 Gerd Gleixner
Gerd Gleixner- Max-Planck-Institut für Biogeochemie, Jena, Germany
We determined the kinetic isotope effect on the serine hydroxymethyltransferase reaction (SHMT), which provides important C1 metabolites that are essential for the biosynthesis of DNA bases, O-methyl groups of lignin and methane. An isotope effect on the SHMT reaction was suggested being responsible for the well-known isotopic depletion of methane. Using the cytosolic SHMT from pig liver, we measured the natural carbon isotope ratios of both atoms involved in the bond splitting by chemical degradation of the remaining serine before and after partial turnover. The kinetic isotope effect 13(VMax/Km) was 0.994 0.006 and 0.995 0.007 on position C-3 and C-2, respectively. The results indicated that the SHMT reaction does not contribute to the 13C depletion observed for methyl groups in natural products and methane. However, from the isotopic pattern of caffeine, isotope effects on the methionine synthetase reaction and on reactions forming Grignard compounds, the involved formation and fission of metal organic bonds are likely responsible for the observed general depletion of “activated” methyl groups. As metal organic bond formations in methyl transferases are also rate limiting in the formation of methane, they may likely be the origin of the known 13C depletion in methane.
Introduction
The stable isotopes of the bioelements (H, C, N, O) are not statistically distributed in natural compounds, but represent specific intramolecular isotope distributions (Schmidt et al., 1995). Many reasons for these distributions are suggested. In 1985, the geochemist E. Galimov proposed thermodynamic reasons. Presuming a total energy equilibration between all atoms of a given system of molecules, which is likely in the “slow” and “energy-rich” rock cycle, Galimov suggested that isotope effects on chemical bond enthalpies explain these isotopic patterns (Galimov, 1985). His calculated “β-factors” were in line with the isotopic patterns of some compounds known at that time (Abelson and Hoering, 1961; Meinschein et al., 1974; Rinaldi et al., 1974); however, they were not in agreement with patterns observed in natural products (Schmidt et al., 1995). Obviously, the presumption that all molecules and all atoms within these molecules are in thermodynamic binding equilibrium is not valid for biological systems. Here, kinetic isotope effects on enzymatic reactions are expected to contribute to isotopic discrimination. This has widely been demonstrated for the primary CO2 fixation reactions (O'Leary, 1981; Winkler et al., 1982; Farquhar, 1983; WinklerSchmidt et al., 1983), and as an example in secondary metabolism, it has been shown that the isotope effect on the pyruvate dehydrogenase reaction is responsible for the general depletion of 13C in metabolites of acetyl-CoA, such as fatty acids or isoprenoids (Park and Epstein, 1960; Whelan et al., 1970; DeNiro and Epstein, 1977; Winkler and Schmidt, 1980; Monson and Hayes, 1982; Melzer and Schmidt, 1987; Schmidt et al., 1995). In addition, it has been demonstrated that the formation of 13C depleted lipids has to be compensated by 13C enrichments in other compounds (Abelson and Hoering, 1961; Epstein, 1968). Furthermore, investigations on the isotopic pattern of glucose and its metabolites (Roßmann et al., 1991; Gleixner et al., 1993) demonstrate the importance of the isotopic pattern of the metabolic precursor and the influence of turnover rates at metabolic branching points (Schmidt et al., 1995) for the observed pattern of a natural product.
The reason for well-known 13C depletions in biological N- and O-methyl groups of lignin (Galimov et al., 1976; Keppler et al., 2004) and other biological compounds like methane (Krueger and Krueger, 1984; Caer et al., 1991; Schmidt et al., 1995; Weilacher et al., 1996), however, involves several possible reactions. A common starting ground is that all depleted methyl groups originate from the C1 metabolism. This pool is fed by the serine hydroxymethyltransferase (SHMT) reaction, and it was suggested that an isotope effect on this reaction could be responsible for the 13C depletion found for CH3-groups in natural products.
SHMT [EC 2.1.2.1] catalyzes the reversible cleavage of serine to glycine and tetrahydrofolic acid (THF) bound formaldehyde (N5, N10-Methylene-THF, Figure 1). Methylene THF is the most important source for the C1 metabolism, which is essential for cell proliferation (Rao et al., 1987; Stover and Schirch, 1993). The pool provides C1 units for the biosynthesis of purines, thymine, formylmethionine tRNA, and methionine, the latter being the precursor of S-adenosylmethionine (SAM), the biological agent for methylations. Correspondingly, SHMT is one of the most ubiquitous enzymes in nature (Schirch, 1982), being found in all organisms (fungi, bacteria, insects, and plants), in different organs of animals (liver, kidney, and brain), and in different cell compartments (cytosol and mitochondria).
