 Shang-Te Danny Hsu1,2*
Shang-Te Danny Hsu1,2* Yun-Tzai Cloud Lee1,2‡
Yun-Tzai Cloud Lee1,2‡ Kornelia M. Mikula3‡Sofia M. Backlund3Igor Tascón3†
Kornelia M. Mikula3‡Sofia M. Backlund3Igor Tascón3† Adrian Goldman4,5
Adrian Goldman4,5 Hideo Iwaï3*
Hideo Iwaï3*- 1Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan
- 2Institute of Biochemical Sciences, National Taiwan University, Taipei, Taiwan
- 3Institute of Biotechnology, University of Helsinki, Helsinki, Finland
- 4Division of Biochemistry, Department of Biosciences, University of Helsinki, Helsinki, Finland
- 5Astbury Centre for Structural Molecular Biology, School of Biomedical Sciences, University of Leeds, West Yorkshire, United Kingdom
Knots have attracted scientists in mathematics, physics, biology, and engineering. Long flexible thin strings easily knot and tangle as experienced in our daily life. Similarly, long polymer chains inevitably tend to get trapped into knots. Little is known about their formation or function in proteins despite >1,000 knotted proteins identified in nature. However, these protein knots are not mathematical knots with their backbone polypeptide chains because of their open termini, and the presence of a “knot” depends on the algorithm used to create path closure. Furthermore, it is generally not possible to control the topology of the unfolded states of proteins, therefore making it challenging to characterize functional and physicochemical properties of knotting in any polymer. Covalently linking the amino and carboxyl termini of the deeply trefoil-knotted YibK from Pseudomonas aeruginosa allowed us to create the truly backbone knotted protein by enzymatic peptide ligation. Moreover, we produced and investigated backbone cyclized YibK without any knotted structure. Thus, we could directly probe the effect of the backbone knot and the decrease in conformational entropy on protein folding. The backbone cyclization did not perturb the native structure and its cofactor binding affinity, but it substantially increased the thermal stability and reduced the aggregation propensity. The enhanced stability of a backbone knotted YibK could be mainly originated from an increased ruggedness of its free energy landscape and the destabilization of the denatured state by backbone cyclization with little contribution from a knot structure. Despite the heterogeneity in the side-chain compositions, the chemically unfolded cyclized YibK exhibited several macroscopic physico-chemical attributes that agree with theoretical predictions derived from polymer physics.
Introduction
Knots always fascinate people and have attracted scientists from all disciplines. Long flexible strings can spontaneously knot themselves upon agitation (Raymer and Smith, 2007). Whereas circular supercoiled DNA in nature can be a true mathematical knot, proteins are linear polymers consisting of 20 different amino acids connected by peptide bonds with open amino (N) and carboxyl (C) termini. Proteins fold into defined three-dimensional (3D) conformations and execute various functions at the molecular level. The apparent complexity of threading events involved in tying a protein knot made them inconceivable to many structural biologists at first (Mansfield, 1994). Nevertheless, systematic surveys of the protein database have identified more than 1,000 knotted protein structures with different knot types and structural complexities. However, in this context, “knot” does not imply a topological knot, which cannot be undone except by breaking the protein backbone, a knot in the sense of knotted tie, or a sailor’s reef knot (Jamroz et al., 2015; Lim and Jackson, 2015). It has been challenging to reconcile experimental and theoretical views of how a polypeptide chain attains an intricately knotted topology (Mallam et al., 2008; Mallam and Jackson, 2012; Beccara et al., 2013; Sulkowska et al., 2013; Lim and Jackson, 2015; Ziegler et al., 2016; Jackson et al., 2017). Now, there have been many experimental studies for better understanding of the protein knotting mechanisms. While the majority of experimental studies showed that knotted proteins fold into the knotted conformations with highly populated folding intermediates along their kinetic folding pathways (Mallam and Jackson, 2007; Andersson et al., 2009; Lim and Jackson, 2015; Wang et al., 2015; Lou et al., 2016; Wang et al., 2016; Dabrowski-Tumanski and Sulkowska, 2017; Jackson et al., 2017; He et al., 2019; Jarmolinska et al., 2019; Rivera et al., 2020), there are also some knotted (or slipknotted) proteins that can fold without populating intermediate states (He et al., 2019; Rivera et al., 2020). Different experimental studies have shown that knotting is rate-limiting (Mallam and Jackson, 2012). Computational approaches have also been used to verify the experimental observations, such as the rugged free energy landscapes of several knotted proteins and multiple intermediates populated along their folding pathways. These computational studies might explain the rate-limiting step of protein knotting (Li et al., 2012; Beccara et al., 2013; Sulkowska and et al., 2013; Faísca, 2015) and lead to various protein knotting mechanisms, such as direct threading, slipknotting, and mousetrapping (Noel et al., 2010; Covino et al., 2014). Only very recently, we combined experimental and computational data to obtain a converged view of how the smallest knotted protein, MJ0366, attains a knotted transition state (Passioni et al., 2021). However, all investigations of protein knots have so far been reported for protein knots with open ends because proteins are synthesized as linear polypeptide chains. As such, proteins are not true mathematical knots but are defined by virtual connections of the N- and C-termini by different mathematical schemes (Taylor, 2000; Lai et al., 2012; Millett et al., 2013).
In this study, we asked whether a backbone knotted protein without open ends could be generated by backbone cyclization. Whereas backbone cyclization of proteins has been widely accepted to stabilize proteins (Iwai and Plückthun, 1999; Scott et al., 1999; Clark and Craik, 2010; Montalbán-López et al., 2012; Borra and Camarero, 2013), disulfide bridges, which were originally considered to stabilize proteins by reducing the entropy of the denatured state as backbone cyclization, have more enthalpic contributions to the stability in the folded state (Mitchinson and Wells, 1989; Betz, 1993). Moreover, backbone cyclization of knotted proteins could create the unique possibility to investigate the unfolded state of proteins with a knot unequivocally. A backbone cyclized knotted protein would conform to the mathematical definition for a truly knotted topology whose path closure does not depend on how the ends are joined together in space. Whereas naturally occurring protein knots have open polypeptide-chain ends and could be disentangled into linear polypeptide chains without knots and entanglements under denaturing conditions, backbone cyclized knotted proteins cannot untie anymore without proteolysis even under denaturing conditions.
Here, we presented the unprecedented characterization of a mathematical backbone protein knot without open peptide-chain ends using various structural and biophysical methods including SAXS, X-ray crystallography, and 15N nuclear relaxation analysis by NMR spectroscopy.
Results
Production of a Knotted Protein With and Without Open Ends
To produce a protein knot without open polypeptide ends, we chose the highly conserved bacterial RNA methyltransferase as a model system, namely YibK from Pseudomonas aeruginosa (PaYibK). YibK contains a trefoil (31) knotted backbone topology. PaYibK also shares 65% sequence identity with one of the most-studied knotted proteins, YibK form Haemophilus influenzae (HiYibK), but contains only one tryptophan (Supplementary Figure S1). (Tkaczuk et al., 2007) We first determined the crystal structure of the wild-type PaYibK in its linear form (PaYibK) to confirm the same trefoil knot structure and the dimeric assembly as found in HiYibK (Figure 1A; Supplementary Table S1; and Supplementary Figure S1). The root mean square deviation (RMSD) was only 0.6 Å between the crystal structures of PaYibK and HiYibK, indicating a highly conserved higher-order structure among YibK proteins throughout evolution including the knot structure (Figure 1A). The N- and C-termini of PaYibK are separated by ca. 8 Å in the crystal structure, which is sufficiently close for the head-to-tail backbone cyclization without disturbing the backbone knot structure. For backbone cyclization of proteins, various strategies have been established, including intein-mediated protein ligation (or expressed protein ligation), enzymatic protein cyclization using enzymes such as sortaseA (SrtA) and asparagine endopeptidase (AEP), and protein trans-splicing (PTS) (Scott et al., 1999; Popp and Ploegh, 2011; Mikula et al., 2017; Iwai et al., 2001). We initially attempted in vivo protein cyclization of PaYibK by PTS using the naturally split DnaE intein (PaYibK_Int) (Figure 1B) (Aranko et al., 2013), but the in vivo spliced product was insoluble (Figure 1C). PTS-based backbone cyclization relies on the self-association of intein fragments that brings the N- and C-termini together during the protein folding, followed by spontaneous auto-catalytic removal of the intein fragments, thereby achieving backbone cyclization (Scott et al., 1999; Iwai et al., 2001). We speculated that the rapid self-association of the split intein fragments could interfere with the folding and knotting of YibK, thereby resulting in insoluble aggregation. The insoluble spliced product could be backbone cyclized YibK without knotting (cYibK_Int) (Figure 1C). Therefore, we used an alternative enzymatic approach with S. aureus sortase A (SrtA), which catalyzes a trans-peptidase reaction between the LPETG motif and an N-terminal tri-glycine peptide (Figure 1D). (Popp and Ploegh, 2011) When a seven-residue linker (PaYibK_sh) was used to link the N and C-termini through SrtA-mediated ligation (Figure 1E), backbone cyclization was inefficient, and accumulation of a covalent dimer (di-lYibK_sh) resulting from intermolecular ligation was more prominent than the monomeric backbone-cyclized form (cYibK_sh; Figure 1F). This finding underscores the need to further optimize the linker length for the enzymatic ligation. By introducing a longer nine-residue linker to the backbone cyclization (PaYibK_lo), a much more efficient backbone cyclization was achieved to produce a higher amount of monomeric cyclized YibK (cYibK_lo hereafter designated as cYibK) than the covalent dimer (di-PaYibK_lo; Figure 1G). We also produced a linear form of YibK (lYibK) as the N-terminal SUMO fusion so that lYibK and cYibK (PaYibK_lo) have the identical protein sequence for the analysis. The backbone cyclization of cYibK manifests in greater mobility during SDS-PAGE as compared with linear YibK (lYibK); the complete cyclization was also confirmed by mass spectrometry (Supplementary Figure S2).
