
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 12 August 2021
Sec. Organic Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.694870
 Kamal M. Dawood1*
Kamal M. Dawood1* Mohamed A. Raslan2
Mohamed A. Raslan2 Ashraf A. Abbas1
Ashraf A. Abbas1 Belal E. Mohamed2
Belal E. Mohamed2 Magda H. Abdellattif3
Magda H. Abdellattif3 Mohamed S. Nafie4
Mohamed S. Nafie4 Mohamed K. Hassan5,6
Mohamed K. Hassan5,6A series of bis-thiazoles 5a–g were synthesized from bis-thiosemicarbazone 3 with hydrazonoyl chlorides 4a–g. Reaction of 3 with two equivalents of α-halocarbonyl compounds 6–8, 10, and 12a–d afforded the corresponding bis-thiazolidines 9, 11, and 13a–d, respectively. Condensation of bis-thiazolidin-4-one 9 with different aromatic aldehydes furnished bis-thiazolidin-4-ones 14a–d. Compounds 5a–g, 9, and 13a,c,d were screened in vitro for their cytotoxic activities in a panel of cancer cell lines. Compounds 5a–c, 5f–g, and 9 exhibited remarkable cytotoxic activities, especially compound 5c with potent IC50 value 0.6 nM (against cervical cancer, Hela cell line) and compound 5f with high IC50 value 6 nM (against ovarian cancer, KF-28 cell line). Compound 5f–induced appreciated apoptotic cell death was measured as 82.76% associated with cell cycle arrest at the G1 phase. The apoptotic pathways activated in KF-28 cells treated with 5a, 5b, and 5f were further investigated. The upregulation of some pro-apoptotic genes, bax and puma, and the downregulation of some anti-apoptotic genes including the Bcl-2 gene were observed, indicating activation of the mitochondrial-dependent apoptosis. Together with the molecular docking studies of compounds 5a and 5b, our data revealed potential Pim-1 kinase inhibition through their high binding affinities indicated by inhibition of phosphorylated C-myc as a downstream target for Pim-1 kinase. Our study introduces a set of bis-thiazoles with potent anti-cancer activities, in vitro.
Thiazole-based heterocycles are highly versatile scaffolds, characterized by pronounced pharmacological importance and widely used by medicinal chemistry researchers for drug design purposes. For example, thiazoles demonstrated antimicrobial, antiviral, anti-diabetic, diuretic, anticonvulsant, antioxidant, anti-HIV, analgesic, anti-inflammatory, neuroprotective, and antitumor activities. These wide and potent activities were extensively published in several review articles (Siddiqui et al., 2011; Leoni et al., 2014; Ayati et al., 2015; Morigi et al., 2015; Rouf and Tanyeli, 2015; Chhabria et al., 2016; Das et al., 2016; Kumar et al., 2016; Agarwal et al., 2018; Scarim et al., 2019; de Oliveira et al., 2020). The anti-cancer activities of thiazole derivatives were also intensively reviewed (Ayati et al., 2019; Sharma et al., 2020). In addition, the thiazole moiety is incorporated in several natural products such as thiamine (vitamin B1), epothilone (anti-cancer drug), bacitracin, and penicillin (antibiotic drugs). Furthermore, the thiazole moiety is an essential component of a wide range of synthetic commercial drugs in the market, such as sulfathiazole (sulfa-drug), niridazole (schistosomicide drug), thiabendazole (antifungal drug), tiazofurin (anti-cancer drug), famotidine (stomach’s anti-ulcer drug), and meloxicam (non-steroidal anti-inflammatory drug), as depicted in Chart 1.
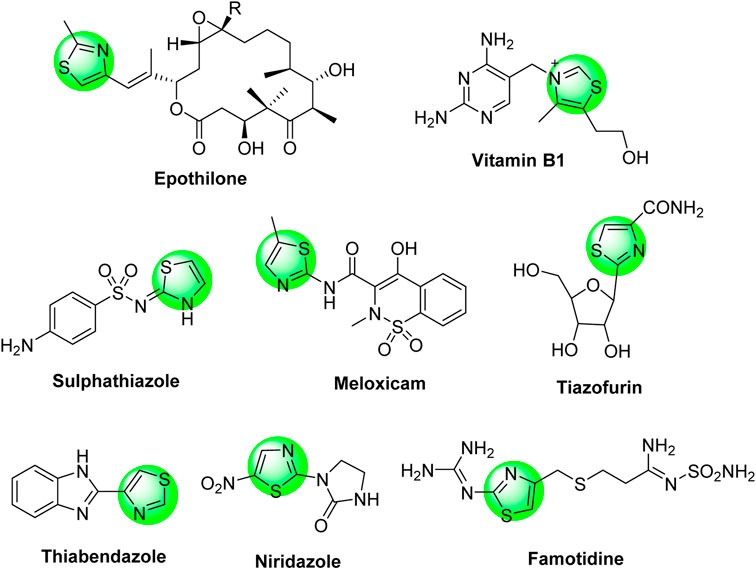
Chart 1. Some thiazole-based synthetic commercial drugs in the market.
On the contrary, 4-thiazolidinone derivatives were also highly active heterocyclic substrates and have featured potent therapeutic inhibitory activities as published in several reviews and research articles (Jain et al., 2012; Tripathi et al., 2014; Kaminsky et al., 2017; Manjal et al., 2017; de Siqueira et al., 2019; Nirwan et al., 2019). Their biological potencies are exemplified as antibacterial, antifungal, antitubercular, anti-cancer, anti-inflammatory, analgesic, anticonvulsant, antibiofilm, antidepressant, antiviral, anti-diabetic, and antiarrhythmic activities. 4-Thiazolidinones were also reported as ingredients of several synthetic drugs such as pioglitazone and epalrestat (anti-diabetic drugs), etozoline (diuretic drug), and darbufelone (anti-inflammatory drug) (Chart 2).
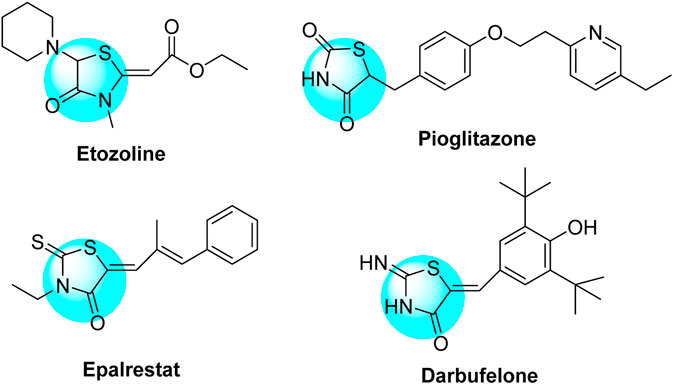
Chart 2. Some 4-thiazolidinone–based synthetic commercial drugs.
Multi-component reaction (MCR) is attracting the attention of chemists for the construction of diverse heterocyclic scaffolds. MCR is a powerful synthetic methodology in which three or more reactive substrates are mixed in one reaction flask to form a new product employing all substrates with minimized by-products and good yields (Paprocki et al., 2018; de Marigorta et al., 2019; Zhi et al., 2019; Kappe, 2020).
Our research interest is focused on the synthesis and biological evaluation of thiazole-based heterocycles (Abdel-Gawad et al., 2010; Dawood and EI-Deftar, 2010; Hegazi et al., 2010; Dawood et al., 2011; Dawood et al., 2013; Dawood and Abu-Deif, 2014; Dawood et al., 2015; Dawood et al., 2015; Dawood, 2019; Mahmoud et al., 2020). Therefore, keeping the above facts in mind and inspired by the reported extraordinary bioactivity behaviors of thiazole and 4-thiazolidinone derivatives, the objective of the current study is to design and synthesize new bis-thiazole structures for examining their potency against several of human cancer cell lines as well as investigating their effects as an inducer of apoptosis on the tested cancer cells.
1,4-Cyclohexanedione-bis-thiosemicarbazone 3 was reported once in the literature in 1982 as an analytical reagent for spectrophotometric estimation of copper, but no more applications of this substrate in organic synthesis were published so far (Ceba et al., 1982). Thus, when 1,4-cyclohexanedione-bis-thiosemicarbazone 3 was treated with a number of hydrazonoyl chlorides 4a–g in ethanol at refluxing temperature using triethylamine as a base, a single product was isolated, in each case, as detected by TLC analysis. Spectral data (1H and 13C NMR, MS and IR) as well as elemental analyses confirmed the assigned bis-thiazole structures 5a–g, Scheme 1. The IR spectra of structures 5a–g showed an NH-stretching absorption at around 3,230–3,300 cm−1, and their mass spectra revealed, in each case, a peak due to their molecular ions (M+). The 1H NMR spectrum of compound 5b, for example, revealed three singlet signals at δ 2.25, 2.51, and 10.41 (D2O-exchangeable) due to the thiazole-CH3, p-tolyl, and thiazole-NH protons, respectively, and two multiplets in the regions 2.73–2.74 and 2.89–2.90 assigned to cyclohexane protons plus two doublet peaks at 7.12 and 7.23 (J = 8.4 Hz) corresponding to the para-substituted aromatic protons. The 13C NMR spectrum of 5b exhibited 14 peaks corresponding to six aliphatic carbon atoms and eight aromatic carbon atoms.
Interestingly, a multi-component one-pot procedure was investigated for the construction of the products 5a–c as outlined in Scheme 1. Thus, when 1,4-cyclohexanedione 1 was treated with two equivalents of both thiosemicarbazide 2 and hydrazonoyl chlorides 4a–c (as representative examples) in a one-pot flask and then dissolved in ethanol and heated at reflux temperature using two equivalents of triethylamine, to give a single product in each case, that was in complete accordance with compounds 5a–c in almost similar yields as the stepwise procedure discussed above.

Scheme 1. Synthesis of 1,4-bis((5-(arylazo)thiazol-2-ylidene)hydrazono)cyclohexane 5.
In the same way, treatment of bis-thiosemicarbazone 3 with ethyl chloroacetate (6) in 1:2 M ratios in refluxing ethanol using triethylamine as a base led to production of a compound that was established as the bis-4-thiazolidinone derivative (9) in 73% yield (Scheme 2) based on its spectral analyses. The IR spectrum of 9 showed two peaks at 3320 and 1712 cm−1 assignable for NH and C=O functions, respectively. Moreover, a peak at m/z 338 due to the molecular ion (M+) appeared in its mass spectrum. The 1H NMR spectrum of compound 9 revealed two multiplet peaks in the regions δ 2.53–2.57 and 2.68–2.73 assignable to cyclohexane protons and a singlet peak at δ 3.81 for thiazolidinone-CH2 protons besides a broad peak at δ 11.72 (D2O-exchangeable) due to thiazolidinone-NH protons. 13C NMR of compound 9 exhibited five peaks at δ 25.35, 32.23, 32.72, 166, and 173.93. Two alternative methods for synthesis of the bis-thiazolidinone derivative 9 were conducted as shown in Scheme 2. Thus, treatment of bis-thiosemicarbazone 3 with chloroacetic acid (7) in ethanol solvent at reflux temperature employing sodium acetate as a base or with ethyl bromoacetate (8) in dimethylformamide and triethylamine as a base yielded the same product in 62 and 68% yields, respectively, which was identical in all aspects to compound 9 obtained above. Similarly, treatment of compound 3 with two equivalents of α-chloroacetylacetone (10), under a typical reaction procedure above, furnished the 1,4-diylidene-bis-thiazolidine derivative 11 in 71% yield.
