
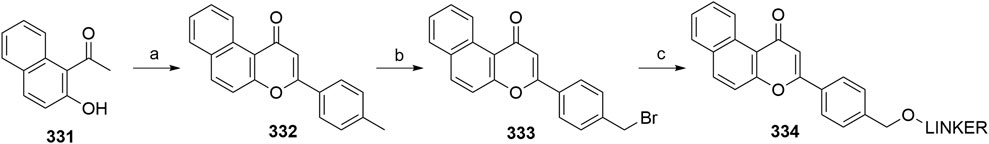
SCHEME 60. Synthesis of β-NF-derived compound 334. Reagents and conditions: a) i. p-toluoyl chloride, pyridine, rt, 1 h; ii. KOH, pyridine, 60°C, 30 min; iii. H2SO4, AcOH, 110°C, 6 h, 69% yield; b) NBS, AIBN, CCl4, 80°C, 12 h, 82% yield; c) OH-linker, NaH, THF, rt, 24 h, 43% yield for linker used (Ohoka et al., 2019).
ITE Derivatives
To synthesise endogenous AhR ligand ITE, l-cysteine methyl ester was acylated with starting material 335 to obtain glyoxylamide 336. The following cyclization was performed using TiCl4 in CH2Cl2, forming thiazoline ester 337. Oxidation with MnO2 yielded ITE (338) (DeLuca et al., 2003), the methyl ester of which was then hydrolyzed and available for coupling with amine linkers to obtain compound 339, available for incorporation into chimeric degraders (Scheme 61) (Ohoka et al., 2019).
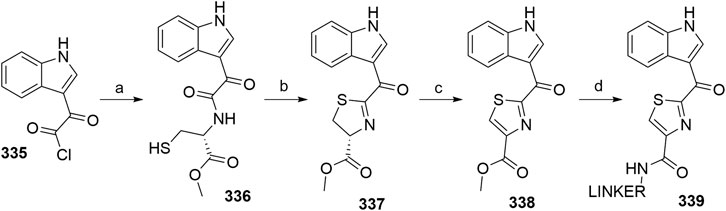
SCHEME 61. Synthesis of ITE (338) and its derivatization for use in degraders. Reagents and conditions: a) l-cysteine methyl ester, Et3N, benzene, 0°C to rt, 20 h, then reflux, 2.5 h, 55% yield; b) TiCl4, CH2Cl2, reflux, 5 h, then rt, 16 h, 25% yield; c) MnO2, CH2Cl2, rt, 3 h, 88% yield (DeLuca et al., 2003); d) i. 4 M KOH (aq), THF, rt, 16 h; ii. linker-NH2, HATU, DIPEA, DMF, rt, 16 h, 57% yield over 2 steps for linker used (Ohoka et al., 2019).
Conclusion
Principles of PROTAC design and tackling the challenges that accompany the field were explored extensively (Maple et al., 2019). To reiterate those findings, we would like to briefly touch on optimization in the early stages of planning chimeric molecules in a way that increases the likelihood of obtaining potent, cell-permeable degraders. In Figure 9, a representative ligand for each of the four most commonly used E3 ligases is presented along with its molecular descriptors. This radar plot can serve as a quick navigational tool to evaluate how the chosen E3 ligase ligand might contribute to the physicochemical properties of final degraders and aid in ligand selection in order to balance out the size, lipophilicity, and related characteristics that affect the success rate of PROTACs. Additionally, a selection of commercially available building blocks for the most widely applied CRBN, VHL, and IAP ligands are collected in Figure 10 and can be used as a quick aid for those starting with E3 ligase ligand synthesis.
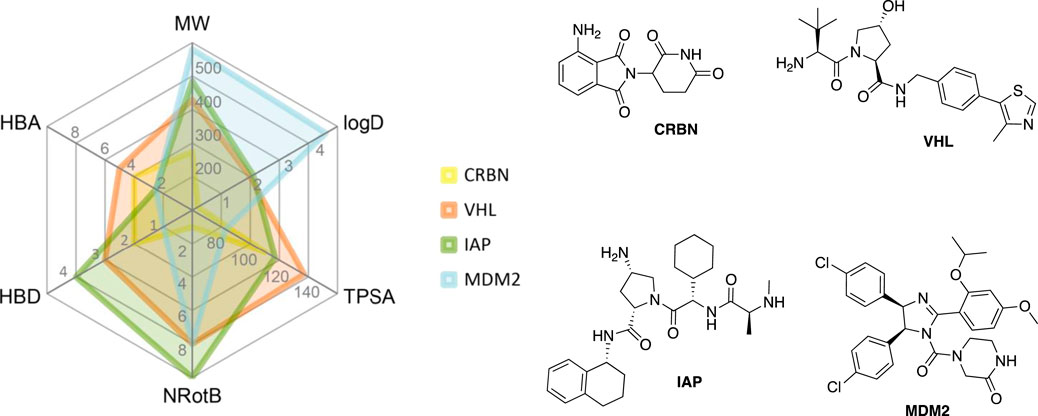
FIGURE 9. Radar plot incorporating the molecular descriptors of representative CRBN, VHL, IAP, and MDM2 ligands. Values were calculated with SwissADME (Daina et al., 2017).
FIGURE 10. CAS numbers of building blocks for commonly utilized CRBN, VHL, and IAP ligands.
This review gives an extensive overview of successful synthetic routes towards functionalized E3 ligase ligands. This enables the reader to better assess which reaction conditions are suitable and which yields can be achieved. Most of the starting materials are either commercially available or can be produced by simple synthesis techniques. Due to the scope of our research, this review may give new impulses in the synthesis laboratories to try out new linker connections or to test novel reactions under proven conditions. Ultimately, it is not only the accessibility and capital efficiency that determine the success of a PROTAC program, but also aspects such as rigidity, hydrolytic and metabolic stability, solubility and cell permeability of the chimeric molecules. This work represents a unique compendium to re-evaluate the many facets involved in the synthesis of such complex molecules.
Author Contributions
AB, CS, RK, MG, and IS analyzed data and wrote the manuscript. MG and IS supervised the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguilar, A., Sun, W., Liu, L., Lu, J., McEachern, D., Bernard, D., et al. (2014). Design of Chemically Stable, Potent, and Efficacious MDM2 Inhibitors that Exploit the Retro-Mannich Ring-Opening-Cyclization Reaction Mechanism in Spiro-oxindoles. J. Med. Chem. 57, 10486–10498. doi:10.1021/jm501541j
Amm, I., Sommer, T., and Wolf, D. H. (2014). Protein Quality Control and Elimination of Protein Waste: The Role of the Ubiquitin-Proteasome System. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1843, 182–196. doi:10.1016/j.bbamcr.2013.06.031
An, S., and Fu, L. (2018). Small-molecule PROTACs: An Emerging and Promising Approach for the Development of Targeted Therapy Drugs. EBioMedicine 36, 553–562. doi:10.1016/j.ebiom.2018.09.005
An, Z., Lv, W., Su, S., Wu, W., and Rao, Y. (2019). Developing Potent PROTACs Tools for Selective Degradation of HDAC6 Protein. Protein Cell 10, 606–609. doi:10.1007/s13238-018-0602-z
Anderson, N. A., Cryan, J., Ahmed, A., Dai, H., McGonagle, G. A., Rozier, C., et al. (2020). Selective CDK6 Degradation Mediated by Cereblon, VHL, and Novel IAP-Recruiting PROTACs. Bioorg. Med. Chem. Lett. 30, 127106. doi:10.1016/j.bmcl.2020.127106
Balaev, A. N., Okhmanovich, K. A., and Fedorov, V. E. (2013). Alternative Synthesis of Lenalidomide. Pharm. Chem. J. 46, 676–678. doi:10.1007/s11094-013-0868-7
Bassi, Z. I., Fillmore, M. C., Miah, A. H., Chapman, T. D., Maller, C., Roberts, E. J., et al. (2018). Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem. Biol. 13, 2862–2867. doi:10.1021/acschembio.8b00705
Bijlmakers, M.-J., Kanneganti, S. K., Barker, J. N., Trembath, R. C., and Capon, F. (2011). Functional Analysis of the RNF114 Psoriasis Susceptibility Gene Implicates Innate Immune Responses to Double-Stranded RNA in Disease Pathogenesis. Hum. Mol. Genet. 20, 3129–3137. doi:10.1093/hmg/ddr215
Bondeson, D. P., Mares, A., Smith, I. E. D., Ko, E., Campos, S., Miah, A. H., et al. (2015). Catalytic In Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 11, 611–617. doi:10.1038/nchembio.1858
Borzilleri, R. M., Kim, K. S., Perez, H. L., Stang, E. M., Williams, D. K., and Zhang, L. (2014). IAP Antagonists. WO 2014/047024 (Accessed March 27, 2014).