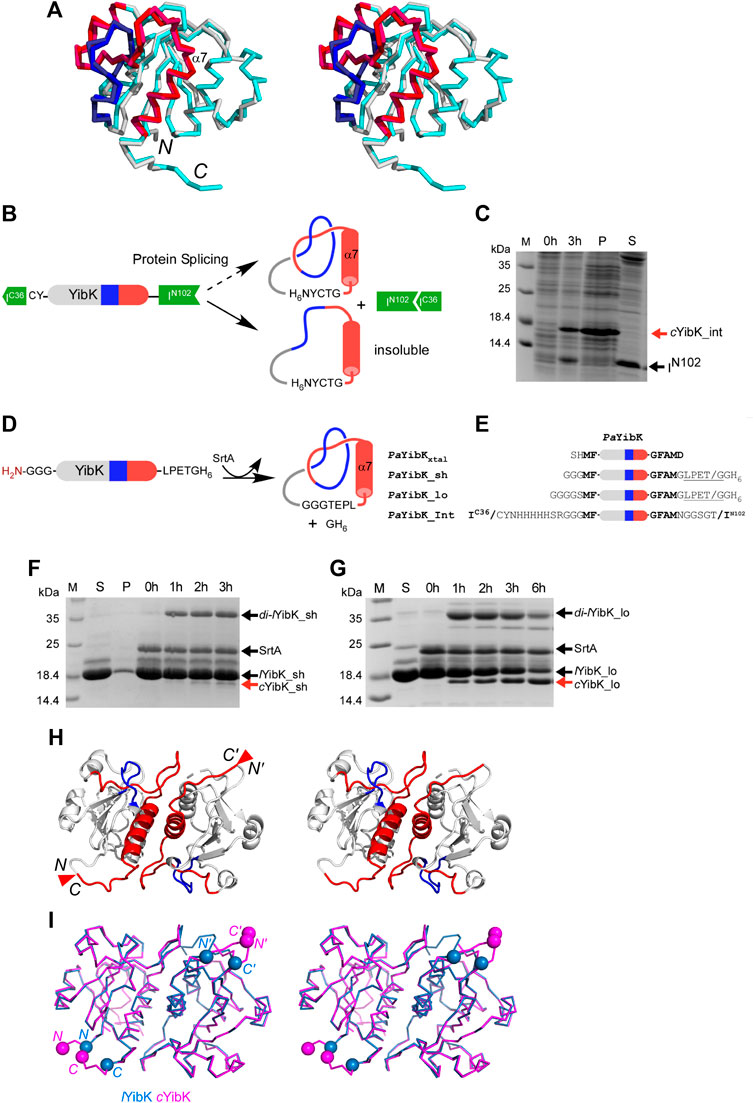
FIGURE 1. Cyclization and structural analysis of a truly knotted PaYibK. (A) A stereo view of the superposition of PaYibK (pdb:6qkv) and HaYibK (pdb:1j85) in ribbon presentation. (B) Schematic representation of the experimental strategy used for cyclization of YibK by the split-intein strategy. (C)In vivo backbone cyclization of YibK by protein trans-splicing. Spliced YibK and N-terminal fragment are indicated; M, Molecular weight marker P, insoluble fraction; S, soluble material. (D) Backbone cyclization of YibK using sortase A (SrtA). (E) Constructs used in backbone cyclization with different lengths of linkers connecting the termini. (F) and (G) Impact of linker lengths on the in vitro sortase ligation efficiency monitored by SDS-PAGE of linear YibK with short (F) and long (G) poly-glycine linkers annotated as lYibK_sh and lYibK_lo, respectively; 0–6 h: ligation reactions harvested at specified time points; SrtA, sortase added post-purification; di-YibK_sh/lo, undesired dimeric ligation products; cYibK_sh/lo, desired cyclized products. The N and C-terminal extensions of YibK for different ligation experiments are shown with their names indicated on the left. (H) A stereo-view of the crystal structure of cYibK_lo. The structures are shown in cartoon representation with the knotting loop and threading C-terminal helix shown in blue and red, respectively. The positions of the path closures between N- and C-termini are indicated by red triangles. (I) Comparison of the crystal structure of linear PaYibK (6qkv; orchid blue) and cYibK (6qh8; magenta). The Cα atoms of the S1 and D155 of PaYibK are shown in orchid blue spheres, and the positions of path closures in cYibK are indicated by magenta triangles as in (H).
Structural Comparison of a Knotted Protein With and Without Open Ends
We determined the crystal structures of cYibK to a resolution of 2.20 Å (Figure 1H and Supplementary Table S1). The root mean square deviation (RMSD) between the backbone Cα atoms of the crystal structures of PaYibK and cYibK was <0.2 Å, confirming that the backbone cyclization did not perturb the 3D structure of PaYibK in the folded state (Figure 1I). The structural similarity between lYibK and cYibK and their dimeric states were confirmed by small angle X-ray scattering (SAXS) in solution state (vide infra).
Functional Assessment of lYibK and cYibK
Next, we assessed the functional impact of cyclization on the cofactor binding activity, which is essential for the RNA methyltransferase activity of YibK. We used isothermal titration calorimetry (ITC) to determine the dissociation constants (Kd) of lYibK and cYibK for S-adenosyl-L-homocysteine (SAH), which is the product of the conserved RNA methylation reaction among all SPOUT family members that utilize S-adenosyl-L-methionine (SAM) as the methyl donor (Tkaczuk et al., 2007). The Kd for SAH was 8.80 ± 0.01 and 8.93 ± 0.03 μM for lYibK and cYibK, respectively, which corroborates our structural analyses showing no appreciable structural perturbation in PaYibK after the backbone cyclization (Supplementary Table S2; Supplementary Figure S4). In contrast, side-chain disulfide bond-mediated cyclization of HiYibK was reported to decrease SAH binding affinity (Kd increased from 20 to 71 µM) (Mallam et al., 2010). Furthermore, our experimental Kd values for SAH binding to cYibK/lYibK were 2 to 3-fold smaller than the reported values for other tRNA methyltransferases, namely HiYibK (Kd = 20 μM) (Mallam and Jackson, 2007) and TrmL from E. coli (Kd = 25 μM) (Liu et al., 2013). ITC analysis revealed that SAH binding to cYibK was enthalpically more favorable than that of lYibK (ΔH = −20.1 vs −16.5 kcal mol−1); the difference in enthalpic changes was compensated by the entropic differences (ΔS), resulting in the comparable net free energies of SAH binding (Figure 2A). The greater entropic loss in cYibK upon SAH binding may be associated with the dimer formation of cYibK.
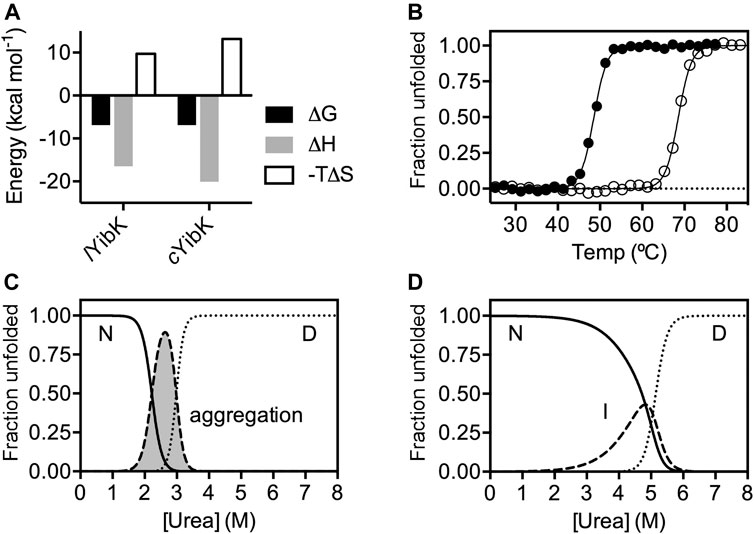
FIGURE 2. The impacts of cyclization on SAH binding and chemical stability of YibK. (A) Thermodynamics parameters associated with SAH binding derived from ITC analyses. (B) Normalized fractional unfolded populations of lYibK (filled circles) and cYibK (open circles) derived from thermal denaturation monitored by far-UV CD spectroscopy. Intrinsic fluorescence-derived normalized fractional populations of native (N), intermediate (I) and denatured (D) states of lYibK (C) and cYibK (D) as a function of urea concentration. For lYibK, fluorescence signal loss due to aggregation was observed and the corresponding population is shaded in gray and indicated.