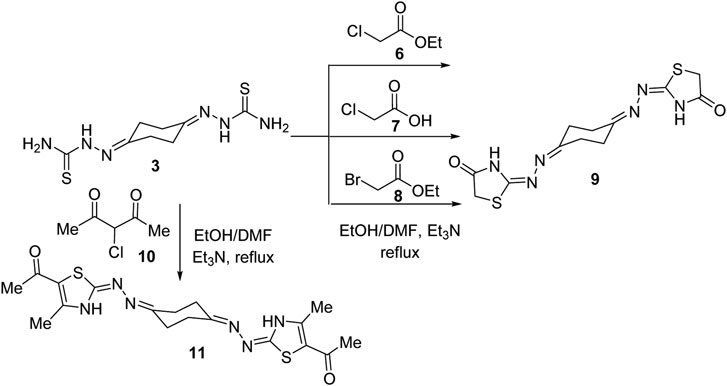
Scheme 2. Synthesis of cyclohexane-1,4-diylidene-bis-thiazolidine derivatives 9 and 11.
The scope of our strategy was extended to encompass synthesis of the new 1,4-bis((4-arylthiazol-2-ylidene)hydrazono)cyclohexane derivative 13. Thus, when bis-thiosemicarbazone 3 was allowed to react with 2-(bromoacetyl)benzothiazole (12a) in a 1:2 M ratio in ethanol at reflux conditions using triethylamine as a base, it furnished a single product that was established as 1,4-bis((4-(2-benzothiazolyl)thiazol-2-ylidene)hydrazono)-cyclohexane (13a) in 72% yield (Scheme 3) on the basis of its elemental analyses and spectroscopic data. The same product was obtained alternatively, in 77% yield, via a one-pot three-component procedure by adding the three substrates: 1,4-cyclohexanedione (1), thiosemicarbazide (2), and 2-(bromoacetyl)benzothiazole (12a) in 1:2:2 M ratios in one flask, dissolved in ethanol and then heated at reflux temperature and catalyzed by triethylamine. The 1H-NMR spectrum of compound 13a exhibited three multiplet peaks in the regions δ 2.58–2.71, 7.43–7.53, and 8.01–8.09 corresponding to the cyclohexane and benzothiazole protons, respectively, in addition to two singlets at δ 7.73 and 11.03 assignable to thiazole-5-CH and thiazole-3-NH protons, respectively. 13C NMR revealed four aliphatic carbon atoms at δ 23.94, 26.18, 29.29, and 31.50 and nine aromatic carbon atoms at 109.32, 122.34, 122.54, 125.15, 126.52, 134.54, 144.65, 153.59, and 154.11 in addition to 162.79 (C=N) and 170.37 (C=O). Similarly, reaction of bis-thiosemicarbazone 3 with three extra examples of α-bromoketones, namely, 2-(bromoacetyl)benzofuran (12b), 3-(bromoacetyl)coumarin-2-one (12c), and 4-chlorophenacyl bromide (12d), under a typical reaction procedure described above, gave the corresponding 1,4-bis((4-arylthiazol-2-ylidene)hydrazono)cyclohexane derivatives 13b–d in high yields as shown in Scheme 3.
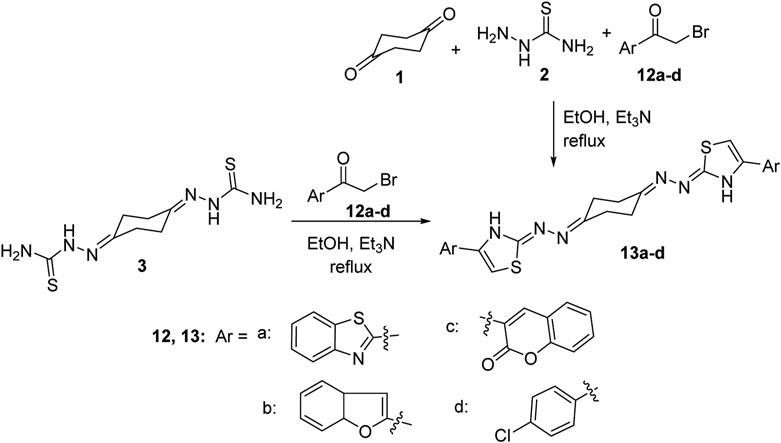
Scheme 3. Synthesis of 1,4-bis((4-arylthiazol-2-ylidene)hydrazono)cyclohexanes 13a–d.
Finally, synthesis of the new bis(thiazolidin-4-one) derivatives 14a–d was also carried out. Thus, condensation reaction of the bis-thiazolidin-4-one derivative 9 with a number of aromatic aldehydes in ethanol heated at reflux conditions, using piperidine as a base, afforded the 1,4-bis((5-arylidene)-4-oxo-thiazolidin-2-ylidene)hydrazono-cyclohexane derivatives 14a–d (Scheme 4). Structures of the obtained products were elucidated from their elemental and spectral data. The 1H NMR spectra of 14a–d were free of the thiazolidinone-5-CH2 protons of 9 at δ 3.81 and exhibited a singlet peak around δ 7.7 due to the methine = CH proton. The stereochemical Z-configuration of the exocyclic C=CH bonds in 14a–d was assigned based on their 1H NMR spectral data where the methine C=CH proton near δ 7.7 is closer to the analogously reported (Z)-5-arylidene-thiazolidin-4-one derivatives (Momose et al., 1991; Paprocki et al., 2018) instead of the E-configuration of the methine C=CH proton that resonates at δ < 7.5 (Karigar et al., 2011).
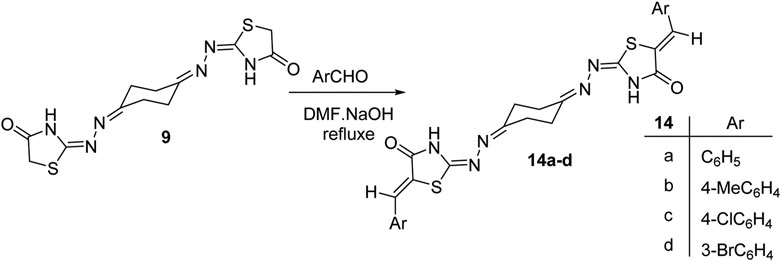
Scheme 4. Synthesis of 1,4-bis((5-arylidene-thiazolidin-2-ylidene)hydrazono)cyclohexanes 14a–d.
The synthesized derivatives were screened for their cytotoxic activities against five cancer cell lines (two breast cancer cell lines: MCF-7 and MDA-MB-231, two ovarian cancer cell lines: A2780 and KF-28, and one cervical cancer cell line: Hela). This study was performed using serial concentrations of 25, 50, 100, 150, and 200 µM of each compound using the MTT assay. Percentages of cell viability of the tested compounds with serial concentration against the tested panel of cell lines are depicted in Figure 1, in which there is a decrease in the cell viability with increasing concentrations in a dose–response pattern. Additionally, as seen in Table 1 and Figure 1, the MTT assay results for ovarian cancer cell lines for compound 5f exhibited a pronounced cytotoxic effect with an IC50 value of 0.0061 µM. In contrast, compounds 5a and 5b were notably cytotoxic with IC50 values of 0.718 and 3.374 µM, respectively, against the KF-28 cell line when treated for 72 h. Additionally, compounds 5f and 5g showed a remarkable cytotoxic activity with IC50 values 2.34 and 7.45 µM, respectively, against the A2780 cell line, when treated for 72 h. Regarding breast cancer cells, compound 5e showed a potent cytotoxic effect against MCF-7 breast cancer cells with an IC50 value of 0.6648 µM (72 h), while compound 5a displayed a notable cytotoxic effect with an IC50 value of 1.51 µM against the MDA-MB-231 cells (treated for 72 h). Finally, the results from cervical cancer, Hela, cells showed a pronounced response when treated with compound 5c with the most significant inhibition potency having IC50 value 0.00065 µM (72 h treatment). Therefore, compounds 5a, 5b, and 5f were thought worthwhile to further investigate their effects as either cytotoxic or antiproliferative effects and whether any of them is an inducer of apoptotic cell death in the representative cancer cell lines KF-28 and A2780.
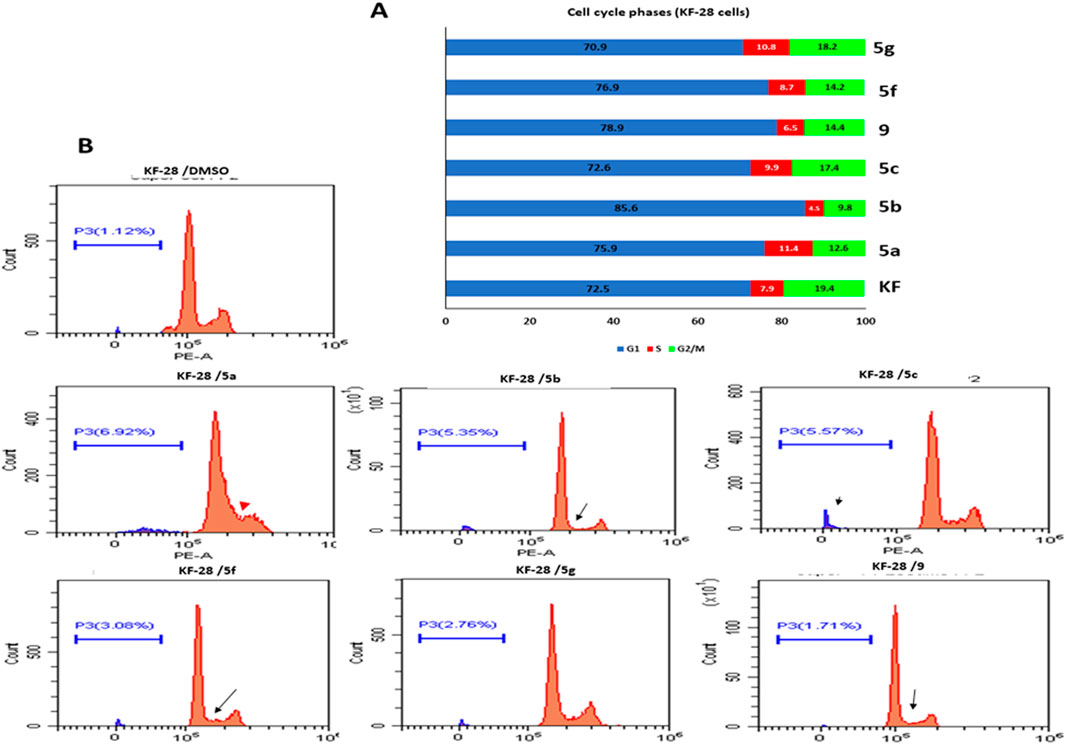
FIGURE 1. Effect of selected compounds on cell cycle phases in KF-28 cell lines. (A) Representative cytographs show the effect of vehicle and compounds 5a–c, 5f, 5g, and 9 on the different cell cycle phases of KF-28 cells when treated with each compound for two days. (B) Collected results show the effect of each compound on different populations at each cell cycle phase.
