 Jyoti Chauhan
Jyoti Chauhan Srinivas R. Maddi2
Srinivas R. Maddi2 Kshatresh Dutta Dubey
Kshatresh Dutta Dubey Subhabrata Sen
Subhabrata Sen- 1Department of Chemistry, School of Natural Sciences, Shiv Nadar University, Greater Noida, India
- 2Acubiosys PVT LTD, TBI, BITS-Pilani Campus, Hyderabad, India
Indoleamine-2,3-dioxygenase (IDO1) and tryptophan dioxygenases are two heme based metalloenzymes that catalyze the tryptophan oxidation reaction by inserting molecular dioxygen to cleave the pyrrole ring. The mechanism of such ring cleavage reaction is of carcinogenic importance as the malignant tumors recruit this mechanism for immune invasion. In the presence study, we have synthesized a Novel C2 aroyl indoles inhibitor, 8d, which shows significant inhibition of 180 nM at IC50 scale. The binding and conformational changes that transpire after inhibitor binding were thoroughly studied by molecular docking and MD simulations. The subsequent QM/MM (Quantum Mechanical/Molecular Mechanical) calculations were used to proposed the mechanism of inhibition. The QM/MM calculations show that the reaction proceeds via multistep processes where the dioxygen insertion to the substrate 8a is the rate determining process. Theoretical mechanism is further supported by mass spectroscopy, and drug metabolism/pharmacokinetics study (DMPK) and metabolic stability of compound 8d was investigated in rat and human liver microsomes.
Introduction
The transformation of l-tryptophan (L-tryp) to kynurenine is an essential chemical reaction which is im-plicated in various biological process and in turn can be exploited as a therapeutic options in cancer, neurodegeneration, aging and immunity (Dunn et al., 2004; Munn and Mellor, 2007; Kershaw et al., 2014; Sas et al., 2018). Indoleamine-2, 3-dioxygenase 1 (IDO1) catalyzes the initial and rate limiting step of this reaction (Pradhan et al., 2017). Despite exhibiting potential as a therapeutic target in myriad diseases, IDO1 is mainly implicated in cancer (Okamoto et al., 2005; Zou, 2005; Sugimoto et al., 2006; Mazarei and Leavitt, 2015; Song et al., 2018).
Various bioactive small molecules belonging to different classes of heterocycles have been reported as IDO1 inhibitors (Dolušić and Frédérick, 2013; Muller et al., 2005). Interestingly among these plethora of reported IDO1 inhibitors only few have hitherto entered clinical trials. Representatively, 1-methyl-d-tryptophan 1a (DIMT) (Indoximod) (Figure 1) is the first clinical candidate, with an IC50 of ∼100 μM against hIDO1 and IC50 of 1.5 mM against HeLa and HT29 cells respectively (Vacchelli et al., 2014). The next to enter clinical trial was Incyte Corporation’s Epocodostat 1b belonging to hydroxiamidine class of compounds which inhibited IDO1 with an IC50 of 72 nM (Figure 1). (Prendergast et al., 2017) Another clinical candidate (phase I), the imidazole Novoximid, 1c evolved from Newlink-Genentech’s combined effort (Figure 1). It inhibits IDO1 and T-REX-293 IDO1 with IC50 of 13 and 75 nM respectively (Li et al., 2017). In 2018, two more molecules, Pfizer-iTeos′ indole derivative, 1d and Bristol Myers Squibb’s quinoline compound 1e entered the clinical trials (Figure 1) (Siu et al., 2017; Wen et al., 2019).
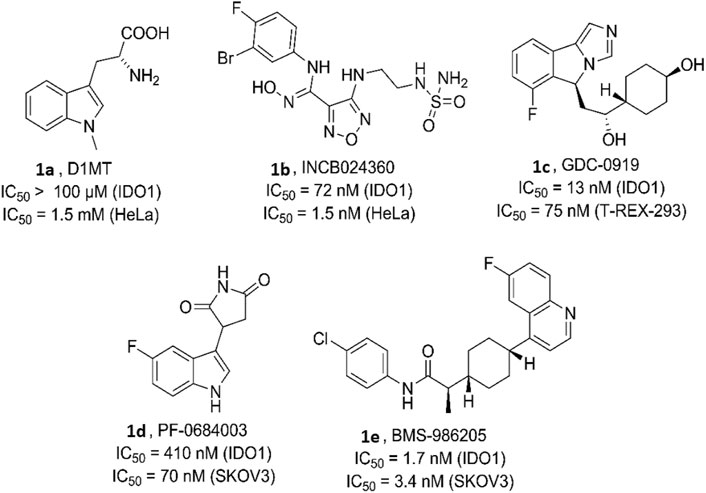
FIGURE 1. Human indoleamine-2, 3-dioxgenase (hIDO) inhibitors as clinical candidates.
Despite recent failures of clinical candidates targeting IDO1, the new emerging up regulation of TDO and the advent of the combination therapy, the interest in IDO1 inhibitors as a potential anticancer drug has not deterred (Van den Eynde et al., 2020). There are still several articles reporting the design, synthesis and in vitro/in vivo evaluation of IDO1 inhibitors. However such “run-of-the-mill” reports does not help in rectifying the shortcomings that led to the road block for the development of IDO1 inhibitors as successful clinical candidates, as it is important to conduct studies that investigate the underlying mechanism of action of these small molecules with the heme protein (IDO1). Critical analysis of the metabolites generated during the reaction could suggest novel molecules potentially better than the present IDO1 inhibitors. There has been reports of thorough investigation on the mechanism by which the two heme enzymes IDO1 and TDO2 catalyzes the degradation of l-tryptophan (Chauhan et al., 2009; Basran et al., 2011). There is also a study published by Röhrig et al. that included investigation of mechanism of action of four known IDO1 lead molecules through computational and experimental approaches (Matin et al., 2006; Paul et al., 2016; Röhrig et al., 2019). Surprisingly, similar studies to understand the interaction of IDO1 or TDO2 with novel inhibitors are missing from the literature. Herein we report our investigation of IDO1 catalyzed degradation of compound 8d, a C2-aroyl indole based novel inhibitor of IDO1, by UV absorbance spectroscopy, Quantum mechanics/molecular mechanics (QM/MM) calculations and molecular dynamic (MD) simulations. The outcome of the computational experimentation was corroborated by the mass spectroscopy results and afforded further substantiation of the mechanism. Additionally, DMPK studies of 8d and two other potent C2-aroyl indoles revealed the druggable attributes of these compounds.
The background of the discovery of compound 8d (Figure 2) as a novel selective IDO1 inhibitor (over TDO2) involved random screening of various inhouse compounds against recombinant protein of human IDO1 enzyme (percentage inhibition at 5 μM concentration (Supplementary Figure S1 [refer SI]). The molecule that provided the best inhibition from this preliminary screening was the C2-aroyl tryptamine derivative 8a that inhibited IDO1 to ∼40% (Figure 2) (Supplementary Figure S1 [refer SI]. A library based on 8a was generated through a novel oxone mediated green oxidative ring opening of tetrahydrocarbolines 9 and 10 (Figure 2) (Supplementary Table S1 and Supplementary Figure S2 [Refer SI]). Percentage inhibition screening of this library at 5 μM concentration of the compounds and subsequent dose response study provided compounds 8a, d and e that induced substantial inhibition to IDO1 with IC50 of 1.45, 0.18 and 1.52 μM respectively compared to 200 μM by 1-LMT and 0.1 μM by NLG-919, which were used as the positive standards in the screening (Figure 2). The IC50 of compounds 8a, d and e against recombinant protein of human TDO2 enzyme were 6.95, 9.16, and 16.82 μM respectively (Figure 2). Based upon the biological studies, compound 8d emerged as the most potent molecule of our molecular library against IDO with reasonable selectivity over TDO. To understand the mechanism of action of our molecules compound 8d was selected for UV-Vis absorption, reaction kinetics and mass spectroscopic investigation. Additionally, compound 8d was used in the QM/MM calculations and molecular dynamic (MD) simulations.
