 Min Zhou1,2
Min Zhou1,2 Xianyi Zhang
Xianyi Zhang Xianglei Kong
Xianglei Kong- 1School of Physics and Electronic Information, Anhui Normal University, Wuhu, China
- 2The State Key Laboratory and Institute of Elemento-Organic Chemistry, Collage of Chemistry, Nankai University, Tianjin, China
- 3Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin, China
A new program for searching global minimum structures of atomic clusters using basin-hopping algorithm based on the xTB method was developed here. The program can be performed with a much higher speed than its replacement directly based on DFT methods. Considering the structural varieties and complexities in finding their global minimum structures, phosphorus cluster cations were studied by the program. The global minimum structures of cationic
Introduction
Clusters bridge atoms, molecules and bulk matter (Johnstom, 2002; Castleman and Jena, 2006; Fehlner et al., 2007; Ha et al., 2019), showing their great potentials for applications in many research fields such as catalysis (Liu and Corma, 2018; Du et al., 2020) and energy storage (VanGelder et al., 2018; VanGelder et al., 2019). They are also characterized by their geometries and electronic structures in many cases (Johnstom, 2002; Fehlner et al., 2007; Ferrando, 2015; Luo et al., 2016; Jena and Sun, 2018; Ha et al., 2019). There are many wonderful examples, including the cage-like fused-ring structure (truncated icosahedron) of C60 fullerene (Kroto et al., 1985), the tetrahedral structure of Au20 (Li et al., 2003), C6v symmetry boron cluster of B36 (Piazza et al., 2014), the borospherene cluster of B40 (Zhai et al., 2014; Li et al., 2017), and the protonated serine octamer (Cooks et al., 2001; Kong et al., 2006; Scutelnic et al., 2018). In lots of cases, the structural information of the clusters can hardly be obtained directly from experiments, and theoretical calculations are very important to provide structural candidates whose predicted properties should be further compared with the experimental results, in order to make the identification stable (Zhang and Glezakou, 2020).
Although the structural determination of small molecules based on density functional theory (DFT) or other methods has become a relatively routine task for computational chemists, the identification of the global minimum structures for clusters, especially those with large sizes, is still a great challenge. The reason is that the complexity of the searching space in their potential energy surface (PES) grows exponentially with the increasing number of atoms inside the clusters. Since the numbers of local minima grow quickly with the size of clusters, the global optimization becomes a very difficult task to overcome. Thus, different search algorithms and methodologies, including the genetic algorithm (GA) (Hartke, 1993; Deaven and Ho, 1995; Hartke, 1995; Daven et al., 1996; Rogan et al., 2013; Kanters and Donald, 2014; Shayeghi et al., 2015; Lazauskas et al., 2017; Rabanal-León et al., 2018; Jäger et al., 2019; Yañez et al., 2019) and relative evolutionary algorithm (EA) (Zhou et al., 2020), the swarm intelligence algorithm (Wang et al., 2012; Jana et al., 2019) and others, have been proposed and applied in the past decades (Zhang and Glezakou, 2020).
It is now accepted that the choosing of a suitable method for a special system based on its properties is very important. Usually, for clusters with tens of atoms, a very detailed investigation on their potential energy surfaces is still too difficult to be performed with reasonable computational cost. The Basin-Hopping (BH) algorithm, has been suggested as a good choice to solve global minima of Lennard–Jones clusters (Wales and Doye, 1997; Wales and Scheraga, 1999). Programs based on the BH algorithm have been developed based on empirical potentials (Zhan et al., 2005; Paz-Borbón et al., 2007) and DFT methods (Yoo et al., 2004.; Bai et al., 2006; Bulusu and Zeng, 2006; Huang et al., 2010; Jiang et al., 2012; Chen et al., 2017; Zhao et al., 2017; Chen et al., 2019). And the self-consistent charge density functional tight binding (SCC-DFTB) methods, including DFTB2, DFTB2-γh, DFTB2-γh + gaus and others, have been also applied (Choi et al., 2013).
Considering the success of the method of GFN-xTB, which was a DFTB3 variant developed by Grimme et al. (2017) (Bannwarth et al., 2019), the current work presents a new BH program named NKCS based on the Python, in conjunction with the xTB method for searching global minima of atomic clusters. Phosphorus clusters are selected to be studied, due to the two facts. The first one is that phosphorus exhibits a variety of structural phases, such as orthorhombic black, rhombohedral, violet, metallic, fibrous red, white, and amorphous. Thus, a better understanding about phosphorus clusters can deepen our knowledge about its structures and properties. The second fact is that although phosphorus cluster ions with wide size distributions have been observed in the laser ablation experiments for a long time (Martin, 1986; Mu et al., 2015; Yang et al., 2016; Kong et al., 2019), its structural studies are still limited for small to medium-sized clusters (Guo et al., 2004; Xue et al., 2010; Mu et al., 2015; Kong et al., 2019). And searching for the global minima in their PES is still a challenging task due to the diverse bonding patterns of the element.