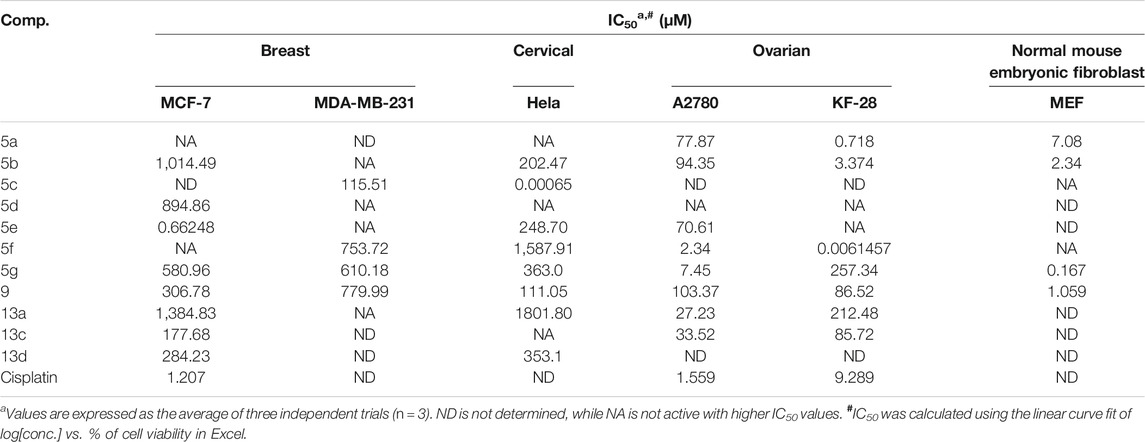
TABLE 1. Summarized IC50 values (µM) of the tested derivatives against MCF-7, Hela, A2780, KF-28, and MDA-MB-231 cancer cell lines.
Our results agree with those of several previous studies (Turan-Zitouni et al., 2016; Kumar and Umadevi, 2018; Borcea et al., 2021), which investigated the cytotoxic activities of analogous bis-thiazole derivatives against various cancer cell lines with promising IC50 values. However, the MTT technique used just indicates the viability under different treatments; therefore, this encouraged us to further investigate the cellular and molecular mechanisms behind such cytotoxicity.
Studying the cell cycle phases by measuring DNA content is a crucial test that helps in evaluating the impact of a given drug(s) on dividing cells and may reflect the mechanism through which this drug(s) affects those cells. Hence, we used the ovarian cancer cells (KF-28) as a representative cellular model to study the effect of the synthesized compounds on the cell cycle. Compounds 5a, 5b, 5c, 5f, 5g, and 9 were added to similar numbers of cells at 200 µM for 48 h, and then, the different cell cycle phases were assessed by flow cytometric analysis.
DNA content analysis of KF-28 cells treated with compounds 5a, 5b, 5c, 5f, 5g, and 9 showed that compound 5a significantly accumulated the cells at the S phase (red arrowhead; 11.4%), while other compounds such as 5b and 9 showed appreciated reduction in the S phase population of the cells tested measured as 4.5 and 6.5%, respectively (indicated by black arrows; Figure 1). Moreover, treating cells with 5a, 5b, 5f, and 9 compounds was associated with relative accumulation at the G1 phase indicated by the elevated percentage of G1 populations measured as 75.9, 85.6, 76.9, and 78%, respectively, compared with 72% at the G1 phase in DMSO-treated control cells. Subsequently, this effect was associated with observable relative reduction in the percentage of G2/M phase populations. Similar results were observed in the A2780 cell line when treated with the same compounds 5a, 5b, 5c, 5f, 5g, and 9 for 48 h even at lower concentrations (100 µM) (Supplementary Figure 2).
Cellular toxicity by specific molecules causing cellular arrest usually ends with cellular death in cancer cells. For instance, taxane family drugs target the microtubules causing arrest at the G2/M phase which ends up with apoptotic cell death (Hassan et al., 2011), while cisplatin causes DNA crosslinking and then arrests dividing cancer cells at G1 to end with apoptotic cell death (Galluzzi et al., 2012). Here, our data revealed that the cytotoxicity indicated by MTT might be due to arresting the cells at a certain cell cycle phase like G1 under 5a, 5b, and 5f compounds. Therefore, to further investigate whether this arrest causes cell death or just reduces cellular proliferation, we moved to studying the possible apoptotic signaling by bis-thiazole derivatives used in this study.
Although the DNA content data showed sub-G0 populations indicating debris as dead cells under treatment with different compounds, such an experiment cannot determine whether this cell death is apoptosis or necrosis. Notably, annexin V/PI staining is an important technique to discriminate the apoptotic cell death from the necrotic one. Therefore, we decided to use such technique to investigate if the cytotoxic effect of the compounds indicated by MTT and the cell cycle experiments is apoptotic cell death or not. For that purpose, two cell lines, KF-28 and A2780, were stained by annexin V/PI after treatment with different compounds vs. DMSO for two days in parallel with cisplatin as a reference drug. Results from annexin staining analysis show appreciated annexin V–stained populations when cells are treated with compounds 5a–c, 5f, and 9. As shown in Figure 2A, the tested compounds, especially 5a, 5b, and 5f, significantly stimulated total apoptotic cell death valued as 38.21, 18.77, and 40.74%, respectively, compared to 0.45% for DMSO-treated controls, in parallel with cisplatin which induced total apoptosis of 25% in KF-28 cells. Similar results were obtained from the other cell line, A2780, treated with the same compounds (Figure 2B).
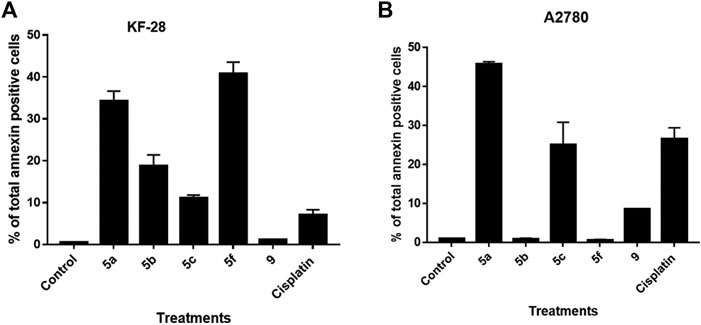
FIGURE 2. Some compounds induce apoptosis in KF-28 and A2780 cells. Average percentage of annexin-positive cells from KF-28 (A) and A2780 (B) cells treated with DMSO and different compounds, 5a, 5b, 5c, 5f, and 9, in parallel with the standard drug, cisplatin (10 μM), treatment for 48 h. Values are presented as mean ± SE.
Our results of the apoptosis induction in the treated KF-28 cells were in accordance with previous research data (Bachmann et al., 2004; Bachmann et al., 2006) which proved the apoptotic activity of the bis-thiazole derivatives in some tested cell lines and confirmed apoptosis as the mechanism of action. Moreover, our data revealed that compound 9 did not induce appreciated apoptosis. Together, this observation with the MTT results indicates that compound 9 is affecting the cellular proliferation rather than killing the cells by apoptosis or necrosis.
Next, we investigated the pathway through which apoptosis is induced by compounds 5a, 5b, and 5f in KF-28 cells. For that purpose, after treating cells with each compound, RNA was extracted, cDNA was synthesized, and RT-PCR was performed to quantify the mRNA expression of pro-apoptotic genes (p53, bax, puma), caspases 3, 8 9, anti-apoptotic gene (Bcl-2), and Pim-1 kinase genes in KF-28 cells.
As shown in Figure 3, treatment of KF-28 cells with compound 5a resulted in a remarkable activation of p53 mRNA relative expression (≈4.07-fold) with a concomitant activation of the puma and bax mRNA levels with a maximum increase of ≈5.08-fold and 5.2-fold, respectively. Compound 5a was able to significantly increase the mRNA levels of caspase 3, 8, 9 genes with a maximum increase of ≈6.2-fold, 2.9-fold, and 5.6-fold, respectively, while it exceptionally inhibited the anti-apoptotic Bcl-2 expression (maximum decrease of ≈0.27-fold) and the Pim-1 kinase expression (maximum decrease of ≈0.31-fold). Additionally, treatment of KF-28 cells with compound 5b significantly activated the level of p53 expression (≈3.41-fold) with consequent activation of the puma and bax expression levels with a maximum increase of ≈4.14-fold and 3.33-fold, respectively, as shown in Figure 3. The bis-thiazole derivative 5a was also able to significantly increase the mRNA levels of caspase 3, 8, 9 genes with a maximum increase of ≈4.2-fold, 2.9-fold, and 4.7-fold, respectively. At the same time, it significantly inhibited the anti-apoptotic Bcl-2 expression (maximum decrease of ≈0.5-fold) and the Pim-1 kinase expression (maximum decrease of ≈0.48-fold). Finally, in the same Figure 3, treatment of KF-28 cells with compound 5f significantly activated the level of p53 gene (≈3.81-fold) with concomitant activation of the puma and bax expression levels with a maximum increase of ≈3.14-fold and 2.33-fold, respectively. Compound 5f was able to effectively increase the mRNA levels of caspase 3, 8, 9 gene expression with a maximum increase of ≈6.9-fold, 5.5-fold, and 6.0-fold, respectively, while it notably inhibited the anti-apoptotic Bcl-2 gene (maximum decrease of ≈0.76-fold) and the Pim-1 kinase mRNA expression (maximum decrease of ≈0.86-fold).
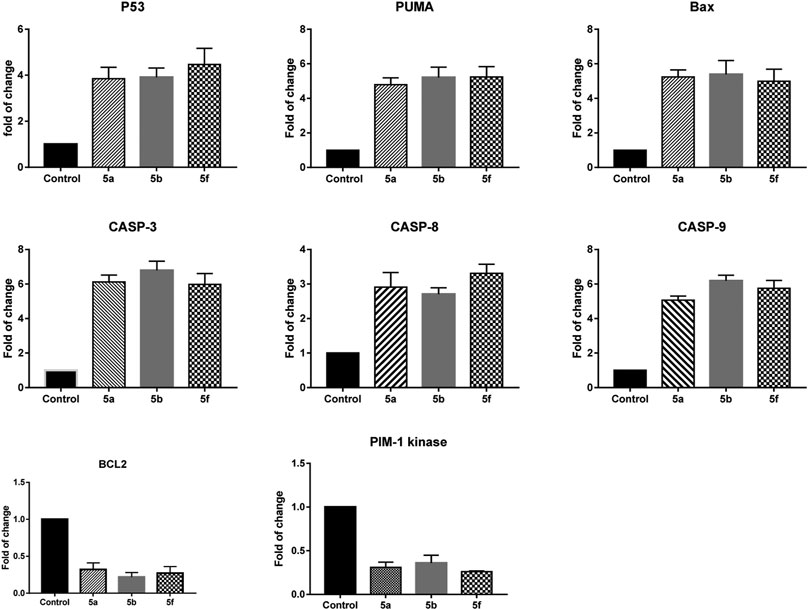
FIGURE 3. Quantitative RT-PCR result analysis of the apoptosis-related genes: p53, bax, puma, caspases 3, 8, and 9, Bcl-2, and Pim-1 kinase, respectively, in KF-28 cells treated with some selected compounds 5a, 5b, and 5f for 48 h of incubation.
Based on the fact that cancer is known as one of the scenarios where too little apoptosis occurs (Wong, 2011), testing the ability of novel drugs for their anti-cancer potency depends mainly on their ability to induce apoptosis. Our data revealed that some of the tested bis-thiazole derivatives used here (5a, 5b, and 5f) were able to induce apoptosis in the representative cancer cell lines. This apoptosis is mainly a mitochondrial-dependent one as indicated by the gene expression experiment. These compounds were able to enhance the pro-apoptotic genes and inhibit the anti-apoptotic ones, transcriptionally.
