 Ahmed M. El-Saghier1
Ahmed M. El-Saghier1 Mohamed El-Naggar2Abdel Haleem M. Hussein3Abu-Bakr A. El-Adasy3
Mohamed El-Naggar2Abdel Haleem M. Hussein3Abu-Bakr A. El-Adasy3 M. Olish3
M. Olish3 Aboubakr H. Abdelmonsef4*
Aboubakr H. Abdelmonsef4*- 1Chemistry Department, Faculty of Science, Sohag University, Sohag, Egypt
- 2Chemistry Department, Faculty of Sciences, University of Sharjah, Sharjah, United Arab Emirates
- 3Chemistry Department, Faculty of Science, Al-Azhar University, Assiut, Egypt
- 4Chemistry Department, Faculty of Science, South Valley University, Qena, Egypt
A new series of quinoline derivatives 5–12 were efficiently synthesized via one-pot multicomponent reaction (MCR) of resorcinol, aromatic aldehydes, β-ketoesters, and aliphatic/aromatic amines under solvent-free conditions. All products were obtained in excellent yields, pure at low-cost processing, and short time. The structures of all compounds were characterized by means of spectral and elemental analyses. In addition, all the synthesized compounds 5–12 were in vitro screened for their antioxidant and antibacterial activity. Moreover, in silico molecular docking studies of the new quinoline derivatives with the target enzymes, human NAD (P)H dehydrogenase (quinone 1) and DNA gyrase, were achieved to endorse their binding affinities and to understand ligand–enzyme possible intermolecular interactions. Compound 9 displayed promising antioxidant and antibacterial activity, as well as it was found to have the highest negative binding energy of -9.1 and -9.3 kcal/mol for human NAD (P)H dehydrogenase (quinone 1) and DNA gyrase, respectively. Further, it complied with the Lipinski’s rule of five, Veber, and Ghose. Therefore, the quinoline analogue 9 could be promising chemical scaffold for the development of future drug candidates as antioxidant and antibacterial agents.
Introduction
Quinolines are very important compounds used for new drug development. They are reported as highly selective cytotoxic (Luo et al., 2009; Bawa et al., 2010; Meshram et al., 2012; Sidoryk et al., 2015), broad-spectrum antimicrobial (including activity against Mycobacterium tuberculosis as well as HIV-1 integrase inhibition activity) (Wang et al., 2019), antileishmanial (Asif, 2014), anticonvulsant, anti-inflammatory, cardiovascular activity (Acharyulu et al., 2008; Kumar, Bawa, and Gupta, 2010), and have antidiabetic effect (Shang, et al., 2018a).
Moreover, natural and synthetic chromene moiety attached to quinolines has important biological activity such as anticancer (El-Maghraby, 2014), anticoagulant, antispasmolytic (El-Maghraby and Aboubakr, 2019), antiangiogenesis (Sangani et al., 2012), antimicrobial (Gómez and Vladimir, 2013), anti-inflammatory (Asif, 2014), anti-invasive (Sidoryk et al., 2015), antioxidant (Shang, et al., 2018b), analgesic, and anticonvulsant agents (Dua et al., 2011). Therefore, many researchers have synthesized these compounds as target structures and were evaluated for their biological activity.
Recently, there is a growing demand for the development of organic reactions in eco-friendly media. Synthetic manipulations have to be made to minimize the use of hazardous chemicals by replacing the traditional organic solvents in reactions and their subsequent workup with other nontoxic and environmentally benign solvents such as water.
For complementing this way and in continuation of our search work (Abdelmonsef and Mosallam, 2020; Haredi Abdelmonsef et al., 2020; Noser et al., 2020; Rashdan et al., 2020; Shehadi et al., 2020; Gomha et al., 2021), we decided to eco-friendly synthesize some quinoline scaffolds and test in vitro their antioxidant and antibacterial activity. Moreover, the compounds were docked into the binding sites of the target enzymes: human NAD (P)H dehydrogenase (quinone 1) and DNA gyrase, respectively. In addition, adsorption, distribution, metabolic, excretion, and toxicity (ADME/T) properties of the newly synthesized compounds were also calculated.
Materials and Methods
Chemistry
All melting points were measured by a Stuart SMP10 Melting point apparatus. IR spectra (KBr) were recorded by using a Bruker spectrometer (ν, cm−1). 1H-NMR, 13C-NMR, and DEPT 135 spectra were recorded on 400 and 100 MHz, DMSO-d6 at AVANCE-III 400 MHz High performance FT-NMR Spectrum BRUKER. Bio Spin International AG-Switzerland at Sohag University. Elemental analysis was carried out at the Microanalytical Research Center, Faculty of Science, Cairo University. All the chemicals were commercially available from Sigma-Aldrich and El-Gomhouria Company, Egypt.
General Procedure for Synthesis of Compounds 5–12
A mixture of resorcinol 1 (1.10 g, 10 mmol), different aromatic aldehydes, namely, benzaldehyde (1.06 g, 10 mmol), 4-chlorobenzaldehyde (1.40 g, 10 mmol), 2-chlorobenzaldehyde (1.40 g, 10 mmol), 4-methylbenzaldehyde (1.20 g, 10 mmol) 2a-d, 2-hydroxybenzaldehyde (1.22 g, 10 mmol) 2e, and/or different β-ketoesters, namely, ethyl cyanoacetate (1.13 g, 10 mmol) 3a, ethyl acetoacetate (1.30 g, 10 mmol) 3b, and/or different aromatic or aliphatic amines, namely, 4-aminophenol (1.09 g, 10 mmol) 4b, benzyl amine (1.07 g, 10 mmol) 4c, was refluxed in an oil bath at 110°C for one hour. After completion of the transformation, the reaction mixture was cooled to RT, and then water was added (50 ml). The product was collected by filtration, then washed with water repeatedly, and recrystallized from appropriate solvents (25 ml) to give compounds 5a-d, 6, 7, 9, 10, 11, and 12.
Ethyl 2-amino-7-hydroxy-4-phenyl-1,4-dihydroquinoline-3-carboxylate (5a)
Yield (90%), color: brown crystals, mp = 115–117°C, IR (KBr) ν cm-1; 3450 (OH), 3325 (NH), 3213, 3201 (NH2), and 1739 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ 1.08 (t, 3H, CH3), 4.18 (q, 2H, CH2), 4.65 (s, 1H, CH-pyridine), 6.22–7.28 (m, 10H, Ar-H+NH2), 8.46 (s, 1H, NH), and 9.05 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δ 14.32, 60.00, 103.72, 112.28, 120.98, 128.24, 129.20, 129.68, 129.95, 131.48, 132.19, 141.32, 152.48, 155.86, 158.28, 167.94, and 173.64. Anal. Calcd. for C18H18N2O3 (310.35): C, 69.66; H, 5.85; N, 9.03%. Found C, 70.00; H, 5.34; N, 9.20%.
Ethyl 2-amino-4-(4-chlorophenyl)-7-hydroxy-1,4-dihydro-quinoline-3-carboxylate (5b)
Yield (97%), color: orange crystals, mp = 138–140°C; IR (KBr) ν cm-1; 3442 (OH), 3332 (NH), 3246, 3231 (NH2), and 1738 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ 1.02 (t, 3H, CH3), 4.40 (q, 2H, CH2), 5.81 (s, 1H, CH-pyridine), 6.29–7.37 (m, 10H, Ar-H+NH2), 8.88 (s, 1H, NH), and 9.01 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δ 14.42, 60.00, 103.92, 112.28, 120.98, 128.24, 129.21, 129.68, 129.95, 131.48, 132.19, 141.32, 152.48, 155.86, 158.28, 167.94, and 173.64. Anal. Calcd. for C18H17ClN2O3 (344.79): C, 62.70; H, 4.57; Cl, 10.28; N, 8.12%. Found C, 62.50; H, 4.73; Cl, 10.37; N, 8.27%.