Methods
The newly developed program NKCS described here is written in Python language. The procedure of the program is shown in Figure 1. It couples the xTB-based local optimization and BH global search algorithm. For all initially generated or distorted structures through the BH processes, xTB-based local optimization was applied. Then these structures were ranked according to their energies. In the last step, the selected isomers were further sent to high-level DFT calculations. The procedure of the program is shown in the middle part of the picture. According to the input parameters set by the user, initial structures of the clusters are randomly built for xTB optimization. Then the BH algorithm are employed to some selected isomers and during the process, the criteria to accept newly distorted isomers are judged according to their energies calculated by the xTB method. At last, some local minima are further selected to perform high-level DFT calculations to identify the global minimum.
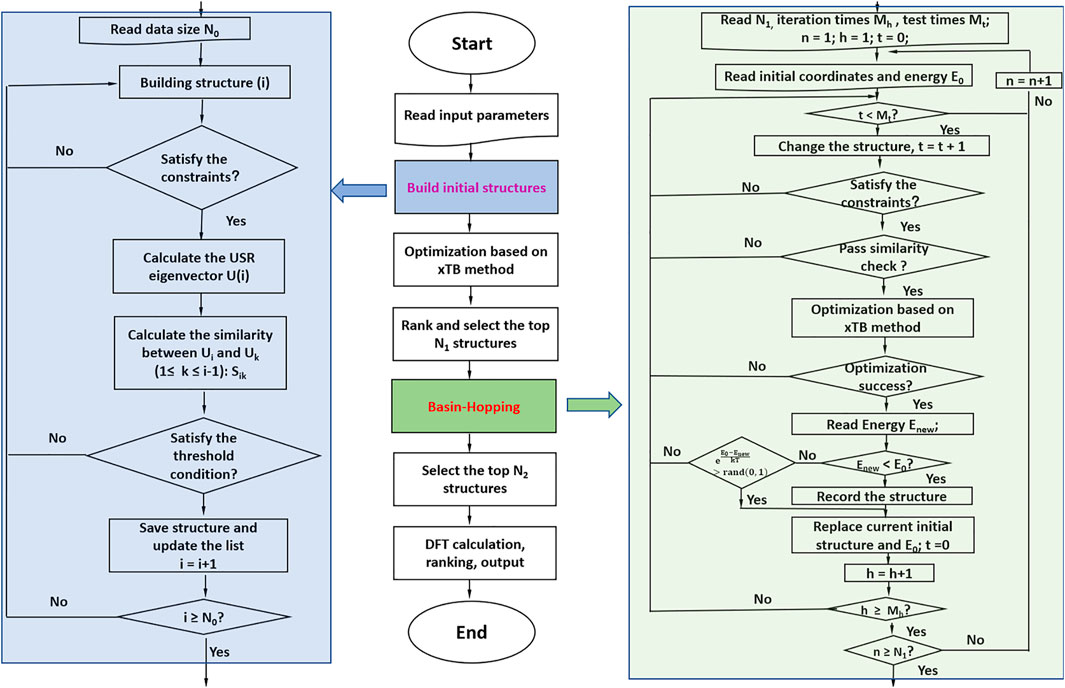
FIGURE 1. The procedure of the whole program is shown in the middle. The left side shows the process of building initial structures, and the right side shows the detail of the basin-hopping algorithm applied here.
To improve the efficiency of the program, the randomly generated or distorted geometries should be instantly checked according to their inter-atomic distances. Unreasonable structures with inter-atomic distances much smaller than the sum of their covalent radii are directly discarded. After that, a similarity check algorithm is applied to avoid duplicated structures. In this process, the ultrafast shape recognition (USR) algorithm is applied to compare the similarities of the randomly generated or distorted structures and the ones stored in the database (Ballester and Richards, 2007a; Ballester and Richards, 2007b; Ballester et al., 2009). For the homo atomic clusters studied here, the previously suggested 12 descriptors were applied here. The set of intra-cluster atomic distances from four locations are considered: the molecular centroid (ctd), the closest atom to ctd (cst), the farthest atom from ctd (fct) and the farthest atom from fct (ftf). So a molecule can be described as:
where
For the randomly constructed structure sets, the xTB method was applied to perform structural optimization and energy calculation. The parametrization in the method covers all spd-block elements and the lanthanides up to Z = 86 and it has been considered as a suitable method for dealing with various clusters with complex electronic structures (Grimme et al., 2017). The BH algorithm was then applied for the selected isomers based on their energies after the xTB optimization. Considering the BH method is one of the individual-based methods (Zhang and Glezakou, 2020), an initialized population with suitable size is applied here to improve its performance in the global optimization. After the selection of initial seed structures for BH algorithm based on their energies calculated by the xTB method, distorted structures are generated from the seeds by the displacement. The reasonability and similarity of the new structure should be checked and then optimized by the xTB method. For the acceptance of the newly distorted structure, the previously suggested criteria by Zhou et al. are applied here (Zhou et al., 2019). And the NKCS program also integrates the interface of Gaussian computing software (Frisch et al., 2009) to perform high-level structure optimization and frequency calculation for the selected isomers by the BH algorithm.
