 Shiqun Xiang
Shiqun Xiang Yinghua Li1
Yinghua Li1 Deguang Huang
Deguang Huang- 1State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, China
- 2School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing, China
A simple and efficient method is explored for the synthesis of 2-hydroxyimino-2-phenylacetonitriles (2) and phthalimides (4), by using nitromethane as nitrogen donors. Both reactions are promoted by Cu(II) system with the participation of dioxygen as an oxidant. The scope of the method has been successfully demonstrated with a total of 51 examples. The flexible and diversified characteristics of reactions are introduced in terms of electronic effect, steric effect, position of substituted groups, and intramolecular charge transfer. Experimental studies suggest that the methyl nitrite could be a precursor in the path to the final products. A possible reaction mechanism is proposed, including the Cu(II)/O2-facilitated transformation of nitromethane to methyl nitrite, the base-induced formation of 2-hydroxyimino-2-phenylacetonitriles, and the base-dioxygen-promoted formation of phthalimides.
Introduction
Nitrogen is one of the most important elements that constitute organisms and plays an important role in life activities of biomolecules. Cyano-oxime compounds and their derivatives have shown tremendous potentials for the development of a variety of biological compounds such as herbicides, drugs, and antibacterial agents (Gerasimchuk et al., 2008; El-Faham et al., 2014). The presence of two important functional groups in a single molecule makes them widely applicable for the synthesis of nitrogenous organic compounds in chemical engineering (Robertson et al., 2005; Aakeröy et al., 2012; Soliman et al., 2017). Of these, 2-hydroxyimino-2-phenylacetonitriles (2) are useful building blocks for the construction of polynitrogen organic substances, especially for the synthesis of peptides and/or nitrogen-containing heterocyclic compounds (Itoh et al., 1975; Kunai et al., 1990; Neel and Zhao, 2018; Alam et al., 2020). A few methods are available for the synthesis of these compounds, including the condensation of phenylketone/benzaldehyde and the cyanidation of their secondary products (Schemes 1A,B) (Ma et al., 1999; Uysal, 2010; Lemercier and Pierce, 2014; Kryshenko et al., 2016; Ushakov et al., 2019), the nitrosylation and nitridation of benzyl-cyanide (Schemes 1C,D) (Dubery et al., 1999; Bohle and Perepichka, 2009; Bohle et al., 2013; Neel and Zhao, 2018), and the nitration and nitrosation of styrene (Schemes 1E,F) (Kunai et al., 1990; Alam et al., 2020). However, these methods suffer from several drawbacks, such as the use of toxic reagents, rigorous reaction conditions, the cost of starting materials, multistep processes, and/or low yields. Thus, a simple and efficient method for the synthesis of compound 2 is desired, consistent with the development of benzyl-cyano-oxime compounds and their related pharmacological and/or material chemistry.