Ethyl 2-amino-4-(2-chlorophenyl)-7-hydroxy-1,4-dihydro-quinoline-3-carboxylate (5c)
Yield (95%), color: reddish crystals, mp = 140–142°C, IR (KBr) ν cm-1; 3420 (OH), 3364 (NH), 3192, 3175 (NH2), and 1741 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ H = 1.21 (t, 3H, CH3), 4.74 (q, 2H, CH2), 5.03 (s, 1H, CH-pyridine), 6.60–7.36 (m, 9H, Ar-H+NH2), 8.22 (s, 1H, NH), and 9.19 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δ 14.35, 56.30, 103.14, 106.32, 114.90, 119.57, 127.27, 128.79, 129.52, 130.52, 133.14, 139.09, 152.92, 156.04, 158.50, 167.67, and 173.16. Anal. Calcd. for C18H17ClN2O3(344.79): C, 62.70; H, 4.57; Cl, 10.28; N, 8.12%. Found C, 62.56; H, 4.39; Cl, 10.30; N, 8.06%.
Ethyl 2-amino-7-hydroxy-4-(p-tolyl)-1,4-dihydroquinoline-3-carboxylate (5d)
Yield (93%), color: reddish crystals, mp = 107–109°C, IR (KBr) ν cm-1; 3351, 3329, 3294, 3260 (OH, NH, NH2), and 1739 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ 1.20 (t, 3H, CH3), 2.32 (s, 3H, CH3), 4.08 (q, 2H, CH2), 5.71 (s, 1H, CH-pyridine), 6.27–7.28 (m, 9H, Ar-H+NH2), 7.58 (s, 1H, NH), and 8.77 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δ 21.01, 21.49, 59.96, 102.89, 112.15, 125.77, 128.66, 128.99, 129.36, 129.63, 137.81, 139.81, 155.82, and 168.18. Dept. 135 NMR (100 MHZ, DMSO-d6): δ (+) 21.02, 21.49, (-) 37.24, (+) 102.88, 105.85, 112.15, 115.77, 128.66, 128.99, 129.36, 129.79, and 139.92. Anal. Calcd. for C19H20N2O3 (324.37): C, 70.35; H, 6.21; N, 8.64%. Found C, 70.68; H, 5.66; N, 8.57%.
6-Amino-10-hydroxy-8,12b-dihydro-7H-chromeno[3,4-c]quinolin-7-one (6)
Yield (89%), color: yellow crystals, mp = 270–272°C, IR (KBr) ν cm-1; 3345, 3305, 3293, 3251 (OH, NH, NH2), and 1668 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ 2.38 (s, 1H, sp3 CH), 5.95 (s, 1H, NH2), 6.21–7.66 (m, 8H, Ar-H+ CH-pyridine), 7.27 (s, 1H, NH), 9.98 (s, 1H, NH), and 11.12 (s, 1H, OH). 13C-NMR (100 MHZ, DMSO-d6): δ 40.95, 44.62, 79.19, 97.70, 115.01, 117.67, 124.09, 125.22, 129.03, 131.54, 135.61, 135.82, 141.10, 141.32, 155.30, and 171.91. Anal. Calcd. for C16H12N2O3 (280.28): C, 68.56; H, 4.32; N, 9.99%. Found C, 68.45; H, 4.67; N, 9.89%.
10-Hydroxy-7-methyl-8,12b-dihydrodibenzo[c,f][2,7]na-phthayridin-6(5H)-one (7)
Yield (95%), color: yellow crystals, mp = 185–187°C, IR (KBr) ν cm-1; 3317, 3212 (OH, NH), and 1662 (C=O-amide). 1H-NMR (400 MHz, DMSO-d6): δ H = 1.87 (s, 3H, CH3), 4.87 (s, 1H, sp3CH CH-pyrane), 6.22–6.94 (m, 8H, Ar-H), 7.73 (s, 1H, NH), and 8.64 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δ 33.68, 42.60, 103.04, 103.41, 106.68, 116.12, 118.88, 119.32, 126.71, 128.55, 128.95, 130.09, 130.43, 153.60, 155.88, 156.12, and 158.96. Anal. Calcd. for C17H14N2O2 (278.31): C, 73.37; H, 5.07; N, 10.0 7%. Found C, 73.41; H, 5.20; N, 10.00%.
10-Hydroxy-8-(4-hydroxyphenyl)-6-methyl-8,12b-dihydro-7H-chromeno[3,4-c]quinolin-7-one (9)
Yield (92%), color: pale brown crystals, mp = 160–162°C, IR (KBr) ν cm-1; 3244 (2OH). 1H-NMR (400 MHz, DMSO-d6): δ 1.94 (s, 3H, CH3), 5.27 (s, 1H, CH-pyridine), 6.76–7.33 (m, 12H, Ar-H), 8.90 (s, 1H, NH), and 11.53 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δ 19.23, 36.24, 102.94, 106.51, 108.85, 116.19, 116.44, 119.94, 121.26, 123.08, 127.19, 127.71, 128.32, 140.60, 142.92, 152.45, 155.15, 156.12, 157.44, and 160.64. MS (relative intensity) m/z: 371 (M, 5.1%), 289 (23%), 165 (35%), 105 (70%), and 44 (100%). Anal. Calcd. for C23H17NO4 (371.39): C, 74.38; H, 4.61; N, 3.77%. Found C, 74.76; H, 4.90; N, 3.70%.
3-Acetyl-4-(4-chlorophenyl)-7-hydroxy-1-(4-hydroxy-phenyl)quinolin-2(1H)-one (10)
Yield (81%), color: dark red crystals, mp = 115–117°C, IR (KBr) ν cm-1; 3335, 3273 (2OH), and 1710 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ 1.95 (s, 3H, CH3), 6.22–6.94 (m, 11H, Ar-H), 8.42 (s, 1H, NH), and 11.56 (s, 2H, 2OH). 13C-NMR (100 MHz, DMSO-d6): δ 19.75, 103.30, 115.78, 115.94, 116.06, 116.23, 123.05, 127.37, 128.82, 129.13, 129.30, 130.27, 135.83, 141.06, 142.74, 148.80, 150.82, 156.15, 157.02, and 172.53. Anal. Calcd. for C23H16ClNO4 (405.83): C, 68.07; H, 3.97; Cl, 8.74; N, 3.45%. Found C, 68.21; H, 3.39; Cl, 8.62; N, 3.47%.
Ethyl 2-amino-1-benzyl-7-hydroxy-4-phenyl-1,4-dihydro-quinoline-3-carboxylate (11)
Yield (89%), color: pink crystals, mp = 122–124°C, IR (KBr) ν cm-1; 3400, 3286, 3273 (OH, NH2), and 1700 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ 1.19 (t, 3H, CH3), 3.93 (q, 2H, CH2), 4.32 (s, 2H, CH2), 5.00 (s, 1H, CH-pyridine), 6.28–7.40 (m, 13H, Ar-H+NH2), and 8.93 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δ 14.13, 49.67, 53.68, 60.73, 100.10, 115.01, 127.67, 127.97, 128.59, 128.60, 128.63, 128.80, 128.70, 128.85, 128.92, 128.95, 129.03, 129.23, 135.61, 136.74, 141.50, 141.65, 154.44, 156.64, and 165.12. Anal. Calcd. for C25H24N2O3 (400.47): C, 74.98; H, 6.04; N, 7.00%. Found C, 74.99; H, 6.01; N, 7.20%.
10-Hydroxy-8-(4-hydroxyphenyl)-6-imino-6H-chromeno[3,4-c]quinolin-7(8H)-one (12)
Yield (97%), color: dark red crystals, mp = 260–262°C, IR (KBr) ν cm-1; 3432, 3238, 3227 (OH, NH2), and 1708 (C=O). 1H-NMR (400 MHz, DMSO-d6): δ 4.31 (s, 1H, CH-pyridine), 6.76–7.43 (m, 12H, Ar-H+NH2), and 9.24 (s, 2H, 2OH). 13C-NMR (100 MHz, DMSO-d6): δ 49.67, 100.10, 113.72, 115.01, 116.46, 116.66, 122.72, 123.65, 123.85, 126.77, 129.03, 129.13, 129.23, 131.33, 135.61, 141.65, 144.73, 154.12, 154.44, 155.80, 156.64, and 165.12. Anal. Calcd. for C22H14N2O4 (370.36): C, 71.35; H, 3.81; N, 7.56%. Found C, 71.22; H, 3.48; N, 7.52%.