Results and Discussion
The NKCS program has been tested with the odd-numbered phosphorus cluster cations here. In order to make the process clear, an example for searching the global minimum of
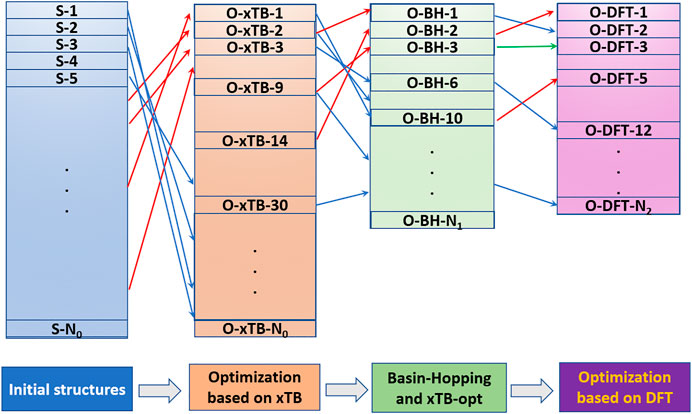
FIGURE 2. The process for searching the global minimum by the NKCS program.
Figures 3, 4 show the optimized global minimum geometries of
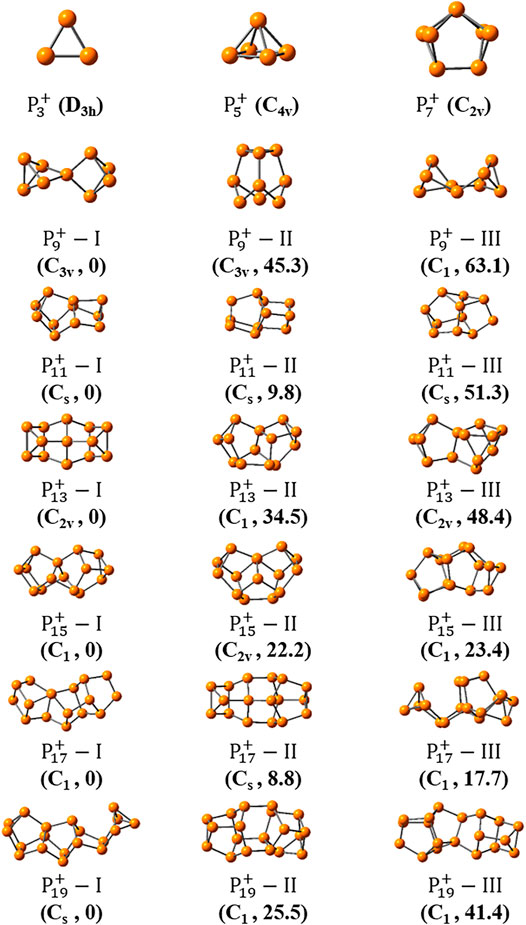
FIGURE 3. The most stable isomers of

FIGURE 4. The three most stable isomers of
As the size increased, the symmetry of the cluster ions decreases. The newly found most stable isomer of
And the most stable isomers of
Briefly, the BH algorithm based on the xTB method has been developed for searching global minima of clusters. Considering the parametrization of xTB method covers all spd-block elements and the functional form of the xTB mostly avoids element-pair-specific parameters, the program developed here has a very wide range of applications. And compared with DFT-based methods, it significantly saves computing time. Medium-sized phosphorus cluster cations were studied here, and new energetically favored structures were identified. Based on these results, some structural rules of these cationic clusters should be further discussed, since it might be helpful to get some general pictures about these energetically preferred structures and structural tendency about large-sized clusters.