Comparison of Folding of lYibK and cYibK
As cYibK and lYibK have the identical primary structure and crystal structure in their native states, we assume their associated free energy levels in the folded states are very similar except for the entropic contribution associated with the decreased degrees of freedom of the fraying ends by closing the ends. To investigate how a path closure to form a truly knotted protein may affect folding stability and kinetics, we assessed the thermal stabilities of lYibK and cYibK by far-UV circular dichroism (CD) spectroscopy and their chemical stabilities by urea-induced chemical denaturation monitored by intrinsic fluorescence. As expected, the apparent melting temperature (Tm) of lYibK was increased by 20 °C for cYibK (Tm = 68.7 vs 48.7 °C; Figure 2B). This observation is in line with other proteins with cyclized peptide backbones, suggesting that backbone cyclization of a knotted protein reduced the conformational entropy of the unfolded state. As the thermal unfolding was not fully reversible, particularly in the case of lYibK, the Tm values derived from the CD analysis could be underestimated. We additionally analyzed the urea-induced equilibrium unfolding of lYibK and cYibK (Figures 2C,D). However, we observed the unexpected loss of intrinsic fluorescence of lYibK between 2 and 3 M urea, which we attributed to the aggregation of lYibK in the analysis (Supplementary Figure S4a). In contrast, cYibK did not show the similar loss of intrinsic fluorescence during urea-induced denaturation (Supplementary Figure S4b). The experimental data were fit to a three-state unfolding model by the singular value decomposition approach without considering the contributions of dimerization (Wang et al., 2015; Wang et al., 2016). Although the chemical denaturation of lYibK was not fully reversible with lYibK, the chemical stability of cYibK was clearly higher than lYibK by >2 M of the transition urea concentration required to unfold lYibK and cYibK, supporting the increased apparent thermal stability of cYibK (Supplementary Table S3; Figures 2C,D).
Additionally, we analyzed the folding kinetics of lYibK and cYibK as a function of urea concentration by monitoring the intrinsic fluorescence of the only endogenous tryptophan residue (W150 according to the nomenclature of lYibK construct) lining the dimer interface (Figure 3). It is noteworthy that the well-investigated HiYibK contains two tryptophan residues, of which W145 is positioned at the same dimer interface as W150 of PaYibK (Supplementary Figure S4). Similar to the reported multiphasic kinetics of HiYibK, lYibK also exhibited two unfolding and refolding phases; the faster phase had a very small m-value associated with the unfolding arm (Figure 3). The slower intrinsic unfolding rate of lYibK (
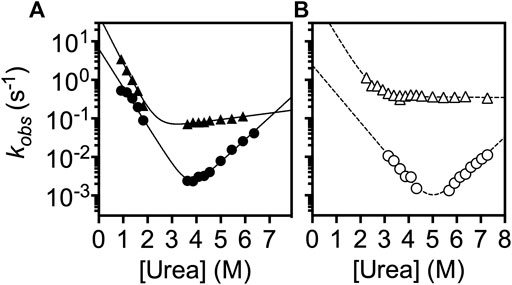
FIGURE 3. Chevron plot analysis of the folding kinetics of lYibK and cYibK. The observed folding rates of lYibK (A) and cYibK (B) are plotted as a function of urea concentration. Two kinetic phases were observed. Circles and triangles correspond to the slow and fast kinetic phases, respectively. Filled and open symbols are used for lYibK and cYibK, respectively. The data were fitted to a simple two-state folding model.
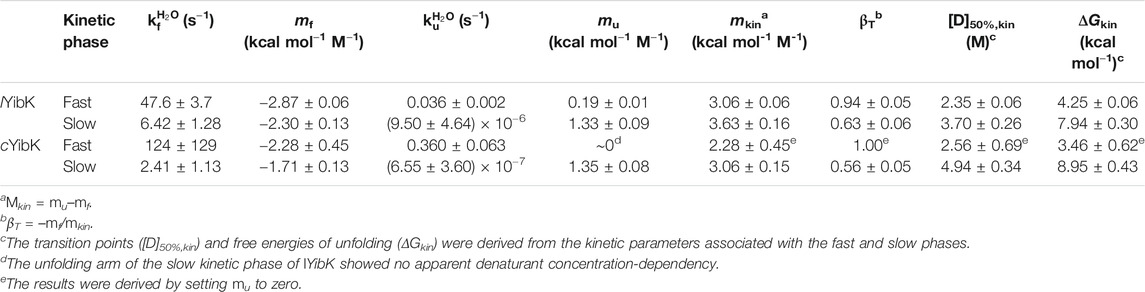
TABLE 1. Kinetic parameters derived from chevron plot analysis of lYibK and cYibK.
Comparison Between lYibK and cYibK Under a Denaturing Condition by NMR
Considering that the chemical compositions (sequences) and 3D structures of cYibK and lYibK essentially are identical with the exception of a peptide bond introduced, we presumed that the native states of both YibKs have similar free energies. We assume that the observed changes in the folding/unfolding pathway of YibK could be caused by the unfolded state of cYibK, which has a significantly reduced conformational space compared with a linear polypeptide due to the circular backbone peptide chain having closed ends and the presence of a knot structure.
As NMR spectroscopy can investigate proteins under various solution conditions including denaturating ones, we used NMR to characterize the unfolded states into both lYibK and cYibK in 7.2 M urea to gain structural insights into the denatured states of lYibK and cYibK (Figure 4). The two-dimensional [15N-1H] correlation spectra for both lYibK and cYibK in 7.2 M urea showed poor chemical shift dispersions along the 1H dimension, characteristic of unfolded and disordered polypeptides (Figure 4A). Furthermore, a large number of crosspeaks exhibited major chemical shift differences between the two [15N-1H] SOFAST-HMQC spectra (Figure 4A). We could obtain near-complete site-specific NMR assignments of the observed [15N-1H] correlations of lYibK and cYibK in 7.2 M using a described protocol (Hsu et al., 2009; Hsieh et al., 2014). As expected, the backbone amide of T162 at the C-terminus of lYibK showed the largest chemical shift difference from that of cYibK owing to the introduction of an additional peptide bond as a result of backbone cyclization. Furthermore, several residues near the N- and C-termini also exhibited significant chemical shift perturbations (Figure 4A; Supplementary Figure S6).
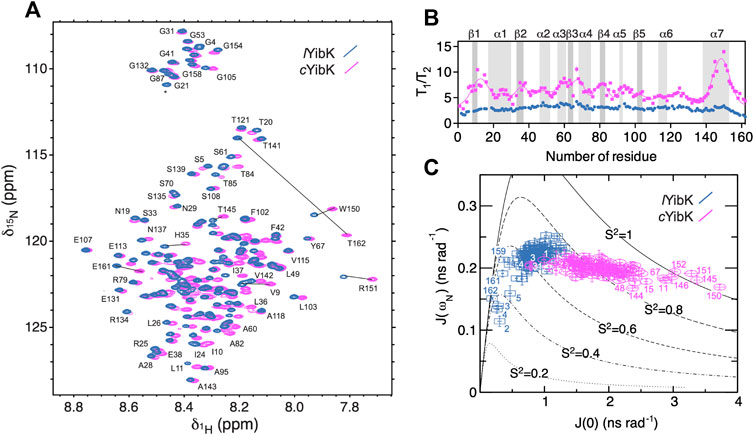
FIGURE 4. NMR spectroscopy and 15N spin relaxation analysis of the chemically denatured lYibK and cYibK. (A) Superimposition of the [15N-1H] correlation spectra of lYibK (orchid blue) and cYibK (magenta) in the presence of 7.2 M urea. The spectra were recorded at a 1H Larmor frequency of 850 MHz, and 298 K. Residues with large chemical shift differences upon cyclization are indicated by solid lines that connect the pairs of crosspeaks. (B)T1/T2 ratios of lYibK and cYibK as a function of residue number. T1/T2 ratios of cYibK were fitted to a sum of multiple Gaussian distributions. Regions that correspond to the β-sheets and α-helices in the native structure are highlighted by dark and light gray, respectively, and are indicated above the panel. (C) Spectral density mapping of the 15N relaxation data expressed as J (ωN) as a function of J (0). Residues that deviate from the cluster distributions are indicated with their residue numbers. Theoretical curves with the assumption of isotropic motions were calculated for different-order parameters, S (Liu et al., 1980), as indicated. All data points are colored using the same scheme as in (A).