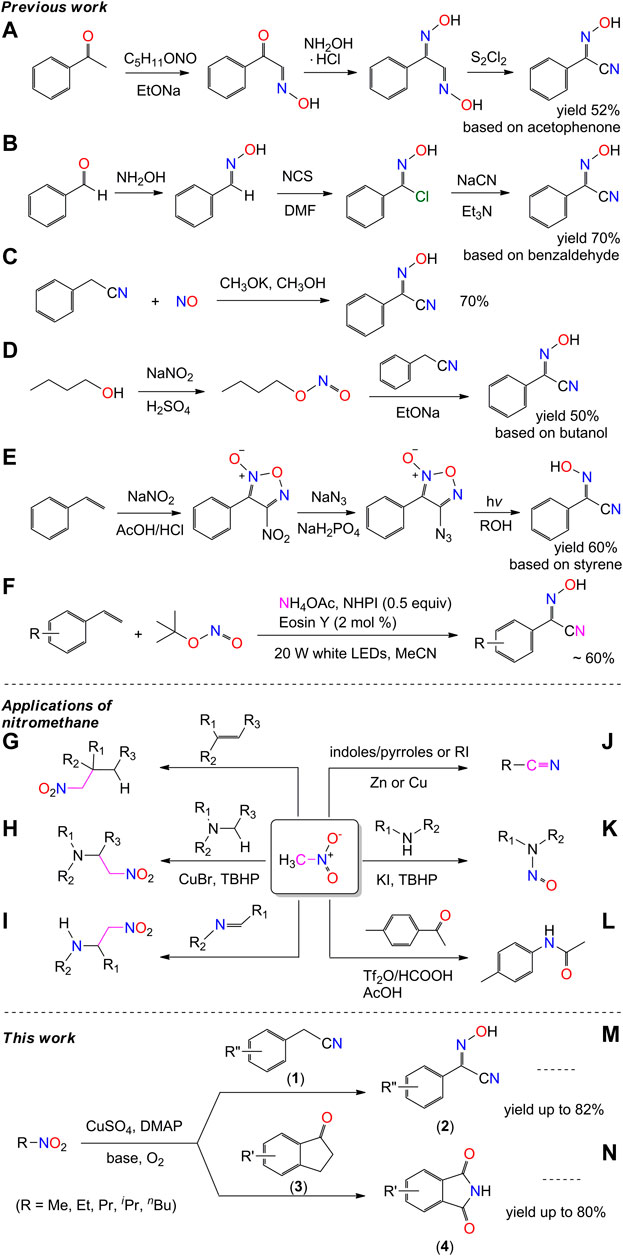
SCHEME 1. Previous work of the synthesis of 2-(hydroxyimino)-2-phenylacetonitrile and the reported applications of nitromethane as nitrogen donor.
In the literature, the direct protocol to enhance the scope of conventional synthesis of N-containing compounds generally involves the development and utilization of nitrogen transfer agents. Many nitrogen donors have been used in the catalytic transformations, e.g., oxidative amination, nitrogenation, and nitrogen heterocyclization. They include diazo/azide (Hennessy and Betley, 2013), isocyanide (Kim and Hong, 2015), cyanide (Wu et al., 2016a), ammonia (Shimokawa et al., 2016), sulfonamide/amide (Wang et al., 2019a), and hydroxylamine (Mudshinge et al., 2020). In contrast, the reactivity of nitromethane as a nitrogen donor was rarely developed, until a recent article in the journal of Science provided a reference of nitromethane activation for use in the Schmidt reaction (Scheme 1L) (Liu et al., 2019). The efficient utilization of nitro compounds as nitrogen donors has attracted considerable attention, and the interest in the activation of nitromethane grows.
Nitromethane is shelf-stable and commercially available. It is commonly used as a solvent for organic synthesis. It is also used as an intermediate in the carbon-chain-growth synthesis reaction owing to the strongly electrophilic properties of the nitro group (Xu et al., 2015; Aksenov et al., 2015; Wu et al., 2016b; Aksenov et al., 2020), for example, nitroaldol reactions (Jacobsen, 2001; Akagawa and Kudo, 2012; Kwiatkowski et al., 2014; Fu et al., 2019), cross dehydrogenative coupling reaction (Li et al., 2006; Basle and Li, 2007), and nitro-Mannich reactions (Schemes 1G–I) (Noble and Anderson, 2013). These reactions took place in the active site of the methyl group, without any change to the skeleton of the nitro group. Moreover, nitromethane is also used as cyano and nitroso sources in the formation of nitriles and nitrosamines (Schemes 1J,K), in the presence of zinc or copper catalysts, respectively (Nagase et al., 2014; Sakai et al., 2015; Chaudhary et al., 2016; Ogiwara et al., 2017; Qiu et al., 2018; Mudithanapelli et al., 2019). However, the substrates of those reactions are limited to heterocycles/aryl iodides and amines.