Biological Study
In-Vitro Antioxidant Assay
The total antioxidant capacity of the compounds was evaluated according to the method described by Prieto et al. (1999). An aliquot of 0.5 ml of sample solution was combined with 4.5 ml of reagent solution (0.6 M sulfuric acid, 28 mM sodium phosphate, and 4 mM ammonium molybdate). In case of blank, 0.5 ml of 45% DMSO (dimethyl sulphoxide) has been used. The tubes incubated in a boiling water bath at 95°C for 90 min. After the samples cooled at RT, the absorbance of the aqueous solution of each sample was measured at 695 nm against blank by using a UV-2450 spectrophotometer (Shimadzu, Japan). The total antioxidant activity was expressed as the absorbance of the sample at 695 nm. The higher absorbance value indicated higher antioxidant activity (Wan et al., 2011).
Antimicrobial Assay
The antimicrobial activity of compounds was tested in vitro against various bacterial strains, Gram-negative (Escherichia coli), and Gram-positive (Staphylococcus haemolyticus, Kocuria kristinae, Enterococcus casseliflavus, and Bacillus subtilis) identified in Al-Azhar University, Regional Center for Mycology and Biotechnology. Amikacin, levofloxacin, and gentamicin were used as standard antibacterial agents obtained from Bioanalyse® Ltd. (Turkey) for the comparison of biological activity of newly synthesized molecules.
The method applied is “modified agar diffusion” (Bauer et al., 1966) using 2.0 mg per disc used to determine the antimicrobial activity. Nutrient agar (ready for use from EDM company,Egypt) inoculated with microbial cell suspension into sterile Petri dishes (200 µl in 20 ml medium). Sterile paper discs of 6 mm diameter saturated with a tested compound placed on the surface of the inoculated agar plates and negative control done using paper discs loaded with 20 µl of DMSO. Incubate overnight (24 h) at 37°C. Inhibition zone was measured at the end of the incubation period.
In Silico Docking Protocol
The molecular docking study of all compounds was carried out to identify their plausible mode of action against the active site residues of the target enzymes.
The 2D structures of the newly synthetic compounds were accurately drawn using ChemDraw Ultra 7.0 software and then converted to SDF format using Open Babel GUI tool (O’Boyle et al., 2011). An in-house library of ten synthesized compounds was generated for further study. The enzymes of NQO1: human NAD (P)H dehydrogenase (quinone 1) (PDB code 1DXO) (Faig et al., 2000) and DNA gyrase (PDB code 1JA6) (Holdgate et al., 1997) were selected as targets for docking simulation. The crystal structures of the targets were retrieved from the RCSB Protein Data Bank web server. The protein files were optimized by removing the ligands and water molecules. The grid box was generated around the active site pocket. Subsequently, the docking process was achieved using PyRx, virtual screening tool of AutoDock 4 software (Dallakyan and Olson, 2015). Among the nine confirmations of these ligand molecules obtained from the docking simulation, the pose with the lowest binding energy was selected for further study (HA and SP, 2016; Hussein et al., 2018). In addition, 7,8-Dihydroxyflavone (tropoflavin) and novobiocin were selected as standard drugs to compare the docking score with that of the synthesized compounds. Discovery Studio 3.5 was then used to visualize the intermolecular interactions between the ligand molecules and enzymes.
The adsorption, distribution, metabolic, excretion, and toxicity (ADME/T) analyses and physicochemical properties of the newly synthesized compounds were also calculated using admetSAR (http://lmmd.ecust.edu.cn/admetsar2) and Molinspiration (https://molinspiration. com/) free Web-based tools.
Results and Discussion
Chemistry
The present study entails the synthesis of a novel series of quinoline analogues 5–12via one-pot multicomponent reaction (MCR) under solvent-free conditions.
Our synthesis begins with heating a mixture of resorcinol 1, aromatic aldehyde, namely, benzaldehyde, 2-chlorobenzaldehyde, 4-chlorobenzaldehyde, 4-methyl benzaldehyde 2a-d, and ethyl cyanoacetate 3a in the presence of ammonium acetate 4a to obtain quinoline derivatives 5a-d under solvent-free conditions (Scheme 1).
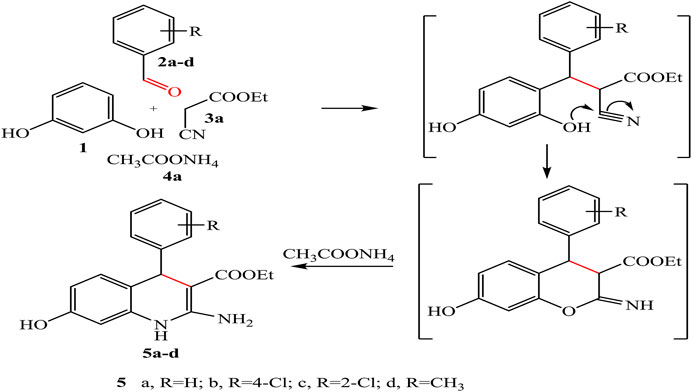
SCHEME 1. Synthesis of quinoline derivative 5a–d under solvent-free conditions.
The chemical structures of the products 5a-d were established on the basis of IR, 1H-NMR, 13C-NMR spectral data, and elemental analysis.
For example, IR spectrum of compound 5d showed new absorption bands at 3,351, 3,329, 3,294, 3,260 cm-1 for OH, NH, and NH2, respectively. The 1H-NMR spectrum of compound 5d revealed triplet signal at 1.20 ppm for –CH2-CH3, singlet signal at 2.32 ppm for -CH3 attached to phenyl ring, quartet signal at 4.08 ppm for -CH2, singlet signal at 5.71 ppm for CH-pyridine, singlet signal at 7.58 ppm for NH group, and singlet signal at 8.77 ppm for OH group, beside the appearance of the amino protons in interference with the aromatic protons at 6.27–7.28 ppm as a singlet. In addition, 13C-NMR spectrum of compound 5d showed signals at 21.02, 21.49, 59.96, and 168.18 ppm assigned to 2CH3 and CH2 and acetyl carbonyl group, respectively. Moreover, a negative signal of the CH2 group was obtained at 61.59 ppm in DEPT 135 spectrum.
By analogy, multicomponent reaction of resorcinol 1, aromatic aldehyde 2e, ethyl cyanoacetate 3a, and ammonium acetate 4a afforded the quinoline derivative 6 (Scheme 2).
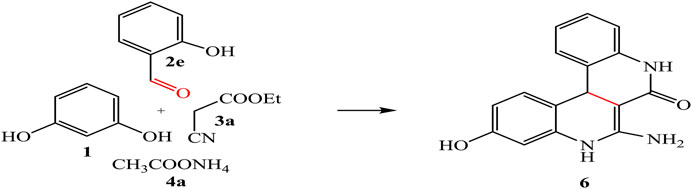
SCHEME 2. Synthesis of quinoline 6.
IR spectrum of compound 6 showed band at 1,668 cm-1 referred to carbonyl group; in addition, its 1H-NMR spectrum exhibited beside the aromatic signals; new singlet signals at 11.12, 9.98, 7.27, 5.95, and 2.38 ppm were consistent with the OH, 2NH, NH2, and sp3CH groups, respectively.
By adding ethyl acetoacetate 3b instead of ethyl cyanoacetate 3a, afforded the quinoline analogue 7 (Scheme 3). The chemical structure of compound 7 was established by elemental analysis and spectroscopic data, where IR spectrum showed amide carbonyl at 1,662 cm-1. 1H-NMR spectrum revealed a singlet signal at 1.87 ppm referred to CH3, 4.87 ppm for sp3CH-pyrane, and OH at 8.64 ppm beside signals at 6.22–6.94 ppm for aromatic protons. Moreover, 13C-NMR spectrum represented a signal at 33.68 ppm referred to CH3, in addition to other signals which confirmed the chemical structure of 7.
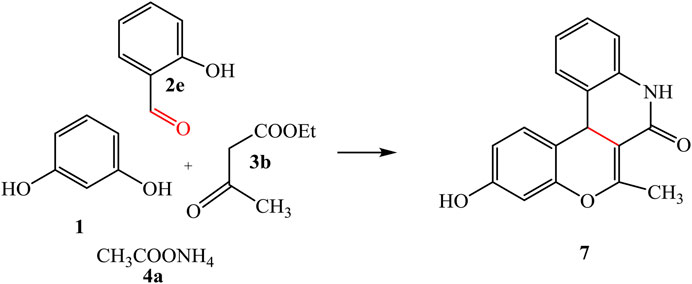
SCHEME 3. Schematic representation of the synthesis of component 7.