For one thing, it is interesting to find that symmetric structures are very important for small-sized clusters of
For the second point, most medium-sized globe minima of
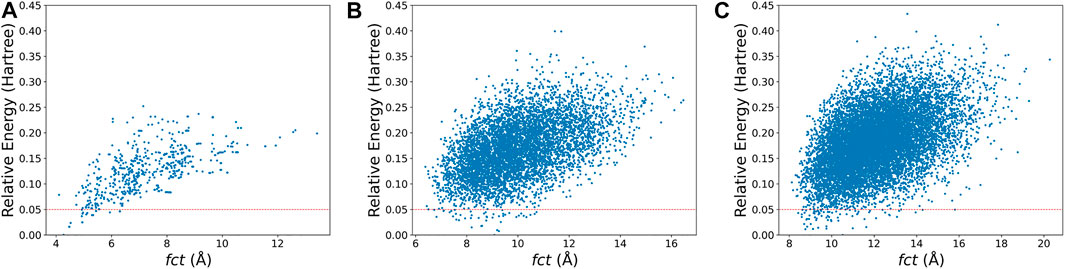
FIGURE 5. The relative energy-fct diagrams for the xTB-optimized structures of the randomly generated populations of (A)
For the third point, it is interesting to found that both isomers of
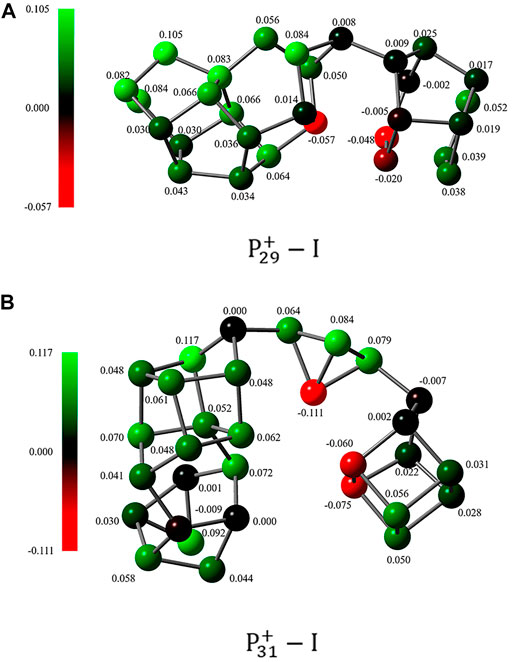
FIGURE 6. The NBO charge distribution of (A)
On the other hand, the short distances between the nonbonding phosphorus atoms indicate that the pnicogen bonds (Zahn, et al., 2011; Scheiner, 2013) inside the clusters may play a very important role. In

FIGURE 7. ELFs of (A)
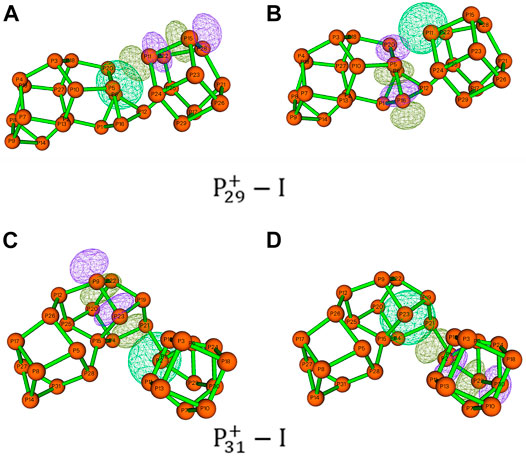
FIGURE 8. Hyperconjugation of LP(P) → σ*(P–P). The natural bonding orbits were obtained by NBO analysis of (A,B)
Both Wiberg bond index (WBI) and QTAIM topological analysis were employed to analyze the bonding nature of these bonds. The calculated total WBI values of the bonds P5···P11 in
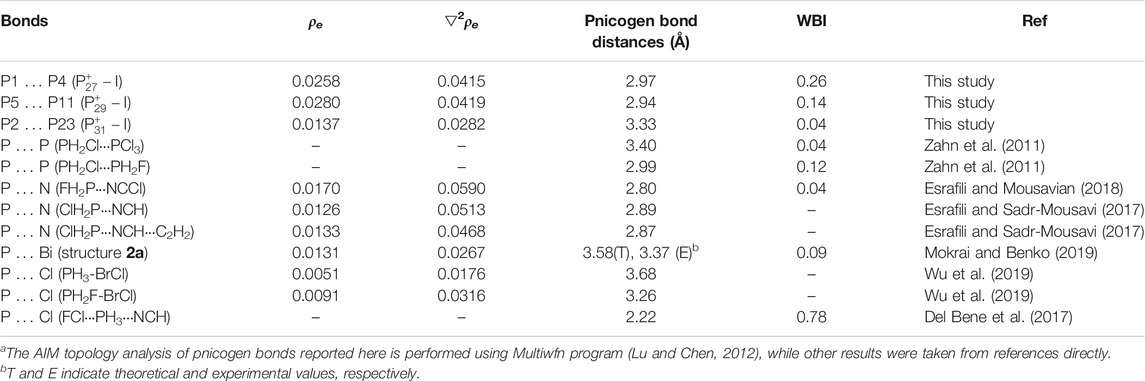
TABLE 1. Electron densities (ρe, a.u.), Laplacian of the electron densities (▽2ρe, a.u.), Pnicogen bond distances (Å), and Wiberg Bond Indexes (WBI) of the pnicogen bonds in
Although pnicogen bonds have been previously reported and studied for different species by many research groups, this is still the first study showing that the pnicogen bonds can exist and play important roles in homoatomic phosphorus clusters without the help of other ligands or heteroatoms. By comparing the pnicogen bonds reported herein with other previously reported pnicogen bonds, the important roles of these interactions in phosphorus clusters can be further reflected. Thus, some typical pnicogen bonds were selected from relative references (Zahn, et al., 2011; Del Bene et al., 2017; Esrafili and Sadr-Mousavi, 2017; Esrafili and Mousavian, 2018; Mokrai, and Benko, 2019; Wu, et al., 2019) and were compared with those bonds reported here. Results were shown in Table 1. The P … P pnicogen bonds reported here have similar bond distances and WBIs with those previously reported (Zahn, et al., 2011) and similar to other type pnicogen bonds including PN, P … Bi and P … Cl interactions. And the bond of P1 … P4 in
Briefly, these results reflected that the intracluster pnicogen bonds can greatly stabilize the cluster, thus play important roles in large-size phosphorus clusters and phosphorus-related materials. On the other hand, the ELF analysis shown in Figure 7 also indicates the possibility of the existence of multiple pnicogen bonds in large-size phosphorus clusters. And a very interesting topic is how the introduce of heteroatom can affect the pnicogen bonds and their most stable structures. So, we hope the result reported here can attract more researchers to focus on this issue.