NMR chemical shift analysis suggested very limited structural differences between urea-denatured lYibK and cYibK (Supplementary Figure S6). Therefore, we performed 15N relaxation analysis to compare their backbone dynamics. Under the denaturing condition, the mean 15N transverse relaxation time (T2) for cYibK was much shorter than that of lYibK in 7.2 M (122 vs. 238 ms; Supplementary Figure S7). We also observed that lYibK exhibited longer and more uniform T2 values across the primary sequence with the exception of the fraying ends because of the unrestricted chain dynamics at the open ends Supplementary Figure S7C). In contrast, cYibK exhibited clusters of fast-relaxing residues across the primary sequence (Supplementary Figure S7C), reminiscent of the previously observed long-range non-native interactions in urea-denatured lysozyme (Klein-Seetharaman et al., 2002). For urea-denatured lysozyme, the non-native interactions reflected the clustering of bulky hydrophobic tryptophan residues. However, for cYibK, not all rapidly relaxing residues contain aromatic side-chains. The T2 relaxation clusters may stem from the internal friction imposed by backbone cyclization that restricted the fraying motions of the N- and C-termini. Differences in dynamics or populations were particularly manifested in the dispersed T1/T2 values for cYibK, whereas a flat profile of a T1/T2 values was observed for lYibK. The T1/T2 values indicated that the differences of backbone dynamics are in the timescale of micro-to milliseconds. In contrast, the heteronuclear 15N{1H}-NOE of lYibK and cYibK, which is sensitive to a faster pico-to-nanosecond timescale, did not show apparent differences except for the termini of lYibK (Supplementary Figure S7c). Thus, the backbone cyclization did not affect the pico-to-nanosecond motions of the individual backbone peptide bonds except for the termini with the conformation restriction imposed by backbone cyclization (Supplementary Figure S7).
We also applied the reduced spectral density mapping approach for studying the dynamics of both denatured lYibK and cYibK (Figure 4C). (Farrow et al., 1995; Peng and Wagner, 1995; Shih et al., 2015) The results identified two distinct clusters of spectral density distributions for urea-denatured lYibK and cYibK, the former located around the theoretical curve for an order parameter S (Liu et al., 1980) of <0.7 and the latter exhibiting a cluster around the S (Liu et al., 1980) value of 0.8 (Figure 4C). Furthermore, several residues located at the C-terminal helix (α7) and some others were located outside the cluster distribution. Collectively, the NMR relaxation dynamics analysis suggested the presence of abundant conformational exchanges. It also implied a broad range of backbone dynamics caused by the backbone cyclization of YibK, restricting the backbone motions and increasing the ruggedness of the free energy landscape of the denatured state of cYibK.
cYibK_Int Under the Denaturing Condition by NMR
The split-intein approach for cyclization of YibK resulted in the insoluble spliced product that could presumably be the cyclized YibK without knotting (cYibK_Int) (Figure 1C). Even though cYibK_Int has an additional hexahistidine-tag and slightly different amino-acid sequence connecting the N- and C-termini (Figure 1E), we decided to purify and investigate cYibK_Int under a denaturing condition by NMR spectroscopy (Supplementary Figure S8). 15N{1H}-NOE data for the N- and C-termini of cYibK_Int are similar to those of cYibK than lYibK bearing flexible termini due to the linear polypeptide chain. This observation confirms the backbone cyclization of cYibK_Int. The average T2 relaxation time for cYibK_Int was shorter than that of lYibK (157 vs. 238 ms) (Supplementary Figure S8c). On the other hand, a flat profile of T1/T2 values for cYibK_Int is closer to lYibK, indicating the absence of conformational exchanges observed with cYibK, presumably because of the absence of a knot structure under the denaturing condition (Supplementary Figure S8C).
Small-Angle X-Ray Scattering Analysis of the Unfolded States of lYibK and cYibK
We previously used SAXS to demonstrate that chemically denatured knotted proteins with open ends exhibited random coil-like behaviors: their radii of gyration (Rg) scale with their chain lengths to the power of 3/5 (Shih et al., 2015). This observation suggests that backbone knotting with open ends does not necessarily lead to significant compaction of the overall chain dimension under highly denaturing conditions (in good solvent). However, these knotted proteins examined by SAXS are not mathematically knotted because of their free termini. To examine how backbone cyclization may affect the polymer properties of a mathematically knotted protein, we compared the SAXS data of lYibK and cYibK by using online size-exclusion chromatography-coupled SAXS (SEC-SAXS) apparatus as described (Lee and Hsu, 2018). Under native conditions, lYibK and cYibK exhibited the same SAXS profiles, with comparable Rg values — 22.17 ± 0.04 and 21.66 ± 0.06 Å—that were in general agreement with the theoretical value based on the crystal structure (19.4 Å; Figure 5). The corresponding Kratky plots showed comparable compactness (similar bell-shape profiles; cf. black and gray curves for cYibK and lYibK in Figure 5B, respectively). In contrast, 7.2 M urea-denatured cYibK showed a significantly smaller Rg value than lYibK under the same condition (27.71 ± 0.15 vs 39.26 ± 0.38 Å; Figures 5A,C). Urea-denatured lYibK exhibited a monotonously increasing Kratky profile typical of a random coil polypeptide, whereas urea-denatured cYibK exhibited a distinct bell-shape profile, albeit much smaller than that of folded cYibK, which indicates the presence of compact residual structure (blue curve in Figure 5B). According to the empirical scaling relationship established from our previous study, the expected Rg value for a chemically denatured lYibK is 38.0 Å, assuming a random coil-like behavior (Figure 5C). (Shih et al., 2015) A reduction of Rg by >10 Å in chemically denatured cYibK equals a decrease of the global dimension by 1/4 and a 3/5 decrease of the excluded volume (assuming that the exclusion volume of the unfolded polypeptide chain is spherical). The substantial conformational compaction further suggests that the conformational entropy of the unfolded state is reduced as a result of the backbone cyclization, which is in line with the stability improvement of cYibK.
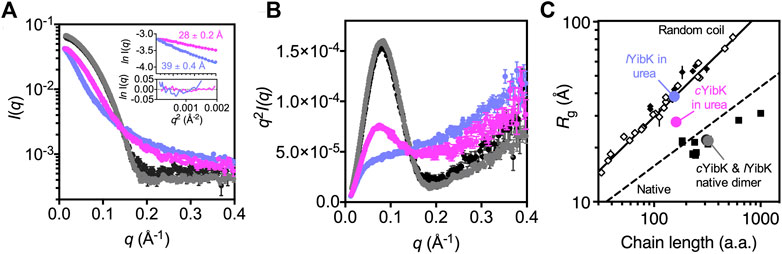
FIGURE 5. Cyclization significantly reduces the global dimension of the denatured state of cYibK. (A) SAXS profiles of lYibK in 0 M urea (gray), cYibK in 0 M urea (black), lYibK in 7 M urea (orchid blue), and cYibK in 7 M urea (magenta). Inset: Guinier plots of lYibK and cYibK in 7 M urea with the corresponding Rg values indicated and fitting residues shown below. (B) Kratky plots of lYibK and cYibK in their native and 7 M urea-denatured states, as shown in (a). Urea-denatured cYibK exhibits a distinct bell-shape profile, indicative of compact residual structures that are absent in urea-denatured lYibK. (C)Rg values of lYibK and cYibK in their native and 7 M urea-denatured states as a function of chain length. The solid and dashed lines indicate the experimentally derived scaling relationship for random-coil and native proteins, respectively. Solid squares and diamonds correspond to reported Rg values of knotted proteins of different topologies and chain lengths. Open diamonds correspond to reported random-coil Rg values of chemically denatured unknotted proteins (Shih et al., 2015).
Discussion
Introduction of a peptide bond between N- and C-termini into a knotted protein unambiguously converts it to a truly mathematical knot without the need to evoke convoluted knot-detecting algorithms, which in some cases have different interpretations of “knots” in proteins. It also eliminates any confusing experimental effects due to fraying of the N- and C-termini in proteins that are “knotted” by the introduction of disulfide bridges, which are largely different due to higher rotational degrees in the side-chains (Betz, 1993; Mallam et al., 2010). Whereas disulfide bonds have a mixture of enthalpic and entropic effects on the protein stability, backbone cyclization connecting the N- and C-termini is generally accepted to stabilize protein by destabilizing the unfolded state (Betz, 1993; Iwai and Plückthun, 1999; Scott et al., 1999; Clark and Craik, 2010; Montalbán-López et al., 2012; Borra and Camarero, 2013).