Brownsey, D. K., Rowley, B. C., Gorobets, E., Gelfand, B. S., and Derksen, D. J. (2021). Rapid Synthesis of Pomalidomide-Conjugates for the Development of Protein Degrader Libraries. Chem. Sci. 12, 4519–4525. doi:10.1039/D0SC05442A
Buckley, D. L., Gustafson, J. L., Van Molle, I., Roth, A. G., Tae, H. S., Gareiss, P. C., et al. (2012a). Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angew. Chem. Int. Ed. 51, 11463–11467. doi:10.1002/anie.201206231
Buckley, D. L., Raina, K., Darricarrere, N., Hines, J., Gustafson, J. L., Smith, I. E., et al. (2015). HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 10, 1831–1837. doi:10.1021/acschembio.5b00442
Buckley, D. L., Van Molle, I., Gareiss, P. C., Tae, H. S., Michel, J., Noblin, D. J., et al. (2012b). Targeting the von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 134, 4465–4468. doi:10.1021/ja209924v
Buetow, L., and Huang, D. T. (2016). Structural Insights into the Catalysis and Regulation of E3 Ubiquitin Ligases. Nat. Rev. Mol. Cel Biol. 17, 626–642. doi:10.1038/nrm.2016.91
Buhimschi, A. D., Armstrong, H. A., Toure, M., Jaime-Figueroa, S., Chen, T. L., Lehman, A. M., et al. (2018). Targeting the C481S Ibrutinib-Resistance Mutation in Bruton's Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 57, 3564–3575. doi:10.1021/acs.biochem.8b00391
Burslem, G. M., and Crews, C. M. (2020). Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114. doi:10.1016/j.cell.2019.11.031
Burslem, G. M., Ottis, P., Jaime-Figueroa, S., Morgan, A., Cromm, P. M., Toure, M., et al. (2018). Efficient Synthesis of Immunomodulatory Drug Analogues Enables Exploration of Structure-Degradation Relationships. ChemMedChem 13, 1508–1512. doi:10.1002/cmdc.201800271
Busbee, P. B., Rouse, M., Nagarkatti, M., and Nagarkatti, P. S. (2013). Use of Natural AhR Ligands as Potential Therapeutic Modalities against Inflammatory Disorders. Nutr. Rev. 71, 353–369. doi:10.1111/nure.12024
Bussiere, D. E., Xie, L., Srinivas, H., Shu, W., Burke, A., Be, C., et al. (2020). Structural Basis of Indisulam-Mediated RBM39 Recruitment to DCAF15 E3 Ligase Complex. Nat. Chem. Biol. 16, 15–23. doi:10.1038/s41589-019-0411-6
Capitosti, S. M., Hansen, T. P., and Brown, M. L. (2003). Facile Synthesis of an Azido-Labeled Thalidomide Analogue. Org. Lett. 5, 2865–2867. doi:10.1021/ol034906w
Casillas, L., Harling, J. D., Miah, A. H., Rackham, M. D., and Smith, I. E. D. (2016). Novel Compounds. WO 2016/172134 A2 (Accessed October 27, 2016).
Chamberlain, P. P., Lopez-Girona, A., Miller, K., Carmel, G., Pagarigan, B., Chie-Leon, B., et al. (2014). Structure of the Human Cereblon-DDB1-Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat. Struct. Mol. Biol. 21, 803–809. doi:10.1038/nsmb.2874
Chaulet, C., Croix, C., Alagille, D., Normand, S., Delwail, A., Favot, L., et al. (2011). Design, Synthesis and Biological Evaluation of New Thalidomide Analogues as TNF-α and IL-6 Production Inhibitors. Bioorg. Med. Chem. Lett. 21, 1019–1022. doi:10.1016/j.bmcl.2010.12.031
Chen, H., Chen, F., Liu, N., Wang, X., and Gou, S. (2018). Chemically Induced Degradation of CK2 by Proteolysis Targeting Chimeras Based on a Ubiquitin-Proteasome Pathway. Bioorg. Chem. 81, 536–544. doi:10.1016/j.bioorg.2018.09.005
Chen, H., Chen, F., Pei, S., and Gou, S. (2019). Pomalidomide Hybrids Act as Proteolysis Targeting Chimeras: Synthesis, Anticancer Activity and B-Raf Degradation. Bioorg. Chem. 87, 191–199. doi:10.1016/j.bioorg.2019.03.035
Chessum, N. E. A., Sharp, S. Y., Caldwell, J. J., Pasqua, A. E., Wilding, B., Colombano, G., et al. (2018). Demonstrating In-Cell Target Engagement Using a Pirin Protein Degradation Probe (CCT367766). J. Med. Chem. 61, 918–933. doi:10.1021/acs.jmedchem.7b01406
Cohen, P., and Tcherpakov, M. (2010). Will the Ubiquitin System Furnish as Many Drug Targets as Protein Kinases?. Cell 143, 686–693. doi:10.1016/j.cell.2010.11.016
Crew, A. P., Hornberger, K. R., Wang, J., Dong, H., Qian, Y., Crews, C. M., et al. (2018a). Compounds and Methods for the Targeted Degradation of Rapidly Accelerated Fibrosarcoma Polypeptides. WO2018119448 A1 (Accesed June 28, 2018).
Crew, A. P., Qian, Y., Dong, H., Wang, J., Hornberger, K. R., and Crews, C. M. (2018b). Tetrahydronaphthalene and Tetrahydroisoquinoline Derivatives as Estrogen Receptor Degraders. WO2018102725 (Accesed June 07, 2018).