By the same way, a multicomponent reaction of resorcinol 1 with aromatic aldehyde 2e, ethyl acetoacetate 3b, and p-aminophenol 4b in the presence of sodium carbonate under solvent-free conditions afforded the quinoline derivative 9. This result was also achieved by the reaction of 3-oxobutanamide 8 with resorcinol 1 and salicylaldehyde 2e in the presence of sodium carbonate which makes the medium alkaline, as declared in Scheme 4.
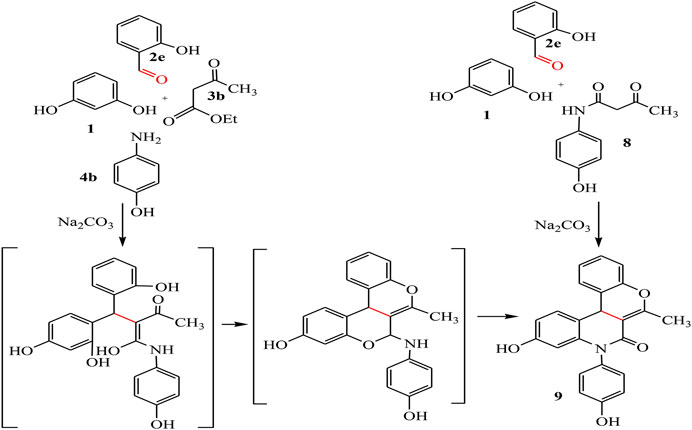
SCHEME 4. Synthetic pathway for compound 9.
IR spectrum confirmed the chemical structure of compound 9 by disappearance of the characteristic bands for acetyl carbonyl and the presence of new bands referred to imide carbonyl at 1,635 cm-1. 1H-NMR spectrum showed a singlet signal for CH3 at 1.94 ppm, at 5.27 ppm a singlet signal referred to CH-pyridine, NH appeared at 8.90 ppm, and OH found at 11.53 ppm 13C-NMR spectrum revealed signals at 19.32 and 160.64 ppm referred to CH3 and imide carbonyl groups, respectively. Mass spectrum confirmed the molecular formula C23H17NO4 by the molecular ion peak at m/z 371.
By the same way, the treatment of resorcinol 1 with 4-chlorobenzaldehyde 2b and 3-oxobutanamide 8 afforded the quinoline derivative 10, as shown in Scheme 5.
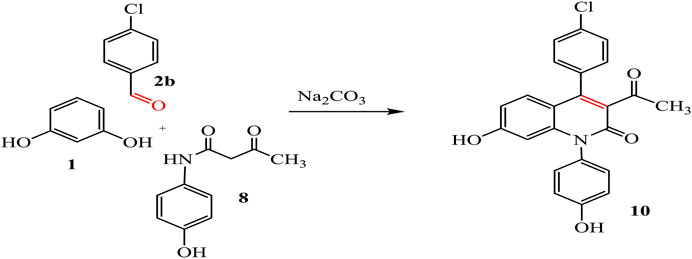
SCHEME 5. Synthesis of quinoline analogue 10.
IR spectrum of compound 10 showed new bands for acetyl and two OH groups at 1,710, 3,335, and 3,273 cm-1, respectively. Its 1H-NMR showed a singlet signal at 1.95 cm-1 for CH3, a singlet signal at 5.02 ppm for CH-pyridine, and two singlet signals at 8.42 and 11.56 ppm referred to two hydroxyl groups, beside multiplet signals of aromatic protons. Further, 13C-NMR spectrum confirmed the presence of methyl group at 19.75 ppm, imide carbonyl at 157 ppm, and acetyl carbonyl at 172.53 ppm.
The reaction of resorcinol 1 with aldehyde, ethyl cyanoacetate, and aromatic amine was also studied to afford the quinoline analogues 11 and 12. For compound 11 stopped at the step of cyclization but compound 12 formed by additional reaction with hydroxyl group on aldehyde with ester and H2O get out. Ester form disappeared from compound 12 while it revealed at compound 11, as represented in Schemes 6, 7.
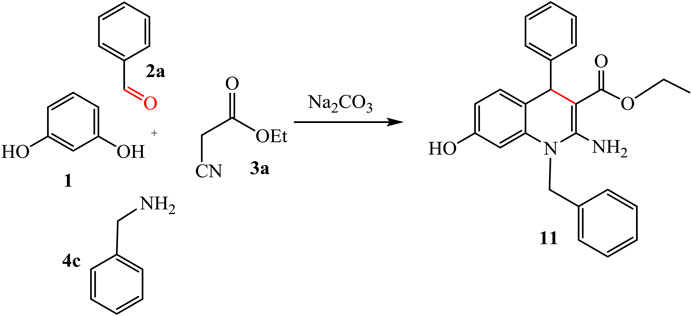
SCHEME 6. Synthesis of compound 11.
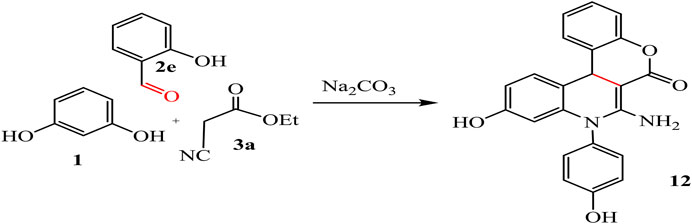
SCHEME 7. Synthesis of compound 12.
The spectral data of the newly synthesized compounds are represented as Supplementary Figures S1–S27 in supplementary information file.
Biological Evaluation
In-Vitro Antioxidant Assay
The total antioxidant activity was determined using the phosphor molybdenum blue complex with a maximum absorption at 695 nm. The data presented in Table 1 showed that the tested compound 9 is the most active as represented in the following order: vit C > 9 > 12 > 5d > 11 > 5b.

TABLE 1. Antioxidant activity of compounds 5–12.
Antimicrobial Evaluation
All the tested compounds showed good activity against bacterial strains tested with inhibition zones in range 9.0–20.0 mm. Amikacin showed inhibition zone 15.0 mm, levofloxacin 14.0–16.0 mm, and gentamicin 20.0 mm, as shown in Table 2. (Supplementary Figure S28 in supplementary information file). The compound 9 with electron donating groups like (-OH) and (-CH3), piperidine, and pyran moieties showed the strongest activity against Gram-negative (Escherichia coli) and Gram-positive (Enterococcus casseliflavus) strains.

TABLE 2. Antibacterial activity of the screened compounds 5–12.
Molecular Docking Protocol
To understand as well as to support the in vitro antioxidant and antibacterial activity of the newly synthesized compounds for the rational design of novel and potential inhibitor molecules, molecular docking studies were performed (Abdelmonsef et al., 2016; Dasari et al., 2017; Rondla et al., 2017; Abdelmonsef, 2019).
Here, in silico molecular docking simulation of standard drugs and the new ten molecules with the active site of the target enzymes 1DXO and 1AJ6 was carried out to evaluate their binding affinities and to understand ligand–enzyme possible intermolecular interactions. The docking energies (ΔGbind) and amino acid interactions for the screened compounds were summarized in Table 3. The 2D and 3D representation of the best docked complexes were represented in Figures 1, 2.