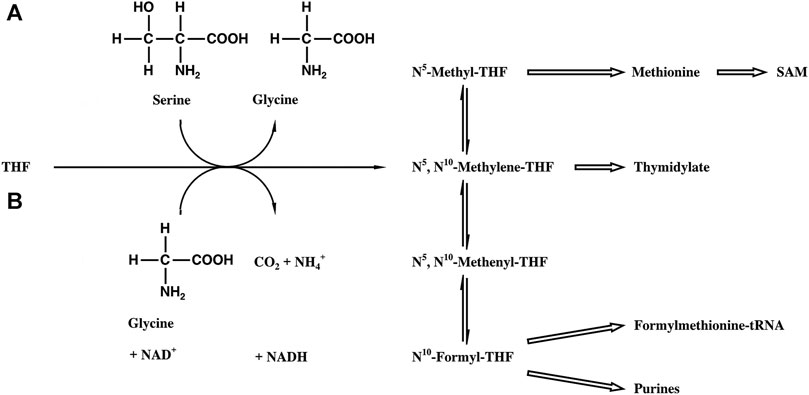
FIGURE 1. Biosynthetic scheme of the THF-bound C1 pool. (A) SHMT reaction. (B) Glycine synthase reaction.
In our experiments, we used the cytosolic SHMT from pig liver, which is a homotetramer with a molar mass of 212 kD. Each subunit has covalently bound pyridoxal 5′-phosphate (PLP), responsible for the yellow color of the enzyme. The enzyme catalyzes in its “closed” form, which is supposed to be the physiological one, the transfer of C1 units to THF, but it also catalyzes in its “open” and probably not physiological form THF independent reactions like aldol cleavages, transaminations, and decarboxylations (Schirch, 1982). For the reaction catalyzed by the “closed” form, three different mechanisms are discussed including a retroaldol cleavage, the formation of a thiohemiacetal, or the direct nucleophilic attack of THF (Matthews and Drummond, 1990). The study of the carbon isotope effect on the SHMT reaction could contribute to the distinction between these discussed enzyme mechanisms, even though from our point of view the role of the enzyme in the depletion of methyl groups would be of primary importance.
Materials and Methods
Chemicals
All ordinary chemicals were of analytical grade and purchased from local suppliers. 5,6 (R,S),7,8-THF C19H23N7O6*2HCl*2H2O was purchased from Boehringer Mannheim GmbH (Mannheim, GER) and l-Serine from Fluka AG (Buchs, CH). 6(S)-THF was produced from racemic folic acid by chemical reduction to 7,8 dihydrofolic acid and by reduction with dihydrofolic acid reductase to 5,6(S),7,8-THF and was donated from V. Schirch, Medical College of Virginia, United States (Stover and Schirch, 1992).
Enzymes
The cytosolic SHMT [EC 2.1.2.1] from pig liver with an activity of 0.43 μkat/mg, dihydrofolate reductase [EC 1.5.1.3], and “trifunctional enzyme” TFE (methylenetetrahydrofolate dehydrogenase [EC 1.5.1.5], methenyltetrahydrofolate cyclohydrolase [EC 3.5.4.9], and formyltetrahydrofolate synthetase [EC 6.3.4.3]) with 0.03 μkat/mg dehydrogenase activity were isolated, and enzyme activity was determined as described previously (Stover and Schirch, 1992). Methylenetetrahydrofolate reductase (MTHFR) was isolated as described previously from pig liver with an activity of 0.14 μkat/ml (NADPH-CH2-H4folate oxidoreductase assay in water, Matthews, 1986). All enzymes were donated by V. Schirch, Medical College of Virginia, United States.
Determination of the Kinetic Isotope Effect on the SHMT Reaction
The competitive method was used throughout (O´Leary, 1980). Here, the enzymatic reaction that is investigated is coupled to a second enzymatic reaction that removes the formed product immediately in order to make the first reaction irreversible. The remaining substrate after partial enzymatic turnover (50%) is analyzed. The irreversibility of the SHMT reaction was guaranteed by enzymatic oxidation or reduction of the methylene THF formed to formyl THF or methyl THF, respectively (Figures 2, 3). The turnover of the reaction was determined on the basis of the absorbance of NADPH at 340 nm. In the oxidative assay 1,000 µl 20 mM KH2PO4 buffer (pH 7.3) containing 45 mM β-mercaptoethanol, 50 µl 10 mM NADP+ solution, 50 µl 24.4 mM 6S-THF solution, 0.13 µkat TFE, and 0.25 nkat SHMT were incubated at 25°C. An aliquot of 300 µl was used in a 1 mm quartz cuvette to measure the baseline at 340 nm (Varian UV/VIS spectrometer). The assay was started in the joint solutions by adding 1 µmol serine. The reductive assay was performed analogously, but 50 µl 12.5 mM NADPH solution and 14 nkat of MTHFR were used.