Crew, A. P., Raina, K., Dong, H., Qian, Y., Wang, J., Vigil, D., et al. (2018). Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 61, 583–598. doi:10.1021/acs.jmedchem.7b00635
Czyzyk-Krzeska, M. F., and Meller, J. (2004). von Hippel-Lindau tumor suppressor: Not only HIF's executioner. Trends Mol. Med. 10, 146–149. doi:10.1016/j.molmed.2004.02.004
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 7, 42717. doi:10.1038/srep42717
Davies, T. G., Wixted, W. E., Coyle, J. E., Griffiths-Jones, C., Hearn, K., McMenamin, R., et al. (2016). Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J. Med. Chem. 59, 3991–4006. doi:10.1021/acs.jmedchem.6b00228
de Wispelaere, M., Du, G., Donovan, K. A., Zhang, T., Eleuteri, N. A., Yuan, J. C., et al. (2019). Small Molecule Degraders of the Hepatitis C Virus Protease Reduce Susceptibility to Resistance Mutations. Nat. Commun. 10, 3468. doi:10.1038/s41467-019-11429-w
DeLuca, F. H., Grzywacz, P., and Sicinski, R. R. (2003). A Concise Synthesis of an AHR Endogenous Ligand with the Indolecarbonylthiazole Skeleton. Heterocycles 60, 1219–1224. doi:10.3987/COM-03-9730
Demizu, Y., Okuhira, K., Motoi, H., Ohno, A., Shoda, T., Fukuhara, K., et al. (2012). Design and Synthesis of Estrogen Receptor Degradation Inducer Based on a Protein Knockdown Strategy. Bioorg. Med. Chem. Lett. 22, 1793–1796. doi:10.1016/j.bmcl.2011.11.086
Ding, Q., Zhang, Z., Liu, J.-J., Jiang, N., Zhang, J., Ross, T. M., et al. (2013). Discovery of RG7388, a Potent and Selective P53-MDM2 Inhibitor in Clinical Development. J. Med. Chem. 56, 5979–5983. doi:10.1021/jm400487c
Dragovich, P. S., Adhikari, P., Blake, R. A., Blaquiere, N., Chen, J., Cheng, Y.-X., et al. (2020). Antibody-mediated Delivery of Chimeric Protein Degraders Which Target Estrogen Receptor Alpha (ERα). Bioorg. Med. Chem. Lett. 30, 126907. doi:10.1016/j.bmcl.2019.126907
Farnaby, W., Koegl, M., Roy, M. J., Whitworth, C., Diers, E., Trainor, N., et al. (2019). BAF Complex Vulnerabilities in Cancer Demonstrated via Structure-Based PROTAC Design. Nat. Chem. Biol. 15, 672–680. doi:10.1038/s41589-019-0294-6
Fischer, E. S., Böhm, K., Lydeard, J. R., Yang, H., Stadler, M. B., Cavadini, S., et al. (2014). Structure of the DDB1-CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 512, 49–53. doi:10.1038/nature13527
Fisher, S. L., and Phillips, A. J. (2018). Targeted Protein Degradation and the Enzymology of Degraders. Curr. Opin. Chem. Biol. 44, 47–55. doi:10.1016/j.cbpa.2018.05.004
Fu, L., and Gribble, G. W. (2013). Efficient and Scalable Synthesis of Bardoxolone Methyl (CDDO-Methyl Ester). Org. Lett. 15, 1622–1625. doi:10.1021/ol400399x
Fulda, S., and Vucic, D. (2012). Targeting IAP Proteins for Therapeutic Intervention in Cancer. Nat. Rev. Drug Discov. 11, 109–124. doi:10.1038/nrd3627
Gadd, M. S., Testa, A., Lucas, X., Chan, K.-H., Chen, W., Lamont, D. J., et al. (2017). Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 13, 514–521. doi:10.1038/nchembio.2329
Galdeano, C., Gadd, M. S., Soares, P., Scaffidi, S., Van Molle, I., Birced, I., et al. (2014). Structure-Guided Design and Optimization of Small Molecules Targeting the Protein-Protein Interaction between the von Hippel-Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with In Vitro Nanomolar Affinities. J. Med. Chem. 57, 8657–8663. doi:10.1021/jm5011258
Gogoi, N., Boruwa, J., and Barua, N. C. (2005). A Total Synthesis of (−)-bestatin Using Shibasaki's Asymmetric Henry Reaction. Tetrahedron Lett. 46, 7581–7582. doi:10.1016/j.tetlet.2005.08.153
Gray, N., Zhang, T., Fischer, E., Verano, A., He, Z., Du, G., et al. (2020). New CRBN Modulators. WO/2020/006262 (Accessed January 02, 2020).
Han, J., Kim, Y.-L., Lee, K.-W., Her, N.-G., Ha, T.-K., Yoon, S., et al. (2013). ZNF313 Is a Novel Cell Cycle Activator with an E3 Ligase Activity Inhibiting Cellular Senescence by Destabilizing p21WAF1. Cell Death Differ 20, 1055–1067. doi:10.1038/cdd.2013.33
Han, T., Goralski, M., Gaskill, N., Capota, E., Kim, J., Ting, T. C., et al. (2017). Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 356, eaal3755. doi:10.1126/science.aal3755
Han, X., Wang, C., Qin, C., Xiang, W., Fernandez-Salas, E., Yang, C.-Y., et al. (2019). Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 62, 941–964. doi:10.1021/acs.jmedchem.8b01631
Hansen, J. D., Correa, M., Alexander, M., Nagy, M., Huang, D., Sapienza, J., et al. (2021). CC-90009: A Cereblon E3 Ligase Modulating Drug that Promotes Selective Degradation of GSPT1 for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 64, 1835–1843. doi:10.1021/acs.jmedchem.0c01489
Hansen, J. D., Correa, M., Nagy, M. A., Alexander, M., Plantevin, V., Grant, V., et al. (2020). Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem. 63, 6648–6676. doi:10.1021/acs.jmedchem.9b01928
Hayhow, T. G., Borrows, R. E. A., Diène, C. R., Fairley, G., Fallan, C., Fillery, S. M., et al. (2020). A Buchwald-Hartwig Protocol to Enable Rapid Linker Exploration of Cereblon E3‐Ligase PROTACs**. Chem. Eur. J. 26, 16818–16823. doi:10.1002/chem.202003137
Henning, N. J., Manford, A. G., Spradlin, J. N., Brittain, S. M., McKenna, J. M., Tallarico, J. A., et al. (2021). Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications. bioRxiv. doi:10.1101/2021.04.15.439993
Hines, J., Lartigue, S., Dong, H., Qian, Y., and Crews, C. M. (2019). MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of P53. Cancer Res. 79, 251–262. doi:10.1158/0008-5472.CAN-18-2918