Pim-1 kinase has been observed to be highly expressed in some solid tumors (Dhanasekaran et al., 2001; Li et al., 2006). Therefore, targeting such molecules may associate with tumor growth inhibition. Our data revealed that the investigated compounds, here, were able to, transcriptionally, inhibit Pim-1. Together, our data are in accordance with those from other research teams (Xia et al., 2009; Zhao et al., 2016; Bataille et al., 2017), who reported that thiazole compounds can target Pim-1 kinase.
There is a lot of research highlighting molecular docking simulations for the anti-cancer activity of thiazole-based derivatives through Pim-1 kinase (Xia et al., 2009; Zhao et al., 2016; Bataille et al., 2017). Therefore, the binding affinities of the tested compounds were screened toward Pim-1 kinase proteins in terms of binding energies and ligand–receptor interactions. Thus, molecular docking simulation of the tested bis-thiazole derivatives toward Pim-1 kinase (PDB = 2OBJ) was investigated at the gene expression level using the RT-PCR in the treated FK-28 cells with compounds 5a, 5b, and 5f as mentioned above (Figure 3).
As seen in Table 2 and Figure 4, compounds 5a and 5b were docked to form two hydrogen bonds (HBs) with the interactive key amino acid residue (Lys 67) with binding energies −11.46 and −12.66 kcal/mol, respectively, compared to the standard Pim-1 kinase inhibitor (AZD-1208), which formed only one HB with the same amino acid Lys 67. All HBs were formed through their nitrogen as HBAs with Lys 67, which is the important amino acid for the biological activity of Pim-1 kinase protein. Other interactions of the two compounds 5a and 5b with the receptor-binding site are arene–arene interactions or HBs with amino acid residues other than Lys 67 which are not important for biological activity but are important for fitting the ligands inside the receptor pocket. The docking studies indicated that both compounds 5a and 5b showed promising binding activity as Pim-1 kinase inhibitors following the RT-PCR results in the treated KF-28 cells in which gene expression levels of Pim-1 kinase were inhibited. This may be the proposed anti-breast cancer mode of action.
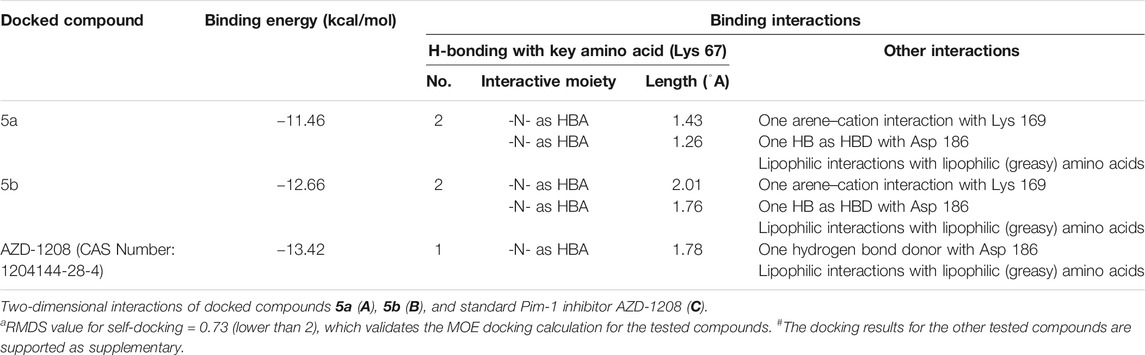
TABLE 2. Ligand–receptor interactions with the binding energy of the docked derivatives# inside Pim-1 kinase (PDB = 2OBJ) using MOE 2015a.
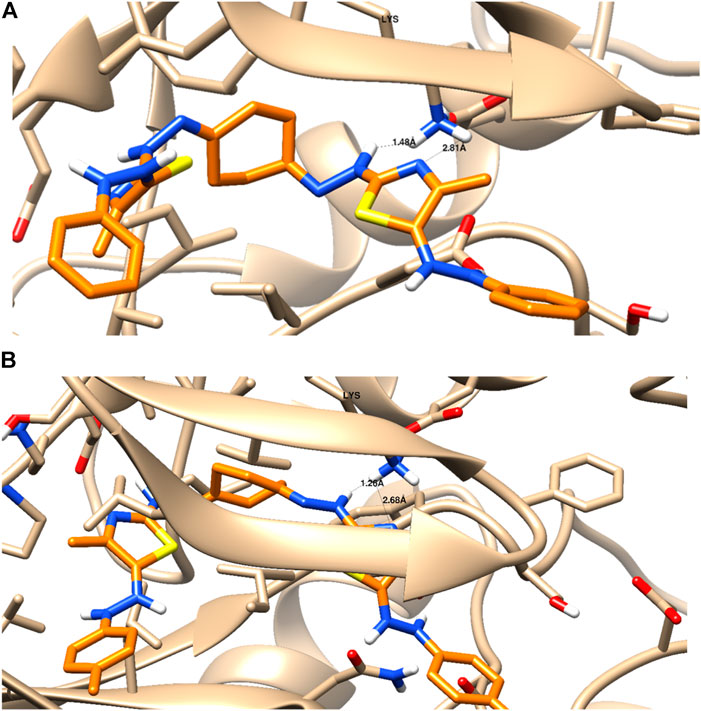
FIGURE 4. Binding disposition and interaction analysis of the docked compounds 5a (A) and 5b (B), inside the Pim-1 kinase protein.
The three active compounds 5a, 5b, and 5f against KF-28 with IC50 values 0.718, 3.37, and 0.006 μM, respectively, were highlighted with the common pharmacophoric regions including the thiazole moiety, aromatic moiety, and hydrogen bond donor/acceptor. Interestingly, compound 5f was found to be the most cytotoxic one with IC50 value 6 nM. This may be greatly affected by the presence of two withdrawing groups (Cl), as shown in Figure 5. The highlighted pharmacophoric regions to which the biological activity may be incorporated are the thiazole moiety, aromatic (phenyl) moiety, HB donor/acceptor, and electron-donating/withdrawing groups.
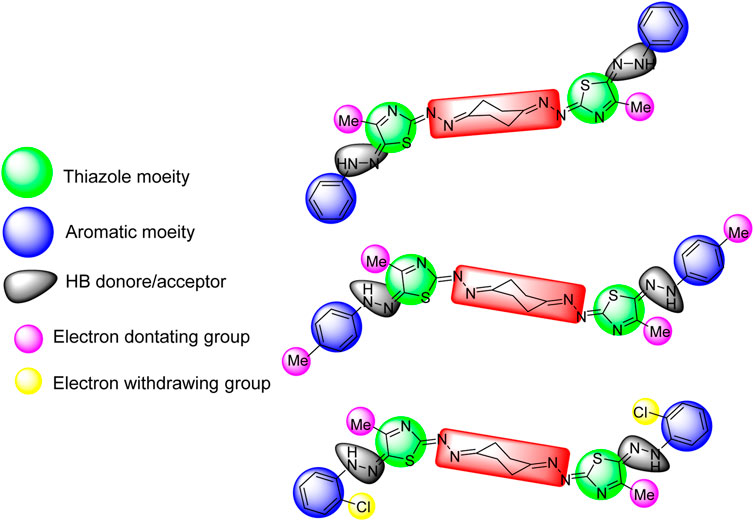
FIGURE 5. Highlighted pharmacophoric regions for the active compounds 5a, 5b, and 5f, respectively (top to down).
Pim-1 has been classified as a weak oncogene. However, a very strong synergism with regard to tumorigenicity occurs between Pim-1 and C-myc. This synergism is observed with bitransgenic mice overexpressing C-myc and Pim-1 in lymphoid tissue (Möröy et al., 1991). To further validate the simulation and docking data which introduced Pim-1 kinase as a potential target molecule for some of the tested compounds, we decided to check the effect of treatment with the selected compounds on the C-myc protein expression as a downstream target molecule known to be phosphorylated and stabilized by Pim-1 as a kinase (Zhang et al., 2008). Western blotting data show that treating KF-28 cells with the compounds 5a–c, 5f, and 9 for two days reflected observable downregulation of the overall C-myc protein under such treatment with 5a, 5b, 5c, and 5f (Figure 6). Taking into consideration the fact that Pim-1 phosphorylates C-myc and controls its stability (Zhang et al., 2008) may conclude that the reduction of C-myc expression under the indicated compounds is due to the inhibition of Pim-1 activity (Figure 3). Therefore, the speculated inhibition of Pim-1 is supported by the above-mentioned qPCR data (Figure 3) and further supported by C-myc downregulation. Our results agree with the reported one which states that some thiazole derivatives are potent inhibitors against protein kinases including Pim-1 kinase (Mohareb et al., 2019).
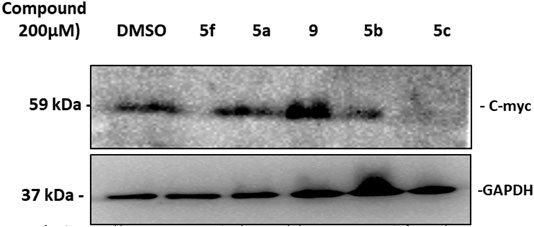
FIGURE 6. Western blot of C-myc expression in KF-28 cells treated either with DMSO or compounds 5a–c, 5f, and 9 for two days. The cell lysates were developed and fractionated by 15% SDS-PAGE, blotted, and probed with anti-C-myc Ab to show its relative expression. Appreciated downregulation of C-myc expression under treatment with 5f, 5b, and 5c is shown. The lower band is GAPDH which was used as a loading control.
All melting points are uncorrected. Compounds prepared by different procedures were characterized by mixed melting points, TLC, and IR. IR spectra (KBr) were recorded on a Fourier transform infrared spectrophotometer (IRAffinity-1S from Shimadzu Corporation). NMR spectra were measured with a Varian Gemini 300 spectrometer (300 MHz 1H NMR), and chemical shifts are given in ppm from TMS. 13C NMR spectra were recorded with a Varian Mercury 300 spectrometer (300 MHz 1H NMR, 75 MHz 13C NMR). The 1H NMR samples were measured in 5 mm NMR tubes with a concentration of about 20 mg of the product in 0.6 ml of solvent (DMSO-d6) and about 75–100 mg of the product in about 0.6 ml of solvent for 13C NMR. Mass spectra were recorded on DI Analysis Shimadzu QP-2010 Plus. Elemental analyses were carried out at the Microanalytical Center, Cairo University. 1,4-Cyclohexanedione, ethyl chloroacetate, chloroacetic acid, ethyl bromoacetate, α-chloroacetylacetone, and thiosemicarbazide were used as purchased from Aldrich. The starting materials 4a–g were prepared as reported in the literature (Eweiss and Osman, 1980; Farag and Algarib, 1988).
To a solution of 1,4-cyclohexanedione (1) (0.11 g, 1 mmol) in absolute ethanol (20 ml), thiosemicarbazide (2) (0.182 g, 2 mmol) was added, and then the reaction mixture was heated under reflux for 10 min with stirring. The reaction mixture was then left to cool to room temperature, and the solid product was collected by filtration, washed with ethanol, dried, and finally recrystallized from ethanol/DMF to give compound 3 as colorless crystals, 0.18 g (65%), mp. 213–214°C [Lit. mp. 213–214°C (Ceba et al., 1982)]; IR (KBr) υ 3,255, 3,201, 3,185 (NH, NH2), 1,597 (C=N) cm−1; 1H NMR (DMSO-d6) δ 2.42–2.50 (m, 4H, cyclohexane-H’s), 2.54–2.74 (m, 4H, cyclohexane-H’s), 7.59 (br. s, 2H, NH2), 8.05 (br. s, 2H, NH2), 9.9–10.11 (br. s, 2H, NH).