Conclusion
A combined algorithm of BH and xTB to locate global minima in potential energy surface of atomic clusters has been developed here. Several strategies, including the similarity check, are considered in the algorithm. The
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
MZ and YX designed the program and performed the calculations. YC participated in the calculation. XZ and XK directed the work, contributed to the interpretation of the data and wrote the paper. All authors have read and approved the content of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (21627810, 11704004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.694156/full#supplementary-material
References
Bai, J., Cui, L.-F., Wang, J., Yoo, S., Li, X., Jellinek, J., et al. (2006). Structural Evolution of Anionic Silicon Clusters SiN(20 ≤N≤ 45). J. Phys. Chem. A. 110, 908–912. doi:10.1021/jp055874s
Ballester, P. J., Finn, P. W., and Richards, W. G. (2009). Ultrafast Shape Recognition: Evaluating a New Ligand-Based Virtual Screening Technology. J. Mol. Graphics Model. 27, 836–845. doi:10.1016/j.jmgm.2009.01.001
Ballester, P. J., and Richards, W. G. (2007a). Ultrafast Shape Recognition for Similarity Search in Molecular Databases. Proc. R. Soc. A. 463, 1307–1321. doi:10.1098/rspa.2007.1823
Ballester, P. J., and Richards, W. G. (2007b). Ultrafast Shape Recognition to Search Compound Databases for Similar Molecular Shapes. J. Comput. Chem. 28, 1711–1723. doi:10.1002/jcc.20681
Bannwarth, C., Ehlert, S., and Grimme, S. (2019). GFN2-xTB-An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-dependent Dispersion Contributions. J. Chem. Theor. Comput. 15, 1652–1671. doi:10.1021/acs.jctc.8b01176
Bulusu, S., and Zeng, X. C. (2006). Structures and Relative Stability of Neutral Gold Clusters: Aun (N=15-19). J. Chem. Phys. 125 (125), 154303. doi:10.1063/1.2352755
Carpenter, J. E., and Weinhold, F. (1988). Analysis of the Geometry of the Hydroxymethyl Radical by the “Different Hybrids for Different Spins” Natural Bond Orbital Procedure. J. Mol. Struct. THEOCHEM 169, 41–62. doi:10.1016/0166-1280(88)80248-3
Castleman, A. W., and Jena, P. (2006). Clusters: a Bridge between Disciplines. Proc. Natl. Acad. Sci. 103, 10552–10553. doi:10.1073/pnas.0601783103
Chen, X., Zhao, Y.-F., Wang, L.-S., and Li, J. (2017). Recent Progresses of Global Minimum Searches of Nanoclusters with a Constrained Basin-Hopping Algorithm in the TGMin Program. Comput. Theor. Chem. 1107, 57–65. doi:10.1016/j.comptc.2016.12.028
Chen, X., Zhao, Y. F., Zhang, Y. Y., and Li, J. (2019). TGMin: An Efficient Global Minimum Searching Program for Free and Surface‐supported Clusters. J. Comput. Chem. 40, 1105–1112. doi:10.1002/jcc.25649
Choi, T. H., Liang, R., Maupin, C. M., and Voth, G. A. (2013). Application of the SCC-DFTB Method to Hydroxide Water Clusters and Aqueous Hydroxide Solutions. J. Phys. Chem. B 117, 5165–5179. doi:10.1021/jp400953a
Cooks, R. G., Zhang, D., Koch, K. J., Gozzo, F. C., and Eberlin, M. N. (2001). Chiroselective Self-Directed Octamerization of Serine: Implications for Homochirogenesis. Anal. Chem. 73, 3646–3655. doi:10.1021/ac010284l
Daven, D. M., Tit, N., Morris, J. R., and Ho, K. M. (1996). Structural Optimization of Lennard-Jones Clusters by a Genetic Algorithm. Chem. Phys. Lett. 256, 195–200. doi:10.1016/0009-2614(96)00406-X
Deaven, D. M., and Ho, K. M. (1995). Molecular Geometry Optimization with a Genetic Algorithm. Phys. Rev. Lett. 75, 288–291. doi:10.1103/PhysRevLett.75.288
Del Bene, J. E., Alkorta, I., Elguero, J., and Sánchez-Sanz, G. (2017). Lone-Pair Hole on P: P···N Pnicogen Bonds Assisted by Halogen Bonds. J. Phys. Chem. A. 121 (6), 1362–1370. doi:10.1021/acs.jpca.6b12553
Du, Y., Sheng, H., Astruc, D., and Zhu, M. (2020). Atomically Precise Noble Metal Nanoclusters as Efficient Catalysts: A Bridge between Structure and Properties. Chem. Rev. 120, 526–622. doi:10.1021/acs.chemrev.8b00726
Esrafili, M. D., and Mousavian, P. (2018). The Strengthening Effect of a Halogen, Chalcogen or Pnicogen Bonding on Halogen-π Interaction: a Comparative Ab Initio Study. Mol. Phys. 116, 526–535. doi:10.1080/00268976.2017.1406166
Esrafili, M. D., and Sadr-Mousavi, A. (2017). Modulating of the Pnicogen-Bonding by a H⋯π Interaction: An Ab Initio Study. J. Mol. Graphics Model. 75, 165–173. doi:10.1016/j.jmgm.2017.04.017
Fehlner, T., Halet, J. F., and Saillard, J. Y. (2007). Molecular Clusters: A Bridge to Solid-State Chemistry. Cambridge: Cambridge University Press.