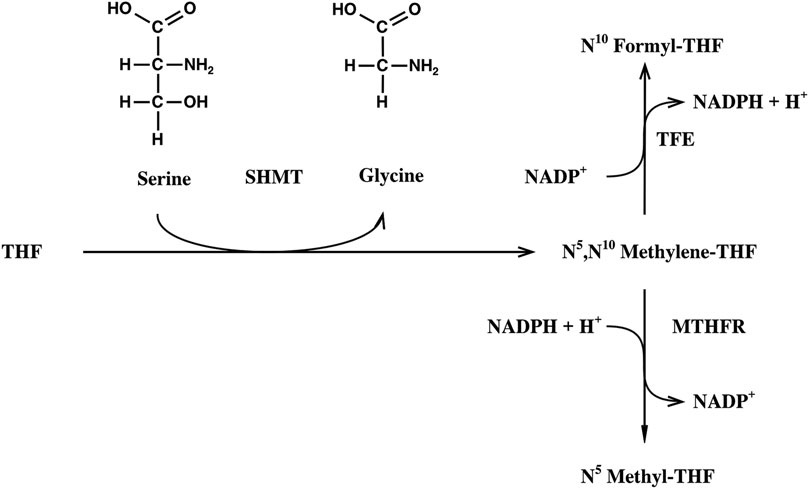
FIGURE 2. Alternative reaction assays for the determination of the kinetic isotope effect on the SHMT reaction. THF, tetrahydofolic acid; SHMT, serine hydroxymethyltransferase; TFE, trifunctional enzyme; MTHR, methylene THF reductase.
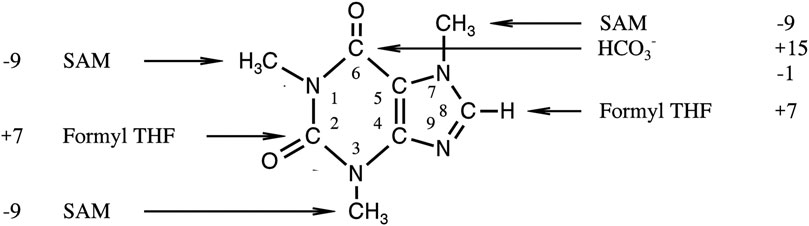
FIGURE 3. Origin of different positions in caffeine and their difference in [‰ (mUr)]PDB to the mean δ13C-value of whole the molecule (see 22).
An aliquot of the reaction mixture (300 µl) was again transferred to a quartz cuvette (1 mm) in order to control the turnover rate on the basis of NADPH absorption (ε340 = 6230 M−1 cm−1). After a turnover of 50–70%, the remaining solution was heated to 100°C to denature the enzymes, and the solution was concentrated to a volume of 500 µl.
The enzymatic experiments were performed at the Medical College of Virginia, United States under the supervision of V. Schirch and R.G. Matthews.
Isolation of Serine
The residual serine was isolated from the assay medium by HPLC (Perkin Elmer) in the Medical College of Virginia. The solution (500 µl) was applied to a LiChroSorb CX 10 µm cation exchange column (outer diameter 4.6 x column length 250 mm, Merck (Darmstadt, GER)). During the isocratic (H3PO4 pH 2.8) separation, serine was monitored through its absorption at 210 nm and finally identified from the collected eluate (1 ml) by its UV spectrum (HP 1050, photodiode array spectrometer). Serine containing fractions (retention time ca. 20 min) were pooled (3–5 ml) and stored at −70°C.
Degradation of Serine, Positional Isotope Analysis, and Calculation of Isotope Effects
For the chemical degradation of isolated serine and the isotopic analysis of its fragments, 2 µl 500 mM KH2PO4 buffer pH 5.8, containing 100 nmol serine, 300 nmol NaIO4 in 2 µl H2O were added in a closed autosampler vial (700 µl); the mixture was incubated for 15 min at 65°C. Under these conditions, periodate degrades serine into CO2 (C-1), formic acid (C-2), and formaldehyde (C-3) (Simon and Floss, 1967; Gleixner, 1994). One microliter of this reaction mixture was transferred to the GC-C-IRMS system (Isochrom 1, VG Isogas (Middelwich, GB)). The reaction products were separated on a PoraPLOT U capillary column (Chrompack (Frankfurt, GER), outer diameter 0.32 mm, column length 5 m) using a temperature program (2 min 110, 0.3°C/min to 140°C) and oxidized by online combustion (CuO, 800°C). The CO2 formed was directly transferred to the IRMS and measured against bottled CO2 monitor gas that was calibrated against NBS 19. All isotope ratios are expressed as 13C-values (Coplen, 2011).