To a mixture of 1,4-cyclohexane-bis-thiosemicarbazone (3) (0.258 g, 1 mmol) and the appropriate hydrazonoyl chlorides 4a–g (2 mmol) in absolute ethanol (10 ml), triethylamine (0.1 ml) was added, and the resulting mixture was refluxed for 6–8 h and then left to cool to room temperature. The solid product formed was collected by filtration, washed with ethanol, dried, and finally recrystallized from the ethanol/DMF mixture to give the corresponding bis-thiazole derivatives 5a–g.
To a mixture of 1,4-cyclohexadione (1) (0.11 g, 1 mmol), thiosemicarbazide (2) (0.182 g, 2 mmol), and the appropriate hydrazonoyl chlorides 4a–c (2 mmol) in absolute ethanol (10 ml), triethylamine (0.1 ml) was added, and then the reaction mixture was heated under reflux for 8 h. The reaction mixture was then left to cool to room temperature for complete precipitation. The solid product was collected by filtration, washed with ethanol, dried, and finally recrystallized from the ethanol/DMF mixture to afford the bis-thiazole derivatives 5a, 5b, and 5c in 77, 76, and 60% yields, respectively.
1,4-Bis((5-phenylazo-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (5a): Brown crystals, yield (0.45 g, 83%), mp. 211–212 °C; IR (KBr) ν 3,229 (NH), 1,600 (C=N) cm−1; MS m/z (%): 77 (100%), 93 (71.2%), 108 (31%), 218 (40%), 542 (M+, 1.4%); 1H NMR (DMSO-d6) δ 2.56 (s, 6H, 2CH3), 2.73–2.75 (m, 4H, cyclohexane-H’s), 2.89–2.91 (m, 4H, cyclohexane-H’s), 6.9–7.33 (m, 10H, ArH), 10.44 (s, 2H, 2NH). Anal. calcd. for C26H26N10S2 (542.68): C, 57.54; H, 4.83; N, 25.81%. Found: C, 57.24; H, 4.64; N, 25.59%.
1,4-Bis((5-(4-tolylazo)-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (5b): Brown crystals, yield (0.25 g, 87%), mp. 221–222 °C; IR (KBr) ν 3,297 (NH), 1,615 (C=N) cm−1; MS m/z (%): 91 (100%), 106 (53%), 232 (28%), 570 (M+, 1%); 1H NMR (DMSO-d6) δ 2.25 (s, 6H, 2CH3-Ar), 2.51 (s, 6H, 2CH3-thiazole), 2.73–2.74 (m, 4H, cyclohexane-H’s), 2.89–2.90 (m, 4H, cyclohexane-H’s), 7.12 (d, 4H, J = 8.4 Hz, ArH), 7.23 (d, 4H, J = 8.1 Hz, ArH), 10.41 (s, 2H, 2NH); 13C NMR (DMSO-d6) δ 16.53, 20.49, 25.96, 30.88, 32.25, 35.89, 114.25, 120.70, 129.83, 131.19, 141.25, 162.42, 171.01, 177.69. Anal. calcd. for C28H30N10S2 (570.74): C, 58.93; H, 5.30; N, 24.54%. Found: C, 58.72; H, 5.10; N, 24.34%.
1,4-Bis((5-(4-chlorophenylazo)-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (5c): Brown crystals, yield (0.33 g, 54%), mp. 269–270 °C; IR (KBr) ν 3,250 (NH), 1,620 (C=N) cm−1; MS m/z (%): 92 (82%), 111 (100%), 252 (42%), 611 (M+, 2%); 1H NMR (DMSO-d6) δ 2.5 (s, 6H, 2CH3), 2.73–2.75 (m, 4H, cyclohexane-H’s), 2.9 (m, 4H, cyclohexane-H’s), 7.30–7.37 (m, 8H, ArH), 10.50 (s, 2H, 2NH). Anal. calcd. for C26H24Cl2N10S2 (611.57): C, 51.06; H, 3.96; N, 22.90%. Found: C, 50.89; H, 3.76; N, 22.65%.
1,4-Bis((5-(4-nitrophenylazo)-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (5d): Green crystals, yield (0.42 g, 66%), mp. >300 °C; IR (KBr) ν 3,271 (NH), 1,627 (C=N) cm−1; MS m/z (%): 55 (100%), 71 (64%), 263 (19.4%), 632 (M+, 5%); 1H NMR (DMSO-d6) δ 2.59 (s, 6H, 2CH3), 2.77 (s, 4H, cyclohexane-H’s), 2.92 (s, 4H, cyclohexane-H’s), 7.44 (d, 4H, J = 7.8 Hz, ArH), 8.19 (d, 4H, J = 8.1 Hz, ArH), 10.98 (s, 2H, 2NH). Anal. calcd. for C26H24N12O4S2 (632.68): C, 49.36; H, 3.82; N, 26.57%. Found: C, 49.06; H, 3.62; N, 26.36%.
1,4-Bis((5-(2-tolylazo)-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (5e): Brown crystals, yield (0.45 g, 78%), mp. 224–225 °C; IR (KBr) ν 3,200 (NH), 1,624 (C=N) cm−1; MS m/z (%): 65 (33%), 77 (19.4%), 91 (100%), 106 (42%), 232 (4%), 339 (4%), 452 (2%), 542 (3%), 570 (M+, 1%); 1H NMR (DMSO-d6) δ 2.34 (s, 6H, 2CH3-Ar), 2.55 (s, 6H, 2CH3-thiazole), 2.75 (s, 4H, cyclohexane-H’s), 2.9 (m, 4H, cyclohexane-H’s), 6.94–7.43 (m, 8H, ArH), 9.39 (s, 2H, 2NH). Anal. calcd. for C28H30N10S2 (570.74): C, 58.93; H, 5.30; N, 24.54%. Found: C, 58.63; H, 5.27; N, 24.34%.
1,4-Bis((5-(2-chlorophenylazo)-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (5f): Brown crystals, yield (0.3 g, 75%), mp. >300 °C; IR (KBr) ν 3,270 (NH), 1,620 (C=N) cm−1; MS m/z (%): 92 (54%), 127 (100%), 217 (12%), 359 (1.3%), 611 (M+, 1.5%); 1H NMR (DMSO-d6) δ 2.58 (s, 6H, 2CH3), 2.73–2.77 (m, 4H, cyclohexane-H’s), 2.91 (m, 4H, cyclohexane-H’s), 7.06 (t, 2H, J = 6.6 Hz, ArH), 7.37 (t, 2H, J = 7.51 Hz, ArH), 7.45 (d, 2H, J = 7.2 Hz, ArH), 7.59 (d, 2H, J = 8.1 Hz, ArH), 9.33 (s, 2H, 2NH). Anal. calcd. for C26H24Cl2N10S2 (611.57): C, 51.06; H, 3.96; N, 22.90%. Found: C, 50.98; H, 3.76; N, 22.70%.
1,4-Bis((5-(2-nitrophenylazo)-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (5g): Brown crystals, yield (0.20 g, 51%), mp. 220–221 °C; IR (KBr) ν 3,379 (NH), 1,651 (C=N) (cm−1); MS m/z (%): 71 (100%), 92 (57%), 230 (19.4%), 263 (49%), 372 (3%), 539 (5%), 630 (M+-2, 3%); 1H NMR (DMSO-d6) δ 2.58 (s, 6H, 2CH3), 2.73–2.76 (m, 4H, cyclohexane-H’s), 2.88–2.91 (m, 4H, cyclohexane-H’s), 7.55–8.05 (m, 8H, ArH), 10.73 (s, 2H, 2NH). Anal. calcd. for C26H24N12O4S2 (632.68): C, 49.36; H, 3.82; N, 26.57%. Found: C, 49.16; H, 3.63; N, 26.38%.
(A) To a mixture of 1,4-cyclohexane-bis-thiosemicarbazone (3) (0.26 g, 1 mmol) and 2 mmol of the appropriate α-halocarbonyl derivatives [ethyl chloroacetate (6), chloroacetic acid (7), and ethyl bromoacetate (8)] in a 1:1 DMF/ethanol mixture (10 ml), triethylamine (0.1 ml) was added, and the resulting mixture was heated at reflux temperature for 8 h and then left to cool to room temperature. The solid product obtained was collected by filtration, washed with ethanol, dried, and finally recrystallized from the ethanol/DMF mixture to give compound 9. Yields of compound 9 obtained from α-chlorocarbonyls 6, 7, and 8 were (0.25 g, 73%), (0.21 g, 62%), and (0.23 g, 68%), respectively.
(B) Reaction of 1,4-cyclohexane-bis-thiosemicarbazone (3) (0.26 g, 1 mmol) and α-chloroacetylacetone (10) (2 mmol) was carried out under typical reaction conditions as in procedure (A) to give compound 11 in 71% yield (0.15 g).
1,4-Bis((4-oxo-thiazolidin-2-ylidene)hydrazono)cyclohexane (9): Yellow crystals, yield (0.25 g, 73%), mp. 279–280 °C; IR (KBr) ν 3,320 (NH), 1712 (C=O), 1,638 (C=N) cm−1; MS m/z (%): 87 (40%), 107 (100%), 223 (11%), 338 (M+, 2%); 1H NMR (DMSO-d6) δ 2.53–2.57 (m, 4H, cyclohexane-H’s), 2.68–2.73 (m, 4H, cyclohexane-H’s), 3.81 (s, 4H, 2CH2CO), 11.72 (s, 2H, 2NH). 13C NMR (DMSO-d6) δ 25.35, 32.23, 32.72, 166, 173.93. Anal. calcd. for C12H14N6O2S2 (338.40): C, 42.59; H, 4.17; N, 24.83%. Found: C, 42.30; H, 4.42; N, 24.63%.
1,4-Bis((5-acetyl-4-methylthiazol-2-ylidene)hydrazono)cyclohexane (11): Yellow crystals, yield (0.15 g, 71%), mp. 280–281 °C; IR (KBr) ν 3,194 (NH), 1,695 (C=O), 1,597 (C=N) cm−1; MS m/z (%): 107 (83%), 113 (77%), 141 (100%), 156 (49%), 233 (24%), 263 (90%), 418 (M+, 8%); 1H NMR (DMSO-d6) δ 3.36 (s, 6H, 2CH3), 4.47 (s, 6H, CH3CO), 2.55–2.65 (m, 8H, cyclohexane-H’s), 11.4 (br. s, 2H, NH). Anal. calcd. for C18H22N6O2S2 (418.53): C, 51.66; H, 5.30; N, 20.8%. Found: C, 51.89; H, 5.22; N, 21.03%.
To a mixture of cyclohexane-1,4-bis-thiosemicarbazone (3) (0.26 g, 1 mmol) and the appropriate α-bromoketone derivatives 12a–d (2 mmol) in absolute ethanol (10 ml), triethylamine (0.1 ml) was added, and the resulting mixture was refluxed for 8 h and then left to cool to room temperature. The solid product was collected by filtration, washed with ethanol, dried, and finally recrystallized from the ethanol/DMF mixture to give the corresponding bis-thiazoles 13a–d.