Here, we produced a trefoil-knotted protein without open ends, a truly mathematical backbone knot in a protein, by post-translational modification by enzymatic ligation (Figure 1). Such a backbone modification is irreversible as opposed to disulfide crosslinking between a pair of engineered cysteines at the N- and C-termini. Furthermore, the path closure by a backbone peptide bond essentially removes the origin of the protein sequence, rendering obsolete the conventional definition of a protein folding topology by the hierarchical arrangements of secondary structure elements. As compared with circular permutation, which was recently used to untie the trefoil-knotted HiYibK (Chuang et al., 2019), and E. coli YbeA (Ko et al., 2019), SrtA-mediated backbone cyclization allowed us to examine the contribution of a true backbone knot from a completely different perspective with clarity. The apparent melting temperature was increased by 20 °C for the knotted cYibK compared with lYibK with open ends (Figure 2). The path closure by a peptide bond after folding also has seemingly remodeled the protein folding/unfolding pathway of the original lYibK and alleviated the aggregation propensity of the folding intermediate observed for lYibK, whereas maintaining the native structure and ligand binding affinity (Figure 2; Supplementary Table S2). Indeed, the backbone cyclization significantly increased the folding rate of the intermediate-to-native state transition, with the corresponding transition state being highly compact and native-like, as evidenced by the βT value being close to 1 (Figure 3; Supplementary Table S3). Furthermore, the unfolding rate of intermediate-to-denatured state transition of cYibK,
To this end, 15N spin relaxation analysis of lYibK showed no appreciable conformational exchange contributions to the three different timescales probed by J (0), J (ωH), and J (ωN), suggesting the absence of interconversion between knotted and unknotted states of lYibK (Supplementary Figure S7). cYibK_Int, which has closed peptide ends and possibly no knot structure, showed a similar profile of T1/T2 to that of lYibK in the 15N spin relaxation analysis. In contrast, the enhanced T2 relaxation observed in cYibK, which has significant contributions to the J (0) term, likely reflects the increased internal friction of the cyclized polypeptide chain in the denatured state, thereby leading to destabilization of the unfolded state (Figure 4). As observed for many backbone cyclized proteins, we think that the increase in folding stability of cYibK could be attributed mainly to the reduced conformational entropy in the denatured state of cYibK.
In line with the stability enhancement, the chemically denatured state of cYibK was significantly more compact than the random coil-like lYibK in 7 M urea (Figure 5). The effect of cyclization on the polymer dimension is well understood in the literature. Kramers’ polymer model predicts that the Rg values of linear and cyclized polymers follow a simple relationship.
where
Materials and Methods
Constructions and Production of Recombinant YibK Variants
For backbone cyclization, P. aeruginosa YibK with a sortase recognition sequence LPETG followed by the C-terminal hexa-histidine (H6) was cloned in pRSF vector as N-terminal SUMO fusion by PCR, resulting in pITRSF1A (PaYibK_sh) and pITRSF3D (PaYibK_lo). In vivo cyclization vector for PaYibK was cloned into a pBAD vector containing the genes of split NpuDnaE-C intein fragment, H6-tag, and NpuDnaE-N intein fragment by using XbaI/KpnI sites, resulting in pJMBAD36(PaYibK_Int) (Iwai et al., 2006). For biophysical characterization of linear PaYibK_lo, plasmid pBHRSF260 was constructed with N-terminally His-tagged SUMO fusion to have the identical sequence as PaYibK_lo construct (Guerrero et al., 2015). Each plasmid was transformed into E. coli strain ER2566 cells (New England Biolabs, Ipswich, United States). The cells were cultured in Luria-Bertani medium supplemented with kanamycin at 25 μg⋅ml−1 until OD600nm reached 0.6 at 37 °C. The recombinant protein overexpression was induced for 4 h with a final concentration of 1 mM IPTG. The cells were harvested by centrifugation and resuspended in binding buffer (50 mM sodium phosphate buffer, pH 8.0, and 300 mM NaCl). The resuspended cells were lyzed at 15,000 psi for 10 min by using Emulsiflex C3, and the supernatant was separated from cell debris by 1 h centrifugation at 38,465 g. The supernatant was loaded onto a pre-packed HisTrap HP column (GE Healthcare Life Sciences, United States). The His-tagged fusion proteins were eluted by a linear gradient of 50–250 mM imidazole and dialyized against 2 L of 50 mM Tris-HCl buffer, pH 7.5, 0.5 mM EDTA, and 0.5 mM DTT overnight at 8 °C (Guerrero et al., 2015). The fusion proteins were digested by Ulp1 protease as described previsouly (Guerrero et al., 2015). The digested fusion proteins were loaded again on pre-equibriated HisTrap HP column and washed to remove the SUMO-tag. The C-terminally His-tagged YibK were eluted by a linear gradient of 50–250 mM imidazole and dialyzed against 50 mM Tris-HCl buffer, pH 7.5 overnight, followed by concentration with a centrifugal device. cYibK by the split intein fusion (cYibK_Int) was produced from the plasmid pJMBAD36. E. coli strain ER2566 cells bearing pJMBAD36 were grown in 2 L of M9 medium supplemented with ampicilin at 100 μg⋅mL−1 at 37 °C and induced for 4 h with a final concentration of 0.02% (w/v) arabinose. The cells were harvested and lyzed at 15,000 psi for 10 min using Emulsiflex C3. The insoluble pellet was collected after discarding the supernatant by 1 h centrifugation at 38,465 g. The pellet was resolubilized in 25 ml of 8M urea with shaking at 350 rpm overnight. The dissolved solution was cleared by 1 h centrifugation at 38,465 g. The supernatant was loaded onto the HisTrap HP column, which was pre-equilibrated with a binding buffer (100 mM sodium phosphate buffer, pH 8.0, 10 mM Tris, and 8 M urea). The His-tagged cyclized YibK (cYibK_Int) was eluted by 100 mM sodium phosphate buffer, pH 5.0, 10 mM Tris, and 8 M urea. The precursor protein in the elution fractions was removed by size-exclusion chromatography with a Superdex 75 16/60 column (GE Healthcare, United States) in 20 mM sodium phosphate buffer, pH 5.0, 8M urea. The fractions containing cYibK_Int were pooled and concentrated for NMR analysis.
SrtA-Mediated Backbone Cyclization
For the backbone cyclization, sortase (SrtA, from Staphylococcus aureus) was added to purified YibK in 1–5 molar ratio and dialyzed against 50 mM Tris-HCl buffer (pH 7.5), 10 mM CaCl2 and 2 mM DTT, for 20 h at room temperature. Finally, the unreacted YibK that contained the His6-tag at the C-terminus was separated from cYibK by incubation with Ni-NTA resin in an open column. The cYibK collected from the flow-through fractions was further polished by size-exclusion chromatography with a Superdex 75 16/60 column (GE Healthcare, United States) in buffer A (50 mM Tris-HCl (pH 7.4), 0.5 mM EDTA and 5 mM DTT).
Protein Crystallography
For the crystal structure of PaYibK, the plasmid (pJMRSF13) encoding YibK gene with N-terminal His-tag and the SUMO fusion was produced and purified as described previously (Guerrero et al., 2015). Crystallization was performed with 9.2 mg/ml solution of PaYibK and 11 mg/ml solution of cYibK. Drops of 200 nl (100 nl protein solution and 100 nl well solution) were placed in 96-well MRC (Molecular Dimensions) crystallization plates using a Mosquito LCP (TTPLabtech, United Kingdom). Initial hits were obtained from the traditional sparse matrix screens with the local modifications (Cudney et al., 1994). The initial hits were further optimized by grid screening. The final growth conditions for diffracting crystals were 0.15 M ammonium sulfate, 0.9 M lithium sulfate, 0.1 M sodium citrate buffer (pH 5.6) for PaYibK, and 0.3 M ammonium sulfate, 0.1 M MES buffer (pH 6.0), 25% polyethylene glycol monomethyl ether (PEG MME) 5,000 for cYibK. 20% glycerol was added on top of the drop of PaYibK for cryoprotection prior to flash-freezing crystals in liquid nitrogen. For cYibK, the 25% PEG MME 5000 present in the crystallization drop served as a sufficient cryoprotectant. Diffraction data for the crystals of PaYibK and cYibK were collected at the beamline ESRF ID14–4, Grenoble, France and I03 at the Diamond Light Source, Oxfordshire, UK, respectively. The diffraction data were then indexed, integrated, and scaled to 2.0 and 2.2 Å resolution for PaYibK and cYibK, respectively, in XDS (Kabsch, 2010). The final crystal parameters and data processing statistics are in Supplementary Table S1. The structures of PaYibK and cYibK were solved by molecular replacement with MolRep from the CCP4 package (Winn et al., 2011). The structure of HiYibK (PDB ID: 1mxi) was used as a search model for molecular replacement. The model was then built using Coot, followed by rounds of refinement with Refmac5 from the CCP4 package and Phenix (Adams et al., 2010). The final refinement was performed with Phenix, and the quality of the final model was validated by using MolProbity (Supplementary Table S1). (Chen et al., 2010) The final refined model of PaYibK was used as a starting model for the molecular replacement to solve the structure of cYibK. The structure was solved, refined and validated as mentioned above (Supplementary Table S1). The final coordinates were deposited in the Protein Data Bank (PDB) with the accession codes 6qkv and 6qh8 for PaYibK and cYibK, respectively.