Copper complexes are often used as oxidants and/or oxidation catalysts in organic catalytic synthesis. As advanced oxidizing agents, copper-oxygen species have been used in the activation of C-H bond of alkyl group (Allen et al., 2013; Thorseth et al., 2013; Solomon et al., 2014; Elwell et al., 2017). On considering our previous work on the C-H bond activation of pyridine (Xiang et al., 2019), we set out to study the application of copper(II)-oxygen species on the activation of nitromethane, from the perspective of applied chemistry and practical use. We herein report a new method for the synthesis of 2-hydroxyimino-2-phenylacetonitrile (2a) and its derivatives, by nitrosation of phenylacetonitriles with nitromethane under dioxygen atmosphere (Scheme 1M). The employment of easily available reagents, environmentally friendly solvents, and mild reaction conditions make this process very practical. Meanwhile, the copper(II)-oxygen facilitated nitrosation reaction is validated with the amination of indanones with nitromethane in yields up to 80% (Scheme 1N). Experimental studies showed that the methyl nitrite could be an intermediate of the activation of nitromethane in our system, through which the final products two and four were obtained by means of nucleophilic reaction and electrophilic reaction, respectively.
Material and Methods
General Information
The single-crystal data of compounds were collected by a Cu/Mo-Kα rotating anode source, using a Supernova diffractometer with the ω-scan method. ESI-MS were obtained using a Bruker Impact II quadrupole time-of-flight mass spectrometer. 1H NMR and 13C NMR spectra were recorded on Bruker Avance III (400 MHz) and JNM ECS400S (400 MHz). Chemical shifts are expressed in δ ppm values with reference to tetramethylsilane (TMS) as an internal standard. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, and m = multiplet. Coupling constants (J) are expressed in Hz. 1H and 13C NMR spectra are provided for all the compounds in the SI. Pyridine was distilled before use. MeNO2 and DMF were distilled over CaH2 and stored over molecular sieve (3Å). THF was distilled with metal sodium over a period of 5 h. All other reagents were of analytical grade, purchased from commercial sources, and used as received.
Warning. The starting materials of nitroalkanes and the nitrointermediates generated during reactions are energetic and should be handled as if they are explosive materials. All the reactions should be conducted behind a blast shield.
General Procedure for the Synthesis of Compounds Shown in Scheme 2
For safety reasons, the feeding order of chemicals should be followed. To a mixture of phenylacetonitriles (0.60 mmol), 4-dimethylaminopyridine (DMAP, 0.40 mmol), NaOH (0.40 mmol), CuSO4.5H2O (0.20 mmol), and THF (1 mL) in a 50 mL Teflon screw-cap sealed tube, MeNO2 (0.20 mmol) was added slowly in air atmosphere. The tube was charged with O2 (1 atm) and the mixture was stirred at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (7:1 v/v) to afford the products in yields up to 82% (2e). The scale-up production of compound 2 was examined with the typical nitrile of phenylacetonitrile. The reaction of PhCH2CN (3.51 g, 30 mmol), DMAP (2.44 g, 20 mmol), NaOH (0.80 g, 20 mmol), CuSO4.5H2O (2.50 g, 10 mmol), THF (30 mL), and MeNO2 (0.61 g, 10 mmol) under the same conditions afforded the product 2a in yield 68% (0.99 g).