Ferrando, R. (2015). Symmetry Breaking and Morphological Instabilities in Core-Shell Metallic Nanoparticles. J. Phys. Condens. Matter 27, 013003. doi:10.1088/0953-8984/27/1/013003
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). “Gaussian 09,” in Revision D.01 (Wallingford, CT: Gaussian, Inc).
Grimme, S., Bannwarth, C., and Shushkov, P. (2017). A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All Spd-Block Elements (Z = 1-86). J. Chem. Theor. Comput. 13, 1989–2009. doi:10.1021/acs.jctc.7b00118
Guo, L., Wu, H., and Jin, Z. (2004). First Principles Study of the Evolution of the Properties of Neutral and Charged Phosphorus Clusters. J. Mol. Struct. THEOCHEM 677, 59–66. doi:10.1016/j.theochem.2004.02.014
Ha, M., Kim, J.-H., You, M., Li, Q., Fan, C., and Nam, J.-M. (2019). Multicomponent Plasmonic Nanoparticles: From Heterostructured Nanoparticles to Colloidal Composite Nanostructures. Chem. Rev. 119, 12208–12278. doi:10.1021/acs.chemrev.9b00234
Hartke, B. (1995). Global Geometry Optimization of Clusters Using a Growth Strategy Optimized by a Genetic Algorithm. Chem. Phys. Lett. 240, 560–565. doi:10.1016/0009-2614(95)00587-T
Hartke, B. (1993). Global Geometry Optimization of Clusters Using Genetic Algorithms. J. Phys. Chem. 97, 9973–9976. doi:10.1021/j100141a013
Huang, W., Sergeeva, A. P., Zhai, H.-J., Averkiev, B. B., Wang, L.-S., and Boldyrev, A. I. (2010). A Concentric Planar Doubly π-aromatic B19− Cluster. Nat. Chem 2, 202–206. doi:10.1038/nchem.534
Jäger, M., Schäfer, R., and Johnston, R. L. (2019). GIGA: a Versatile Genetic Algorithm for Free and Supported Clusters and Nanoparticles in the Presence of Ligands. Nanoscale 11, 9042–9052. doi:10.1039/C9NR02031D
Jana, G., Mitra, A., Pan, S., Sural, S., and Chattaraj, P. K. (2019). Modified Particle Swarm Optimization Algorithms for the Generation of Stable Structures of Carbon Clusters, Cn (N = 3-6, 10). Front. Chem. 7, 65–77. doi:10.3389/fchem.2019.00485
Jena, P., and Sun, Q. (2018). Super Atomic Clusters: Design Rules and Potential for Building Blocks of Materials. Chem. Rev. 118, 5755–5870. doi:10.1021/acs.chemrev.7b00524
Jiang, M., Zeng, Q., Zhang, T., Yang, M., and Jackson, K. A. (2012). Icosahedral to Double-Icosahedral Shape Transition of Copper Clusters. J. Chem. Phys. 136, 104501. doi:10.1063/1.3689442
Kanters, R. P. F., and Donald, K. J. (2014). Cluster: Searching for Unique Low Energy Minima of Structures Using a Novel Implementation of a Genetic Algorithm. J. Chem. Theor. Comput. 10, 5729–5737. doi:10.1021/ct500744k
Kong, X., Mu, L., Zhou, M., and Yang, S. (2019). “Phosphorus Clusters and Quantum Dots,” in Fundamentals and Applications of Phosphorous Nanomaterial. Editor H.-F. Ji (New York: ACS Symposium Series volume ACS Books), 79–102. doi:10.1021/bk-2019-1333.ch005
Kong, X., Tsai, I.-A., Sabu, S., Han, C.-C., Lee, Y. T., Chang, H.-C., et al. (2006). Progressive Stabilization of Zwitterionic Structures in [H(Ser)2-8]+ Studied by Infrared Photodissociation Spectroscopy. Angew. Chem. Int. Ed. 45, 4130–4134. doi:10.1002/anie.200600597
Kroto, H. W., Heath, J. R., O’Brien, S. C., Curl, R. F., and Smalley, R. E. (1985). C60: Buckminsterfullerene. Nature 318, 162–163. doi:10.1038/318162a0
Lazauskas, T., Sokol, A. A., and Woodley, S. M. (2017). An Efficient Genetic Algorithm for Structure Prediction at the Nanoscale. Nanoscale 9, 3850–3864. doi:10.1039/C6NR09072A