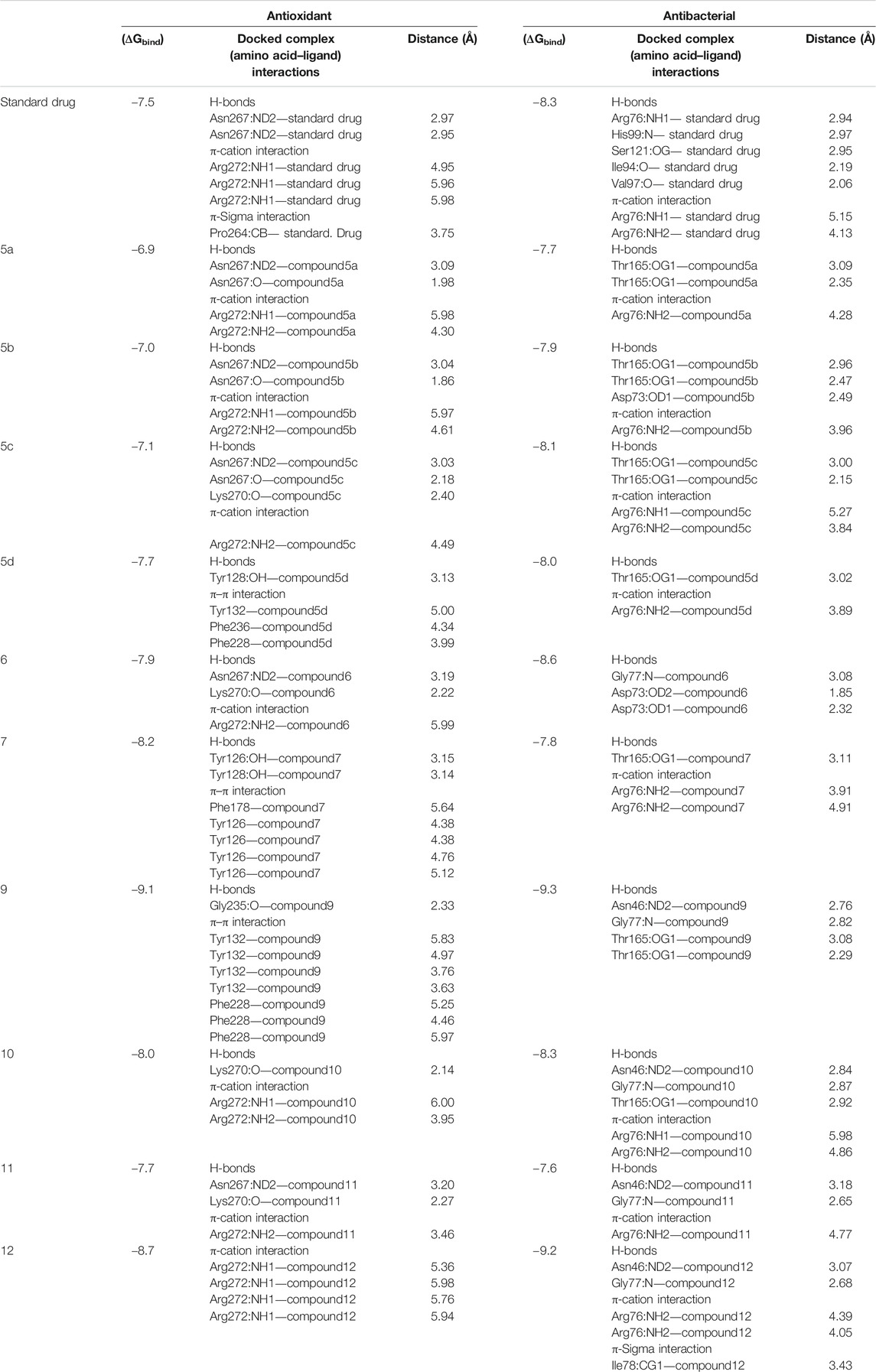
TABLE 3. The binding energies (ΔGbind) of the docked standard drugs and compounds 5–12 and their intermolecular interactions with the active site of the target enzymes.
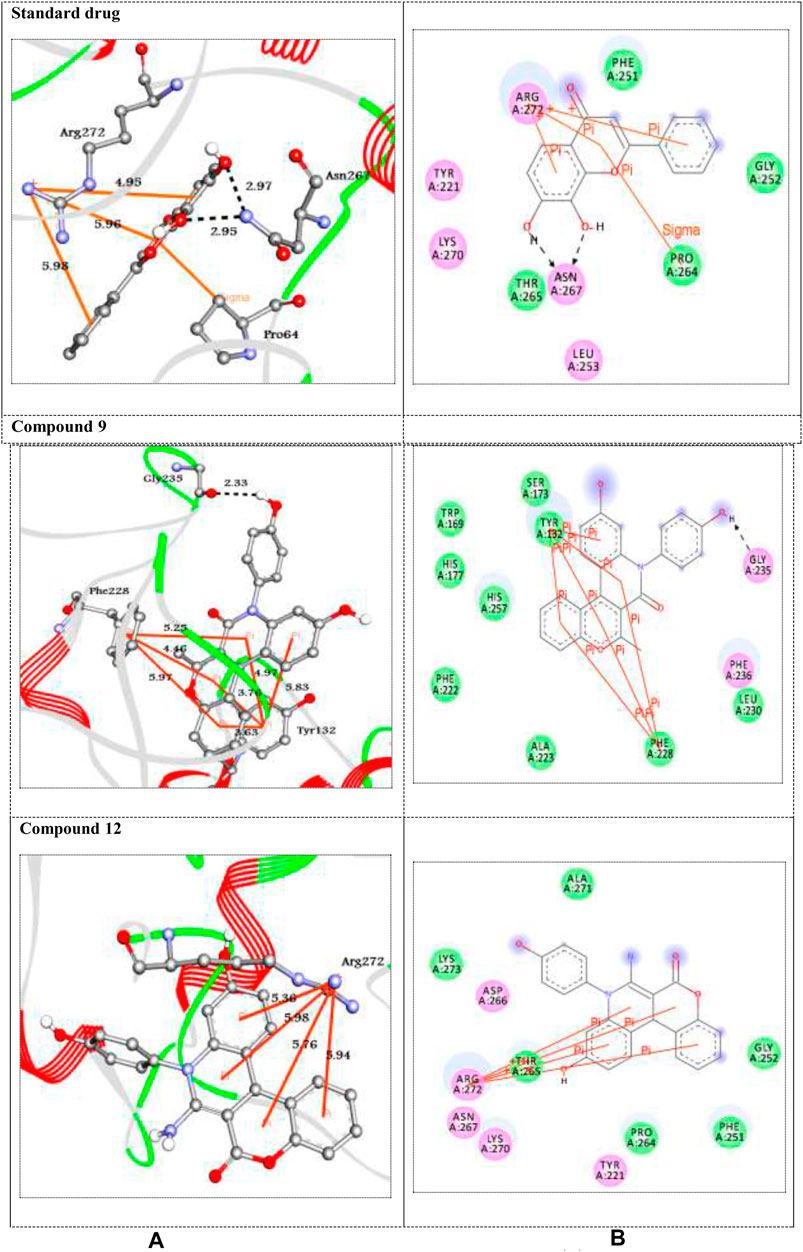
FIGURE 1. (A) 3D and (B) 2D representations of standard drug and the best docked compounds with 1DXO. H-bonds are represented in black dotted lines while pi-interactions are shown in orange lines. Colored balls with 3-letter code represented the amino acid residues of the target 1DXO.