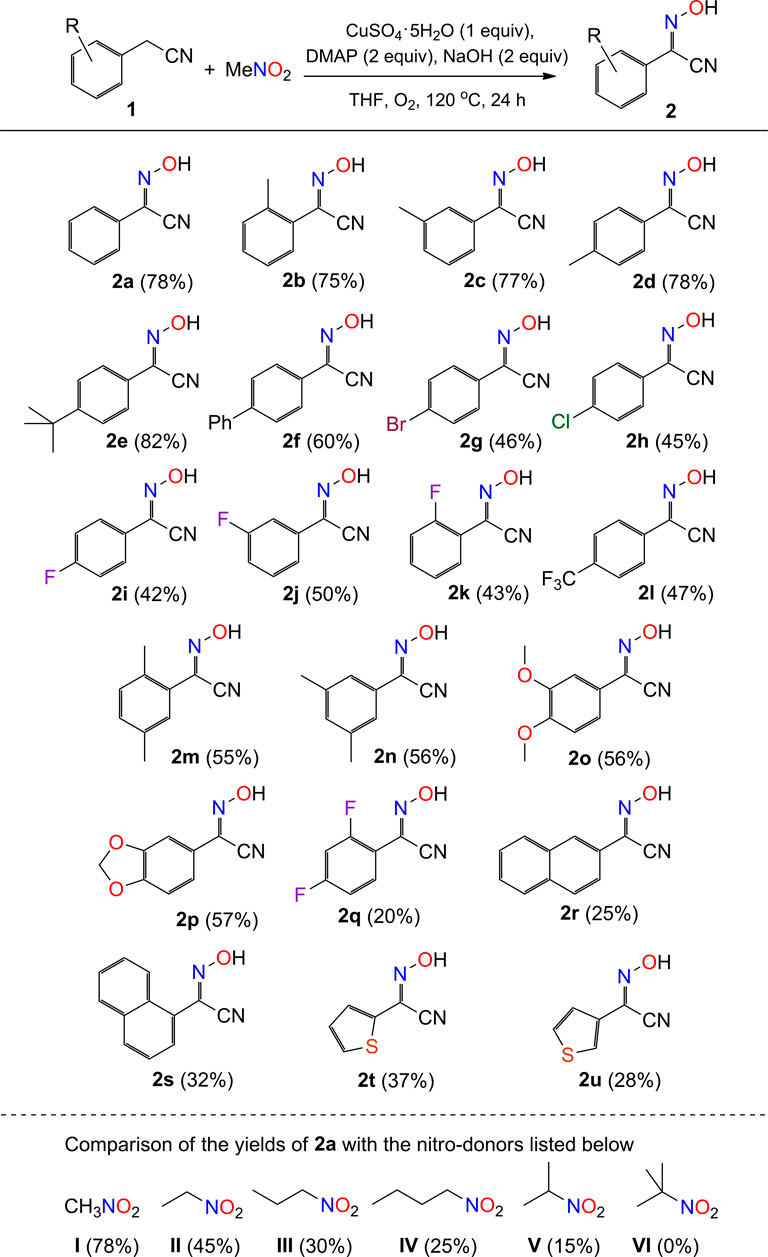
SCHEME 2. Scope of phenylacetonitriles with respect to compound 2a. aReaction conditions: substrate 1 (0.6 mmol), MeNO2 (0.2 mmol), CuSO4.5H2O (0.2 mmol), DMAP (0.4 mmol), NaOH (0.4 mmol), THF (1 mL), O2 (1 atm), 120 °C, and 24 h.
General Procedure for the Synthesis of Compounds Shown in Scheme 3
Part a. For safety reasons, the reaction procedure should be strictly followed. A mixture of indanones (0.20 mmol), DMAP (0.40 mmol), tBuOK (0.40 mmol), and CuSO4.5H2O (0.20 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and MeNO2 (1 mL) was added dropwise while stirring. After the addition was completed, the mixture was warmed up, charged with O2 (1 atm), and stirred at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with dichloromethane/methanol (20:1 v/v) to afford the products in yields up to 80% (4e). The scale-up production of compound 4 was examined with the typical starting material of 1-indanone. The reaction of 1-indanone (1.32 g, 10 mmol), DMAP (2.44 g, 20 mmol), tBuOK (2.24 g, 20 mmol), CuSO4.5H2O (2.50 g, 10 mmol), and MeNO2 (30 mL) under the same conditions shown above afforded the product 4a in yield 65% (0.96 g).