Chemical Denaturation Monitored by Intrinsic Fluorescence Spectroscopy
Urea-induced equilibrium unfolding of lYibK and cYibK was monitored by intrinsic fluorescence as described (Wang et al., 2015; Wang et al., 2016). Briefly, 41 aliquots of protein solution (at a final concentration of 2 μM buffered in buffer A) were prepared in a series of urea concentrations (0–7 M) with a linear increment step of 2.5% generated by a two-channel liquid syringe dispenser (Hamilton, United States). The samples were incubated at 25 °C overnight before fluorescence measurements with a fluorimeter (JASCO FP8500, Japan). The samples were excited at 280 nm and emission spectra between 300 and 450 nm were collected. The results underwent singular value decomposition analysis with MatLab (MATLAB and Statistics Toolbox release 2012b; The MathWorks, United States) to determine the number of states associated with the unfolding processes, followed by fitting to a three-state folding equilibrium model with Prism (GraphPad, United States) as described (Wang et al., 2014; Wang et al., 2015; Wang et al., 2016; Lee et al., 2017).
Thermal Denaturation Monitored by Far-UV CD Spectroscopy
The protein solutions were diluted to 10–15 μM in buffer A with a total volume of 0.3 ml, and transferred into a 1 mm path-length quartz cuvette (Hellma, Germany) for far-UV CD measurements. The CD signals between 195 and 260 nm were collected as a function of temperature between 25 and 80 °C with an interval of 2°C by using a CD spectrometer (J-815, JASCO, Japan). The spectra bandwidth was set to 1 nm with a data interval of 0.5 nm, and an averaging time of 1 s. The melting temperatures (Tm) of lYibK and cYibK were derived by global-fitting the CD spectra as a function of temperature to a two-state model as described (Wang et al., 2014; Lee et al., 2017).
Isothermal Titration Calorimetry
ITC analysis of SAH binding to lYibK and cYibK was monitored by using MicroCal VP-ITC (Malvern, United Kingdom) as described (Zhao et al., 2015). Stock solutions of YibK variants were dialyzed overnight against buffer B (50 mM Tris-HCl (pH 7.4), 0.5 mM EDTA, and 0.1 mM TCEP) to remove DTT before ITC measurements. The dialysis buffer was used to prepare the stock solution of the titrant, SAH (Sigma-Aldrich, United States) at a concentration of 0.5 mM. An amount of 20 μM lYibK or 13 μM cYibK was used in the sample cells for ITC measurements. The resulting isotherms were processed by using NITPIC followed by data fitting with SEDPHAT (Zhao et al., 2015).
Folding Kinetics Monitored by Intrinsic Fluorescence Spectroscopy
Chevron plot analyses of the folding kinetics of lYibK and cYibK involved using a combination of stopped-flow and manual mixing measurements as described (Wang et al., 2015; Wang et al., 2016). Briefly, 10 μM native or 7 M urea-denatured protein stock solution was mixed with 10-fold excess denaturing or refolding buffer (buffer A with different concentrations of urea) and the kinetic traces of total fluorescence emission excited at 280 nm and cutoff by a 320 nm cutoff filter were fit to a linear combination of 2–4 exponential functions depending on the experimental conditions. For the slowest refolding rate of cYibK, manual mixing of 7 M urea-denatured cYibK with refolding buffer at a 1:10 mixing ratio was performed before fluorescence measurement (excitation 280 nm and emission 325 nm with a bandwidth of 5 nm) with a fluorimeter (JASCO FP8500, Japan).
Small-Angle X-Ray Scattering
SEC-SAXS experiments were performed on beamline BL23A at the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan) with the capacity to separate aggregated particles on a silica-based size-exclusion column (Bio SEC-3, Agilent, United States). SAXS signals were detected by using a Pilatus detector (1M-F) and processed by an in-house developed program to obtain the SAXS profiles (Kohn et al., 2004; Jeng et al., 2010; Lee and Hsu, 2018). The SAXS data were collected for momentum transfer q ranging from 0.005 to 0.434 Å−1, with X-ray wavelength 1.03 Å and 13 keV. The beam geometry was set to 0.5 × 0.5 mm2. During the HPLC separation before SAXS measurements, the mobile phase consisted of buffer A (with and without 7 M urea) with the addition of 2% glycerol to prevent radiation damage. The protein solutions were concentrated to 10 mg/ml with the same mobile phase buffer immediately before SAXS measurements.
Nuclear Magnetic Resonance Spectroscopy
Uniformly 15N-labeled and 20% 13C-labeled cYibK and lYibK were prepared by using M9 medium containing 1 g/L [15N] ammonium chloride as a nitrogen source and a mixture of 0.6 g/L [13C] D-glucose and 2.4 g/L [U-12C] D-glucose as a carbon source, as described (Iwai and Fiaux, 2007; Heikkinen et al., 2021). The NMR samples were fully denatured by 7.2 M urea in buffer A containing 10% D2O (v/v) at a protein concentration ca. 0.3 mM. A suite of triple resonance experiments in addition to the [15N-1H] band-selective optimized flip-angle short transient heteronuclear multi-quantum correlation (SOFAST-HMQC) were recorded at 298 K on 850 MHz NMR spectrometer equipped with a cryogenic triple resonance probe (Bruker, Germany) for backbone resonance assignments following the strategy described previously (Hsu et al., 2009; Hsieh et al., 2014). Near-complete backbone resonance assignments (HN, N, C’, Cα, Cβ, and Hα) were achieved for both cYibK and lYibK. The assignments were deposited in the Biological Magnetic Resonance Bank (BMRB) under accession numbers 27685 and 27686 for cYibK and lYibK, respectively. 15N spin relaxation NMR measurements for longitudinal (R1) and transverse (R2) relaxation rates and the heteronuclear Overhauser effect (hetNOE) were as described (Hsu et al., 2009). Eight longitudinal relaxation delays (20, 60, 120, 200, 300, 500, 800, and 1,000 ms) and nine transverse relaxation delays (16, 32, 64, 96, 128, 160, 192, 224, and 256 ms) were used for both cYibK and lYibK, and the data were collected as pseudo-3D spectra with the relaxation delays incremented in an interleaved manner to minimize heating effects. The R1 and R2 rates were extracted by fitting the peak intensities of the individual residues in the 15N-1H correlation spectra to a single exponential decay function by using the relaxation analysis module in Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, United States).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.wwpdb.org/, 6qkv, 6qh8; https://bmrb.io/, 27685, 27686.
Author Contributions
HI and STDH conceptualized and planned the project. IT, KM, YL established and produced the recombinant proteins. KM and IT crystallized proteins and solved the structures. HI, SB, YL, and STDH recorded and analyzed NMR data. SAXS and biophysical characterizations were performed and analyzed by YL and STDH. HI, AG, and STDH wrote the manuscript with input from all the authors.