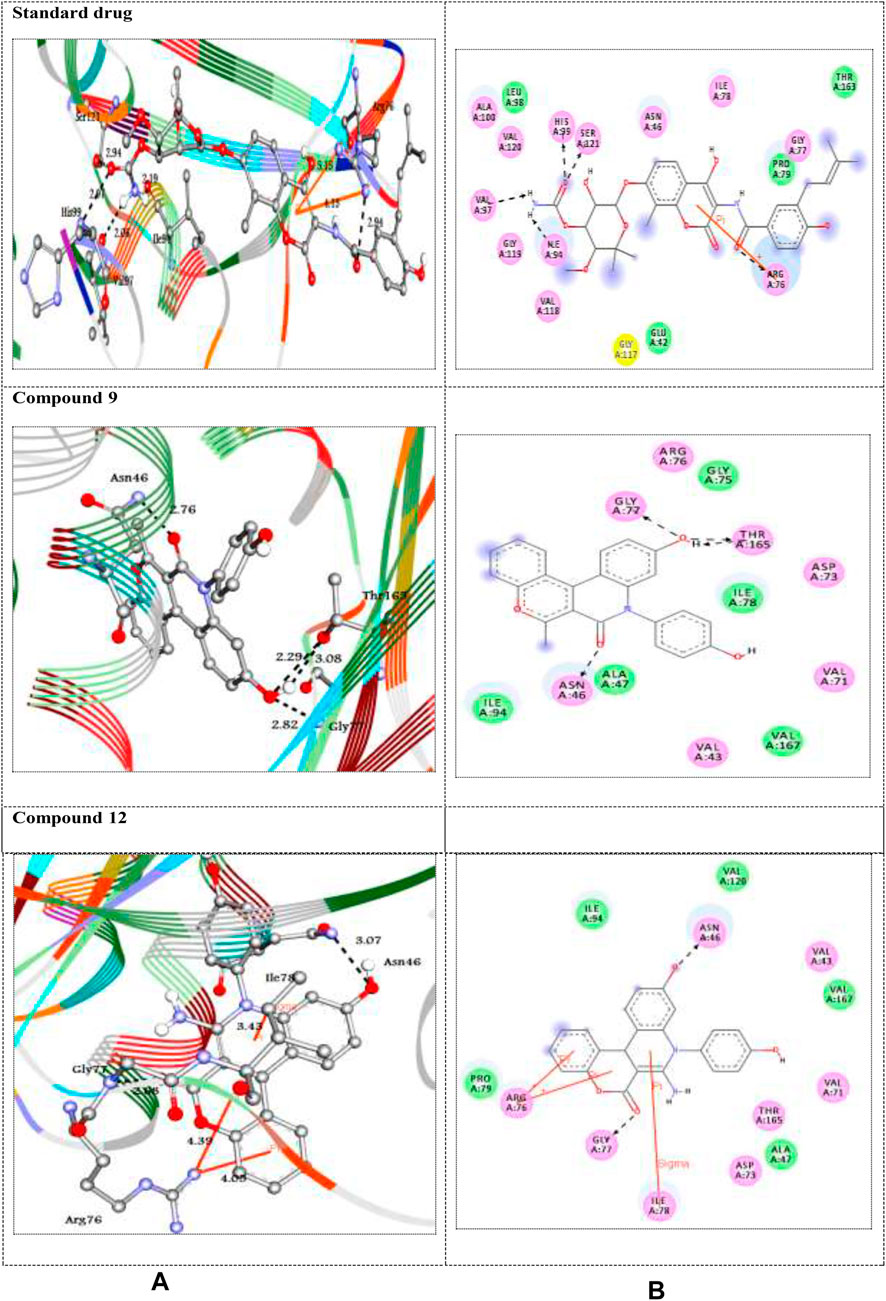
FIGURE 2. (A) 3D and (B) 2D representations of standard drug and the best docked compounds standard drug with 1JA6. H-bonds are represented in black dotted lines while pi-interactions are shown in orange lines. Colored balls with 3-letter code represented the amino acid residues of the target 1JA6.
Antioxidant Activity
Human NAD (P)H dehydrogenase (quinone 1) is an enzyme that combats the oxidative stress conditions (Dinkova-Kostova and Talalay, 2010; Atia and Abdullah, 2020) as a gene highly expressed in human adipocytes and performing its antioxidant activity (Palming et al., 2007). In the present study, the docking studies were performed against the crystal structure of human NAD (P)H dehydrogenase (quinone 1) with PDB code 1DXO. All the docked compounds were fit on the enzyme active site with the docking scores (ΔGbind) of the range -9.1–-6.9 kcal/mol; in addition, the standard drug exhibited binding energy (ΔGbind) = -7.5 kcal/mol, (Table 3). Compound 9 with the highest binding energy (-9.1 kcal/mol) docked to the target enzyme 1DXO through hydrogen bond and π–π stacking interactions with the amino acid residues Gly235, Tyr132, and Phe228 at the distances 2.33, 5.83, 4.97, 3.76, 3.63, 5.25, 4.46, and 5.97 Å, respectively. In addition, compound 12 with − 8.7 kcal/mol showed four π-cation interactions with the residue Arg272 at distances 5.36, 5.98, 5.76, and 5.94 Å, respectively (Figure 1). On the other hand, the standard drug (tropoflavin) with the binding energy − 7.5 kcal/mol binds with the target enzyme through similar amino acid residues Asn267, Arg272, and Pro264at 2.97, 2.95, 4.95, 5.95, 5.98, and 3.75 Å, respectively. The rest of compounds are shown in supplementary file section as Supplementary Figure S30.
Antibacterial Activity
The DNA gyrase is a topoisomerase enzyme that controls the DNA’s topological transition (Samadpour and Merrikh, 2018). In addition, the enzyme DNA gyrase has been considered as an essential for bacterial survival that catalyzes ATP-dependent negative super-coiling of bacterial chromosome (Reece et al., 1991; Tanitame et al., 2004). In this regard, in the present work, DNA gyrase has been selected as antibacterial drug target. The molecular docking simulation of the compounds 5–12 was carried out to identify their binding pattern with bacterial DNA gyrase. The compounds were observed to have the binding energies (ΔGbind) ranging from -9.3 to -7.7 kcal/mol; in addition the standard drug exhibited binding energy (ΔGbind) = -8.3 kcal/mol, as shown in Table 3. The screened compounds 5–12 docked to the target enzyme through various intermolecular interactions as hydrogen bond and π- stacking. Compound 9 has the best docking score (-9.3 kcal/mol) and exhibited four hydrogen bond interactions with the active site residues Asn46, Gly77, and Thr165 at the distances 2.76, 2.82, 3.08, and 2.29 Å, respectively. Moreover, the analogue 12 with -9.2 kcal/mol, showed intermolecular interactions through two hydrogen bond, two π-cation, and π-sigma at the distances of 3.07, 2.68, 4.39, 4.05, and 3.43 Å, respectively (Figure 2). On the other hand, the standard drug (novobiocin) with the binding energy -8.3 kcal/mol docked to the target through similar residues Arg76, His99, Ser121, Ile94, and Val97 at distances 2.94, 2.97, 2.95, 2.19, 2.06, 5.15, and 4.13 Å, respectively (Figure 2). The other docked compounds with the target enzyme are shown in Supplementary Figure S2 (Supplementary file section).
Structure Activity Relationship Analysis
From the obtained results, we can conclude that the compound 9 with electron donating groups like (-OH) and (-CH3), piperidine, and pyran moieties showed the best docking score (ΔGbind) toward both target enzymes NQO1 and DNA gyrase (Haredi Abdelmonsef et al., 2020; Gomha et al., 2021). Comparing the standard drugs (tropoflavin and novobiocin), it has been found that they possess the same functional groups (-OH), (-CH3), and pyran moieties. The docking scores of the synthesized quinoline molecules were in agreement with the experimental results which showed that the compound 9 could be used as potent inhibitor of NQO1 and DNA gyrase enzymes. Overall, the newly synthesized quinoline scaffolds have potential antioxidant and antibacterial activity and could be optimized to use as potent lead compounds as antioxidant and antibacterial agents.
ADMET/Pharmacokinetic Prediction Studies
In silico ADME/T and druglikeness prediction of the molecules 5–12, in addition to the standard drugs (tropoflavin and novobiocin), was computationally calculated in terms of absorption, distribution, metabolic, excretion, and toxicity via admetSAR (Cheng et al., 2012) and Molinspiration Web-based servers. The ADME/T analysis for different synthesized molecules was found to be in acceptable ranges (Table 4). All compounds have molecular weight in the range of 279.30–405.84 g/mol (<500). The % oral intestinal drug absorption of all compounds was in the acceptable range (>80), indicating their possibilities in oral drug formulation for the treatment of bacterial infections. In addition, the new compounds exhibited little chance to cross the blood–brain barrier. The topological surface areas (TPSA) were found to be in the acceptable range (<140). In addition, H-bond acceptors (HBA) and donors (HBD) were found to be in the range of 3–6 and 2–4, respectively. Moreover, the newly synthesized compounds had high numbers of rotatable bonds (0–5), which indicates that they are flexible. Finally, the evaluation of toxicity and carcinogenic profiles for the compounds 5–12 declared that they are nontoxic and noncarcinogenic. Overall, the druglikeness study revealed that the new compounds fulfill the requirements of Lipinski’s rule of five (Ro5) (Lipinski et al., 1997), Veber (Veber et al., 2002), and Ghose (Ghose et al., 2012) without any violations, suggesting that these compounds theoretically have ideal oral bioavailability. From all these results, we can conclude that all molecules exhibited good solubility and oral bioavailability.