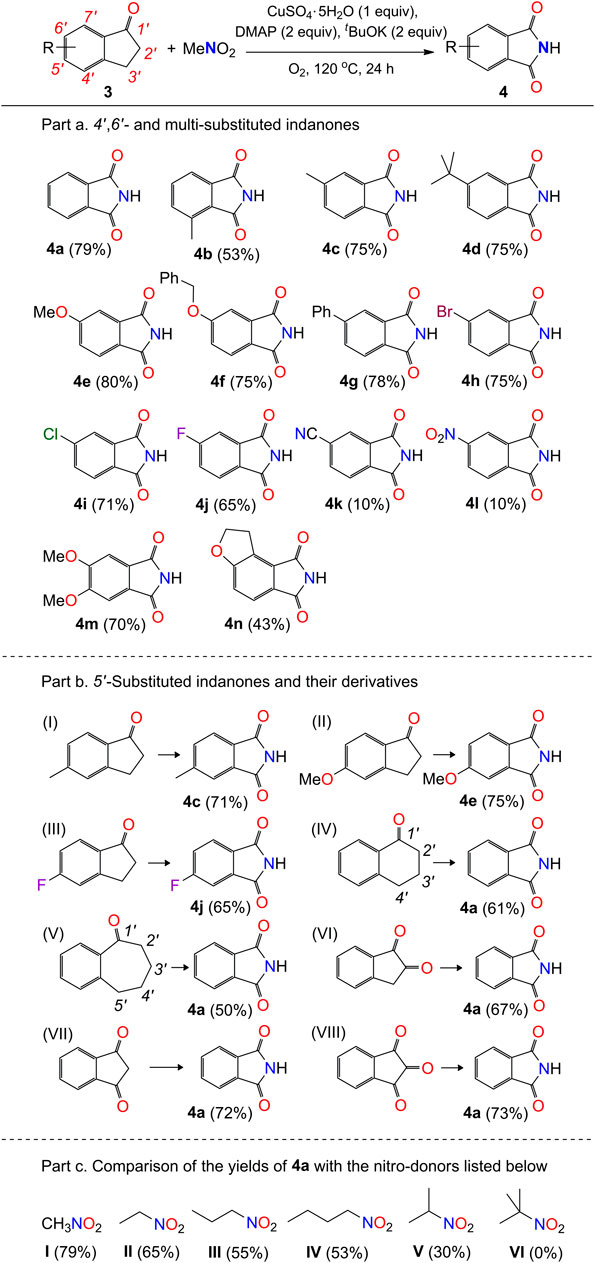
SCHEME 3. Scope of indanones with respect to compound 4a. aReaction conditions: substrate 3 (0.2 mmol), MeNO2 (1 mL), CuSO4.5H2O (0.2 mmol), DMAP (0.4 mmol), tBuOK (0.4 mmol), O2 (1 atm), 120 °C, and 24 h.
Part b. The above procedure (part a) was modified for the preparation of compounds listed in Part b, with the use of corresponding 1-carbonyl-benzohetercyclic compounds (0.2 mmol) as substrates.
Part c. The above procedure (part a) was used for the preparation of compound 4a with the use of nitroethane, nitropropane, 1-nitrobutane, 2-nitropropane, and 2-methyl-2-nitropropane as solvents (1 mL), respectively.