Hoffmann, M., Kasserra, C., Reyes, J., Schafer, P., Kosek, J., Capone, L., et al. (2013). Absorption, Metabolism and Excretion of [14C]pomalidomide in Humans Following Oral Administration. Cancer Chemother. Pharmacol. 71, 489–501. doi:10.1007/s00280-012-2040-6
Hu, J., Hu, B., Wang, M., Xu, F., Miao, B., Yang, C.-Y., et al. (2019). Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J. Med. Chem. 62, 1420–1442. doi:10.1021/acs.jmedchem.8b01572
Huang, D., Shen, C., Wang, W., Huang, L., Ni, F., and Li, J. (2016). New Synthesis Route for the Preparation of Pomalidomide. Synth. Commun. 46, 1343–1348. doi:10.1080/00397911.2016.1189574
Ishoey, M., Chorn, S., Singh, N., Jaeger, M. G., Brand, M., Paulk, J., et al. (2018). Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem. Biol. 13, 553–560. doi:10.1021/acschembio.7b00969
Ito, T., Ando, H., and Handa, H. (2011). Teratogenic Effects of Thalidomide: Molecular Mechanisms. Cell. Mol. Life Sci. 68, 1569–1579. doi:10.1007/s00018-010-0619-9
Itoh, Y., Ishikawa, M., Kitaguchi, R., Okuhira, K., Naito, M., and Hashimoto, Y. (2012). Double Protein Knockdown of cIAP1 and CRABP-II Using a Hybrid Molecule Consisting of ATRA and IAPs Antagonist. Bioorg. Med. Chem. Lett. 22, 4453–4457. doi:10.1016/j.bmcl.2012.04.134
Itoh, Y., Ishikawa, M., Naito, M., and Hashimoto, Y. (2010). Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 132, 5820–5826. doi:10.1021/ja100691p
Itoh, Y., Kitaguchi, R., Ishikawa, M., Naito, M., and Hashimoto, Y. (2011). Design, Synthesis and Biological Evaluation of Nuclear Receptor-Degradation Inducers. Bioorg. Med. Chem. 19, 6768–6778. doi:10.1016/j.bmc.2011.09.041
Jiang, B., Wang, E. S., Donovan, K. A., Liang, Y., Fischer, E. S., Zhang, T., et al. (2019). Development of Dual and Selective Degraders of Cyclin‐Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. 58, 6321–6326. doi:10.1002/anie.201901336
Jiang, Z.-Y., Lu, M.-C., and You, Q.-D. (2016). Discovery and Development of Kelch-like ECH-Associated Protein 1. Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction Inhibitors: Achievements, Challenges, and Future Directions. J. Med. Chem. 59, 10837–10858. doi:10.1021/acs.jmedchem.6b00586
Jin, Y.-H., Lu, M.-C., Wang, Y., Shan, W.-X., Wang, X.-Y., You, Q.-D., et al. (2020). Azo-PROTAC: Novel Light-Controlled Small-Molecule Tool for Protein Knockdown. J. Med. Chem. 63, 4644–4654. doi:10.1021/acs.jmedchem.9b02058
Kester, R. F., Donnell, A. F., Lou, Y., Remiszewski, S. W., Lombardo, L. J., Chen, S., et al. (2013). Optimization of Benzodiazepinones as Selective Inhibitors of the X-Linked Inhibitor of Apoptosis Protein (XIAP) Second Baculovirus IAP Repeat (BIR2) Domain. J. Med. Chem. 56, 7788–7803. doi:10.1021/jm400732v
Kleiger, G., and Mayor, T. (2014). Perilous Journey: a Tour of the Ubiquitin-Proteasome System. Trends Cel Biol. 24, 352–359. doi:10.1016/j.tcb.2013.12.003
Kounde, C. S., Shchepinova, M. M., Saunders, C. N., Muelbaier, M., Rackham, M. D., Harling, J. D., et al. (2020). A Caged E3 Ligase Ligand for PROTAC-Mediated Protein Degradation with Light. Chem. Commun. 56, 5532–5535. doi:10.1039/D0CC00523A
Krönke, J., Fink, E. C., Hollenbach, P. W., MacBeth, K. J., Hurst, S. N., Udeshi, N. D., et al. (2015). Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188. doi:10.1038/nature14610
Krönke, J., Udeshi, N. D., Narla, A., Grauman, P., Hurst, S. N., McConkey, M., et al. (2014). Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 343, 301–305. doi:10.1126/science.1244851
Lai, A. C., and Crews, C. M. (2017). Induced Protein Degradation: an Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov. 16, 101–114. doi:10.1038/nrd.2016.211
Lai, A. C., Toure, M., Hellerschmied, D., Salami, J., Jaime-Figueroa, S., Ko, E., et al. (2016). Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 55, 807–810. doi:10.1002/anie.201507634
Lebraud, H., Wright, D. J., Johnson, C. N., and Heightman, T. D. (2016). Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2, 927–934. doi:10.1021/acscentsci.6b00280
Lee, B. W., Lee, J. H., Jang, K. C., Kang, J. E., Kim, J. H., Park, K.-M., et al. (2003). Diastereoselective Synthesis of Syn-Aminoalcohols via Contributing CH-π Interaction: Simple Synthesis of (−)-bestatin. Tetrahedron Lett. 44, 5905–5907. doi:10.1016/S0040-4039(03)01394-7
Lee, H., Puppala, D., Choi, E.-Y., Swanson, H., and Kim, K.-B. (2007). Targeted Degradation of the Aryl Hydrocarbon Receptor by the PROTAC Approach: A Useful Chemical Genetic Tool. ChemBioChem 8, 2058–2062. doi:10.1002/cbic.200700438
Levine, A. J. (1997). p53, the Cellular Gatekeeper for Growth and Division. Cell 88, 323–331. doi:10.1016/S0092-8674(00)81871-1
Li, W., Gao, C., Zhao, L., Yuan, Z., Chen, Y., and Jiang, Y. (2018). Phthalimide Conjugations for the Degradation of Oncogenic PI3K. Eur. J. Med. Chem. 151, 237–247. doi:10.1016/j.ejmech.2018.03.066
Li, Y., Yang, J., Aguilar, A., McEachern, D., Przybranowski, S., Liu, L., et al. (2019). Discovery of MD-224 as a First-In-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 62, 448–466. doi:10.1021/acs.jmedchem.8b00909
Liang, C., Shi, X., and Fan, C. (2017). Pathological and Diagnostic Implications of DCAF16 Expression in Human Carcinomas Including Adenocarcinoma, Squamous Cell Carcinoma, and Urothelial Carcinoma. Int. J. Clin. Exp. Pathol. 10, 8585–8591. ISSN:1936-2625/IJCEP0052079.