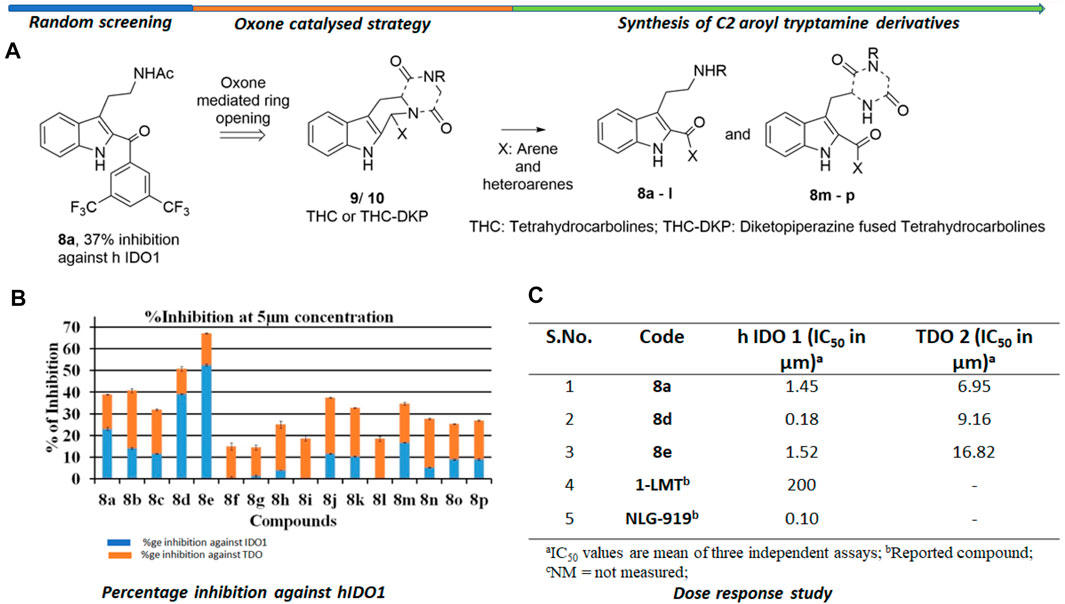
FIGURE 2. Background study of the evolution of compound 8d as a IDO one inhibitor: (A) Design and synthesis of C2 aroyl tryptamine derivatives 8b → p; (B) Percentage inhibition induced by compounds 8a → p on IDO1 at 5 μM concentration; (C) Dose response study of compounds 8a, d, and e. For X and R, please refer to the supplementary information, Supplementary Figure S2.
Materials and Methods
Experimental Section
Materials and Instruments
All reactions were carried out under N2 atmosphere as specified. Column chromatography was performed on Silica gel (60–120 mesh), and reactions was monitored by thin layer chromatography (TLC, Silica gel 60 F254), using UV light to visualize the course of the reaction. 1H NMR and 13C NMR spectra were recorded with tetramethylsilane as an internal standard at ambient temperature unless otherwise indicated with Bruker instruments at 400 MHz for 1H NMR and 100 MHz for 13C NMR spectroscopy. Splitting patterns are designated as singlet (s), broad singlet (br, s), doublet (d), triplet (t). Splitting patterns that could not be interpreted or easily visualized are designated as multiplet (m). Mass spectrometry analysis was done with a 6,540 Accurate-Mass QTOF LC-MS system (Agilent Technologies) equipped with an Agilent 6540 LC system. Accurate mass analysis calibration was carried out by ESI-low concentration tuning mix solution provided by Agilent technologies, United States. High performance liquid chromatography (HPLC) experiments were carried out on Agilent 1,290 infinity series of LC system (Agilent Technologies, United States) with photodiode-array/ELSD detector to monitor the purity of compounds. High performance liquid chromatography (HPLC) experiments were carried out on a Waters Alliance System (Milford, MA) consisting of e2695 separation module and a 2,998 photodiode-array detector. The HPLC system was controlled with EMPOWER software (Waters Corporation, Milford, MA) obtained by the Department of Chemistry, School of Natural Sciences, Shiv Nadar University, Uttar Pradesh 201,314, India.
General procedure for preparation of the target compounds 8a - p.
The oxone (1.2 equiv.) was added to a solution of 9a - n or 10a - d (1.0 equiv.) and KBr (0.05 equiv.) in MeCN/H2O (10/1) at 0°C (see SI for the preparation of (9a - n or 10a—d). The reaction mixture was allowed to warm to r. t. and was stirred for another 2–12 h. Once TLC confirmed the complete consumption of the starting material, the reaction was quenched with aqueous saturated sodium bicarbonate (aq. sat. NaHCO3) and aqueous saturated sodium sulfite (aq. sat. Na2SO3) followed by diluting it with ethyl acetate (EtOAc). The organic fractions were collected, and the aqueous phase was extracted twice with ethyl acetate. The combined organic fractions were washed with brine, dried over anhydrous magnesium sulfate (anh. MgSO4), filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (EtOAc/hexane = 1:10 → 1:1) to provide the desired C2-aroyl compounds. A comprehensive protocol for the synthesis of compounds 8a—p is discussed in SI.
Enzymatic Assay In-Vitro
The enzyme culture materials Potassium phosphate, dipotassium hydrogen phosphate, 1-methyl-l-tryptophan, l-kynurenine, l-tryptophan, catalase, trichloroacetic acid (TCA), p-Dimethylaminobenzaldehyde (Ehrlich’s reagent) and sodium dithionate were purchased from sigma Aldrich and methylene blue, dimethyl sulfoxide (DMSO), glacial acetic acid, ascorbic acid, glycerol and tris-HCL buffer were purchased from Sisco Research Laboratories, India. The recombinant human IDO/TDO protein were purchased from Biovision (Catalog no.: P1096 human recombinant IDO1 and CSB-YP023351HU human recombinant TDO2). All the other HPLC grade solvent were purchased from Merck Pvt. Ltd. The IDO/TDO assays were performed according to the reported procedures.1 and 2, the details of which is mentioned in the SI.
UV-Vis Absorption
All the measurements of binding of compound and enzyme were recorded at room temperature using an Agilent Cary 8454 UV-Vis diode array spectrophotometer with 1 cm light path. The experiments were performed in 50 mM potassium phosphate buffer pH- 6.5 with IDO1 protein concentration of 1 µM and compound concentration of 25 µM was incubate for 3 h at 37°C. The deoxy-form of protein was prepared by pre-purged with nitrogen gas and adding sodium dithionate (∼5 fold excess) in protein with potassium phosphate system.
Inhibition Kinetics
The kinetic mode of inhibition of highest active compound against indolamine-2, 3-dioxygenase one was determined by series of test solution in which the concentrations of respective substrate l-tryptophan (1–4 mM) was varied in presence of different concentrations of the inhibitors (0–1.5 µM). The IDO1 activity assay was performed to measure the formation of N-formylkynurenine by absorbance plate reader at 492 nm. The mode of inhibition of the screened compounds was determined from the Lineweaver-Burk plots, which are the double reciprocal plots of enzyme reaction velocities (1/V) vs. substrate concentrations (1/[S]), where V and [S] represent the initial rate of enzymatic reaction and l-tryptophan concentration respectively.