1,4-Bis((4-(2-benzothiazolyl)thiazol-2-ylidene)hydrazono)cyclohexane (13a): Green solid, yield (0.31 g, 72%), mp. >300 °C; IR (KBr) ν 3,250 (NH), 1,635 (C=N) cm−1; 1H NMR (DMSO-d6) δ 2.58–2.71 (m, 8H, cyclohexane-H's), 7.43 (t, 2H, J = 7.2 Hz, ArH), 7.53 (t, 2H, J = 7.5 Hz, ArH), 7.73 (s, 2H, thiazole-2-CH), 8.01 (d, 2H, J = 8.1 Hz, ArH’s), 8.09 (d, 2H, J = 7.8 Hz, ArH’s), 11.03 (s, 2H, 2NH) ppm. 13C NMR (DMSO-d6) δ 23.94, 26.18, 29.29, 31.50, 109.32, 122.34, 122.54, 125.15, 126.52, 134.54, 144.65, 153.59, 154.11, 162.79, 170.37 ppm. Anal. calcd. for C26H20N8S4 (572.07): C, 54.52; H, 3.52; N, 19.56%. Found: C, 54.42; H, 3.46; N, 19.46%.
1,4-Bis((4-(2-benzofuryl)thiazol-2-ylidene)hydrazono)cyclohexane (13b): Brown powder, yield (0.40 g, 74%), mp. 269–270 °C; IR (KBr) ν 3,325 (NH), 1,612 (C=N) cm−1; MS m/z (%): 102 (48%), 145 (25.6%), 216 (100%), 305 (14%), 419 (2%), 461 (4.3%), 538 (M+, 4.4%); 1H NMR (DMSO-d6) δ 2.55–2.72 (m, 8H, cyclohexane-H’s), 7.03 (s, 2H, ArH), 7.23–7.32 (m, 6H, ArH), 7.55 (d, 2H, J = 8.1 Hz, ArH), 7.64 (d, 2H, J = 7.8 Hz, ArH), 10.79 (br. s, 2H, 2NH). Anal. calcd. for C28H22N6O4S2 (538.64): C, 62.44; H, 4.12; N, 15.60%. Found: C, 62.50; H, 4.25; N, 15.33%.
1,4-Bis((4-(3-coumarinyl)thiazol-2-ylidene)hydrazono)cyclohexane (13c): Yellow solid, yield (0.25 g, 83%), mp. 241–242 °C; IR (KBr) ν 3,250 (NH), 1,697 (C=O), 1,620 (C=N) cm−1; MS m/z (%): 102 (47%), 244 (100%), 452 (0.7%), 487 (3%), 523 (3%), 593 (M+-1, 4%); 1H NMR (DMSO-d6) δ 2.57–2.73 (m, 8H, cyclohexane-H’s), 7.35–7.45 (m, 4H, ArH), 7.62 (t, 2H, J = 7.2 Hz, ArH), 7.71 (s, 2H, CH-thiazole), 7.81 (d, 2H, J = 7.8 Hz, ArH), 8.54 (s, 2H, chromenone-H), 10.75 (s, 2H, 2NH). Anal. calcd. for C30H22N6O4S2 (594.66): C, 60.59; H, 3.73; N, 14.13%. Found: C, 60.87; H, 3.63; N, 14.08%.
1,4-Bis((4-(4-chlorophenyl)thiazol-2-ylidene)hydrazono)cyclohexane (13d): Green solid, yield (0.4 g, 76%), mp. 230–231 °C; IR (KBr) ν 3,327 (NH), 1,609 (C=N) cm−1; MS m/z (%): 89 (48%), 168 (52%), 210 (100%), 408 (2%), 433 (3%), 482 (2%), 527 (M+, 1%); 1H NMR (DMSO-d6) δ 2.73 (s, 4H, cyclohexane-H’s), 2.88 (s, 4H, cyclohexane-H’s), 7.29 (s, 2H, thiazole-CH), 7.44 (d, 4H, J = 8.4 Hz, ArH), 7.86 (d, 4H, J = 8.4 Hz, ArH), 10.65 (br. s, 2H, 2NH). Anal. calcd. for C24H20Cl2N6S2 (527.49): C, 54.65; H, 3.82; N, 15.93%. Found: C, 54.55; H, 3.75; N, 15.89%.
To a mixture of the bis-thiazolidinone derivative 9 (0.34 g, 1 mmol) and the appropriate aromatic aldehyde derivatives (2 mmol) in DMF/EtOH (10 ml, 1:1), sodium hydroxide (0.08 g) was added, and the resulting mixture was refluxed for 8 h and then left to cool to room temperature. The solid product obtained was collected by filtration, washed with ethanol, dried, and finally recrystallized from the ethanol/DMF mixture to give 14a–d.
1,4-Bis((5-benzylidene-4-oxo-thiazolidin-2-ylidene)hydrazono)cyclohexane (14a): Yellowish-red crystals, yield (0.2 g, 77%), mp. >300 °C; IR (KBr) ν 3,124 (NH), 1,697 (C=O), 1,597 (C=N) cm−1; 1H NMR (DMSO-d6) δ 2.66–2.73 (m, 4H, cyclohexane-H’s), 2.79–2.88 (m, 4H, cyclohexane-H’s), 7.07–7.66 (m, 10H, ArH& CH = Ar), 12.2 (br. s, 2H, 2NH). Anal. calcd. for C26H22N6O2S2 (514.62): C, 60.68; H, 4.31; N, 16.33%. Found: C, 60.59; H, 4.52; N, 16.30%.
1,4-Bis((5-(4-methylbenzylidene)-4-oxo-thiazolidin-2-ylidene)hydrazono)cyclohexane (14b): Yellow crystals, yield (0.16 g, 58%), mp. 269–270 °C; IR (KBr) ν 3,232 (NH), 1,690 (C=O), 1,630 (C=N) cm−1; MS m/z (%): 107 (44.6%), 148 (100%), 218 (13%), 325 (19%), 514 (2%), 542 (M+, 6%); 1H NMR (DMSO-d6) δ 2.36 (s, 6H, CH3-Ar), 2.25–2.63 (m, 4H, cyclohexane-H’s), 2.65–2.81 (m, 4H, cyclohexane-H’s), 7.35 (d, 4H, J = 7.5 Hz, ArH), 7.57–7.76 (m, 6H, ArH& CH = Ar), 12.45 (br. s, 2H, 2NH). Anal. calcd. for C28H26N6O2S2 (542.68): C, 61.97; H, 4.83; N, 15.49%. Found: C, 61.91; H, 4.75; N, 15.39%.
1,4-Bis((5-(4-chlorobenzylidene)-4-oxo-thiazolidin-2-ylidene)hydrazono)cyclohexane (14c): Orange-red crystals, yield (0.15 g, 51%), mp. > 300°C; IR (KBr) ν 3,124 (NH), 1,697 (C=O), 1,635 (C=N) cm−1; MS m/z (%): 55 (100%), 71 (44%), 139 (32%), 278 (13.4%), 435 (1.5%), 581 (M+-2, 2%), 587 (M++4, 2%); 1H NMR (DMSO-d6) δ 2.66 (m, 4H, cyclohexane-H’s), 2.82 (m, 4H, cyclohexane-H’s), 7.56–7.66 (m, 10H, ArH& CH = Ar), 12.5 (br. s, 2H, 2NH). Anal. calcd. for C26H20Cl2N6O2S2 (583.51): C, 53.52; H, 3.45; N, 14.4%. Found: C, 53.45; H, 3.41; N, 14.31%.
1,4-Bis((5-(4-bromobenzylidene)-4-oxo-thiazolidin-2-ylidene)hydrazono)cyclohexane (14d): Orange-red crystals, yield (0.15 m, 45%), mp. 290–291°C; IR (KBr) ν 3,210 (NH), 1,697 (C=O), 1,643 (C=N) cm−1; MS m/z (%): 89 (100%), 133 (39%), 214 (8%), 284 (4%), 481 (0.5%), 566 (6%), 673 (M++1, 5%);1H NMR (DMSO-d6) δ 2.50–2.72 (m, 4H, cyclohexane-H’s), 2.79–2.87 (m, 4H, cyclohexane-H’s), 7.47–7.81 (m, 10H, ArH and CH = Ar), 12.55 (s, 2H, 2NH). Anal. calcd. for C26H20Br2N6O2S2 (672.41): C, 46.44; H, 3.00; N, 12.50%. Found: C, 46.30; H, 3.25; N, 12.25%.
All biological assays were carried out at Faculty of Science, Port Said University, Egypt, as well as at Department of Chemistry, Faculty of Science, Suez Canal University, Ismailia, Egypt.
The human ovarian serous carcinoma cell line KF-28 was kindly provided by Prof. Yoshihiro Kikuchi (Department of Obstetrics and Gynecology, National Defense Medical College, Saitama, Japan), and A2780 cell line was purchased from the ECACC (ECACC 93112519). The cells were maintained in RPMI-1640 (Sigma-Aldrich, United States). The cervical cancer cell line, Hela cells, breast cancer cell line, MCF-7, and MDA-MB-231 were purchased from the ATCC and cultured in DMEM. Both types of media were supplemented with 2 mM L-glutamine (Lonza, Belgium), 10% FBS (Sigma, St. Louis, MO, United States), and 1% penicillin/streptomycin (Lonza, Belgium). All cells were incubated at 37°C in 5% CO2 atmosphere (NuAire). All cancer cell lines were cultured according to the routine tissue culture work as discussed by the Freshney group (Freshney, 2010). The cells were plated at a density of 5,000 cells in triplicates in a 96-well plate. On the next day, the cells were treated with the indicated compound(s) at the indicated concentrations in a final volume of 100 μL media. Cell viability was assessed after 72 h using MTT solution (Promega) (Mosmann, 1983). 20 μL of the reagent was added to each well, the plate was incubated for 3 h, fluorescence was subsequently measured (570 nm) using a plate reader, and then the IC50 values were calculated using Excel (Tantawy et al., 2020).
The cells were seeded into six-well culture plates (3–5 × 105 cells/well) and incubated overnight at 37°C, under 5% CO2. The cells were then treated with indicated compounds for 48 h. Next, media supernatants and the cells were collected and washed with ice-cold PBS. Next, the cells were stained with PI (10 mg/ml) for 30 min and analyzed for DNA contents using flow cytometry analysis using an FL2 (λex/em 535/617 nm) signal detector (ACEA Novocyte™ flow cytometer, ACEA Biosciences, Inc., San Diego, CA, United States). For each sample, 12,000 events are acquired. Cell cycle distribution is calculated using a CytoFLex machine and analyzed by CytExpert software. Flow cytometric methodologies (annexin V/PI staining with cell cycle analyses) are carried out as previously described (Nafie et al., 2020a; Nafie et al., 2020b; Gad et al., 2020).
The cells were grown in 10 cm2 culture dishes to a monolayer with a confluence of 90% in triplicates. After different treatments, the cells were washed twice with PBS and then pelleted by centrifugation for 5 min at 1800 rpm at 4°C. The cells were lysed by Trizol reagent (Invitrogen), and then total RNA was purified with the phenol/chloroform extraction method. Concentration and purity of RNA samples were measured using NanoDrop. The RNA purity was assessed as OD260/OD230 (≥1.5) using NanoDrop. gDNA was eliminated by DNase. The RNA from three biological samples was then pooled for mRNA analysis, cDNA was synthesized using the iScript kit (Bio-Rad, United States) according to the manufacturer’s recommendation, and RT-PCRs consisted of 8.34 µL Fluocycle® II SYBR® (Euroclone, Milan, Italy), 0.5 µL of both 10 µM forward and reverse primers, 1 µL cDNA, and 6.34 µL of H2O. All reactions were performed for 35 cycles using the following temperature profile: 95°C for 5 min (initial denaturation), 95°C for 15 min (denaturation), 55°C for 30 min (annealing), and 72°C for 30 min (extension) (Nafie et al., 2020a; Sarhan et al., 2020). Then, the Ct values were collected, with the folds of changes between all the samples. The primers used are listed in Table 3.