The kinetic isotope effect on the SHMT reaction 13(VMax/Km) was determined from the isotope ratio of serine in position C-3 before and after partial turnover (R0 and RS) and the turnover fraction f were used:
Results
Principle of the Determination of the Kinetic Isotope Effect on the SHMT Reaction
The assay developed was based on the enzymatic assay of Vanoni et al. (Vanoni et al., 1990) for the determination of the secondary tritium isotope effect on the SHMT reaction. Serine was split by SHMT into glycine and methylene THF. The latter was either immediately reduced by methylene THF reductase and NADPH to methyl THF, or oxidized by TFE and NADP+ to formyl THF (Figure 2). The activity of the auxiliary enzymes was always far higher than that of the SHMT, in order to guarantee that this reaction was rate limiting and the whole reaction sequence irreversible. Results obtained with both alternatives were identical; however, as handling of TFE and NADP+ was more convenient, most assays were performed with the SHMT and TFE method. The isotope effect was determined on the basis of the isotopic pattern analysis of the substrate at 0% and of the remaining substrate after partial turnover (50–70%); for this purpose, the residual serine was isolated from the medium by cation exchange. The amino acid was submitted to NaIO4 fission, and the isotope ratios of the fission products were determined by GC-C-IRMS.
Check for Possible Isotope Effects on the Method Itself
The kinetics, yield, and reproducibility of the serine degradation by periodate were controlled on mmolar samples of pure serine measuring the amount of CO2 formed. This product was cryogenically purified and determined gas volumetrically. Between 92 and 99% yield were found. Independent of the sample size (nmol - mmol), an identical isotope ratio of the CO2 was found (0.2–0.5‰ (mUr) difference).
Furthermore, the degradation method was checked for isotope effects from an isotope balance, comparing the total isotope ratios before and after degradation. After serine degradation in nmol amounts, the mean isotope ratio of the fragments was identical to the measured isotope ratio of the whole molecule (Table 1). Finally, in order to check the isolation method for isotope effects, serine was degraded before and after chromatographic purification from incubation medium, and the isotope ratios of the fragments were compared (Table 1). Differences of 2 and 7‰ (mUr) were found for positions C-1 and C-2, respectively, but no difference for position C-3, the only relevant one in regard to the one carbon pool. Therefore, the method developed is suitable for the determination of the kinetic isotope effect on the SHMT reaction.

TABLE 1. δ13C-values of serine and of its molecule positions before and after isolation from incubation medium.
Results and Calculation of the Kinetic Isotope Effect on the SHMT Reaction
The mean calculated kinetic isotope effect (KIE) on position C-3 of serine was 13(VMax/Km) = 0.994 0.006 (Table 2), which is in the range of a secondary isotope effect.
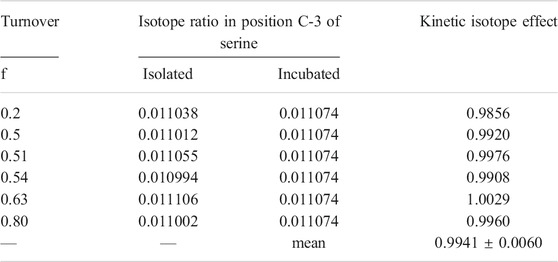
TABLE 2. Isotope ratio of position C-3 in serine before and after incubation with SHMT and calculated kinetic isotope effect on the reaction.
For the calculation of the KIE on positions C-1 and C-2, the measured δ13C-value shifts of these positions after the reaction were corrected for the shifts observed during the separation step (Table 3). The resulting KIE on position C-1 and C-2 of serine were 13(VMax/Km) = 0.995 0.005 and 0.995 0.007, respectively. Hence the SHMT reaction does not appear to contribute to the observed carbon isotope discrimination.
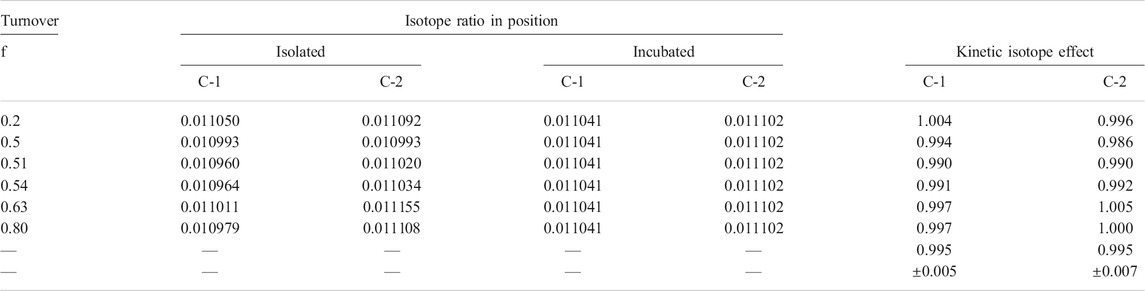
TABLE 3. Isotope ratio of position C-1 and C-2 in serine before and after incubation with SHMT and calculated kinetic isotope effect on the reaction. Isotope ratios of the isolated products were linear corrected for the isotope effect of the separation step.