Experimental Procedure for the Synthesis of Compounds Shown in Scheme 4
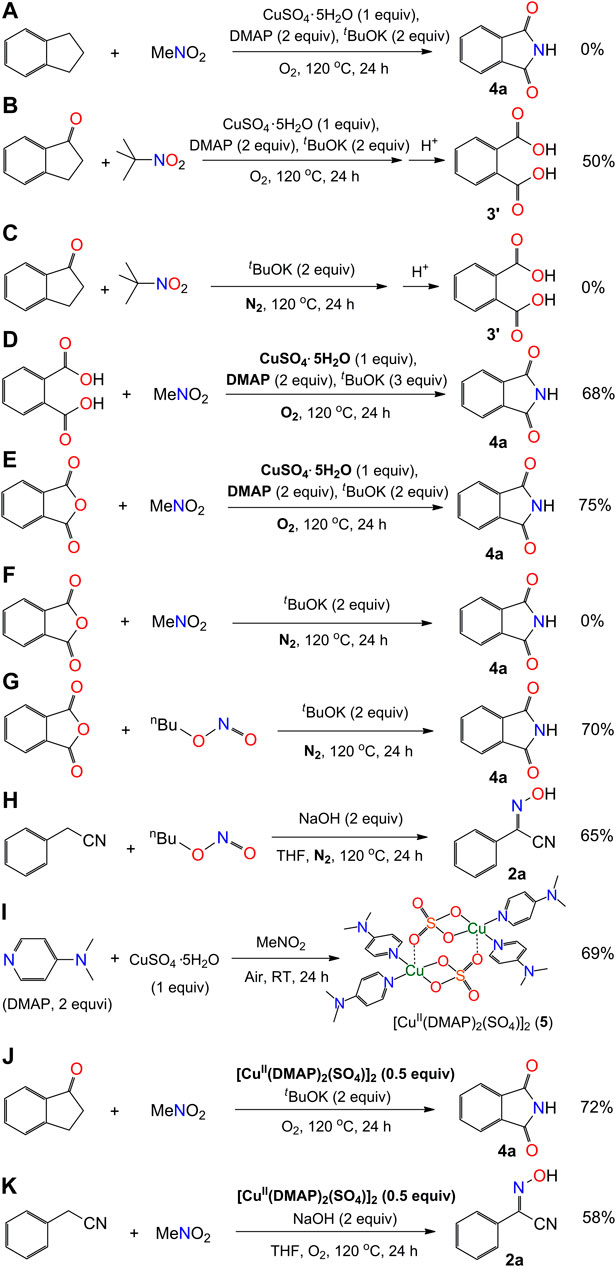
SCHEME 4. Mechanistic exploration of the synthesis of phthalimide and 2-hydroxyimino-2-phenylacetonitrile.
For Safety Reasons, the Feeding Order of Chemicals Should Be Followed
a. A mixture of indanone (23.6 mg, 0.20 mmol), DMAP (48.8 mg, 0.40 mmol), tBuOK (44.8 mg, 0.40 mmol), and CuSO4.5H2O (50.0 mg, 0.20 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and MeNO2 (1 mL) was added dropwise while stirring. After the addition was completed, the mixture was warmed up to room temperature. The tube was charged with O2 (1 atm) and the mixture was stirred at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The residue was checked with the techniques of Thin-Layer Chromatography (TLC) and 1H NMR, but no signal of compound 4a was found.
b. A mixture of 1-indanone (26.4 mg, 0.20 mmol), DMAP (48.8 mg, 0.40 mmol), tBuOK (44.8 mg, 0.40 mmol), and CuSO4.5H2O (50.0 mg, 0.20 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and tBuNO2 (1 mL) was added dropwise while stirring. After the addition was completed, the mixture was warmed up to room temperature. The tube was charged with O2 (1 atm) and the mixture was stirred at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL), acidified by acetic acid to pH = 3, filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with dichloromethane/methanol (10:1 v/v) to afford the product 3′ in yield 50% (16.6 mg).
c. A mixture of 1-indanone (26.4 mg, 0.20 mmol) and tBuOK (44.8 mg, 0.40 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and tBuNO2 (1 mL) was added dropwise while stirring. After the addition was completed, the mixture was warmed up to room temperature. The tube was charged with N2 (1 atm) and the mixture was stirred at 120 °C for 24 h. The reaction mixture was cooled, diluted with dichloromethane (20 mL), acidified by acetic acid to pH = 3, filtered through a pad of silica gel, and concentrated under reduced pressure. The residue was checked with the techniques of TLC and 1H NMR, but no signal of compound 3′ was found.