Mass Spectroscopy Analysis
Metabolic analysis assays were done using an Ultra-performance liquid chromatography (UPLC), an Agilent 6,540 accurate–mass Q-TOF LC/MS (Agilent Technologies, United States). Liquid chromatographic separation was performed at room temperature of 18°C, using a UPLC C18 analytical column (kinetex C18 100 A Column, 4.6 × 50 mm, 2.6 micron). MS analyses were performed under the following operation parameters: dry gas temperature 350°C, dry gas (N2) flow rate 10 L/min, nebulizer pressure 30 psi, Vcap 4,000 and fragmentor voltage 120 V. Mass spectra were acquired in the positive ion mode as well as negative ion mode by scanning from 50 to 1,500 in the mass to charge ratio (m/z).
The assay was performed in 50 mM Tris-HCl buffer (pH- 8.0), 1.0 mM sodium ascorbate (pH- 7.0), 1.0 μg/ml of IDO enzyme protein and 1 mM of inhibitor 8d was added in to the deep well plate and incubate for 3 h at 37°C. After incubation, 100 µL of acetonitrile (HPLC grade) was added and the samples were centrifuged for 15 min at 13,000 rpm at 4°C in Amicon® Ultra-15 centrifugal filter devices. The 200 µL of supernatant was then analyzed by Q-TOF LC/MS. For standardization of protocol same conditions was performed with 1.0 mM l-tryptophan to analyze the formation of N-formyl-kynurenine and N-kynurenine. The preparation of stock solution of inhibitor 8d was prepared in DMSO.
Drug Metabolism and Pharmacokinetics (DMPK Studies)
For the DMPK studies of the compound 8a-d we used rat as well as human plasmas. Rat plasma was purchased from Hylasco Biotechnology Pvt. Ltd.™ Hyderabad, India. We have not performed any in vivo experiments for this manuscript neither for animals nor for humans. Hence, we did not require any approval from the ethics committee. Also please note the blood plasma was obtained from the blood of Dr Srinivas R Maddi, who donated it and is the co-author of this article. The DMPK studies were supplemented by kinetic solubility, Log p and Log D determinations, PAMPA permeability, Metabolic and Plasma stability in rat and human, and finally the plasma folding in rat and human. A thorough discussion of each step is mentioned in the supplementary information.
Computational Section
Computational Setup
Since the crystal structure of the hIDO in presence of the inhibitors (8a–8e) were not available, we started with the Molecular Docking of the inhibitors 8a to 8e. The initial coordinates of the -IDO were imported from the protein data bank (PDB: 2DOT). The crystal structure contains 4-phenylimidazole as a bound ligand, therefore, we used the same binding site during docking of drugs 8a-e. During docking, we found the docking score of 8d as the best among all inhibitors. Therefore we chose the best pose of 8d for the further Molecular Dynamics simulations. Prior to MD simulations, all missing loops were modeled by MODELLER program, while we used Autodock Vina for all molecular docking calculations using UCSF Chimera (Pettersen et al., 2004).
During the MD simulations setup, the missing hydrogens and other heavy atoms were added by the leap module of the Amber20 program. For the protein residues we used Amber ff14SB forcefield while the parameters for the substrate was prepared using the antechamber module of the amber using Amber generalized forcefields for organic molecules (GAFF2). The partial charged of the substrate was calculated by the RESP charge fitting of a Quantum Mechanical (QM) optimized geometry at HF/6–31 g* level of theory (Bayly et al., 1993; Cornell et al., 1993). Parameters for the heme-bound histidine were prepared using the MCPB (Metal Center Parameter Builder Program) implemented in the Amber MD package. The pKa value of histidine, Aspartate and Glutamate were checked by propKa 3.0, and we found that the pKa of all these residues were well under 6.5. Therefore, these residues were kept at their default protonation state. However, Histidine residues were further checked for either as HIE (protonation at e position) or HID (protonation at δ position), depending upon the possible interactions with nearby polar group. A Na + ions was added into the protein surface to neutralize the total charge of the system. Finally, the resulting system was solvated in a rectangular box of TIP3P waters extending up to minimum cutoff of 10 Å from the protein boundary which resulted overall system size containing 47,531 atoms including protein, water and a Na + ion.
Following system setup and parametrization of the complex, the resulting geometries of the systems were minimized to remove the poor contacts and relax the proteins. The systems were then gently annealed from 10 to 300 K under the NVT ensemble for 50 ps? Subsequently, the systems were maintained for 1 ns in the NPT ensemble at a target temperature of 300 K and with the target pressure of 1.0 atm. using the Langevin thermostat (Jorgensen et al., 1983) and Berendsen barostat (Izaguirre et al., 2001) with collision frequency of 2 ps and pressure relaxation time of 1 ps. This was followed by further equilibrations for ∼3 ns for each system. Finally, a productive MD run was performed for 100 ns for the complex. The 100 ns simulation was performed using a multi-trajectory approach in which we performed two consecutive simulations each of 50 ns. At every 50 ns we restarted the simulation for the next 50 ns with a random velocity. During all MD simulations, the hydrogen bonds were constrained using SHAKE (Berendsen et al., 1984) and particle mesh Ewald (PME) (Ryckaert et al., 1977) was used to treat long-range electrostatic interactions. All MD simulations were performed with the GPU version of Amber 20 package (Darden et al., 1993).
Quantum Mechanical/Molecular Mechanical Setup
The equilibrated snapshot from the MD simulations were used for the QM/MM calculations. All water molecules beyond 6Å of the protein were stripped from the MD snapshot. The QM region during the QM/MM calculations consists of truncated heme-porphyrin, O2, the imidazole group of proximal histidine, and the substrate. The atomic coordinates for the QM region can be found in the supplementary information (SI) document. The protein and water residues as close as 8 Å of the QM regions were treated as active atoms to account for their effect due to electrostatic and van der Waals interactions. All QM/MM calculations were performed using ChemShell (Sherwood et al., 2003; Salomon-Ferrer et al., 2013) combining Turbomole (Metz et al., 2014) for the QM part and DL_POLY (Ahlrichs et al., 1989) for the MM part using the Amber force field. The electronic embedding scheme (Smith and Forester, 1996) was used to account for the polarizing effect of the enzyme environment on the QM region. Hydrogen link atoms with the charge-shift model (Sherwood et al., 2003; Salomon-Ferrer et al., 2013) was applied to treat the QM−MM boundary. In QM/MM geometry optimizations, the QM region was treated using the hybrid UB3LYP functional with two basis sets, B1 and B2, where B1 stands for LACVP* and B2 is for def2-TZVP. For geometry optimization and frequency calculations, we used B1, i.e., LACVP basis set. All of the QM/MM transition states (TSs) were located by relaxed potential energy surface (PES) scans followed by full TS optimizations using the P-RFO optimizer implemented in the HDLC code. The energies were further corrected with the large all-electron def2-TZVP basis set. The zero-point energy (ZPE) was calculated for all species, and all of the final energies are reported as UB3LYP/B2+ZPE. The feasibility of these protocol has already been validated in several other studies by us in similar heme metalloenzymes (Dubey et al., 2017; Dubey and Shaik, 2018; Dubey and Shaik, 2019; Kalita et al., 2020). Previous studies (Capece et al., 2012; Shin et al., 2018) show Fe+3 and triplet state as the ground state for the hIDO mediated reactions, therefore we used the same oxidation and spin state in our calculations.