Li, J., Li, X., Zhai, H. J., and Wang, L. S. (2003). Au20: A Tetrahedral Cluster. Science 299, 864–867. doi:10.1126/science.1079879
Li, W.-L., Chen, X., Jian, T., Chen, T.-T., Li, J., and Wang, L.-S. (2017). From Planar boron Clusters to Borophenes and Metalloborophenes. Nat. Rev. Chem. 1, 0071. doi:10.1038/s41570-017-0071
Liu, L., and Corma, A. (2018). Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 118, 4981–5079. doi:10.1021/acs.chemrev.7b00776
Lu, T., and Chen, F. (2012). Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 33, 580–592. doi:10.1002/jcc.22885
Luo, Z., Castleman, A. W., and Khanna, S. N. (2016). Reactivity of Metal Clusters. Chem. Rev. 116, 14456–14492. doi:10.1021/acs.chemrev.6b00230
Martin, T. P. (1986). Compound Clusters. Z. Phys. D - Atoms, Mol. Clusters 3, 211–217. doi:10.1007/BF01384809
Mokrai, R., Barrett, J., Apperley, D. C., Batsanov, A. S., Benkő, Z., and Heift, D. (2019). Weak Pnictogen Bond with Bismuth: Experimental Evidence Based on Bi−P Through‐Space Coupling. Chem. Eur. J. 25, 4017–4024. doi:10.1002/chem.201900266
Mu, L., Yang, S., Bao, X., Yin, H., and Kong, X. (2015). Medium-sized Phosphorus Cluster cationsP+2m+1(6 ≤ M ≤ 32) Studied by Collision-Induced Dissociation Mass Spectrometry. J. Mass. Spectrom. 50, 1352–1357. doi:10.1002/jms.3705
Paz-Borbón, L. O., Mortimer-Jones, T. V., Johnston, R. L., Posada-Amarillas, A., Barcaro, G., and Fortunelli, A. (2007). Structures and Energetics of 98 Atom Pd-Pt Nanoalloys: Potential Stability of the Leary Tetrahedron for Bimetallic Nanoparticles. Phys. Chem. Chem. Phys. 9, 5202–5208. doi:10.1039/b707136a
Piazza, Z. A., Hu, H.-S., Li, W.-L., Zhao, Y.-F., Li, J., and Wang, L.-S. (2014). Planar Hexagonal B36 as a Potential Basis for Extended Single-Atom Layer boron Sheets. Nat. Commun. 5, 3113. doi:10.1038/ncomms4113
Rabanal-León, W. A., Tiznado, W., Osorio, E., and Ferraro, F. (2018). Exploring the Potential Energy Surface of Small lead Clusters Using the Gradient Embedded Genetic Algorithm and an Adequate Treatment of Relativistic Effects. RSC Adv. 8, 145–152. doi:10.1039/C7RA11449D
Reed, A. E., Curtiss, L. A., and Weinhold, F. (1988). Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 88, 899–926. doi:10.1021/cr00088a005
Rogan, J., Varas, A., Valdivia, J. A., and Kiwi, M. (2013). A Strategy to Find Minimal Energy Nanocluster Structures. J. Comput. Chem. 34, 2548–2556. doi:10.1002/jcc.23419
Scheiner, S. (2013). The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 46, 280–288. doi:10.1021/ar3001316
Scutelnic, V., Perez, M. A. S., Marianski, M., Warnke, S., Gregor, A., Rothlisberger, U., et al. (2018). The Structure of the Protonated Serine Octamer. J. Am. Chem. Soc. 140, 7554–7560. doi:10.1021/jacs.8b02118
Shayeghi, A., Götz, D., Davis, J. B. A., Schäfer, R., and Johnston, R. L. (2015). Pool-BCGA: a Parallelised Generation-free Genetic Algorithm for the Ab Initio Global Optimisation of Nanoalloy Clusters. Phys. Chem. Chem. Phys. 17, 2104–2112. doi:10.1039/C4CP04323E
VanGelder, L. E., Kosswattaarachchi, A. M., Forrestel, P. L., Cook, T. R., and Matson, E. M. (2018). Polyoxovanadate-alkoxide Clusters as Multi-Electron Charge Carriers for Symmetric Non-aqueous Redox Flow Batteries. Chem. Sci. 9, 1692–1699. doi:10.1039/c7sc05295b
VanGelder, L. E., Pratt, H. D., Anderson, T. M., and Matson, E. M. (2019). Surface Functionalization of Polyoxovanadium Clusters: Generation of Highly Soluble Charge Carriers for Nonaqueous Energy Storage. Chem. Commun. 55, 12247–12250. doi:10.1039/C9CC05380H