Discussion
The measured isotope effects on the SHMT reaction have to be discussed in order to identify the rate determining step of the SHMT reaction, and to interpret the strong 13C depletions known for certain C1 metabolites (Schmidt et al., 1995).
Concerning the first aspect, it has to be pointed out that the isotope effect found on the two carbon atoms involved in the bond cleavage catalyzed by the SHMT reaction, as well as that on the C-1 (not involved in the bond cleavage) are identical and close to unity (KIE for C-1 = 0.994, C-2 = 0.995, C-3 = 0.995). This result is in line with findings of Vanoni et al. (Vanoni et al., 1990) who did not observe any secondary tritium isotope effect on the reaction. Both results therefore suggest that the C-C bond splitting is not rate limiting for the reaction, and that substrate dissociation is slower than the chemical steps of the reaction sequence, resulting in a high forward commitment (cf) to catalysis. Given the “closed” conformation of the SHMT, as proposed for this reaction, a high cf is reasonable. Therefore, the results cannot contribute to a further distinction of the alternative chemical mechanisms discussed for the enzyme reaction.
More informative and important as to our main point of view is the fact that the SHMT reaction obviously does not contribute to the observed 13C depletions of methyl groups in natural products. This finding has therefore to be discussed taking additionally into account the observed isotope pattern of caffeine, which we described elsewhere (Weilacher et al., 1996). In the core of this heteroaromatic purine alkaloid (Figure 3), two carbon atoms derive from the THF-bound C1 pool and three others, the methyl groups, from the SAM-bound C1 pool. The latter C atoms were depleted of 13C relative to those from the THF-bound C1 pool by 16‰ (mUr) (Figure 3). This discrimination, combined with our results on the isotope effect on the SHMT reaction, indicate unequivocally an isotope effect in the transfer of C1 units from the THF-bound C1 to the SAM-bound C1 pool. A kinetic isotope effect ≥1.017 for a corresponding reaction can be estimated from the δ13C-values of the representatives in the two C1 pools.
This conclusion must have a general importance for isotope fractions in C1 metabolism. Methane liberated in wetlands and swamps can be depleted up to -110‰ (mUr) (Hoefs, 1987). Quite remarkable 13C depletions are also reported for methane produced by bacteria (Fuchs et al., 1979). For bacterial methanogenesis, two sources are known, namely, CO2 and acetate (Thauer et al., 1985; Wolfe et al., 1990, see Figure 4). The pathway of methane biosynthesis from CO2 has in certain phases a strong analogy to the reduction of C1 units for the THF pool; however, for energetic reasons, the CO2 is bound to tetrahydromethanopterin, a cofactor with a lower redox potential than THF.
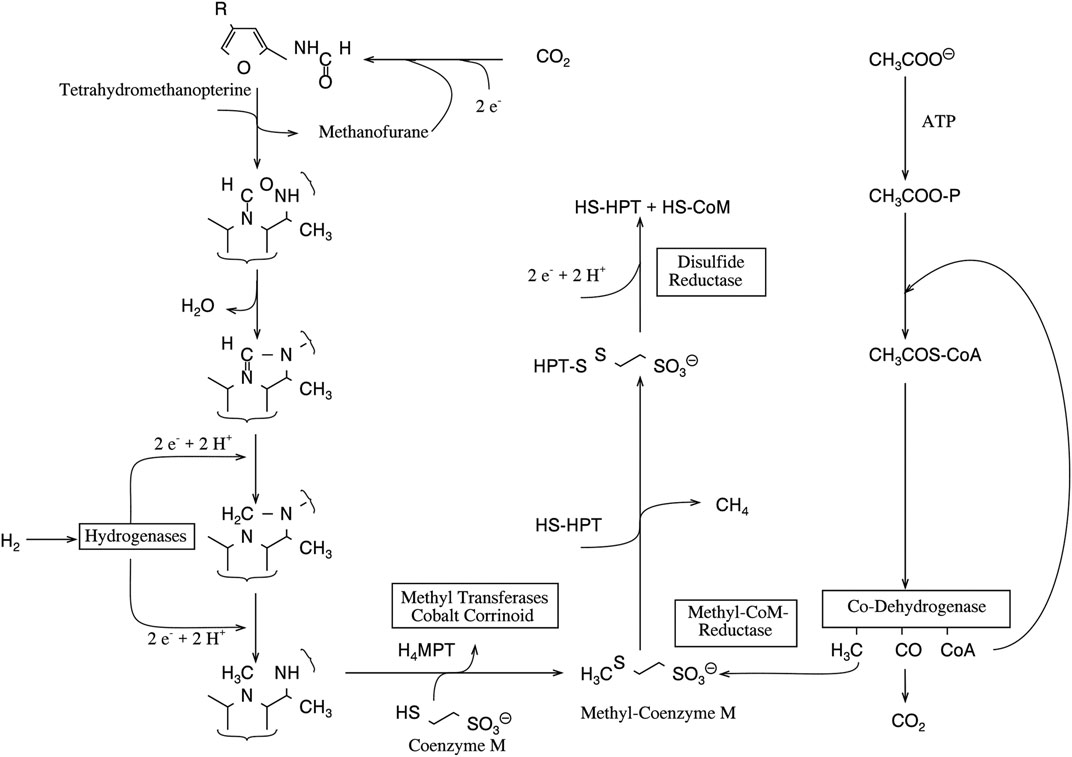
FIGURE 4. Biological methanogenesis starting from CO2 and acetate (adapted after Jaun, 1993).