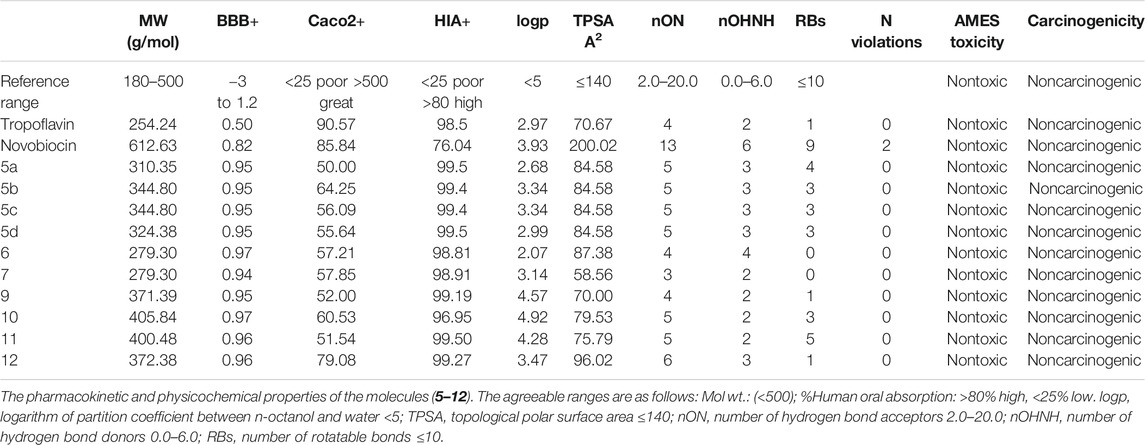
TABLE 4. List of ADME/T and physicochemical properties of standard drugs and compounds 5–12.
Conclusion
In conclusion, we described a rapid, efficient, and low-cost method for synthesis of some quinoline analogues by using four components under solvent-free conditions. In addition, all synthesized compounds were in vitro screened for their antioxidant and antibacterial activity. Further, in silico molecular docking studies were achieved to support the biological experiments. The compound 9 displayed promising antioxidant and antibacterial activity, which was well supported by the in silico binding score, which showed it to have the highest binding energy of -9.1 and -9.3 kcal/mol against the target enzymes 1DXO and 1AJ6, respectively. In addition, compound 9 obeyed the Lipinski’s rule of five, Veber, and Ghose . The experimental and in silico findings indicated that compound 9 could be used as a promising inhibitor of enzymes NQO1 and DNA gyrase.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author Contributions
All authors contributed to writing and editing of the manuscript. All authors were responsible for synthesis and characterization of compounds. AA did the virtual screening approach. All the authors agreed on the final version to be submitted.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Chemistry Department, Faculty of Science, Assiut Al-Azhar University and Sohag University for facilitating the publication of this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.679967/full#supplementary-material
References
Abdelmonsef, A. H. (2019). Computer-Aided Identification of Lung Cancer Inhibitors through Homology Modeling and Virtual Screening. Egypt. J. Med. Hum. Genet. 20, 1–14. doi:10.1186/s43042-019-0008-3
Abdelmonsef, A. H., Dulapalli, R., Dasari, T., Padmarao, L. S., Mukkera, T., and Vuruputuri, U. (2016). Identification of Novel Antagonists for Rab38 Protein by Homology Modeling and Virtual Screening. Comb Chem High Throughput Screen. 19 (10). doi:10.2174/1386207319666161026153237
Abdelmonsef, A. H., and Mosallam, A. M. (2020). Synthesis, In Vitro Biological Evaluation and In Silico Docking Studies of New Quinazolin‐2,4‐dione Analogues as Possible Anticarcinoma Agents. J. Heterocycl Chem. 57, 1637–1654. doi:10.1002/jhet.3889
Acharyulu, P. V. R., Dubey, P. K., Reddy, P. V. V. P., and Suresh, T. (2008). Synthesis of New 4(3H)-Quinazolinone Derivatives under Solvent-free Conditions Using PEG-400. Arkivoc. 2008 (11), 104–111. doi:10.3998/ark.5550190.0009.b10
Asif, M. (2014). Chemical Characteristics, Synthetic Methods, and Biological Potential of Quinazoline and Quinazolinone Derivatives. Int. J. Med. Chem. 2014, 1–27. doi:10.1155/2014/395637
Atia, A., and Abdullah, A. (2020). NQO1 Enzyme and its Role in Cellular Protection; an Insight. Iberoamerican J. Med. 02, 306–313. doi:10.5281/zenodo.3877528
Bauer, A. W., Kirby, W. M. M., Sherris, J. C., and Turck, M. (1966). Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol. 45 (4), 493–496. doi:10.1308/rcsann.2013.95.7.532
Bawa, S., Kumar, S., Drabu, S., and Kumar, R. (2010). Structural Modifications of Quinoline-Based Antimalarial Agents: Recent Developments. J. Pharm. Bioall Sci. 2 (2), 64. doi:10.4103/0975-7406.67002
Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., et al. (2012). AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 52 (11), 3099–3105. doi:10.1021/ci300367a
Dallakyan, S., and Olson, A. J. (2015). Small-Molecule Library Screening by Docking with PyRx. Chem. Biol. 1263, 243–250. doi:10.1016/B978-0-12-394447-4.10004-510.1007/978-1-4939-2269-7_19
Dasari, T., Kondagari, B., Dulapalli, R., Abdelmonsef, A. H., Mukkera, T., Padmarao, L. S., et al. (2017). Design of Novel lead Molecules against RhoG Protein as Cancer Target - a Computational Study. J. Biomol. Struct. Dyn. 35 (14), 3119–3139. doi:10.1080/07391102.2016.1244492
Dinkova-Kostova, A. T., and Talalay, P. (2010). NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector Arch. Biochem. Biophys. 501 (1), 116–123. doi:10.1016/j.abb.2010.03.019
Dua, R., Shrivastava, S., Sonwane, S. K., and Srivastava, S. K. (2011). Pharmacological Significance of Synthetic Heterocycles Scaffold : A Review. Advan. Biol. Res. 5 (3), 120–144. doi:10.1023/B:SOLA.0000013030.09729.38
El-Maghraby, A. M., and Aboubakr, H. A. (2019). Synthesis, Characterization and Insilico Molecular Docking Studies of Novel Chromene Derivatives as Rab23 Inhibitors. Egypt. J. Chem. 63 (4), 1341–1358. doi:10.21608/ejchem.2019.15013.1911
El-Maghraby, A. M. (2014). Green Chemistry: New Synthesis of Substituted Chromenes and Benzochromenes via Three-Component Reaction Utilizing Rochelle Salt as Novel Green Catalyst. Org. Chem. Int. 2014 (Scheme 1), 1–6. doi:10.1155/2014/715091
Faig, M., Bianchet, M. A., Talalay, P., Chen, S., Winski, S., Ross, D., et al. (2000). Structures of Recombinant Human and Mouse NAD(P)H:Quinone Oxidoreductases: Species Comparison and Structural Changes with Substrate Binding and Release. Proc. Natl. Acad. Sci. 97 (7), 3177–3182. doi:10.1073/pnas.97.7.3177
Ghose, A. K., HerbertzHudkins, T., Hudkins, R. L., Dorsey, B. D., and Mallamo, J. P. (2012). Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 3 (1), 50–68. doi:10.1021/cn200100h
Gómez, C. M. M., and Vladimir, V. K. (2013). “Recent Developments on Antimicrobial Quinoline Chemistry,” in Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education. Editor A. Méndez-Vilas (FORMATEX), December, 666–677.