Wales, D. J., and Doye, J. P. K. (1997). Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. J. Phys. Chem. A. 101, 5111–5116. doi:10.1021/jp970984n
Wales, D. J., and Scheraga, H. A. (1999). Global Optimization of Clusters, Crystals, and Biomolecules. Science 285, 1368–1372. doi:10.1126/science.285.5432.1368
Wang, Y., Lv, J., Zhu, L., and Ma, Y. (2012). CALYPSO: A Method for crystal Structure Prediction. Comput. Phys. Commun. 183, 2063–2070. doi:10.1016/j.cpc.2012.05.008
Wu, J., Yan, H., Zhong, A., Chen, H., Jin, Y., and Dai, G. (2019). Theoretical and Conceptual DFT Study of Pnicogen- and Halogen-Bonded Complexes of PH2X---BrCl. J. Mol. Model. 25, 28. doi:10.1007/s00894-018-3905-3
Xue, T., Luo, J., Shen, S., Li, F., and Zhao, J. (2010). Lowest-energy structures of cationic P2m+1+ (m=1-12) clusters from first-principles simulated annealing. Chem. Phys. Lett. 485, 26–30. doi:10.1016/j.cplett.2009.12.019
Yañez, O., Báez-Grez, R., Inostroza, D., Rabanal-León, W. A., Pino-Rios, R., Garza, J., et al. (2019). AUTOMATON: A Program That Combines a Probabilistic Cellular Automata and a Genetic Algorithm for Global Minimum Search of Clusters and Molecules. J. Chem. Theor. Comput. 15, 1463–1475. doi:10.1021/acs.jctc.8b00772
Yang, S., Mu, L., and Kong, X. (2016). Collision-induced dissociation mass spectrometry of phosphorus cluster anionsP2m+1−(3 ≤ m ≤ 20). Int. J. Mass Spectrom. 399-400, 27–32. doi:10.1016/j.ijms.2016.02.006
Yoo, S., Zhao, J., Wang, J., and Zeng, X. C. (2004). Endohedral Silicon Fullerenes SiN(27 ≤N≤ 39). J. Am. Chem. Soc. 126, 13845–13849. doi:10.1021/ja046861f
Zahn, S., Frank, R., Hey‐Hawkins, E., and Kirchner, B. (2011). Pnicogen Bonds: A New Molecular Linker?. Chem. Eur. J. 17, 6034–6038. doi:10.1002/chem.201002146
Zhai, H.-J., Zhao, Y.-F., Li, W.-L., Chen, Q., Bai, H., Hu, H.-S., et al. (2014). Observation of an all-boron fullerene. Nat. Chem 6, 727–731. doi:10.1038/NCHEM.1999
Zhan, L., Chen, J. Z. Y., Liu, W.-K., and Lai, S. K. (2005). Asynchronous multicanonical basin hopping method and its application to cobalt nanoclusters. J. Chem. Phys. 122, 244707. doi:10.1063/1.1940028
Zhang, J., and Glezakou, V. A. (2020). Global optimization of chemical cluster structures: Methods, applications, and challenges. Int. J. Quan. Chem. 121, e26553. doi:10.1002/qua.26553
Zhao, Y., Chen, X., and Li, J. (2017). TGMin: A global-minimum structure search program based on a constrained basin-hopping algorithm. Nano Res. 10, 3407–3420. doi:10.1007/s12274-017-1553-z
Zhou, C., Ieritano, C., and Hopkins, W. S. (2019). Augmenting Basin-Hopping With Techniques From Unsupervised Machine Learning: Applications in Spectroscopy and Ion Mobility. Front. Chem. 7, 519. doi:10.3389/fchem.2019.00519
Keywords: global optimization, atomic clusters, basin-hopping algorithm, phosphorus cluster cations, xTB method, pnicogen bond
Citation: Zhou M, Xu Y, Cui Y, Zhang X and Kong X (2021) Search for Global Minimum Structures of
Received: 12 April 2021; Accepted: 15 July 2021;
Published: 26 July 2021.
Edited by:
Ambrish Kumar Srivastava, Deen Dayal Upadhyay Gorakhpur University, IndiaReviewed by:
Ruby Srivastava, Centre for Cellular and Molecular Biology (CCMB), IndiaWei-Ming Sun, Fujian Medical University, China
Copyright © 2021 Zhou, Xu, Cui, Zhang and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianyi Zhang, eHl6aGFuZ0BhaG51LmVkdS5jbg==; Xianglei Kong, a29uZ3hpYW5nbGVpQG5hbmthaS5lZHUuY24=