Lohbeck, J., and Miller, A. K. (2016). Practical Synthesis of a Phthalimide-Based Cereblon Ligand to Enable PROTAC Development. Bioorg. Med. Chem. Lett. 26, 5260–5262. doi:10.1016/j.bmcl.2016.09.048
Lu, J., Qian, Y., Altieri, M., Dong, H., Wang, J., Raina, K., et al. (2015). Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 22, 755–763. doi:10.1016/j.chembiol.2015.05.009
Luecke-Johansson, S., Gralla, M., Rundqvist, H., Ho, J. C., Johnson, R. S., Gradin, K., et al. (2017). A Molecular Mechanism to Switch the Aryl Hydrocarbon Receptor from a Transcription Factor to an E3 Ubiquitin Ligase. Mol. Cel. Biol. 37, e00630–16. doi:10.1128/MCB.00630-16
Luo, M., Spradlin, J. N., Boike, L., Tong, B., Brittain, S. M., McKenna, J. M., et al. (2021). Chemoproteomics-enabled Discovery of Covalent RNF114-Based Degraders that Mimic Natural Product Function. Cel Chem. Biol. 28, 559–566. doi:10.1016/j.chembiol.2021.01.005
Ma, Q., and Baldwin, K. T. (2000). 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced Degradation of Aryl Hydrocarbon Receptor (AhR) by the Ubiquitin-Proteasome Pathway. J. Biol. Chem. 275, 8432–8438. doi:10.1074/jbc.275.12.8432
Man, H.-W., Corral, L. G., Stirling, D. I., and Muller, G. W. (2003). α-Fluoro-substituted Thalidomide Analogues. Bioorg. Med. Chem. Lett. 13, 3415–3417. doi:10.1016/s0960-894x(03)00778-9
Manford, A. G., Rodríguez-Pérez, F., Shih, K. Y., Shi, Z., Berdan, C. A., Choe, M., et al. (2020). A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 183, 46–61.e21. doi:10.1016/j.cell.2020.08.034
Maniaci, C., Hughes, S. J., Testa, A., Chen, W., Lamont, D. J., Rocha, S., et al. (2017). Homo-PROTACs: Bivalent Small-Molecule Dimerizers of the VHL E3 Ubiquitin Ligase to Induce Self-Degradation. Nat. Commun. 8, 830. doi:10.1038/s41467-017-00954-1
Maple, H. J., Clayden, N., Baron, A., Stacey, C., and Felix, R. (2019). Developing Degraders: Principles and Perspectives on Design and Chemical Space. Med. Chem. Commun. 10, 1755–1764. doi:10.1039/C9MD00272C
Mares, A., Miah, A. H., Smith, I. E. D., Rackham, M., Thawani, A. R., Cryan, J., et al. (2020). Extended Pharmacodynamic Responses Observed upon PROTAC-Mediated Degradation of RIPK2. Commun. Biol. 3, 140. doi:10.1038/s42003-020-0868-6
Michael, D., and Oren, M. (2003). The P53-Mdm2 Module and the Ubiquitin System. Semin. Cancer Biol. 13, 49–58. doi:10.1016/S1044-579X(02)00099-8
Momand, J., Jung, D., Wilczynski, S., and Niland, J. (1998). The MDM2 Gene Amplification Database. Nucleic Acids Res. 26, 3453–3459. doi:10.1093/nar/26.15.3453
Naito, M., Ohoka, N., Shibata, N., and Tsukumo, Y. (2019). Targeted Protein Degradation by Chimeric Small Molecules, PROTACs and SNIPERs. Front. Chem. 7, 849. doi:10.3389/fchem.2019.00849
Naro, Y., Darrah, K., and Deiters, A. (2020). Optical Control of Small Molecule-Induced Protein Degradation. J. Am. Chem. Soc. 142, 2193–2197. doi:10.1021/jacs.9b12718
Nemoto, H., Ma, R., Suzuki, I., and Shibuya, M. (2000). A New One-Pot Method for the Synthesis of α-Siloxyamides from Aldehydes or Ketones and its Application to the Synthesis of (−)-Bestatin. Org. Lett. 2, 4245–4247. doi:10.1021/ol006816m
Nietzold, F., Rubner, S., and Berg, T. (2019). The Hydrophobically-Tagged MDM2-P53 Interaction Inhibitor Nutlin-3a-HT Is More Potent against Tumor Cells Than Nutlin-3a. Chem. Commun. 55, 14351–14354. doi:10.1039/C9CC07795B
Nunes, J., McGonagle, G. A., Eden, J., Kiritharan, G., Touzet, M., Lewell, X., et al. (2019). Targeting IRAK4 for Degradation with PROTACs. ACS Med. Chem. Lett. 10, 1081–1085. doi:10.1021/acsmedchemlett.9b00219
Ohoka, N., Morita, Y., Nagai, K., Shimokawa, K., Ujikawa, O., Fujimori, I., et al. (2018). Derivatization of Inhibitor of Apoptosis Protein (IAP) Ligands Yields Improved Inducers of Estrogen Receptor α Degradation. J. Biol. Chem. 293, 6776–6790. doi:10.1074/jbc.RA117.001091
Ohoka, N., Nagai, K., Hattori, T., Okuhira, K., Shibata, N., Cho, N., et al. (2014). Cancer Cell Death Induced by Novel Small Molecules Degrading the TACC3 Protein via the Ubiquitin-Proteasome Pathway. Cell Death Dis 5, e1513. doi:10.1038/cddis.2014.471
Ohoka, N., Nagai, K., Shibata, N., Hattori, T., Nara, H., Cho, N., et al. (2017a). SNIPER(TACC3) Induces Cytoplasmic Vacuolization and Sensitizes Cancer Cells to Bortezomib. Cancer Sci. 108, 1032–1041. doi:10.1111/cas.13198
Ohoka, N., Okuhira, K., Ito, M., Nagai, K., Shibata, N., Hattori, T., et al. (2017b). In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem. 292, 4556–4570. doi:10.1074/jbc.M116.768853
Ohoka, N., Tsuji, G., Shoda, T., Fujisato, T., Kurihara, M., Demizu, Y., et al. (2019). Development of Small Molecule Chimeras that Recruit AhR E3 Ligase to Target Proteins. ACS Chem. Biol. 14, 2822–2832. doi:10.1021/acschembio.9b00704
Ohtake, F., Baba, A., Takada, I., Okada, M., Iwasaki, K., Miki, H., et al. (2007). Dioxin Receptor Is a Ligand-dependent E3 Ubiquitin Ligase. Nature 446, 562–566. doi:10.1038/nature05683
Okuhira, K., Demizu, Y., Hattori, T., Ohoka, N., Shibata, N., Kurihara, M., et al. (2016). Molecular Design, Synthesis, and Evaluation of SNIPER(ER) that Induces Proteasomal Degradation of ERα. Methods Mol. Biol. 1366, 549–560. doi:10.1007/978-1-4939-3127-9_42
Okuhira, K., Demizu, Y., Hattori, T., Ohoka, N., Shibata, N., Nishimaki-Mogami, T., et al. (2013). Development of Hybrid Small Molecules that Induce Degradation of Estrogen Receptor-Alpha and Necrotic Cell Death in Breast Cancer Cells. Cancer Sci. 104, 1492–1498. doi:10.1111/cas.12272
Okuhira, K., Ohoka, N., Sai, K., Nishimaki-Mogami, T., Itoh, Y., Ishikawa, M., et al. (2011). Specific Degradation of CRABP-II via cIAP1-Mediated Ubiquitylation Induced by Hybrid Molecules that Crosslink cIAP1 and the Target Protein. FEBS Lett. 585, 1147–1152. doi:10.1016/j.febslet.2011.03.019
Papatzimas, J. W., Gorobets, E., Maity, R., Muniyat, M. I., MacCallum, J. L., Neri, P., et al. (2019). From Inhibition to Degradation: Targeting the Antiapoptotic Protein Myeloid Cell Leukemia 1 (MCL1). J. Med. Chem. 62, 5522–5540. doi:10.1021/acs.jmedchem.9b00455