Results and Discussion
UV-Vis Absorption Study
To unravel the mechanism by which our ligands interact with IDO1, we utilized the UV-Vis absorbance studies to understand the ligand-binding ability of compound 8d with IDO1 enzyme. For better comparison the next best potent inhibitors compounds 8a and e were also included. It is known that the heme group in the IDO1 enzyme undergoes change in optical properties such as optical rotation and absorption wavelength (in the UV-Vis region) based on the oxidation state of the metal of the heme part and on the nature of the ligands it is bound to (Soret, 1883). Our investigation correlates the IC50 values of inhibition of IDO1 enzyme by ligands 8a, d and e with their binding ability at the active site of the enzyme. It further demonstrated that heme binding ligands like ours increases the stability of the enzyme under the experimental condition. Accordingly, the UV-Vis spectra of ferric-IDO1 and deoxy ferrous-IDO1 were measured in presence and absence of ligands 8a, d and e (Figure 3 1) and b). In absence of the ligands the UV-VIS spectra of ferric-IDO1 in potassium phosphate buffer at pH - 6.5 exhibited a soret peak at 406 nm, which remained stable up to 3 h (Figure 3A). Interestingly, in presence of the ligands at 25 µM the soret peak of Fe3+-IDO1 remain unchanged at 406 nm with slight increase in intensity for 8a and 8d and decrease for 8e (Figure 3A). This alteration of intensity of the soret band further suggest that the change in absorbance intensity of ligands with ferric-IDO1 (8d > 8a > 8e) can directly correlates with their inhibition potency (8d > 8a > 8e) (Figure 2) against IDO1 enzyme. In Figure 3B, the deoxy ferrous-IDO1 exhibited a soret peak at 414 nm. In the presence of compounds 8a, d and e the soret band shifted to 407 nm which nearly coincided with the soret band of Fe3+-IDO1 (Figure 3A). This suggested that not only the ligands bound to the deoxy ferrous-IDO1 but also reacted with it to oxidize it back to the Fe3+ form. With compound 8a the soret band also shifted to 430 nm, and according to reported literature this is typical a six coordinate low spin heme species complex bind with ligands (Figure 3B). (Terentis et al., 2002) Although proof of coordination of the compounds to the heme iron requires some further studies, these preliminary results strongly support the binding of all the three ligands 8a, d and e to IDO1 (Figure 3).
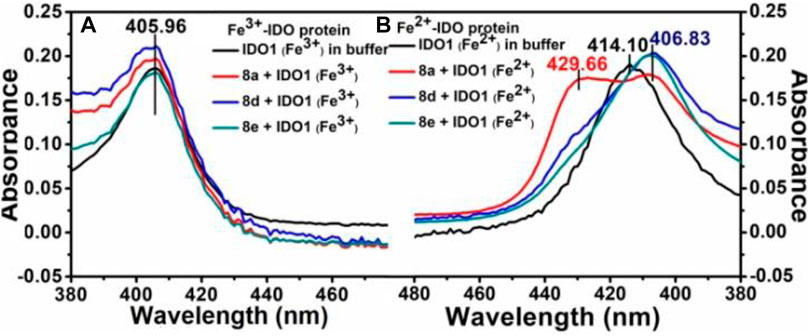
FIGURE 3. UV-Vis spectroscopic analyses of compounds 8a, d and e with hIDO1, Experiment on enzyme kinetics to understand the mode of inhibition.
Next, It was important to understand the mode of inhibition of compound 8d with IDO1. We performed enzyme kinetics experiments and obtained the Lineweaver-Burke plots (Figure 4). The plots of 1/V vs. 1/[S] divulged that 8d inhibited IDO1 enzyme in a competitive manner (Figure 4). V and [S] depicted the initial rate of the reaction and the substrate concentration respectively. It is noteworthy, that the observed competitive mode of inhibition is with respect to IDO1. It may not be same for with respect to O2 (the other substrate in the L-Trp catabolism pathway). For this reason in order to determine the true mode of inhibition of IDO1 by 8d additional kinetics measurements are required.
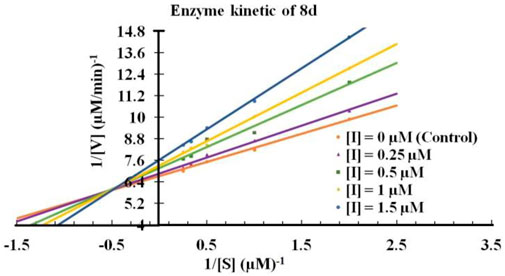
FIGURE 4. Lineweaver-Burke plot of compound 8d against IDO enzyme. Molecular dynamics simulation of compound 8d with IDO1.
To get a microscopic insight of the substrate and enzyme interactions, we further enumerate the binding mode and crucial interactions between the inhibitor 8d and IDO using molecular dynamics (MD) simulation in deoxy and resting state of heme. In both of the simulations the inhibitor 8d stayed close to the catalytic center and occupied the axial position of heme prosthetic group. The binding stability of 8d was mainly due to a stable stacking interaction with one of the pyrrole rings of heme-porphyrin and aromatic side chain of 8days. A thorough inspection of MD trajectories showed that Tyr115, Ser224, and Leu373 surrounded the drug and provided an additional stability to the protein-drug complex (Figure 5). As can be seen from Figure 5, these residues form the active site architecture and provide a tight packing to the substrate via polar and hydrophobic interactions.
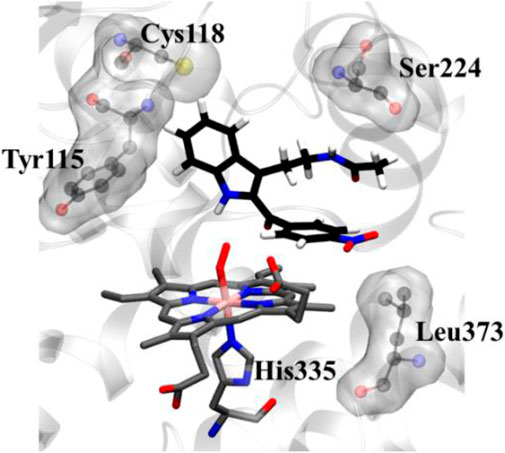
FIGURE 5. Key protein residues that forms the binding cleft for compound 8d. The hydrogen atoms for the heme moiety are hidden for the sake of clarity. QM/MM calculations to investigate the IDO1 catalyzed degradation of compound 8d.
In addition, the indole ring of compound 8d was also in the close contact with the Cys118 which may have act as a base for the catalytic reaction. A previous structural comparison of TDO with IDO indicated a Cysteine residue (C118) in IDO as a catalytic analogue of H55 of TDO (Uyttenhove et al., 2003). Incidentally, this C118 residue was far away from the catalytic center in the crystal structure of IDO-1 which had raised doubt on the role of Cysteine as a catalytic mechanism of IDO-1. However to our surprise, this Cys118 residue comes close to the substrate during the MD simulations which indicated that the presence of substrate may have instigated a local active site rearrangement. The role of Cysteine 118 could not be fully downplayed in the catalysis mechanism of IDO-1 and whether it really played a role in the catalysis is an open question (Figure 5). As classical MD simulations cannot describe the reaction mechanism, subsequently the QM/MM calculations were performed to study the mechanism of the catalysis as discussed in the next section.
To understand the degradation of ligand 8d with IDO1, the reaction mechanism (as depicted in Scheme 1) was modeled using hybrid QM/MM calculations (refer Materials and Methods) for a representative snapshot from the MD simulations (Figure 6). This reaction mechanism was based on the putative mechanism of the catabolism of l-tryptophan by the heme protein IDO1 as reported in the literature and from our initial UV-Vis absorption studies, which revealed the change in the oxidation state of the Fe moiety in IDO1 (Chauhan et al., 2009; Basran et al., 2011). The strategy involves enzyme dynamics at room temperature during the reaction while generating the quantum mechanical accuracy for the description of the active site atoms. This approach has been utilized successfully to model reaction mechanics in a diverse range of enzyme reaction.