CO2 was also the only source for methane in previous investigations of our group on simulated rumen fermentation (Metges et al., 1990). From the molar balance of the substrate and the fermentation products, it was possible to calculate the amount and the δ13C-value of the primarily produced CO2, which was partially reduced for the formation of methane. Its turnover and the δ13C-values of the remaining CO2 and the formed methane (−9‰ (mUr) and −70‰ (mUr), respectively) yielded a discrimination corresponding to an “isotope effect” of 1.06 for the bacterial methanogenesis. This value is much higher than the isotope effect estimated for the transfer of C1 units from the THF to the SAM pool, but still below the isotope effect reported for the catechole-O-methyltransferase reaction (1.09) and the methionine synthase reaction (1.087) (Gray et al., 1979; Hegazi et al., 1979; Romek et al., 2017).
Nevertheless, all these findings indicate that isotope discriminations accompanying the formation or the transfer of methyl groups and not the formation of THF-bound C1 units cause the depletion. This has recently been shown for methionine synthase but is still unknown for the enzymes catalyzing the methyl transfer to coenzyme M for methanogenesis (Figure 4). All these enzymes have a cobalamin as prosthetic group and bind methyl units to this core metal via metal organic bonds. The proposal, that the observed 13C discrimination takes place in this bond fission, is supported by the finding that the release of hydrocarbons from Grignard compounds is also accompanied by a large carbon isotope effect (Holm, 1993; Vogler and Hayes, 1978). In consequence, we suggest that further investigations on the origin of the 13C depletion in methane focus on corresponding reactions.
Author's Note
This manuscript is dedicated to the 90th birthday of H.-L. Schmidt.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
GG performed the research and wrote the publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
I thank H.-L. Schmidt, V. Schirch, and R.G. Matthews for their support performing the experiment and comments on earlier versions of the MS and J. Weissflog for graphical support.
References
Abelson, P. H., and Hoering, T. C. (1961). Carbon Isotope Fractionation in Formation of Amino Acids by Photosynthetic Organisms. Proc. Natl. Acad. Sci. 47, 623–632. doi:10.1073/pnas.47.5.623
Caer, V., Trierweiler, M., Martin, G. J., and Martin, M. L. (1991). Determination of Site-specific Carbon Isotope Ratios at Natural Abundance by Carbon-13 Nuclear Magnetic Resonance Spectroscopy. Anal. Chem. 63, 2306–2313. doi:10.1021/ac00020a021
Coplen, T. B. (2011). Guidelines and Recommended Terms for Expression of Stable-Isotope-Ratio and Gas-Ratio Measurement Results. Rapid Commun. Mass. Spectrom. 25, 2538–2560. doi:10.1002/rcm.5129
DeNiro, M. J., and Epstein, S. (1977). Mechanism of Carbon Isotope Fractionation Associated with Lipid Synthesis. Science 197, 261–263. doi:10.1126/science.327543
Epstein, S. (1968). Proc. Symp. On CO2 Chemical, Biochemical, and Physiological Aspects (Haverford PA: NASA SP-188), 5–14.