Pettersson, M., and Crews, C. M. (2019). PROteolysis TArgeting Chimeras (PROTACs) - Past, Present and Future. Drug Discov. Today Tech. 31, 15–27. doi:10.1016/j.ddtec.2019.01.002
Pfaff, P., Samarasinghe, K. T. G., Crews, C. M., and Carreira, E. M. (2019). Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 5, 1682–1690. doi:10.1021/acscentsci.9b00713
Ponomaryov, Y., Krasikova, V., Lebedev, A., Chernyak, D., Varacheva, L., and Chernobroviy, A. (2015). Scalable and green Process for the Synthesis of Anticancer Drug Lenalidomide. Chem. Heterocycl Comp. 51, 133–138. doi:10.1007/s10593-015-1670-0
Powell, C. E., Gao, Y., Tan, L., Donovan, K. A., Nowak, R. P., Loehr, A., et al. (2018). Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 61, 4249–4255. doi:10.1021/acs.jmedchem.7b01655
Qiu, X., Sun, N., Kong, Y., Li, Y., Yang, X., and Jiang, B. (2019). Chemoselective Synthesis of Lenalidomide-Based PROTAC Library Using Alkylation Reaction. Org. Lett. 21, 3838–3841. doi:10.1021/acs.orglett.9b01326
Raina, K., Lu, J., Qian, Y., Altieri, M., Gordon, D., Rossi, A. M. K., et al. (2016). PROTAC-induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 113, 7124–7129. doi:10.1073/pnas.1521738113
Rana, S., Bendjennat, M., Kour, S., King, H. M., Kizhake, S., Zahid, M., et al. (2019). Selective Degradation of CDK6 by a Palbociclib Based PROTAC. Bioorg. Med. Chem. Lett. 29, 1375–1379. doi:10.1016/j.bmcl.2019.03.035
Remillard, D., Buckley, D. L., Paulk, J., Brien, G. L., Sonnett, M., Seo, H.-S., et al. (2017). Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. Int. Ed. 56, 5738–5743. doi:10.1002/anie.201611281
Reynders, M., Berouti, M., Simoneschi, D., Matsuura, B. S., Marzio, A., Pagano, M., et al. (2020). PHOTACs Enable Optical Control of Protein Degradation. Sci. Adv. 6, eaay5064. doi:10.1126/sciadv.aay5064
Righi, G., D'Achille, C., Pescatore, G., and Bonini, C. (2003). New Stereoselective Synthesis of the Peptidic Aminopeptidase Inhibitors Bestatin, Phebestin and Probestin. Tetrahedron Lett. 44, 6999–7002. doi:10.1016/S0040-4039(03)01799-4
Rimmler, G., Alker, A., Bosco, M., Diodone, R., Fishlock, D., Hildbrand, S., et al. (2016). Practical Synthesis of MDM2 Antagonist RG7388. Part 2: Development of the Cu(I) Catalyzed [3 + 2] Asymmetric Cycloaddition Process for the Manufacture of Idasanutlin. Org. Process. Res. Dev. 20, 2057–2066. doi:10.1021/acs.oprd.6b00319
Robb, C. M., Contreras, J. I., Kour, S., Taylor, M. A., Abid, M., Sonawane, Y. A., et al. (2017). Chemically Induced Degradation of CDK9 by a Proteolysis Targeting Chimera (PROTAC). Chem. Commun. 53, 7577–7580. doi:10.1039/C7CC03879H
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric Molecules that Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. 98, 8554–8559. doi:10.1073/pnas.141230798
Sayo, N., Yamasaki, T., Kumobayashi, H., Yuasa, Y., and Sotoguchi, T. (1996). Process for Preparing Optically Active Allophenylnorstatin Derivatives, and Intermediates for Use Therein. EP0729939A2 (Accesed September 04 1996).
Schapira, M., Calabrese, M. F., Bullock, A. N., and Crews, C. M. (2019). Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discov. 18, 949–963. doi:10.1038/s41573-019-0047-y
Scheepstra, M., Hekking, K. F. W., van Hijfte, L., and Folmer, R. H. A. (2019). Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 17, 160–176. doi:10.1016/j.csbj.2019.01.006
Schneekloth, A. R., Pucheault, M., Tae, H. S., and Crews, C. M. (2008). Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett. 18, 5904–5908. doi:10.1016/j.bmcl.2008.07.114
Schneekloth, J. S., Fonseca, F. N., Koldobskiy, M., Mandal, A., Deshaies, R., Sakamoto, K., et al. (2004). Chemical Genetic Control of Protein Levels: Selective In Vivo Targeted Degradation. J. Am. Chem. Soc. 126, 3748–3754. doi:10.1021/ja039025z
Shah, R. R., Redmond, J. M., Mihut, A., Menon, M., Evans, J. P., Murphy, J. A., et al. (2020). Hi-JAK-ing the Ubiquitin System: The Design and Physicochemical Optimisation of JAK PROTACs. Bioorg. Med. Chem. 28, 115326. doi:10.1016/j.bmc.2020.115326
Shang, S., Willems, A. V., and Chauhan, S. S. (2018). A Practical Diastereoselective Synthesis of (−)-bestatin. J. Pep Sci. 24, e3067. doi:10.1002/psc.3067
Shibata, N., Miyamoto, N., Nagai, K., Shimokawa, K., Sameshima, T., Ohoka, N., et al. (2017a). Development of Protein Degradation Inducers of Oncogenic BCR ‐ ABL Protein by Conjugation of ABL Kinase Inhibitors and IAP Ligands. Cancer Sci. 108, 1657–1666. doi:10.1111/cas.13284
Shibata, N., Nagai, K., Morita, Y., Ujikawa, O., Ohoka, N., Hattori, T., et al. (2017b). Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 61, 543–575. doi:10.1021/acs.jmedchem.7b00168
Shimokawa, K., Shibata, N., Sameshima, T., Miyamoto, N., Ujikawa, O., Nara, H., et al. (2017). Targeting the Allosteric Site of Oncoprotein BCR-ABL as an Alternative Strategy for Effective Target Protein Degradation. ACS Med. Chem. Lett. 8, 1042–1047. doi:10.1021/acsmedchemlett.7b00247
Shu, L., Gu, C., Fishlock, D., and Li, Z. (2016). Practical Synthesis of MDM2 Antagonist RG7388. Part 1: A Cu(II)-Catalyzed Asymmetric [3 + 2] Cycloaddition. Org. Process. Res. Dev. 20, 2050–2056. doi:10.1021/acs.oprd.6b00320
Silva, M. C., Ferguson, F. M., Cai, Q., Donovan, K. A., Nandi, G., Patnaik, D., et al. (2019). Targeted Degradation of Aberrant Tau in Frontotemporal Dementia Patient-Derived Neuronal Cell Models. eLife 8, e45457. doi:10.7554/eLife.45457
Spradlin, J. N., Hu, X., Ward, C. C., Brittain, S. M., Jones, M. D., Ou, L., et al. (2019). Harnessing the Anti-cancer Natural Product Nimbolide for Targeted Protein Degradation. Nat. Chem. Biol. 15, 747–755. doi:10.1038/s41589-019-0304-8
Steinebach, C., Lindner, S., Udeshi, N. D., Mani, D. C., Kehm, H., Köpff, S., et al. (2018). Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem. Biol. 13, 2771–2782. doi:10.1021/acschembio.8b00693
Steinebach, C., Ng, Y. L. D., Sosič, I., Lee, C.-S., Chen, S., Lindner, S., et al. (2020a). Systematic Exploration of Different E3 Ubiquitin Ligases: an Approach towards Potent and Selective CDK6 Degraders. Chem. Sci. 11, 3474–3486. doi:10.1039/D0SC00167H