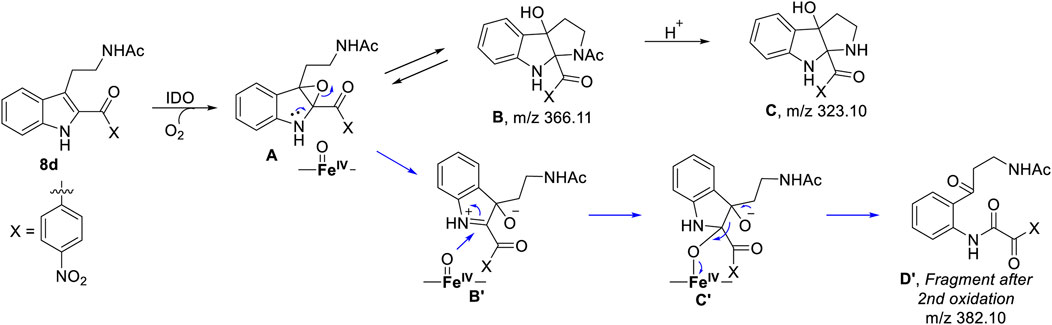
SCHEME 1. Probable mechanism of the degradation of compound 8d based on UV-Vis spectroscopy, molecular dynamics simulation and QM/MM calculations.
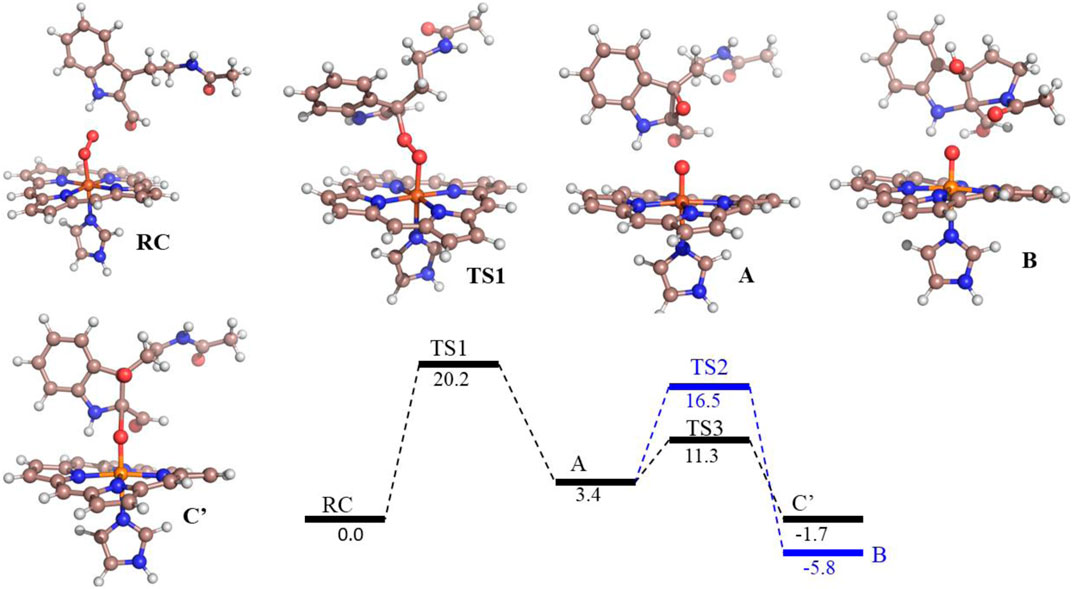
FIGURE 6. Potential energy profile of the reaction of compound 8d with hIDO1. Energies (in kcal/mol) are represented as QM(B1+ZPE)/MM followed by QM(B2+ZPE)/MM levels of theory where B1 is B3LYP-D3/LACVP* and B2 is B3LYP-D3/def2-TZVP Note that propionate group of heme, and benzoate group of substrate is truncated.
To get the energy profile of the reaction, we started the PES scanning from 8d. The energy profile of this reaction along with the key intermediates are shown in Scheme 1/Figure 6. As it can be observed that, the reaction could begin with the conversion of compound 8d (Figure 6), to the intermediate A (Figure 6) via a transition state TS1 (Figure 6). The energy barrier of 20.2 kcal/mol, for this step is significantly high and therefore could be the rate determining process. The calculations indicated that the formation of A is endothermic and can easily transform to B or C′ through low transition state barriers of 16.5 kcal/mol and 11.3 kcal/mol respectively. Therefore, the oxygen insertion from dioxyheme could be a stepwise process composed of two stages. At the initial stage, the reaction could have started with the bridging of molecular oxygen with the inhibitor 8d which formed an epoxy indole intermediate at the cost of high energy. In the second step this epoxy-indole intermediate underwent the second oxygen insertion to ultimately afford the product. Surprisingly, as mentioned in previous section, we could not identify the involvement of cysteine residue as a possible base in the catalytic machinery. In summary, the theoretical calculations, predicted that the catalysis could have proceeded in a multistep sequence via different intermediates A, B and C′, where the epoxidation could be the rate determining step (Figure 6) (Sugimoto et al., 2006).
Mass spectroscopy studies of the metabolism of compound 8d by IDO1.
As depicted in Figure 6 above, we understand that compound 8d and l-tryptophan could initiate a competition between them for binding to the active site of IDO1 enzyme (deemed by QM/MM calculations. This encouraged us to corroborate our the predicted mechanism of the catabolism of 8d with IDO1 via mass spectroscopic analysis (LC-MS). Accordingly IDO1 was incubated with the ligand in tris-HCl buffer (pH 8.0) and ascorbic acid at 37°C for 3 h (refer method and material section). An aliquot of the reaction mixture was then analyzed through QTOF LC-MS. In the LC-MS analysis a peak eluting with m/z 382 was observed in addition to the peaks with m/z 366 and 323. The peak with m/z 366 is consistent with insertion of a single oxygen atom into the substrate (m/z 8d = 350.11) (refer Figure 7). Using selected ion monitoring, the elution profile revealed a peak with m/z 175 (RT 2.69 min) (refer Figure 7) which could have been generated by the loss of C2 aroyl moiety from intermediate C (refer Scheme 1 and Figure 7). We further speculated that the peak with m/z 382 corroborates with the formation of D′, the dioxygenated ring opened analogue of N-formyl kynuramine (NFK) (refer Scheme 1 and Figure 7). We further anticipated that the peak with m/z 366 may have originated from the initial transformation of compound 8d to the 2, 3-epoxide derivative A (as the rate determining step by QM/MM calculations [Figure 6]) which could have immediately converted to the cyclic amino acetal intermediate B (same m/z as the epoxide A). The formation of B could be further established either from the peak with m/z 323 (refer Scheme 1 and Figure 7) which could be either assigned to the deacetylated intermediate C or by the MS-MS of intermediate B (refer Figure 7) (corroborated by MS-MS).