Funding
This work was supported by the Ministry of Science and Technology, Taiwan (105-2113-M-001-005, 106-2113-M-001-004 and 107-2628-M-001-005-MY3) and Academia Sinica, Taiwan to S.T.D.H; Sigrid Juselius Foundation and Academy of Finland (131413, 137995, 277335) to H.I., and Mobility to Finland under the bilateral agreement of the Academy of Finland (308239) and the National Research Council of Taiwan to S.T.D.H and Y.T.C.L. The Finnish Biological NMR center and crystallization facility are supported by Biocenter Finland, FINStruct, and the Helsinki Institute of Life Science (HiLIFE-INFRA). The Helsinki University Library supported the funding for open access publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank J. Mutanen, Dr Y.-Q. Yeh, Dr U. Jeng, and T.M. Kauppala for their contribution and assistance, and S. Jääskeläinen and B. Haas for their technical assistance in cloning and protein production. We thank the Biophysics Facility of the Institute of Biological Chemistry, Academia Sinica, the National Synchrotron Radiation Research Center BL23A bioSAXS beamline, crystallization facility at the Institute of Biotechnology, and Finnish Biological NMR center.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.663241/full#supplementary-material
References
Beccara, S. a., Skrbic, T., Covino, R., Micheletti, C., and Faccioli, P. (2013). Folding Pathways of a Knotted Protein with a Realistic Atomistic Force Field. Plos Comput. Biol. 9, e1003002. doi:10.1371/journal.pcbi.1003002
Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Cryst. 66, 213–221. doi:10.1107/s0907444909052925
Andersson, F. I., Pina, D. G., Mallam, A. L., Blaser, G., and Jackson, S. E. (2009). Untangling the Folding Mechanism of the 52-knotted Protein UCH-L3. FEBS J. 276, 2625–2635. doi:10.1111/j.1742-4658.2009.06990.x
Aranko, A. S., Oeemig, J. S., Kajander, T., and Iwaï, H. (2013). Intermolecular Domain Swapping Induces Intein-Mediated Protein Alternative Splicing. Nat. Chem. Biol. 9, 616–622. doi:10.1038/nchembio.1320
Arrighi, V., Gagliardi, S., Dagger, A. C., Semlyen, J. A., Higgins, J. S., and Shenton, M. J. (2004). Conformation of Cyclics and Linear Chain Polymers in Bulk by SANS. Macromolecules 37, 8057–8065. doi:10.1021/ma049565w
Betz, S. F. (1993). Disulfide Bonds and the Stability of Globular Proteins. Protein Sci. 2, 1551–1558. doi:10.1002/pro.5560021002
Borra, R., and Camarero, J. A. (2013). Recombinant Expression of Backbone-Cyclized Polypeptides. Biopolymers 100, 502–509. doi:10.1002/bip.22306
Capraro, D. T., and Jennings, P. A. (2016). Untangling the Influence of a Protein Knot on Folding. Biophysical J. 110, 1044–1051. doi:10.1016/j.bpj.2016.01.017
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Cryst. 66, 12–21. doi:10.1107/s0907444909042073
Chuang, Y.-C., Hu, I.-C., Lyu, P.-C., and Hsu, S.-T. D. (2019). Untying a Protein Knot by Circular Permutation. J. Mol. Biol. 431, 857–863. doi:10.1016/j.jmb.2019.01.005
Clark, R. J., and Craik, D. J. (2010). Invited Reviewnative Chemical Ligation Applied to the Synthesis and Bioengineering of Circular Peptides and Proteins. Biopolymers 94, 414–422. doi:10.1002/bip.21372
Covino, R., Skrbic, T., Beccara, S. A., Faccioli, P., and Micheletti, C. (2014). The Role of Non-native Interactions in the Folding of Knotted Proteins: Insights from Molecular Dynamics Simulations. Biomolecules 4, 1–19. doi:10.3390/biom4010001
Cudney, R., Patel, S., Weisgraber, K., Newhouse, Y., and McPherson, A. (1994). Screening and Optimization Strategies for Macromolecular Crystal Growth. Acta Cryst. D 50, 414–423. doi:10.1107/s0907444994002660
Dabrowski-Tumanski, P., and Sulkowska, J. (2017). To Tie or Not to Tie? That Is the Question. Polymers 9, 454. doi:10.3390/polym9090454
Faísca, P. F. N. (2015). Knotted Proteins: A Tangled Tale of Structural Biology. Comput. Struct. Biotechnol. J. 13, 459–468. doi:10.1016/j.csbj.2015.08.003
Farrow, N. A., Zhang, O., Szabo, A., Torchia, D. A., and Kay, L. E. (1995). Spectral Density Function Mapping Using 15N Relaxation Data Exclusively. J. Biomol. NMR 6, 153–162. doi:10.1007/bf00211779
Fersht, A. (1999). Structure and Mechanism in Protein Science : A Guide to Enzyme Catalysis and Protein Folding, xxi. New York: W. H. Freeman, 631.
Guerrero, F., Ciragan, A., and Iwaï, H. (2015). Tandem SUMO Fusion Vectors for Improving Soluble Protein Expression and Purification. Protein Expr. Purif. 116, 42–49. doi:10.1016/j.pep.2015.08.019
He, C., Li, S., Gao, X., Xiao, A., Hu, C., Hu, X., et al. (2019). Direct Observation of the Fast and Robust Folding of a Slipknotted Protein by Optical Tweezers. Nanoscale 11, 3945–3951. doi:10.1039/c8nr10070e
Heikkinen, H. A., Backlund, S. M., and Iwaï, H. (2021). NMR Structure Determinations of Small Proteins Using Only One Fractionally 20 % 13C- and Uniformly 100 % 15N-Labeled Sample. Molecules 26, 747. doi:10.3390/molecules26030747
Higgins, J. S., Dodgson, K., and Semlyen, J. A. (1979). Studies of Cyclic and Linear Poly(dimethyl Siloxanes): 3. Neutron Scattering Measurements of the Dimensions of Ring and Chain Polymers. Polymer 20, 553–558. doi:10.1016/0032-3861(79)90164-2
Hsieh, S.-J. M., Mallam, A. L., Jackson, S. E., and Hsu, S.-T. D. (2014). Backbone NMR Assignments of a Topologically Knotted Protein in Urea-Denatured State. Biomol. NMR Assign 8, 283–285. doi:10.1007/s12104-013-9501-7
Hsu, S.-T. D., Cabrita, L. D., Fucini, P., Dobson, C. M., and Christodoulou, J. (2009). Structure, Dynamics and Folding of an Immunoglobulin Domain of the Gelation Factor (ABP-120) from Dictyostelium discoideum. J. Mol. Biol. 388, 865–879. doi:10.1016/j.jmb.2009.02.063
Iwai, H., and Fiaux, J. (2007). Use of Biosynthetic Fractional 13C-Labeling for Backbone NMR Assignment of Proteins. J. Biomol. NMR 37, 187–193. doi:10.1007/s10858-006-9124-8
Iwai, H., Lingel, A., and Plückthun, A. (2001). Cyclic Green Fluorescent Protein Produced In Vivo Using an Artificially Split PI-PfuI Intein from Pyrococcus Furiosus. J. Biol. Chem. 276, 16548–16554. doi:10.1074/jbc.m011639200
Iwai, H., and Plückthun, A. (1999). Circular β-lactamase: Stability Enhancement by Cyclizing the Backbone. FEBS Lett. 459, 166–172. doi:10.1016/s0014-5793(99)01220-x
Iwai, H., Züger, S., Jin, J., and Tam, P.-H. (2006). Highly Efficient Proteintrans-Splicing by a Naturally Split DnaE Intein fromNostoc Punctiforme. FEBS Lett. 580, 1853–1858. doi:10.1016/j.febslet.2006.02.045
Jackson, S. E., Suma, A., and Micheletti, C. (2017). How to Fold Intricately: Using Theory and Experiments to Unravel the Properties of Knotted Proteins. Curr. Opin. Struct. Biol. 42, 6–14. doi:10.1016/j.sbi.2016.10.002
Jamroz, M., Niemyska, W., Rawdon, E. J., Stasiak, A., Millett, K. C., Sułkowski, P., et al. (2015). KnotProt: a Database of Proteins with Knots and Slipknots. Nucleic Acids Res. 43, D306–D314. doi:10.1093/nar/gku1059
Jarmolinska, A. I., Perlinska, A. P., Runkel, R., Trefz, B., Ginn, H. M., Virnau, P., et al. (2019). Proteins' Knotty Problems. J. Mol. Biol. 431, 244–257. doi:10.1016/j.jmb.2018.10.012
Jeng, U.-S., Su, C. H., Su, C.-J., Liao, K.-F., Chuang, W.-T., Lai, Y.-H., et al. (2010). A Small/wide-Angle X-Ray Scattering Instrument for Structural Characterization of Air-Liquid Interfaces, Thin Films and Bulk Specimens. J. Appl. Cryst. 43, 110–121. doi:10.1107/s0021889809043271
Klein-Seetharaman, J., Oikawa, M., Grimshaw, S. B., Wirmer, J., Duchardt, E., Ueda, T., et al. (2002). Long-Range Interactions Within a Nonnative Protein. Science 295, 1719–1722. doi:10.1126/science.1067680
Ko, K.-T., Hu, I.-C., Huang, K.-F., Lyu, P.-C., and Hsu, S.-T. D. (2019). Untying a Knotted SPOUT RNA Methyltransferase by Circular Permutation Results in a Domain-Swapped Dimer. Structure 27, 1224–1233. doi:10.1016/j.str.2019.04.004
Kohn, J. E., Millett, I. S., Jacob, J., Zagrovic, B., Dillon, T. M., Cingel, N., et al. (2004). Random-coil Behavior and the Dimensions of Chemically Unfolded Proteins. Proc. Natl. Acad. Sci. 101, 12491–12496. doi:10.1073/pnas.0403643101