d. A mixture of o-phthalic acid (33.2 mg, 0.20 mmol), DMAP (48.8 mg, 0.40 mmol), tBuOK (44.8 mg, 0.40 mmol), and CuSO4.5H2O (50.0 mg, 0.20 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and MeNO2 (1 mL) was added dropwise while stirring. After the addition was completed, the mixture was warmed up to room temperature. The tube was charged with O2 (1 atm) and stirred at 120 °C for 24 h. The reaction mixture was cooled, diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with dichloromethane/methanol (20:1 v/v) to afford the product 4a in yield 68% (20.0 mg).
e. A mixture of o-phthalic anhydride (29.6 mg, 0.20 mmol), DMAP (48.8 mg, 0.40 mmol), tBuOK (44.8 mg, 0.40 mmol), and CuSO4.5H2O (50.0 mg, 0.20 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and MeNO2 (1 mL) was added dropwise while stirring. The same procedure of (d) was followed for the processing of solution and the purification of crude product. The yield of phthalimide (4a) is 75% (22.0 mg).
f. A mixture of o-phthalic anhydride (29.6 mg, 0.20 mmol) and tBuOK (44.8 mg, 0.40 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and tBuNO2 (1 mL) was added dropwise while stirring. After the addition was completed, the mixture was warmed up to room temperature. The tube was charged with N2 (1 atm) and the mixture was stirred at 120 °C for 24 h. The reaction mixture was cooled, diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The residue was checked with the techniques of TLC and 1H NMR, but no signal of compound 4a was found.
g. A mixture of o-phthalic anhydride (29.6 mg, 0.20 mmol) and tBuOK (44.8 mg, 0.40 mmol) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and nBuONO (1 mL) was added dropwise while stirring. After the addition was completed, the mixture was warmed up to room temperature. The tube was charged with N2 (1 atm) and stirred at 120 °C for 24 h. The reaction mixture was cooled, diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with dichloromethane/methanol (20:1 v/v) to afford the product 4a in yield 70% (20.6 mg).
h. To a mixture of phenylacetonitrile (70.3 mg, 0.60 mmol), NaOH (16.0 mg, 0.40 mmol), and THF (1 mL) in a 50 mL Teflon screw-cap sealed tube, nBuONO (20.6 mg, 0.20 mmol) was added slowly in air atmosphere. The tube was charged with N2 (1 atm) and the mixture was stirred at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (7:1 v/v) to afford 2-(hydroxyimino)-2-phenylacetonitrile (2a) in yield 65% (19.0 mg).
i. A mixture of 1-indanone (26.4 mg, 0.20 mmol), tBuOK (44.8 mg, 0.40 mmol), and compound 5 (80.8 mg, 0.10 mmol, green crystalline solid) in a 50 mL Teflon screw-cap sealed tube was stirred for 5 min in air atmosphere. The mixture was cooled to 0 °C and MeNO2 (1 mL) was added dropwise while stirring. The same procedure of (d) was followed for the processing of solution and the purification of crude product. The yield of phthalimide (4a) is 72% (21.2 mg).
j. To a mixture of phenylacetonitrile (70.3 mg, 0.60 mmol), NaOH (16.0 mg, 0.40 mmol), compound 5 (80.8 mg, 0.10 mmol, green crystalline solid), and THF (1 mL) in a 50 mL Teflon screw-cap sealed tube, MeNO2 (12.2 mg, 0.20 mmol) was added slowly in air atmosphere. The tube was charged with O2 (1 atm) and the mixture was stirred at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL), filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (7:1 v/v) to afford the product 2a in yield 58% (17.0 mg).
The Synthetic Procedure for Compound 5
A mixture of CuSO4.5H2O (25.0 mg, 0.10 mmol) and DMAP (24.4 mg, 0.20 mmol) was stirring in dried MeNO2 (10 mL) for 30 min to give a dark green solution. The mixture was filtered and the filtrate was added Et2O to deposit the product as some green crystalline solid, which was collected, washed with THF/Et2O (0.5/1 mL), and dried (28 mg, 69%). Anal. Calcd. for C28H40Cu2N8O8S2: C, 41.63; H, 4.99; N, 13.87. Found (%): C, 41.16; H, 4.94; N, 14.03. The crystal suitable for X-ray crystallography was grown by diffusion of Et2O into a solution of compound 5 in DMF.