Steinebach, C., Sosič, I., Lindner, S., Bricelj, A., Kohl, F., Ng, Y. L. D., et al. (2019). A MedChem Toolbox for Cereblon-Directed PROTACs. Med. Chem. Commun. 10, 1037–1041. doi:10.1039/C9MD00185A
Steinebach, C., Voell, S. A., Vu, L. P., Bricelj, A., Sosič, I., Schnakenburg, G., et al. (2020b). A facile synthesis of ligands for the von Hippel-Lindau E3 ligase. Synthesis 52, 2521–2527. doi:10.1055/s-0040-1707400
Stewart, S. G., Braun, C. J., Ng, S.-L., Polomska, M. E., Karimi, M., and Abraham, L. J. (2010). New Thalidomide Analogues Derived through Sonogashira or Suzuki Reactions and Their TNF Expression Inhibition Profiles. Bioorg. Med. Chem. 18, 650–662. doi:10.1016/j.bmc.2009.12.001
Su, S., Yang, Z., Gao, H., Yang, H., Zhu, S., An, Z., et al. (2019). Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 62, 7575–7582. doi:10.1021/acs.jmedchem.9b00871
Sun, X., Gao, H., Yang, Y., He, M., Wu, Y., Song, Y., et al. (2019a). PROTACs: Great Opportunities for Academia and Industry. Sig Transduct Target. Ther. 4, 64. doi:10.1038/s41392-019-0101-6
Sun, Y., Ding, N., Song, Y., Yang, Z., Liu, W., Zhu, J., et al. (2019b). Degradation of Bruton's Tyrosine Kinase Mutants by PROTACs for Potential Treatment of Ibrutinib-Resistant Non-hodgkin Lymphomas. Leukemia 33, 2105–2110. doi:10.1038/s41375-019-0440-x
Sun, Y., Zhao, X., Ding, N., Gao, H., Wu, Y., Yang, Y., et al. (2018). PROTAC-induced BTK Degradation as a Novel Therapy for Mutated BTK C481S Induced Ibrutinib-Resistant B-Cell Malignancies. Cell Res 28, 779–781. doi:10.1038/s41422-018-0055-1
Tong, B., Luo, M., Xie, Y., Spradlin, J. N., Tallarico, J. A., McKenna, J. M., et al. (2020a). Bardoxolone Conjugation Enables Targeted Protein Degradation of BRD4. Sci. Rep. 10, 15543. doi:10.1038/s41598-020-72491-9
Tong, B., Spradlin, J. N., Novaes, L. F. T., Zhang, E., Hu, X., Moeller, M., et al. (2020b). A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL. ACS Chem. Biol. 15, 1788–1794. doi:10.1021/acschembio.0c00348
Toure, M., and Crews, C. M. (2016). Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. 55, 1966–1973. doi:10.1002/anie.201507978
Van Molle, I., Thomann, A., Buckley, D. L., So, E. C., Lang, S., Crews, C. M., et al. (2012). Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1α protein-protein interface. Chem. Biol. 19, 1300–1312. doi:10.1016/j.chembiol.2012.08.015
Varala, R., and Adapa, S. R. (2005). A Practical and Efficient Synthesis of Thalidomide via Na/Liquid NH3Methodology1. Org. Process. Res. Dev. 9, 853–856. doi:10.1021/op050129z
Varfolomeev, E., Blankenship, J. W., Wayson, S. M., Fedorova, A. V., Kayagaki, N., Garg, P., et al. (2007). IAP Antagonists Induce Autoubiquitination of C-IAPs, NF-κB Activation, and TNFα-dependent Apoptosis. Cell 131, 669–681. doi:10.1016/j.cell.2007.10.030
Vassilev, L. T., Vu, B., Graves, B., Carvajal, D., Podlaski, F., Filipovic, Z., et al. (2004). In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of MDM2. Science 303, 844–848. doi:10.1126/science.1092472
Vince, J. E., Wong, W. W.-L., Khan, N., Feltham, R., Chau, D., Ahmed, A. U., et al. (2007). IAP Antagonists Target cIAP1 to Induce TNFα-dependent Apoptosis. Cell 131, 682–693. doi:10.1016/j.cell.2007.10.037
Vogelstein, B., Lane, D., and Levine, A. J. (2000). Surfing the P53 Network. Nature 408, 307–310. doi:10.1038/35042675
Vu, B., Wovkulich, P., Pizzolato, G., Lovey, A., Ding, Q., Jiang, N., et al. (2013). Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 4, 466–469. doi:10.1021/ml4000657
Wang, B., Wu, S., Liu, J., Yang, K., Xie, H., and Tang, W. (2019). Development of Selective Small Molecule MDM2 Degraders Based on Nutlin. Eur. J. Med. Chem. 176, 476–491. doi:10.1016/j.ejmech.2019.05.046
Wang, X., Feng, S., Fan, J., Li, X., Wen, Q., and Luo, N. (2016). New Strategy for Renal Fibrosis: Targeting Smad3 Proteins for Ubiquitination and Degradation. Biochem. Pharmacol. 116, 200–209. doi:10.1016/j.bcp.2016.07.017
Weng, G., Shen, C., Cao, D., Gao, J., Dong, X., He, Q., et al. (2021). PROTAC-DB: an Online Database of PROTACs. Nucleic Acids Res. 49, D1381–D1387. doi:10.1093/nar/gkaa807
Wu, H., Yang, K., Zhang, Z., Leisten, E. D., Li, Z., Xie, H., et al. (2019). Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 62, 7042–7057. doi:10.1021/acs.jmedchem.9b00516
Xia, L.-W., Ba, M.-Y., Liu, W., Cheng, W., Hu, C.-P., Zhao, Q., et al. (2019). Triazol: a Privileged Scaffold for Proteolysis Targeting Chimeras. Future Med. Chem. 11, 2919–2973. doi:10.4155/fmc-2019-0159
Xiao, D., Wang, Y. j., Wang, H. l., Zhou, Y. b., Li, J., Lu, W., et al. (2020). Design and Synthesis of New Lenalidomide Analogs via Suzuki Cross‐coupling Reaction. Arch. Pharm. 353, 1900376. doi:10.1002/ardp.201900376
Xue, G., Wang, K., Zhou, D., Zhong, H., and Pan, Z. (2019). Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 141, 18370–18374. doi:10.1021/jacs.9b06422
Yang, J., Li, Y., Aguilar, A., Liu, Z., Yang, C.-Y., and Wang, S. (2019). Simple Structural Modifications Converting a Bona Fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem. 62, 9471–9487. doi:10.1021/acs.jmedchem.9b00846
Yeung, S. Y., Kampmann, S., Stubbs, K. A., Skelton, B. W., Kaskow, B. J., Abraham, L. J., et al. (2011). Novel Thalidomide Analogues with Potent NFκB and TNF Expression Inhibition. Med. Chem. Commun. 2, 1073. doi:10.1039/c1md00184a
Zengerle, M., Chan, K.-H., and Ciulli, A. (2015). Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10, 1770–1777. doi:10.1021/acschembio.5b00216
Zhang, F., Wu, Z., Chen, P., Zhang, J., Wang, T., Zhou, J., et al. (2020a). Discovery of a New Class of PROTAC BRD4 Degraders Based on a Dihydroquinazolinone Derivative and Lenalidomide/pomalidomide. Bioorg. Med. Chem. 28, 115228. doi:10.1016/j.bmc.2019.115228
Zhang, X., Crowley, V. M., Wucherpfennig, T. G., Dix, M. M., and Cravatt, B. F. (2019). Electrophilic PROTACs that Degrade Nuclear Proteins by Engaging DCAF16. Nat. Chem. Biol. 15, 737–746. doi:10.1038/s41589-019-0279-5