Farquhar, G. (1983). On the Nature of Carbon Isotope Discrimination in C4 Species. Funct. Plant Biol. 10, 205–226. doi:10.1071/pp9830205
Fuchs, G., Thauer, R., Ziegler, H., and Stichler, W. (1979). Carbon Isotope Fractionation by Methanobacterium Thermoautotrophicum. Arch. Microbiol. 120, 135–139. doi:10.1007/bf00409099
Gleixner, G., Danier, H. J., Werner, R. A., and Schmidt, H. L. (1993). Correlations between the 13C Content of Primary and Secondary Plant Products in Different Cell Compartments and that in Decomposing Basidiomycetes. Plant Physiol. 102, 1287–1290. doi:10.1104/pp.102.4.1287
Gray, C. H., Coward, J. K., Schowen, K. B., and Schowen, R. L. (1979). alpha.-Deuterium and Carbon-13 Isotope Effects for a Simple, Intermolecular Sulfur-To-Oxygen Methyl-Transfer Reaction. Transition-State Structures and Isotope Effects in Transmethylation and Transalkylation. J. Am. Chem. Soc. 101, 4351–4358. doi:10.1021/ja00509a051
Hegazi, M. F., Borchardt, R. T., and Schowen, R. L. (1979). J. Am. Chem. Soc. 101, 4358–4365. doi:10.1021/ja00509a052
Holm, T. (1993). Carbon-13 Kinetic Isotope Effects in the Reactions of Grignard Reagents with Benzophenone. J. Am. Chem. Soc. 115, 916–918. doi:10.1021/ja00056a015
Jaun, B. (1993). Das Nickelzentrum in Coenzym F430. Nachr. Chem. Tech. Lab. 41, 1092–1098. doi:10.1002/nadc.19930411006
Keppler, F., Kalin, R. M., Harper, D. B., McRoberts, W. C., and Hamilton, J. T. G. (2004). Carbon Isotope Anomaly in the Major Plant C1 Pool and its Global Biogeochemical Implications. Biogeosciences 1, 123–131. doi:10.5194/bg-1-123-2004
Krueger, D. A., and Krueger, H. W. (1984). Comparison of Two Methods for Determining intramolecular13C/12C Ratios of Acetic Acid. Biol. Mass. Spectrom. 11, 472–474. doi:10.1002/bms.1200110907
Matthews, R. G. (1986). [58] Methylenetetrahydrofolate Reductase from Pig Liver. Methods Enzymol. 122, 372–381. doi:10.1016/0076-6879(86)22196-5
Matthews, R. G., and Drummond, J. T. (1990). Providing One-Carbon Units for Biological Methylations: Mechanistic Studies on Serine Hydroxymethyltransferase, Methylenetetrahydrofolate Reductase, and Methyltetrahydrofolate-Homocysteine Methyltransferase. Chem. Rev. 90, 1275–1290. doi:10.1021/cr00105a010
Meinschein, W. G., Rinaldi, G. G. L., Hayes, J. M., and Schoeller, D. A. (1974). Intramolecular Isotopic Order in Biologically Produced Acetic Acid. Biol. Mass. Spectrom. 1, 172–174. doi:10.1002/bms.1200010306
Melzer, E., and Schmidt, H. L. (1987). Carbon Isotope Effects on the Pyruvate Dehydrogenase Reaction and Their Importance for Relative Carbon-13 Depletion in Lipids. J. Biol. Chem. 262, 8159–8164. doi:10.1016/s0021-9258(18)47543-6
Metges, C., Kempe, K., and Schmidt, H.-L. (1990). Dependence of the Carbon-Isotope Contents of Breath Carbon Dioxide, Milk, Serum and Rumen Fermentation Products on the δ13C Value of Food in Dairy Cows. Br. J. Nutr. 63, 187–196. doi:10.1079/bjn19900106
Monson, K. D., and Hayes, J. M. (1982). Carbon Isotopic Fractionation in the Biosynthesis of Bacterial Fatty Acids. Ozonolysis of Unsaturated Fatty Acids as a Means of Determining the Intramolecular Distribution of Carbon Isotopes. Geochimica et Cosmochimica Acta 46, 139–149. doi:10.1016/0016-7037(82)90241-1
O'Leary, M. H. (1981). Carbon Isotope Fractionation in Plants. Phytochemistry 20, 553–567. doi:10.1016/0031-9422(81)85134-5
Park, R., and Epstein, S. (1960). Carbon Isotope Fractionation during Photosynthesis. Geochimica et Cosmochimica Acta 21, 110–126. doi:10.1016/s0016-7037(60)80006-3
Rao, N. A., Ramesh, K. S., Ratnam, M., Rao, D. N., Vijaylakshmi, D., and Bhaskaran, N ( (1987). J. Sci. Ind. Res. 46, 248–260.