TABLE 3. Primer sequences in forward and reverse.
Cell lysates were prepared by lysing cells in RIPA buffer (10 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% Na deoxycholate), supplemented with protease inhibitor cocktail (Sigma). Protein concentration of whole cell lysates was determined by the BSA assay using the BSA kit (Pierce), and then equal protein amounts were heated to 95 °C for 5 min with sodium dodecyl sulfate (SDS) sample buffer (25 ml glycerol, 31.2 ml Tris buffer, 7.5 ml SDS, a dash of bromophenol blue/100 ml) and run on 15% SDS polyacrylamide gel electrophoresis (SDS-PAGE). Protein samples were then blotted onto PVDF membranes (Immobilon P, Watford, United Kingdom). The membranes were incubated in blocking solution (5% non-fat milk in PBS) for 1 h and then in primary antibody (anti-human C-myc pAb (Abcam; at dilution of 2:1,000) or GAPDH mAb (Santa Cruz; at dilution of 20:1,000)) overnight. After 3 × 10 min washes in TBS (0.1% Tween-20 in PBS), the membrane was incubated for 1 h at room temperature with horseradish-peroxidase–linked (HRP) secondary anti-rabbit or anti-mouse Ab (1:1,000 dilution in PBS). Signals on the membrane were developed using ECL reagent (Amersham, CA, United States) and then were imaged with the Chemidoc system (Bio-Rad, United States).
For elucidation of the virtual mechanism of binding, the molecular docking study toward the Pim-1 kinase active site was carried out. All synthesized derivatives were chemically and energetically optimized. Additionally, the protein structure (PDB = 2OBJ, https://www.rcsb.org/structure/2OBJ) was also optimized following the routine work as discussed by Poli et al. (Poli et al., 2016). MOE 2014 was used as the validated molecular docking calculation, and Chimera software was finally used as the visualized software for the analysis of drug–target interactions.
In this work, we successfully prepared 1,4-cyclohexanedione-bis-thiosemicarbazone and studied its synthetic utility as a key compound for preparing four different series of new 1,4-cyclohexane–based bis-thiazole derivatives via either stepwise reaction or the one-pot multi-component procedure. In addition, most of the obtained compounds were screened in vitro for their cytotoxic activities in five cancer cell lines. Two of the tested compounds (5c and 5f) exhibited pronounced cytotoxic activities with high IC50 values 0.6 and 6 nM against the Hela and KF-28 cell lines, respectively. Taken together, the tested compounds exhibited remarkable cytotoxic activities against the tested cancer cell lines with apoptosis induction through inhibition of Pim-1 kinase. Therefore, compounds 5c and 5f are worthy to be further tested in vivo for developing chemotherapeutic anti-cancer agents.
The original contributions presented in the study are included in the article/Supplementary Material, and further inquiries can be directed to the corresponding author.
KD and AA suggested the idea, interpreted the spectral data, and wrote the manuscript. KD, AA, and MA discussed the results and edited the language. MN and MH carried out the biological experiments, interpreted and wrote the results of the biological part, and revised the language. BM conducted the chemical experiments. MR performed the arrangement of the manuscript, wrote the draft form, and validated the article. MA commented on the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MA is indebted to Taif University Researches Supporting Project Number (TURSP-2020/91), Taif University, Taif, Saudi Arabia.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.694870/full#supplementary-material
Abdel-Gawad, H., Mohamed, H. A., Dawood, K. M., and Badria, F. A.-R. (2010). Synthesis and Antiviral Activity of New Indole-Based Heterocycles. Chem. Pharm. Bull. 58 (11), 1529–1531. doi:10.1248/cpb.58.1529
Agarwal, S., Kalal, P., Gandhi, D., and Prajapat, P. (2018). Thiazole Containing Heterocycles with CNS Activity. Curr. Drug. Discov. Technol. 15, 178–195. doi:10.2174/1570163814666170724170152
Ayati, A., Emami, S., Asadipour, A., Shafiee, A., and Foroumadi, A. (2015). Recent Applications of 1,3-thiazole Core Structure in the Identification of New lead Compounds and Drug Discovery. Eur. J. Med. Chem. 97, 699–718. doi:10.1016/j.ejmech.2015.04.015
Ayati, A., Emami, S., Moghimi, S., and Foroumadi, A. (2019). Thiazole in the Targeted Anticancer Drug Discovery. Future Med. Chem. 11 (16), 1929–1952. doi:10.4155/fmc-2018-0416
Bachmann, M., Hennemann, H., Xing, P. X., Hoffmann, I., and Möröy, T. (2004). The Oncogenic Serine/Threonine Kinase Pim-1 Phosphorylates and Inhibits the Activity of Cdc25C-Associated Kinase 1 (C-TAK1). J. Biol. Chem. 279 (46), 48319–48328. doi:10.1074/jbc.m404440200
Bachmann, M., Kosan, C., Xing, P. X., Montenarh, M., Hoffmann, I., and Möröy, T. (2006). The Oncogenic Serine/threonine Kinase Pim-1 Directly Phosphorylates and Activates the G2/M Specific Phosphatase Cdc25C. Int. J. Biochem. Cel Biol. 38 (3), 430–443. doi:10.1016/j.biocel.2005.10.010
Bataille, C. J. R., Brennan, M. B., Byrne, S., Davies, S. G., Durbin, M., Fedorov, O., et al. (2017). Thiazolidine Derivatives as Potent and Selective Inhibitors of the PIM Kinase Family. Bioorg. Med. Chem. 25 (9), 2657–2665. doi:10.1016/j.bmc.2017.02.056
Borcea, A.-M., Ionu, I., Crian, O., and Oniga, O. (2021). An Overview of the Synthesis and Antimicrobial, Antiprotozoal, and Antitumor Activity of Thiazole and Bisthiazole Derivatives. Molecules 26 (3), 624. doi:10.3390/molecules26030624
Ceba, M. R., Nevado, J. J. B., and García, J. M. (1982). 1,4-Cyclohexanedione Bisthiosemicarbazone as an Analytical Reagent for Spectrophotometric Estimation of Copper. Microchemical J. 27 (3), 380–388. doi:10.1016/0026-265X(82)90027-3
Chhabria, M. T., Patel, S., Modi, P., and Brahmkshatriya, P. S. (2016). Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives. Curr. Top. Med. Chem. 16 (26), 2841–2862. doi:10.2174/1568026616666160506130731
Das, D., Sikdar, P., and Bairagi, M. (2016). Recent Developments of 2-aminothiazoles in Medicinal Chemistry. Eur. J. Med. Chem. 109, 89–98. doi:10.1016/j.ejmech.2015.12.022
Dawood, K., and El-Deftar, M. (2010). Microwave-assisted Synthesis of 2-substituted 4-Biarylyl-1,3-Thiazoles by Carbon-Carbon Cross-Coupling in Water. Synthesis 2010, 1030–1038. doi:10.1055/s-0029-1218662
Dawood, K. M., Abdel-Gawad, H., Mohamed, H. A., and Badria, F. A. (2011). Synthesis, Anti-HSV-1, and Cytotoxic Activities of Some New Pyrazole- and Isoxazole-Based Heterocycles. Med. Chem. Res. 20 (7), 912–919. doi:10.1007/s00044-010-9420-4
Dawood, K. M., and Abu-Deif, H. K.-A. (2014). Synthesis and Antimicrobial Evaluation of Some New 1,2-Bis-(2-(n-Arylimino)-1,3-Thiazolidin-3-Yl)ethane Derivatives. Chem. Pharm. Bull. 62 (5), 439–445. doi:10.1248/cpb.c14-00031
Dawood, K. M. (2019). Bis‐thiourea Derivatives and Their Utility in Synthesis of Mono‐heterocyclic, Bis‐heterocyclic, and Fused Heterocyclic Systems. J. Heterocyclic Chem. 56 (6), 1701–1721. doi:10.1002/jhet.3540
Dawood, K. M., Elamin, M. B., and Faraga, A. M. (2015).Microwave-assisted Synthesis of 2-Acetyl-5-Arylthiophenes and 4-(5-Arylthiophen-2-Yl)thiazoles via Suzuki Coupling in Water. Arkivoc 2015, 50–62. doi:10.3998/ark.5550190.p009.018
Dawood, K. M., Eldebss, T. M. A., El-Zahabi, H. S. A., Yousef, M. H., and Metz, P. (2013). Synthesis of Some New Pyrazole-Based 1,3-thiazoles and 1,3,4-thiadiazoles as Anticancer Agents. Eur. J. Med. Chem. 70, 740–749. doi:10.1016/j.ejmech.2013.10.042
Dawood, K. M., Eldebss, T. M. A., El-Zahabi, H. S. A., and Yousef, M. H. (2015). Synthesis and Antiviral Activity of Some New Bis-1,3-Thiazole Derivatives. Eur. J. Med. Chem. 102, 266–276. doi:10.1016/j.ejmech.2015.08.005
de Marigorta, E. M., Santos, J. M. d. L., Ochoa de Retana, A. M., Vicario, J., and Palacios, F. (2019). Multicomponent Reactions (MCRs): a Useful Access to the Synthesis of Benzo-Fused γ-lactams. Beilstein J. Org. Chem. 15 (1), 1065–1085. doi:10.3762/bjoc.15.104
de Oliveira, V. V. G., Aranda de Souza, M. A., Cavalcanti, R. R. M., de Oliveira Cardoso, M. V., Leite, A. C. L., da Silva Junior, V. A., et al. (2020). Study of In Vitro Biological Activity of Thiazoles on Leishmania (Leishmania) Infantum. J. Glob. Antimicrob. Resist. 22, 414–421. doi:10.1016/j.jgar.2020.02.028
de Siqueira, L. R. P., de Moraes Gomes, P. A. T., de Lima Ferreira, L. P., de Melo Rêgo, M. J. B., and Leite, A. C. L. (2019). Multi-target Compounds Acting in Cancer Progression: Focus on Thiosemicarbazone, Thiazole and Thiazolidinone Analogues. Eur. J. Med. Chem. 170, 237–260. doi:10.1016/j.ejmech.2019.03.024
Dhanasekaran, S. M., Barrette, T. R., Ghosh, D., Shah, R., Varambally, S., Kurachi, K., et al. (2001). Delineation of Prognostic Biomarkers in Prostate Cancer. Nature 412 (6849), 822–826. doi:10.1038/35090585
Eweiss, N. F., and Osman, A. (1980). Synthesis of Heterocycles. Part II. New Routes to Acetylthiadiazolines and Alkylazothiazoles. J. Heterocycl. Chem. 17 (8), 1713–1717. doi:10.1002/jhet.5570170814
Farag, A. M., and Algharib, M. S. (1988). Synthesis and Reactions of C-(2-thenoyl)-N-arylformhydrazidoyl Bromides. Org. Preparations Procedures Int. 20 (5), 521–526. doi:10.1080/00304948809356298
Freshney, R. I. (2010). Culture of Animal Cells. New York: John Wiley & Sons, 463–479. doi:10.1002/9780470649367
Gad, E. M., Nafie, M. S., Eltamany, E. H., Hammad, M. S. A. G., Barakat, A., and Boraei, A. T. A. (2020). Discovery of New Apoptosis-Inducing Agents for Breast Cancer Based on Ethyl 2-Amino-4,5,6,7-Tetra Hydrobenzo[b]Thiophene-3-Carboxylate: Synthesis, In Vitro, and In Vivo Activity Evaluation. Molecules 25 (11), 2523. doi:10.3390/molecules25112523
Galluzzi, L., Senovilla, L., Vitale, I., Michels, J., Martins, I., Kepp, O., et al. (2012). Molecular Mechanisms of Cisplatin Resistance. Oncogene 31 (15), 1869–1883. doi:10.1038/onc.2011.384
Hassan, M. K., Watari, H., Christenson, L., Bettuzzi, S., and Sakuragi, N. (2011). Intracellular Clusterin Negatively Regulates Ovarian Chemoresistance: Compromised Expression Sensitizes Ovarian Cancer Cells to Paclitaxel. Tumor Biol. 32 (5), 1031–1047. doi:10.1007/s13277-011-0207-0
Hegazi, B., Mohamed, H. A., Dawood, K. M., and Badria, F. A.-R. (2010). Cytotoxicity and Utility of 1-indanone in the Synthesis of Some New Heterocycles. Chem. Pharm. Bull. 58 (4), 479–483. doi:10.1248/cpb.58.479
Jain, A. K., Vaidya, A., Ravichandran, V., Kashaw, S. K., and Agrawal, R. K. (2012). Recent Developments and Biological Activities of Thiazolidinone Derivatives: a Review. Bioorg. Med. Chem. 20 (11), 3378–3395. doi:10.1016/j.bmc.2012.03.069
Kaminsky, D., Kryshchyshyn, A., and Lesyk, R. (2017). 5-Ene 4thiazolidinones An Efficient Tool in Medicinal Chemistry. Eur. J. Med. Chem. 140, 542–594. doi:10.1016/j.ejmech.2017.09.031
Kappe, C. O. (2000). Recent Advances in the Biginelli Dihydropyrimidine Synthesis. New Tricks from an Old Dog. Acc. Chem. Res. 33 (12), 879–888. doi:10.1021/ar000048h
Karigar, A. A., Himaja, M., Mali, S. V., Jagadeesh, K. P., and Sikarwar, M. S. (2011). One-Pot Synthesis and Antitubercular Activity of 2-Amino-5-Aryl-5h-Thiazolo[4,3-B]-1,3,4-Thiadiazoles. Internat. Res. J. Pharm. 2 (1), 153–158.