Zhang, X., He, Y., Zhang, P., Budamagunta, V., Lv, D., Thummuri, D., et al. (2020b). Discovery of IAP-Recruiting BCL-XL PROTACs as Potent Degraders across Multiple Cancer Cell Lines. Eur. J. Med. Chem. 199, 112397. doi:10.1016/j.ejmech.2020.112397
Zhang, X., Xu, F., Tong, L., Zhang, T., Xie, H., Lu, X., et al. (2020c). Design and Synthesis of Selective Degraders of EGFRL858R/T790M Mutant. Eur. J. Med. Chem. 192, 112199. doi:10.1016/j.ejmech.2020.112199
Zhao, B., and Burgess, K. (2019). TrkC-Targeted Kinase Inhibitors and PROTACs. Mol. Pharmaceutics 16, 4313–4318. doi:10.1021/acs.molpharmaceut.9b00673
Zhao, Q., Lan, T., Su, S., and Rao, Y. (2019). Induction of Apoptosis in MDA-MB-231 Breast Cancer Cells by a PARP1-Targeting PROTAC Small Molecule. Chem. Commun. 55, 369–372. doi:10.1039/C8CC07813K
Zheng, N., and Shabek, N. (2017). Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 86, 129–157. doi:10.1146/annurev-biochem-060815-014922
Zhou, B., Hu, J., Xu, F., Chen, Z., Bai, L., Fernandez-Salas, E., et al. (2018). Discovery of a Small-Molecule Degrader of Bromodomain and Extra-terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. 61, 462–481. doi:10.1021/acs.jmedchem.6b01816
Zhu, Y. X., Braggio, E., Shi, C.-X., Bruins, L. A., Schmidt, J. E., Van Wier, S., et al. (2011). Cereblon Expression Is Required for the Antimyeloma Activity of Lenalidomide and Pomalidomide. Blood 118, 4771–4779. doi:10.1182/blood-2011-05-356063
Zoppi, V., Hughes, S. J., Maniaci, C., Testa, A., Gmaschitz, T., Wieshofer, C., et al. (2019). Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 62, 699–726. doi:10.1021/acs.jmedchem.8b01413
Zou, X. J., Yang, W. L., Zhu, J. Y., and Deng, W. P. (2020). Catalytic Enantioselective Formal Synthesis of MDM2 Antagonist RG7388 and its Analogues. Chin. J. Chem. 38, 435–438. doi:10.1002/cjoc.201900530
Glossary
AhR arylhydrocarbon receptor
AIBN azobisisobutyronitrile
AR androgen receptor
bHLH basic helix-loop-helix
BIPHEP bis(diphenylphosphino)biphenyl
BRD bromodomain-containing protein
CDI carbonyldiimidazole
BTK Bruton’s tyrosine kinase
cIAP cellular inhibitor of apoptosis
CLIPTACs in-cell click-formed proteolysis targeting chimeras
COMU (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium-hexafluorophosphate
CPME cyclopentylmethylether
CRABPs cellular retinoic acid binding proteins
CRBN cereblon
CRL cullin/RING E3 ubiquitin ligase
DBAD di-tert-butyl azodicarboxylate
DBU diazabicycloundecene
DCAF DDB1-and-CUL4-associated factor
DCC N,N′-Dicyclohexylcarbodiimide
DDB damage-specific DNA binding protein
DEACM diethylamino coumarin
DIAD diisopropyl azodicarboxylate
DIPEA N,N-diisopropylethylamine
DMA N,N-dimethylacetamide
DMAP 4-(N,N-dimethylamino)pyridine
DMNB 4,5-dimethoxy-2-nitro-benzyl
DTBAD di-tert-butyl azodicarboxylate
E1 ubiquitin-activating enzyme
E2 ubiquitin-conjugating enzyme
E3 ubiquitin ligase
EDC 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
Ent enantiomeric
ER Estrogen receptor
FKBP12 peptidyl-prolyl cis-trans isomerase
Fmoc fluorenylmethyloxycarbonyl
FNIP1 lolliculin-interacting protein 1
HATU 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
HCTU 2-(6-chloro-1H-benzotriazol-1-yl)-1,1,3,3-tetramethylaminium-hexafluorophosphate
HECT homologous to E6AP C-terminus
HIF hypoxia-inducible factor
HMBC heteronuclear multiple bond correlation
HOAt 1-hydroxy-7-azabenzotriazole
HOBt 1-hydroxybenzotriazole
IAP inhibitor of apoptosis proteins
IKZF transcription factors Ikaros (IKZF1) and Aiolos (IKZF3)
IMiDs immunomodulatory imide drugs
ITE 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester
KEAP1 Kelch-like ECH-associated protein 1
mCPBA meta-chloroperoxybenzoic acid
MDM2 mouse double minute 2 homologue
ML-IAP melanoma IAP
MTBE methyl tert-butyl ether
MW microwave
NF naphthoflavone
NMP N-methylpyrrolidone
NPOM 6-nitropiperonyloxymethyl
NRF2 nuclear factor erythroid 2–related factor 2
NS3 nonstructural protein 3, hepatitis C virus protein
PARP1 poly(ADP-ribose)-polymerase 1;
PAS Per-Arnt-Sim
Pd/C palladium on activated carbon
Phosferrox phosphinoferrocenyloxazoline
PHOTACs photochemically targeting chimeras
POI protein of interest
PROTACs proteolysis targeting chimeras
PyBOP benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
RBR RING-between-RING
Rbx-1 RING-box protein 1
RING really interesting new gene
RNF114 RING-type zinc-finger protein 114
ROS reactive oxygen species
rt room temperature
SCFβ-TRCP Skp1–Cullin–F box complex
SFC supercritical fluid chromatography
Smac/DIABLO second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI
SNIPERs specific and nongenetic IAP-dependent protein erasers
TBAF tetra-N-butylammonium fluoride
TBAI tetra-N-butylammonium iodide
TBDPSCl tert-butyl(chloro)diphenylsilane
TBSCl tert-butyl(chloro)dimethylsilane
TBTU 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate
TCDD 2,3,7,8-tetrachlorodibenzo-p-dioxin
TEMPO 2,2,6,6-tetramethylpiperidine-1-oxyl
Teoc-OSu 1-[2-(trimethylsilyl)ethoxycarbonyloxy]pyrrolidin-2,5-dione
Tf/triflic trifluoromethanesulfonyl
TIPS triisopropylsilyl
Tle 2-amino-3,3-dimethylbutyric acid
TPPO triphenylphosphine oxide
TrkC tropomyosin receptor kinase C
Trt/trityl triphenylmethyl
UIM ubiquitin-interacting motif
UPS ubiquitin proteasome system
VHL von-Hippel–Lindau
XIAP chromosome-linked IAP
Keywords: bifunctional molecules, cereblon, drug synthesis, E3 ligase ligands, linker attachment, PROTACs, von Hippel–Lindau
Citation: Bricelj A, Steinebach C, Kuchta R, Gütschow M and Sosič I (2021) E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front. Chem. 9:707317. doi: 10.3389/fchem.2021.707317
Received: 09 May 2021; Accepted: 04 June 2021;
Published: 05 July 2021.
Edited by:
Hany S. Ibrahim, Egyptian Russian University, EgyptReviewed by:
Andrea Testa, Amphista Therapeutics Limited, United KingdomMichael Mathieson and Gregor Meier, Amphista Therapeutics Limited, UK, in collaboration with reviewer AT
Yosuke Demizu, National Institute of Health Sciences (NIHS), Japan
Copyright © 2021 Bricelj, Steinebach, Kuchta, Gütschow and Sosič. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Gütschow, Z3VldHNjaG93QHVuaS1ib25uLmRl; Izidor Sosič, aXppZG9yLnNvc2ljQGZmYS51bmktbGouc2k=