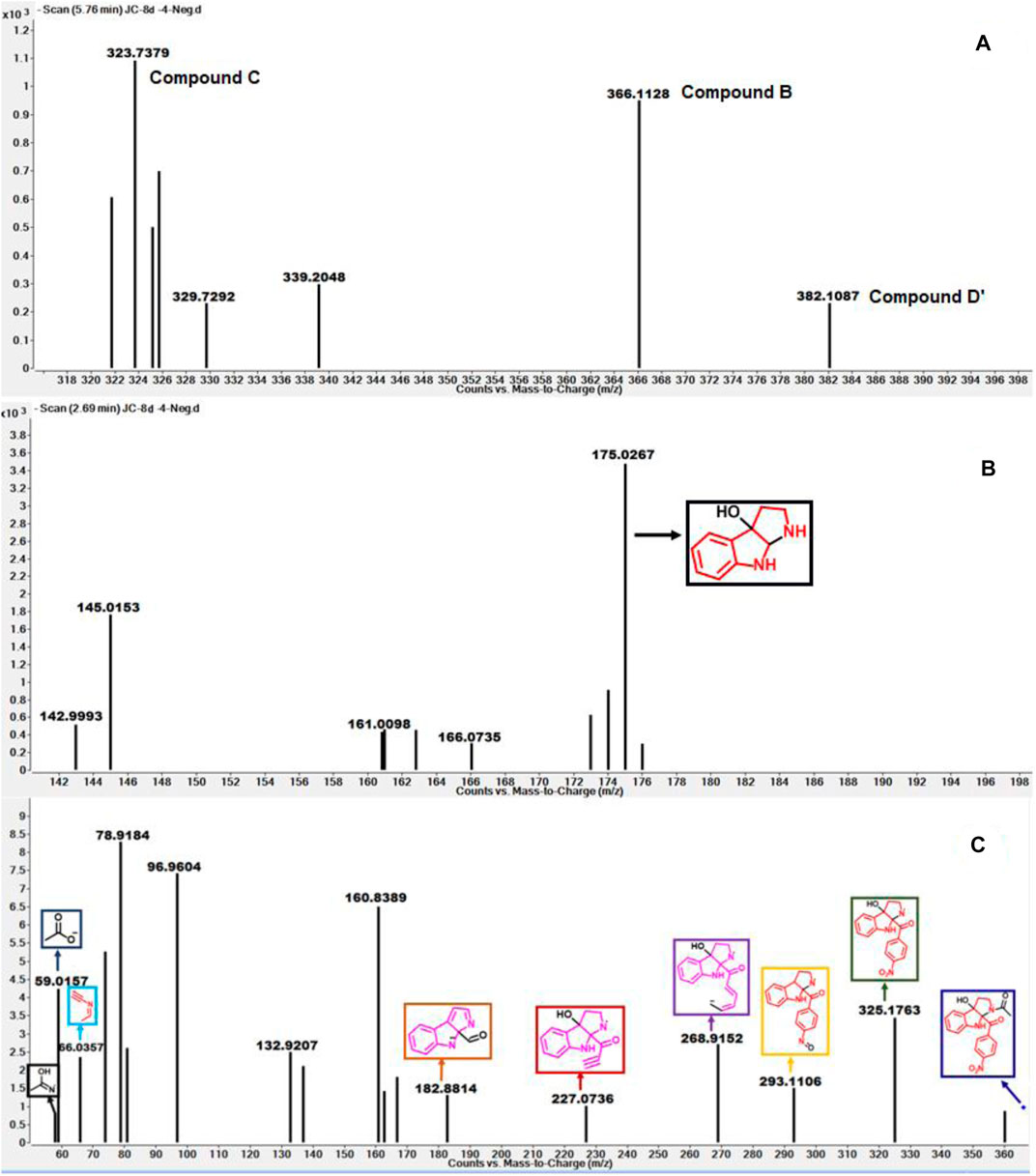
FIGURE 7. (A) The mass analysis of reaction of compound 8d with IDO1 through QTof LC-MS; (B) LC-MS of 8d with IDO1 at Rt = 2.69°min, (C) Fragmentation pattern via LC-MS/MS of compound B in negative mode.
Hence this detailed analysis of the mechanism of metabolization of compound 8d by IDO1 highlighted that during this reaction driven by oxygen, intermediate A was formed via insertion of a single oxygen atom into the compound 8d (Scheme 1). Mechanistically it emphasized that the initial attack by the ferrous superoxide intermediate from IDO1 (formed as the primary requirement of the process) at C2 of 8d afforded the epoxide A. Furthermore competitive reactions on A happened which included 1) an intramolecular cyclization of the N-acetylamine to open up the epoxide ring to provide C and 2) second oxidation by IDO-Fe(IV)-oxide to afford the ring opened N1-acyl-N2-acetylkynuramine derivative D′ (Scheme 1). This investigation clarifies the degradation pathway by which indole based ligands metabolizes in presence of IDO1. This information will not only help to modify the lead compound 8d into an appropriate preclinical candidate but will also guide to design appropriate new indole based lead molecules. It is noteworthy that the literature reports till date indicated that the reaction always occurred in an intermolecular fashion where the oxidized enzyme played the key role in the metabolism of l-tryptophan (the substrate) (Chauhan et al., 2009; Basran et al., 2011; Röhrig et al., 2019). It is the delivery of the oxygen atom from the oxidized enzyme that initiates the reaction at the C2-C3 position of the indole nucleus of the substrate and for our case, the ligand 8d (Scheme 1). Ultimate cleavage of the C2-C3 bond to afford metabolite D′ or intramolecular cyclization of the tethered amide to afford metabolite C was prompted by the transfer of oxygen atom from the oxidized heme to the ligand/substrate (Scheme 1).
Drug Metabolism/Pharmacokinetics Studies of Compounds 8a, d, and e
Understanding the drug metabolism and pharmacokinetics (DMPK) properties of molecules is critical to decide its ability to become drug candidates. Hence the kinetic solubility, partition/distribution coefficient and PAMPA permeability of compounds 8a, d and e were measured (Table 1). The experiments demonstrated that 8a, d, and e showed very good solubility and permeability. The data in Table 1 indicated that these compounds may be amenable toward oral absorption in vivo pharmacokinetic studies. The partition and distribution studies of these compounds revealed a reasonable balance between hydrophilic and lipophilic property. Metabolic stability in different liver microsomes is presented in Table 2. 8a was unstable and showed high clearance, where as 8d and 8e were found to be moderately stable. They showed 33 and 58% hepatic extraction ratio which is 30–70% of human hepatic blood flow. The animal microsomal data among rat, dog and monkey indicated high clearance for compound 8a and low to moderate clearance for 8d and e. Plasma stability data of compounds 8a, 8d, and 8e in human plasma is also presented in Table 2. More than 85% of all the three compounds were retained in the plasma even after 120 min, which meant they are stable in the plasma. The rat and human plasma protein binding is presented in Table 2. Compound 8a binds highly to human plasma followed by rat plasma with fraction unbound of 0.64, 0.49 and 2.7 and 1.6 respectively. On the contrary, compounds 8d and 8e exhibited moderate binding in human and rat plasma with fraction unbound of 9.5, 2.1 and 17.9 and 4.1 respectively. The stability and recovery of all three compounds in plasma was good across the species tested. Overall the DMPK properties of compound 8d make it the most compatible for a lead candidate.

TABLE 1. Drug Metabolism and Pharmacokinetic (DMPK) Studies of compounds 8a, d and e.