Rinaldi, G., Meinschein, W. G., and Hayes, J. M. (1974). Intramolecular Carbon Isotopic Distribution in Biologically Produced Acetoin. Biol. Mass. Spectrom. 1, 415–417. doi:10.1002/bms.1200010609
Romek, K. M., Krzemińska, A., Remaud, G. S., Julien, M., Paneth, P., and Robins, R. J. (2017). Insights into the Role of Methionine Synthase in the Universal 13 C Depletion in O - and N -methyl Groups of Natural Products. Arch. Biochem. Biophys. 635, 60–65. doi:10.1016/j.abb.2017.10.012
Schmidt, H.-L., Kexel, H., Butzenlechner, M., Schwarz, S., Gleixner, G., Thimet, S., et al. (1995). in Stable Isotopes in the Biosphere. Editors E. Wada, T. Yoneyama, M. Minagawa, T. Ando, and B. D. Fry (Kyoto: Kyoto University Press), 17–35.
Simon, H., and Floss, H. G. (1967). Anwendung von Isotopen in der organischen Chemie und Biochemie, Band 1, Bestimmung von Isotopenverteilungen in markierten Verbindungen. Berlin: Springer-Verlag.
Stover, P., and Schirch, V. (1992). Synthesis of (6S)-5-Formyltetrahydropteroyl-Polyglutamates and Interconversion to Other Reduced Pteroylpolyglutamate Derivatives. Anal. Biochem. 202, 82–88. doi:10.1016/0003-2697(92)90210-x
Stover, P., and Schirch, V. (1993). The Metabolic Role of Leucovorin. Trends Biochem. Sci. 18, 102–106. doi:10.1016/0968-0004(93)90162-g
Thauer, R. K., Brandis-Heep, A., Diekert, G., Gilles, H.-H., Graf, E.-G., Jaenchen, R., et al. (1985). “Nickel Enzymes in Anaerobic Metabolism,” in Environmental Regulation of Microbial Metabolism. Editors I. S. Kulaev, E. A. Dawes, and D. W. Tempest (New York: Academic Press), 231–239. doi:10.1016/b978-0-12-428580-4.50026-5
Vanoni, M. A., Lee, S., Floss, H. G., and Matthews, R. G. (1990). Stereochemistry of Reduction of Methylenetetrahydrofolate to Methyltetrahydrofolate Catalyzed by Pig Liver Methylenetetrahydrofolate Reductase. J. Am. Chem. Soc. 112, 3987–3992. doi:10.1021/ja00166a040
Vogler, E. A., and Hayes, J. M. (1978). The Synthesis of Carboxylic Acids with Carboxyl Carbons of Precisely Known Stable Isotopic Composition. Int. J. Appl. Radiat. Isotopes 29, 297–300. doi:10.1016/0020-708x(78)90058-3
Weilacher, T., Gleixner, G., and Schmidt, H.-L. (1996). Carbon Isotope Pattern in Purine Alkaloids a Key to Isotope Discriminations in C1 Compounds. Phytochemistry 41, 1073–1077. doi:10.1016/0031-9422(95)00757-1
Whelan, T., Sackett, W. M., and Benedict, C. R. (1970). Carbon Isotope Discrimination in a Plant Possessing the C4 Dicar☐ylic Acid Pathway. Biochem. Biophysical Res. Commun. 41, 1205–1210. doi:10.1016/0006-291x(70)90214-7
Winkler, F. J., Kexel, H., Kranz, C., and Schmidt, H.-L. (1982). in Stable Isotopes. Editors H.-L. Schmidt, H. Förstel, and K. Heinzinger (Amsterdam: Elsevier Scientific Publishing Company), 83–89.
Winkler, F. J., and Schmidt, H.-L. (1980). Einsatzmglichkeiten der13C-Isotopen-Massenspektrometrie in der Lebensmitteluntersuchung. Z. Lebensm Unters Forch 171, 85–94. doi:10.1007/bf01140746
WinklerSchmidt, F. J. H.-L., Wirth, E., Latzko, E., Lenhart, B., and Ziegler, H. (1983). Physiol. Vég. 21, 889–895.
Keywords: intramolecular isotope distribution, carbon isotope (δ13C), kinetic isotope effect (KIE), serine, methane
Citation: Gleixner G (2022) Insights Into the Known 13C Depletion of Methane—Contribution of the Kinetic Isotope Effects on the Serine Hydroxymethyltransferase Reaction. Front. Chem. 9:698067. doi: 10.3389/fchem.2021.698067
Received: 27 April 2021; Accepted: 24 November 2021;
Published: 05 January 2022.
Edited by:
Guigen Li, Texas Tech University, United StatesReviewed by:
Florenci Vicent González, University of Jaume I, SpainMarkus Greule, Heidelberg University, Germany
Copyright © 2022 Gleixner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerd Gleixner, Z2VyZC5nbGVpeG5lckBiZ2MtamVuYS5tcGcuZGU=