Gomha, S. M., Hyam, A. A., Doaa Zh, H., Aboubakr, H. A., and Mohamed, E-N. (2021). Thiazole-Based Thiosemicarbazones: Synthesis, Cytotoxicity Evaluation and Molecular Docking Study. Drug Des Devel Ther. Vol. 15 (February), 659–677. doi:10.2147/DDDT.S291579
Ha, A., and Sp, L. (2016). Human Rab8b Protein as a Cancer Target - an In Silico Study. J. Comput. Sci. Syst. Biol. 9 (4). doi:10.4172/jcsb.1000231
Haredi Abdelmonsef, A., Eldeeb Mohamed, M., El-Naggar, M., Temairk, H., and Mohamed Mosallam, A. (2020). Novel Quinazolin-2,4-Dione Hybrid Molecules as Possible Inhibitors against Malaria: Synthesis and In Silico Molecular Docking Studies. Front. Mol. Biosci. 7, 105. doi:10.3389/fmolb.2020.00105
Holdgate, G. A., Tunnicliffe, A., Ward, W. H. J., Weston, S. A., Rosenbrock, G., Barth, P. T., et al. (1997). The Entropic Penalty of Ordered Water Accounts for Weaker Binding of the Antibiotic Novobiocin to a Resistant Mutant of DNA Gyrase: A Thermodynamic and Crystallographic Study. Biochemistry 36 (32), 9663–9673. doi:10.1021/bi970294+
Hussein, M. A., Ola, H. Z., Abo-bakr, H. A. M., Rizk, S. A., Shaimaa, M., Amany, S. K., et al. (2018). Synthesis, Molecular Docking and Insecticidal Activity Evaluation of Chromones of Date Palm Pits Extract against Culex Pipiens (Diptera: Culicidae). Int. J. Mosquito Res. 5 (4), 22–32.
Kumar, S., Bawa, S., and Gupta, H. (2009). Biological Activities of Quinoline Derivatives. Mini Rev Med Chem. 9 (14), 1648–1654. doi:10.2174/138955709791012247
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (1997). Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Develop Ment Settings. Adv. Drug Deliv. Rev. 23 (August), 3–25. doi:10.1016/S0169-409X(00)00129-0
Luo, Z. G., Cheng, C. Z., Fang, W., Hong, Q. H., Cun, X. W., Hong, G. D., et al. (2009). Synthesis and Biological Activities of Quinoline Derivatives as HIV-1 Integrase Inhibitors. Chem. Res. Chin. Universities 25 (6), 841–845.
Meshram, H. M., Chennakesava Reddy, B., Aravind Kumar, D., Kalyan, M., Ramesh, P., Kavitha, P., et al. (2012). Synthesis and Cytotoxicity of New Quinoline Derivatives. Indian J. Chem. - Section B Org. Med. Chem. 51 (9), 1411–1416.
Noser, A. A., El-Naggar, M., Donia, T., and Abdelmonsef, A. H. (2020). Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition. Molecules 25 (20), 4780. doi:10.3390/molecules25204780
O’Boyle, N. M., Michael, B., Craig, A. J., Chris, M., Tim, V., and Geoffrey, R. H. (2011). Open Babel: An Open Chemical Toolbox. J. Cheminformatics 3 (10), 33. doi:10.1186/1758-2946-3-33
Palming, J., Sjöholm, K., Jernås, M., Lystig, T. C., Gummesson, A., Romeo, S., et al. (2007). The Expression of NAD(P)H:Quinone Oxidoreductase 1 Is High in Human Adipose Tissue, Reduced by Weight Loss, and Correlates with Adiposity, Insulin Sensitivity, and Markers of Liver Dysfunction. Dysfunction 92 (6), 2346–2352. doi:10.1210/jc.2006-2476
Prieto, P., Pineda, M., and Aguilar, M. (1999). Spectrophotometric Quantitation of Antioxidant Capacity through the Formation of a Phosphomolybdenum Complex: Specific Application to the Determination of Vitamin E. Anal. Biochem. 269 (2), 337–341. doi:10.1006/abio.1999.4019
Rashdan, H. R. M., Rashdan, H. R. M., Aboubakr, H. A., Ihsan, A. S., Sobhi, M. G., Abdel Mohsen, M. S., et al. (2020). Synthesis, Molecular Docking Screening and Anti-proliferative Potency Evaluation of Some New Imidazo[2,1-b]Thiazole Linked Thiadiazole Conjugates. Molecules 25 (21), 4997. doi:10.3390/molecules25214997
Reece, R. J., Maxwell, A., and Wang, James. C. (1991). DNA Gyrase: Structure and Function. Crit. Rev. Biochem. Mol. Biol. 26 (3–4), 335–375. doi:10.3109/10409239109114072
Rondla, R., PadmaRao, L. S., Ramatenki, V., Haredi-Abdel-Monsef, A., Potlapally, S. R., and Vuruputuri, U. (2017). Selective ATP Competitive Leads of CDK4: Discovery by 3D-QSAR Pharmacophore Mapping and Molecular Docking Approach. Comput. Biol. Chem. 71 (December), 224–229. doi:10.1016/j.compbiolchem.2017.11.005
Samadpour, A. N., and Merrikh, H. (2018). DNA Gyrase Activity Regulates DnaA-dependent Replication Initiation inBacillus Subtilis. Mol. Microbiol. 108 (2), 115–127. doi:10.1111/mmi.13920
Sangani, C., Shah, N., Patel, M., and Patel, R. (2012). Microwave Assisted Synthesis of Novel 4h-Chromene Derivatives Bearing Phenoxypyrazole and Their Antimicrobial Activity Assess. Jscs. 77 (9), 1165–1174. doi:10.2298/jsc120102030s
Shang, X.-F., Morris-Natschke, S. L., Liu, Y.-Q., Guo, X., Xu, X.-S., Goto, M., et al. (2018a). Biologically Active Quinoline and Quinazoline Alkaloids Part I. Med. Res. Rev. 38, 775–828. doi:10.1002/med.21466
Shang, X.-F., Morris-Natschke, S. L., Liu, Y.-Q., Guo, X., Xu, X.-S., Goto, M., et al. (2018b). Biologically Active Quinoline and Quinazoline Alkaloids Part II. Med. Res. Rev. 38, 1614–1660. doi:10.1002/med.21492
Shehadi, I. A., Rashdan, H. R. M., and Abdelmonsef, A. H. (2020). Homology Modeling and Virtual Screening Studies of Antigen MLAA-42 Protein: Identification of Novel Drug Candidates against Leukemia-An In Silico Approach. Comput. Math. Methods Med. 2020, 1–12. doi:10.1155/2020/8196147
Sidoryk, K., Świtalska, M., Jaromin, A., Cmoch, P., Bujak, I., Kaczmarska, M., et al. (2015). The Synthesis of Indolo[2,3-b]Quinoline Derivatives with a Guanidine Group: Highly Selective Cytotoxic Agents. Eur. J. Med. Chem. 105, 208–219. doi:10.1016/j.ejmech.2015.10.022
Tanitame, A., Oyamada, Y., Ofuji, K., Fujimoto, M., Iwai, N., Hiyama, Y., et al. (2004). Synthesis and Antibacterial Activity of a Novel Series of Potent DNA Gyrase Inhibitors. Pyrazole Derivatives. J. Med. Chem. 47 (14), 3693–3696. doi:10.1021/jm030394f
Veber, D. F., Stephen, R. J., Hung-Yuan, C., Brian, R. S., Keith, W. W., and Kenneth, D. (2002). Molecular Properties that Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 45 (12), 2615–2623. doi:10.1021/jm020017n
Wan, C., Yu, Y., Zhou, S., Liu, W., Tian, S., and Cao, S. (2011). Antioxidant Activity and Free Radical-Scavenging Capacity of Gynura Divaricata Leaf Extracts at Different Temperatures. Pharmacogn Mag. 7, 40–45. doi:10.4103/0973-1296.75900
Keywords: multicomponent reaction, chromenoquinolines, antioxidant, antibacterial, molecular docking
Citation: El-Saghier AM, El-Naggar M, Hussein AHM, El-Adasy A-BA, Olish M and Abdelmonsef AH (2021) Eco-Friendly Synthesis, Biological Evaluation, and In Silico Molecular Docking Approach of Some New Quinoline Derivatives as Potential Antioxidant and Antibacterial Agents. Front. Chem. 9:679967. doi: 10.3389/fchem.2021.679967
Received: 16 March 2021; Accepted: 14 May 2021;
Published: 10 June 2021.
Edited by:
Simone Brogi, the University of Pisa, ItalyReviewed by:
Dusan Misic, Wroclaw University of Environmental and Life Sciences, PolandMarcus Scotti, Federal University of Paraíba, Brazil
Copyright © 2021 El-Saghier, El-Naggar, Hussein, El-Adasy, Olish and Abdelmonsef. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aboubakr H. Abdelmonsef, YWJvdWJha3IuYWhtZWRAc2NpLnN2dS5lZHUuZWc=