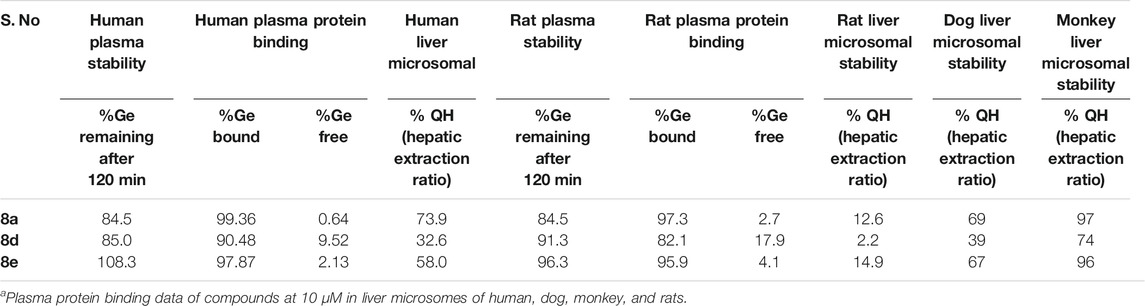
TABLE 2. Metabolic stability of compound 8a, d, and e in rat, dog, monkey and human liver microsomes.a
Conclusion
Here in we have investigated the mechanism of inhibition of C2-aroyl indoles, a novel class of molecules against IDO1 through UV-Vis spectroscopy, MD simulation and QM/MM calculations. Ligand 8d inhibits IDO1 substantially with an IC50 of 180 nM. UV absorption spectroscopy demonstrated that under the erobic reaction condition the heme binding ligands like 8a, d and e stabilizes the ferric IDO1 more than the deoxy ferrous IDO1. Based on this and from the literature reports of l-tryptophan degradation by IDO a probable reaction mechanism was proposed that involved epoxidation of 8d by IDO1 in presence of single oxygen, leading to either an ultimate ring opening of the indole moiety to provide the o-aminoacetophenone derivative D′ or intramolecular cyclization of the exocyclic amine to afford the hexahydropyrrolo [2, 3-b]indole E. The MD simulations and molecular docking calculations rationalized the strong binding of the inhibitor 8d, while the QM/MM calculations supported the putative mechanism of this reactions. This was supported by mass spectroscopic analysis of the reaction mixture over time. The reaction aliquots were analyzed to identify their structure and corroborate the predicted mechanism. The advantage of indole based ligands as inhibitors are their established legacy as IDO1 antagonist. However the collateral disadvantage is the competition that the ligand may face from l-tryptophan which is the original substrate of the enzyme. Through the computational calculations and experimental analysis we have demonstrated that ligand 8d (structurally similar to tryptophan the original substrate of IDO1) is a suitable lead molecule with appropriate DMPK properties that can be developed into a preclinical candidate. This is presently ongoing in our lab.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
JC contributed to the synthesis, purification, characterization of all compounds. and prepared the original manuscript along with SS and KD. KD performed all computational calculations. JC and SM performed the biological activity research. SS, JC and SM analyzed the experimental results. SS, KD and JC drafted the first and second version of the manuscript. All authors discussed, edited, and approved the final version.
Funding
This research was financially supported by grant BT/SBIRI/1655/37/18 (BIRAC-SBIRI). KDD acknowledges Department of Biotechnology for Ramaligamswami re-entry grant (BT/RLS/Re-entry/2017).
Conflict of Interest
Author SM is employed by Acubiosys Pvt. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.691319/full#supplementary-material
References
Ahlrichs, R., Bär, M., Häser, M., Horn, H., and Kölmel, C. (1989). Electronic Structure Calculations on Workstation Computers: the Program System Turbomole. Chem. Phys. Lett. 162, 165–169. doi:10.1016/0009-2614(89)85118-8
Basran, J., Efimov, I., Chauhan, N., Thackray, S. J., Krupa, J. L., Eaton, G., et al. (2011). The Mechanism of Formation ofN-Formylkynurenine by Heme Dioxygenases. J. Am. Chem. Soc. 133, 16251–16257. doi:10.1021/ja207066z
Bayly, C. I., Cieplak, P., Cornell, W., and Kollman, P. A. (1993). A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: the RESP Model. J. Phys. Chem. 97, 10269–10280. doi:10.1021/j100142a004
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., Haak, J. R., and Haak, J. R. (1984). Molecular Dynamics with Coupling to an External bath. J. Chem. Phys. 81, 3684–3690. doi:10.1063/1.448118
Capece, L., Lewis-Ballester, A., Yeh, S.-R., Estrin, D. A., and Marti, M. A. (2012). Complete Reaction Mechanism of Indoleamine 2,3-Dioxygenase as Revealed by QM/MM Simulations. J. Phys. Chem. B 116, 1401–1413. doi:10.1021/jp2082825
Chauhan, N., Thackray, S. J., Rafice, S. A., Eaton, G., Lee, M., Efimov, I., et al. (2009). Reassessment of the Reaction Mechanism in the Heme Dioxygenases. J. Am. Chem. Soc. 131, 4186–4187. doi:10.1021/ja808326g
Cornell, W. D., Cieplak, P., Bayly, C. I., and Kollman, P. A. (1993). Application of RESP Charges to Calculate Conformational Energies, Hydrogen Bond Energies, and Free Energies of Solvation. J. Am. Chem. Soc. 115, 9620–9631. doi:10.1021/ja00074a030
Darden, T., York, D., and Pedersen, L. (1993). Particle Mesh Ewald: AnN⋅Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
Dolušić, E., and Frédérick, R (2013). Indoleamine 2,3-dioxygenase Inhibitors: a Patent Review (2008-2012) Inhibitors . A Patent Rev. 23, 1367-1381. doi:10.1517/13543776.2013.827662
Dubey, K. D., and Shaik, S. (2018). Choreography of the Reductase and P450BM3 Domains toward Electron Transfer Is Instigated by the Substrate. J. Am. Chem. Soc. 140, 683–690. doi:10.1021/jacs.7b10072
Dubey, K. D., and Shaik, S. (2019). Cytochrome P450-The Wonderful Nanomachine Revealed through Dynamic Simulations of the Catalytic Cycle. Acc. Chem. Res. 52, 389–399. doi:10.1021/acs.accounts.8b00467
Dubey, K. D., Wang, B., Vajpai, M., and Shaik, S. (2017). MD Simulations and QM/MM Calculations Show that Single-Site Mutations of Cytochrome P450BM3 Alter the Active Site's Complexity and the Chemoselectivity of Oxidation without Changing the Active Species. Chem. Sci. 8, 5335–5344. doi:10.1039/c7sc01932g
Dunn, G. P., Old, L. J., and Schreiber, R. D. (2004). The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 21, 137–148. doi:10.1016/j.immuni.2004.07.017
Izaguirre, J. A., Catarello, D. P., Wozniak, J. M., and Skeel, R. D. (2001). Langevin Stabilization of Molecular Dynamics. J. Chem. Phys. 114, 2090–2098. doi:10.1063/1.1332996
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
Kalita, S., Shaik, S., Kisan, H. K., and Dubey, K. D. (2020). A Paradigm Shift in the Catalytic Cycle of P450: The Preparatory Choreography during O2 Binding and Origins of the Necessity for Two Protonation Pathways. ACS Catal. 10, 11481–11492. doi:10.1021/acscatal.0c02775
Kershaw, M. H., Westwood, J. A. C., Slaney, C. Y., and Darcy, P. K. (2014). Clinical Application of Genetically Modified T Cells in Cancer Therapy. Clin. Trans. Immunol. 3, e16. doi:10.1038/cti.2014.7
Li, F., Zhang, R., Li, S., and Liu, J. (2017). Ido1: An Important Immunotherapy Target in Cancer Treatment. Int. Immunopharmacology 47, 70–77. doi:10.1016/j.intimp.2017.03.024
Matin, A., Streete, I. M., Jamie, I. M., Truscott, R. J. W., and Jamie, J. F. (2006). A Fluorescence-Based Assay for Indoleamine 2,3-dioxygenase. Anal. Biochem. 349, 96–102. doi:10.1016/j.ab.2005.10.039