Kramers, H. A. (1946). The Behavior of Macromolecules in Inhomogeneous Flow. J. Chem. Phys. 14, 415–424. doi:10.1063/1.1724163
Lai, Y.-L., Chen, C.-C., and Hwang, J.-K. (2012). pKNOT v.2: the Protein KNOT Web Server. Nucleic Acids Res. 40, W228–W231. doi:10.1093/nar/gks592
Lee, Y.-T. C., and Hsu, S.-T. D. (2018). A Natively Monomeric Deubiquitinase UCH-L1 Forms Highly Dynamic but Defined Metastable Oligomeric Folding Intermediates. J. Phys. Chem. Lett. 9, 2433–2437. doi:10.1021/acs.jpclett.8b00815
Lee, Y. T. C., Chang, C-Y, Chen, S-Y, Pan, Y-R, Ho, M-R, and Hsu, S.T.D, (2017). Entropic Stabilization of a Deubiquitinase Provides Conformational Plasticity and Slow Unfolding Kinetics Beneficial for Functioning on the Proteasome. Sci. Rep. 7, 45174. doi:10.1038/srep45174
Li, W., Terakawa, T., Wang, W., and Takada, S. (2012). Energy Landscape and Multiroute Folding of Topologically Complex Proteins Adenylate Kinase and 2ouf-Knot. Proc. Natl. Acad. Sci. 109, 17789–17794. doi:10.1073/pnas.1201807109
Lim, N. C. H., and Jackson, S. E. (2015). Molecular Knots in Biology and Chemistry. J. Phys. Condens. Matter 27, 354101. doi:10.1088/0953-8984/27/35/354101
Liu, L., Liu, C., and Alberts, B. (1980). Type II DNA Topoisomerases: Enzymes that Can Unknot a Topologically Knotted DNA Molecule via a Reversible Double-Strand Break. Cell 19, 697–707. doi:10.1016/s0092-8674(80)80046-8
Liu, R.-J., Zhou, M., Fang, Z.-P., Wang, M., Zhou, X.-L., and Wang, E.-D. (2013). The tRNA Recognition Mechanism of the Minimalist SPOUT Methyltransferase, TrmL. Trml. Nucleic Acids Res. 41, 7828–7842. doi:10.1093/nar/gkt568
Lou, S.-C., Wetzel, S., Zhang, H., Crone, E. W., Lee, Y.-T., Jackson, S. E., et al. (2016). The Knotted Protein UCH-L1 Exhibits Partially Unfolded Forms under Native Conditions that Share Common Structural Features with its Kinetic Folding Intermediates. J. Mol. Biol. 428, 2507–2520. doi:10.1016/j.jmb.2016.04.002
Mallam, A. L., and Jackson, S. E. (2007). A Comparison of the Folding of Two Knotted Proteins: YbeA and YibK. J. Mol. Biol. 366, 650–665. doi:10.1016/j.jmb.2006.11.014
Mallam, A. L., and Jackson, S. E. (2012). Knot Formation in Newly Translated Proteins Is Spontaneous and Accelerated by Chaperonins. Nat. Chem. Biol. 8, 147–153. doi:10.1038/nchembio.742
Mallam, A. L., and Jackson, S. E. (2007). The Dimerization of an α/β-Knotted Protein Is Essential for Structure and Function. Structure 15, 111–122. doi:10.1016/j.str.2006.11.007
Mallam, A. L., Morris, E. R., and Jackson, S. E. (2008). Exploring Knotting Mechanisms in Protein Folding. Proc. Natl. Acad. Sci. 105, 18740–18745. doi:10.1073/pnas.0806697105
Mallam, A. L., Rogers, J. M., and Jackson, S. E. (2010). Experimental Detection of Knotted Conformations in Denatured Proteins. Proc. Natl. Acad. Sci. 107, 8189–8194. doi:10.1073/pnas.0912161107
Mansfield, M. L. (1994). Are There Knots in Proteins?. Nat. Struct. Mol. Biol. 1, 213–214. doi:10.1038/nsb0494-213
Mikula, K. M., Tascón, I., Tommila, J. J., and Iwaï, H. (2017). Segmental Isotopic Labeling of a Single-Domain Globular Protein without Any Refolding Step by an Asparaginyl Endopeptidase. FEBS Lett. 591, 1285–1294. doi:10.1002/1873-3468.12640
Millett, K. C., Rawdon, E. J., Stasiak, A., and Sułkowska, J. I. (2013). Identifying Knots in Proteins. Biochem. Soc. Trans. 41, 533–537. doi:10.1042/bst20120339
Mitchinson, C., and Wells, J. A. (1989). Protein Engineering of Disulfide Bonds in Subtilisin BPN'. Biochemistry 28, 4807–4815. doi:10.1021/bi00437a043
Montalbán-López, M., Sánchez-Hidalgo, M., Cebrián, R., and Maqueda, M. (2012). Discovering the Bacterial Circular Proteins: Bacteriocins, Cyanobactins, and Pilins. J. Biol. Chem. 287, 27007–27013. doi:10.1074/jbc.r112.354688
Noel, J. K., Sulkowska, J. I., and Onuchic, J. N. (2010). Slipknotting upon Native-like Loop Formation in a Trefoil Knot Protein. Proc. Natl. Acad. Sci. 107, 15403–15408. doi:10.1073/pnas.1009522107
Passioni, C., Puri, S., Wang, I., Chen, S. Y., Camilloni, C., and Hsu, S. T. D. (2021). Converging Experimental and Computational Views of the Knotting Mechanism of a Small Knotted Protein. Biophys. J. in press. doi:10.1016/j.bpj.2021.03.032
Peng, J. W., and Wagner, G. (1995). Frequency Spectrum of NH Bonds in Eglin C from Spectral Density Mapping at Multiple Fields. Biochemistry 34, 16733–16752. doi:10.1021/bi00051a023
Popp, M. W.-L., and Ploegh, H. L. (2011). Making and Breaking Peptide Bonds: Protein Engineering Using Sortase. Angew. Chem. Int. Ed. 50, 5024–5032. doi:10.1002/anie.201008267
Raymer, D. M., and Smith, D. E. (2007). Spontaneous Knotting of an Agitated String. Proc. Natl. Acad. Sci. 104, 16432–16437. doi:10.1073/pnas.0611320104
Rivera, M., Hao, Y., Maillard, R. A., and Baez, M, (2020). Mechanical Unfolding of a Knotted Protein Unveils the Kinetic and Thermodynamic Consequences of Threading a Polypeptide Chain. Sci. Rep. 10, 9562. doi:10.1038/s41598-020-66258-5
Scott, C. P., Abel-Santos, E., Wall, M., Wahnon, D. C., and Benkovic, S. J. (1999). Production of Cyclic Peptides and Proteins In Vivo. Proc. Natl. Acad. Sci. 96, 13638–13643. doi:10.1073/pnas.96.24.13638
Shih, P.-M., Wang, I., Lee, Y.-T. C., Hsieh, S.-J., Chen, S.-Y., Wang, L.-W., et al. (2015). Random-coil Behavior of Chemically Denatured Topologically Knotted Proteins Revealed by Small-Angle X-Ray Scattering. J. Phys. Chem. B 119, 5437–5443. doi:10.1021/acs.jpcb.5b01984
Sulkowska, J. I., Noel, J. K., Ramírez-Sarmiento, C. A., Rawdon, E. J., Millett, K. C., Onuchic, J. N., et al. (2013). Knotting Pathways in Proteins. Biochem. Soc. Trans. 41, 523–527. doi:10.1042/BST20120342
Taylor, W. R. (2000). A Deeply Knotted Protein Structure and How it Might Fold. Nature 406, 916–919. doi:10.1038/35022623
Tkaczuk, K. L., Dunin-Horkawicz, S., Purta, E., and Bujnicki, J. M. (2007). Structural and Evolutionary Bioinformatics of the SPOUT Superfamily of Methyltransferases. BMC Bioinformatics 8, 73. doi:10.1186/1471-2105-8-73
Wang, I., Chen, S.-Y., and Hsu, S.-T. D. (2016). Folding Analysis of the Most Complex Stevedore's Protein Knot. Sci. Rep. 6, 31514. doi:10.1038/srep31514
Wang, I., Chen, S.-Y., and Hsu, S.-T. D. (2015). Unraveling the Folding Mechanism of the Smallest Knotted Protein, MJ0366. J. Phys. Chem. B 119, 4359–4370. doi:10.1021/jp511029s
Wang, I., Kowalski, M. P., Langley, A. R., Rodriguez, R., Balasubramanian, S., Hsu, S.-T. D., et al. (2014). Nucleotide Contributions to the Structural Integrity and DNA Replication Initiation Activity of Noncoding Y RNA. Biochemistry 53, 5848–5863. doi:10.1021/bi500470b
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of theCCP4 Suite and Current Developments. Acta Crystallogr. D Biol. Cryst. 67, 235–242. doi:10.1107/s0907444910045749
Zhao, H., Piszczek, G., and Schuck, P. (2015). SEDPHAT - A Platform for Global ITC Analysis and Global Multi-Method Analysis of Molecular Interactions. Methods 76, 137–148. doi:10.1016/j.ymeth.2014.11.012
Keywords: Knotted proteins, NMR spectrocopy, protein trans-splicing, enzymatic ligation, protein dynamics, protein stability and folding
Citation: Hsu S-TD, Lee Y-TC, Mikula KM, Backlund SM, Tascón I, Goldman A and Iwaï H (2021) Tying up the Loose Ends: A Mathematically Knotted Protein. Front. Chem. 9:663241. doi: 10.3389/fchem.2021.663241
Received: 02 February 2021; Accepted: 20 April 2021;
Published: 24 May 2021.
Edited by:
Assaf Friedler, Hebrew University of Jerusalem, IsraelReviewed by:
Sophie Elizabeth Jackson, University of Cambridge, United KingdomStefan G. D. Rüdiger, Utrecht University, Netherlands
Copyright © 2021 Hsu, Lee, Mikula, Backlund, Tascón, Goldman and Iwaï. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shang-Te Danny Hsu, c3Roc3VAZ2F0ZS5zaW5pY2EuZWR1LnR3; Hideo Iwaï, aGlkZW8uaXdhaUBoZWxzaW5raS5maQ==
†Present Address: BIOFISIKA Institute (UPV/EHU, CSIC), Barrio Sarriena S/n 48940 Leioa, Bizkaia, Basque Country, Spain
‡These authors have contributed equally to this work