Kaur Manjal, S., Kaur, R., Bhatia, R., Kumar, K., Singh, V., Shankar, R., et al. (2017). Synthetic and Medicinal Perspective of Thiazolidinones: a Review. Bioorg. Chem. 75, 406–423. doi:10.1016/j.bioorg.2017.10.014
Kumar, P. S., and Umadevi, P. (2018). Novel Bis (1,2,4-oxadiazolyl) Fused Thiazole Derivatives: Synthesis and Anticancer Activity. Russ. J. Gen. Chem. 88 (12), 2611–2615. doi:10.1134/S107036321812023X
Kumar, S., Patil, M. T., Kataria, R., and Salunke, D. B., 2016. “11. Thiazole: A Privileged Scaffold in Drug Discovery.” in Chemical Drug Design, Editors GK Gupta, and V. Kumar (De Gruyter), pp.243–282. doi:10.1515/9783110368826-013
Leoni, A., Locatelli, A., Morigi, R., and Rambaldi, M. (2014). Novel Thiazole Derivatives: a Patent Review (2008 - 2012; Part 1). Expert Opin. Ther. Patents 24 (2), 201–216. doi:10.1517/13543776.2014.858121
Li, Y.-Y., Popivanova, B. K., Nagai, Y., Ishikura, H., Fujii, C., and Mukaida, N. (2006). Pim-3, a Proto-Oncogene with Serine/threonine Kinase Activity, Is Aberrantly Expressed in Human Pancreatic Cancer and Phosphorylates Bad to Block Bad-Mediated Apoptosis in Human Pancreatic Cancer Cell Lines. Cancer Res. 66 (13), 6741–6747. doi:10.1158/0008-5472.CAN-05-4272
Mahmoud, H. K., Abbas, A. A., and Gomha, S. M. (2020). Synthesis, Antimicrobial Evaluation and Molecular Docking of New Functionalized Bis(1,3,4-Thiadiazole) and Bis(thiazole) Derivatives. Polycyclic Aromatic Comp., 1–13. doi:10.1080/10406638.2019.1709085
Mohareb, R. M., Abouzied, A. S., and Abbas, N. S. (2019). Synthesis and Biological Evaluation of Novel 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2- Yl Derivatives Derived from Dimedone with Anti-tumor, C-Met, Tyrosine Kinase and Pim-1 Inhibitions. Anticancer. Agents. Med. Chem. 19 (12), 1438–1453. doi:10.2174/1871520619666190416102144
Momose, Y., Meguro, K., Ikeda, H., Hatanaka, C., Oi, S., and Sohda, T. (1991). Studies on Antidiabetic Agents. X. Synthesis and Biological Activities of Pioglitazone and Related Compounds. Chem. Pharm. Bull. 39 (6), 1440–1445. doi:10.1248/cpb.39.1440
Morigi, R., Locatelli, A., Leoni, A., and Rambaldi, M. (2015). Recent Patents on Thiazole Derivatives Endowed with Antitumor Activity. Recent Pat. Anticancer. Drug. Discov. 10 (3), 280–297. doi:10.2174/1574892810666150708110432
Möröy, T., Verbeek, S., Ma, A., Achacoso, P., Berns, A., and Alt, F. (1991). E Mu N and E Mu LMyc Cooperate with E Mu Pim-1 to Generate Lymphoid Tumors at High Frequency in Double-Transgenic Mice. Oncogene 6 (11), 1941–1948.
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65 (1-2), 55–63. doi:10.1016/0022-1759(83)90303-4
Nafie, M. S., Amer, A. M., Mohamed, A. K., and Tantawy, E. S. (2020a). Discovery of Novel Pyrazolo[3,4-B]pyridine Scaffold-Based Derivatives as Potential PIM-1 Kinase Inhibitors in Breast Cancer MCF-7 Cells. Bioorg. Med. Chem. 28 (24), 115828. doi:10.1016/j.bmc.2020.115828
Nafie, M. S., Arafa, K., Sedky, N. K., Alakhdar, A. A., and Arafa, R. K. (2020b). Triaryl Dicationic DNA Minor-Groove Binders with Antioxidant Activity Display Cytotoxicity and Induce Apoptosis in Breast Cancer. Chemico-Biological Interactions 324, 109087. doi:10.1016/j.cbi.2020.109087
Nirwan, S., Chahal, V., and Kakkar, R. (2019). Thiazolidinones: Synthesis, Reactivity, and Their Biological Applications. J. Heterocyclic Chem. 56 (4), 1239–1253. doi:10.1002/jhet.3514
Paprocki, D., Madej, A., Koszelewski, D., Brodzka, A., and Ostaszewski, R. (2018). Multicomponent Reactions Accelerated by Aqueous Micelles. Front. Chem. 6, 502–523. doi:10.3389/fchem.2018.00502
Poli, G., Martinelli, A., and Tuccinardi, T. 2016. Reliability Analysis and Optimization of the Consensus Docking Approach for the Development of Virtual Screening Studies. J. Enzyme Inhib. Med. Chem. 31, 167–173. doi:10.1080/14756366.2016.1193736
Rouf, A., and Tanyeli, C. (2015). Bioactive Thiazole and Benzothiazole Derivatives. Eur. J. Med. Chem. 97, 911–927. doi:10.1016/j.ejmech.2014.10.058
Sarhan, A. A. M., Boraei, A. T. A., Barakat, A., and Nafie, M. S. (2020). Discovery of Hydrazide-Based Pyridazino[4,5-B]indole Scaffold as a New Phosphoinositide 3-kinase (PI3K) Inhibitor for Breast Cancer Therapy. RSC Adv. 10 (33), 19534–19541. doi:10.1039/d0ra02798g
Scarim, C. B., Jornada, D. H., Machado, M. G. M., Ferreira, C. M. R., Dos Santos, J. L., and Chung, M. C. (2019). Thiazole, Thio and Semicarbazone Derivatives against Tropical Infective Diseases: Chagas Disease, Human African Trypanosomiasis (HAT), Leishmaniasis, and Malaria. Eur. J. Med. Chem. 162, 378–395. doi:10.1016/j.ejmech.2018.11.013
Sharma, P. C., Bansal, K. K., Sharma, A., Sharma, D., and Deep, A. (2020). Thiazole-containing Compounds as Therapeutic Targets for Cancer Therapy. Eur. J. Med. Chem. 188, 112016. doi:10.1016/j.ejmech.2019.112016
Siddiqui, N., Arya, S. K., Ahsan, W., and Azad, B. (2011). Diverse Biological Activities of Thiazoles: A Retrospect. Internat. J. Drug Develop. Res. 3 (4), 55–67.
Tantawy, E. S., Amer, A. M., Mohamed, E. K., Abd Alla, M. M., and Nafie, M. S. (2020). Synthesis, Characterization of Some Pyrazine Derivatives as Anti-cancer Agents: In Vitro and In Silico Approaches. J. Mol. Struct. 1210, 128013. doi:10.1016/j.molstruc.2020.128013
Tripathi, A. C., Gupta, S. J., Fatima, G. N., Sonar, P. K., Verma, A., and Saraf, S. K. (2014). 4-Thiazolidinones: The Advances Continue…. Eur. J. Med. Chem. 72, 52–77. doi:10.1016/j.ejmech.2013.11.017
Turan-Zitouni, G., Altıntop, M. D., Özdemir, A., Kaplancıklı, Z. A., Çiftçi, G. A., and Temel, H. E. (2016). Synthesis and Evaluation of Bis-Thiazole Derivatives as New Anticancer Agents. Eur. J. Med. Chem. 107, 288–294. doi:10.1016/j.ejmech.2015.11.002
Wong, R. S. (2011). Apoptosis in Cancer: from Pathogenesis to Treatment. J. Exp. Clin. Cancer Res. 30 (1), 87. doi:10.1186/1756-9966-30-87
Xia, Z., Knaak, C., Ma, J., Beharry, Z. M., McInnes, C., Wang, W., et al. (2009). Synthesis and Evaluation of Novel Inhibitors of Pim-1 and Pim-2 Protein Kinases. J. Med. Chem. 52 (1), 74–86. doi:10.1021/jm800937p
Zhang, Y., Wang, Z., Li, X., and Magnuson, N. S. (2008). Pim Kinase-dependent Inhibition of C-Myc Degradation. Oncogene 27 (35), 4809–4819. doi:10.1038/onc.2008.123
Zhao, H., Cui, G., Jin, J., Chen, X., and Xu, B. (2016). Synthesis and Pin1 Inhibitory Activity of Thiazole Derivatives. Bioorg. Med. Chem. 24 (22), 5911–5920. doi:10.1016/j.bmc.2016.09.049
Keywords: docking, apoptosis, cytotoxic, bis-thiazoles, hydrazonoyl chlorides, Pim-1 kinase
Citation: Dawood KM, Raslan MA, Abbas AA, Mohamed BE, Abdellattif MH, Nafie MS and Hassan MK (2021) Novel Bis-Thiazole Derivatives: Synthesis and Potential Cytotoxic Activity Through Apoptosis With Molecular Docking Approaches. Front. Chem. 9:694870. doi: 10.3389/fchem.2021.694870
Received: 13 April 2021; Accepted: 07 July 2021;
Published: 12 August 2021.
Edited by:
Upendra Sharma, Institute of Himalayan Bioresource Technology (CSIR), IndiaCopyright © 2021 Dawood, Raslan, Abbas, Mohamed, Abdellattif, Nafie and Hassan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamal M. Dawood, ZHJfZGF3b29kQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.