Mazarei, G., and Leavitt, B. R. (2015). Indoleamine 2,3 Dioxygenase as a Potential Therapeutic Target in Huntington's Disease. Jhd 4, 109–118. doi:10.3233/JHD-159003
Metz, S., Kästner, J., Sokol, A. A., Keal, T. W., and Sherwood, P. (2014). C Hem S Hell-A Modular Software Package for QM/MM Simulations. Wires Comput. Mol. Sci. 4, 101–110. doi:10.1002/wcms.1163
Muller, A. J., Malachowski, W. P., and Prendergast, G. C. (2005). Indoleamine 2,3-dioxygenase in Cancer: Targeting Pathological Immune Tolerance with Small-Molecule Inhibitors. Expert Opin. Ther. Targets 9, 831–849. doi:10.1517/14728222.9.4.831
Munn, D. H., and Mellor, A. L. (2007). Indoleamine 2,3-dioxygenase and Tumor-Induced Tolerance. J. Clin. Invest. 117, 1147–1154. doi:10.1038/cti.2014.710.1172/jci31178
Okamoto, A., Nikaido, T., Ochiai, K., Takakura, S., Saito, M., Aoki, Y., et al. (2005). Indoleamine 2,3-Dioxygenase Serves as a Marker of Poor Prognosis in Gene Expression Profiles of Serous Ovarian Cancer Cells. Clin. Cancer Res. 11, 6031–6039. doi:10.1158/1078-0432.CCR-04-2671
Paul, S., Roy, A., Deka, S. J., Panda, S., Trivedi, V., and Manna, D. (2016). Nitrobenzofurazan Derivatives of N′-hydroxyamidines as Potent Inhibitors of Indoleamine-2,3-Dioxygenase 1. Eur. J. Med. Chem. 121, 364–375. doi:10.1016/j.ejmech.2016.05.061
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Pradhan, N., Paul, S., Deka, S. J., Roy, A., Trivedi, V., and Manna, D. (2017). Identification of Substituted 1H -Indazoles as Potent Inhibitors for Immunosuppressive Enzyme Indoleamine 2,3-Dioxygenase 1. ChemistrySelect 2, 5511–5517. doi:10.1002/slct.201700906
Prendergast, G. C., Malachowski, W. P., DuHadaway, J. B., and Muller, A. J. (2017). Discovery of Ido1 Inhibitors: From Bench to Bedside. Cancer Res. 77, 6795–6811. doi:10.1158/0008-5472.CAN-17-2285
Röhrig, U. F., Reynaud, A., Majjigapu, S. R., Vogel, P., Pojer, F., and Zoete, V. (2019). Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (Ido1). J. Med. Chem. 62, 8784–8795. doi:10.1021/acs.jmedchem.9b00942
Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. C. (1977). Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J. Comput. Phys. 23, 327–341. doi:10.1016/0021-9991(77)90098-5
Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S., and Walker, R. C. (2013). Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh EwaldJ. Chem. Theor. Comput. 9, 3878–3888. doi:10.1021/ct400314y
Sas, K., Szabó, E., and Vécsei, L. (2018). Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 23, 191–218. doi:10.3390/molecules23010191
Sherwood, P., de Vries, A. H., Guest, M. F., Schreckenbach, G., Catlow, C. R. A., French, S. A., et al. (2003). QUASI: A General Purpose Implementation of the QM/MM Approach and its Application to Problems in Catalysis. J. Mol. Struct. THEOCHEM 632, 1–28. doi:10.1016/S0166-1280(03)00285-9
Shin, I., Ambler, B. R., Wherritt, D., Wherritt, D., Griffith, W. P., Maldonado, A. C., et al. (2018), Stepwise O-Atom Transfer in Heme-Based Tryptophan Dioxygenase: Role of Substrate Ammonium in Epoxide Ring Opening. J. Am. Chem. Soc. 140, 4372–4379doi:10.1021/jacs.8b00262
Siu, L. L., Gelmon, K., Chu, Q., Pachynski, R., Alese, O., Basciano, P., et al. (2017). BMS-986205, an Optimized Indoleamine 2,3-dioxygenase 1 (Ido1) Inhibitor, Is Well Tolerated with Po-Tent Pharmacodynamic (PD) Activity, Alone and in Combination with Nivolumab (Nivo) in Advanced Cancers in a Phase 1/2a Trial. Cancer Res. 77, 1538–7445. doi:10.1158/1538-7445CT116aaaa
Smith, W., and Forester, T. R. (1996). DL_POLY_2.0: a General-Purpose Parallel Molecular Dynamics Simulation Package. J. Mol. Graphics 14, 136–141. doi:10.1016/S0263-7855(96)00043-4
Song, M., Chen, X., Chen, X., Wang, L., and Zhang, Y. (2018). Future of Anti-PD-1/pd-L1 Applications: Combinations with Other Therapeutic Regimens. Chin. J. Cancer Res. 30, 157–172. doi:10.21147/j.issn.1000-9604.2018.02.01
Soret, J.-L. (1883). Analyse Spectrale − Sur le spectre d’absorption du sang dans la partie violette et ultra-violette. C. R. Hebd. Seá Nces Acad. Sci. 1883 (97), 1269–1270.
Sugimoto, H., Oda, S.-i., Otsuki, T., Hino, T., Yoshida, T., and Shiro, Y. (2006). Crystal Structure of Human Indoleamine 2,3-dioxygenase: Catalytic Mechanism of O2 Incorporation by a Heme-Containing Dioxygenase. Proc. Natl. Acad. Sci. 103, 2611–2616. doi:10.1073/pnas.0508996103
Terentis, A. C., Thomas, S. R., Takikawa, O., Littlejohn, T. K., Truscott, R. J. W., Armstrong, R. S., et al. (2002). The Heme Environment of Recombinant Human Indoleamine 2,3-Dioxygenase. J. Biol. Chem. 277, 15788–15794. doi:10.1074/jbc.M200457200
Uyttenhove, C., Pilotte, L., Théate, I., Stroobant, V., Colau, D., Parmentier, N., et al. (2003). Evidence for a Tumoral Immune Resistance Mechanism Based on Tryptophan Degradation by Indoleamine 2,3-dioxygenase. Nat. Med. 9, 1269–1274. doi:10.1038/nm934
Vacchelli, E., Aranda, F., Eggermont, A., Sautès-Fridman, C., Tartour, E., Kennedy, E. P., et al. (2014). Trial Watch: Ido Inhibitors in Cancer Therapy. Oncoimmunology 3, e957994–1. doi:10.1080/2162402X.2020.177762510.4161/21624011.2014.957994
Van den Eynde, B. J., van Baren, N., and Baurain, J.-F. (2020). Is There a Clinical Future for Ido1 Inhibitors after the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 4, 241–256. doi:10.1146/annurev-cancerbio-030419-033635
Wen, H., Liu, Y., Wang, S., Wang, T., Zhang, G., Chen, X., et al. (2019). Design and Synthesis of Indoleamine 2,3-Dioxygenase 1 Inhibitors and Evaluation of Their Use as Anti-tumor Agents. Molecules 24, 2124–2151. doi:10.3390/molecules24112124
Keywords: molecular dynamic simulation, QM/ MM calculation, mass spectroscopic analysis, indoleamine (2, 3)-dioxygenase, C2 substituted indoles
Citation: Chauhan J, Maddi SR, Dubey KD and Sen S (2021) Developing C2-Aroyl Indoles as Novel Inhibitors of IDO1 and Understanding Their Mechanism of Inhibition via Mass Spectroscopy, QM/MM Calculations and Molecular Dynamics Simulation. Front. Chem. 9:691319. doi: 10.3389/fchem.2021.691319
Received: 06 April 2021; Accepted: 25 May 2021;
Published: 15 July 2021.
Edited by:
Nino Russo, University of Calabria, ItalyReviewed by:
Vicent Moliner, University of Jaume I, SpainWenzhen Lai, Renmin University of China, China
Copyright © 2021 Chauhan, Maddi, Dubey and Sen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kshatresh Dutta Dubey, a3NoYXRyZXNoLmR1YmV5QHNudS5lZHUuaW4=; Subhabrata Sen, c3ViaGFicmF0YS5zZW5Ac251LmVkdS5pbg==