NMR Data
2-(Hydroxyimino)-2-phenylacetonitrile (2a) (Alam et al., 2020)
Yield, 78% (22.8 mg); white powder; melting point, 120–122 °C (literature 120–122 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.89 (s, 1H), 7.81 (brs, 2H), 7.62 (brs, 3H); 13C NMR (100 MHz, DMSO-d6): δ 131.7, 131.4, 130.0, 129.8, 126.0, 110.6.
2-(Hydroxyimino)-2-(2-methyl)-phenylacetonitrile (2b) (Alam et al., 2020)
Yield, 75% (24.1 mg); white powder; melting point, 132–134 °C; 1H NMR (400 MHz, DMSO-d6): δ 13.85 (s, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.49 (t, J = 7.1 Hz, 1H), 7.41–7.45 (m, 2H), 2.51 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 137.0, 132.1, 131.8, 130.9, 129.7, 129.3, 127.2, 111.4, 21.1; HRMS m/z (ESI) [M + Na+] calculated for C9H8N2O1Na, 183.0534; found, 183.0559.
2-(Hydroxyimino)-2-(3-methyl)-phenylacetonitrile (2c) (Alam et al., 2020)
Yield, 77% (24.7 mg); white powder; melting point, 124–126 °C; 1H NMR (400 MHz, DMSO-d6): δ 13.75 (s, 1H), 7.53 (s, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.35 (d, J = 7.6 Hz, 1H), 2.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 138.8, 131.6, 131.2, 129.5, 129.3, 125.9, 122.8, 110.2, 20.9.
2-(Hydroxyimino)-2-(4-methyl)-phenylacetonitrile (2d) (Alam et al., 2020)
Yield, 78% (25.0 mg); white powder; melting point, 150–152 (literature 150–152 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.70 (s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 2.40 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 141.4, 131.6, 130.4, 127.3, 126.0, 110.7, 21.4.
4-(tert-Butyl)-N-hydroxybenzimidoyl Cyanide (2e) (Alam et al., 2020)
Yield, 82% (33.1 mg); pale yellow powder; melting point, 158–160 °C (literature 158–160 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.58 (s, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 1.16 (s, 9H); 13C NMR (100 MHz, DMSO-d6): δ 153.8, 131.1, 126.9, 126.2, 125.4, 110.2, 34.7, 30.9.
2-(Hydroxyimino)-2-(4-phenyl)-phenylacetonitrile (2f) (Alam et al., 2020)
Yield, 60% (26.4 mg); white powder; melting point, 140–142 °C (literature 141–143 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.83 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.81 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 7.2 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.42 (t, J = 7.2 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 142.4, 138.9, 130.9, 129.1, 128.6, 128.2, 127.5, 126.8, 126.1, 110.2; HRMS m/z (ESI) [M + H+]: calculated for C14H11N2O1, 223.0872; found, 223.0866.
2-(Hydroxyimino)-2-(4-bromo)-phenylacetonitrile (2g) (Gao et al., 2018)
Yield, 46% (20.7 mg); white powder; melting point, 127–129 °C (literature 123–125 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.94 (s, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 132.3, 130.4, 128.8, 127.4, 124.3, 109.9; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C8H4N2O1BrNa2, 268.9297; found, 268.9295.
2-(Hydroxyimino)-2-(4-chloro)-phenylacetonitrile (2h) (Neel and Zhao, 2018)
Yield, 45% (16.3 mg); white powder; melting point, 137–139 °C (literature 139–141 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.95 (s, 1H), 7.74 (d, J = 8.6 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 135.5, 130.3, 129.4, 128.5, 127.3, 109.9.
2-(Hydroxyimino)-2-(4-fluoro)-phenylacetonitrile (2i) (Gao et al., 2018)
Yield, 42% (13.8 mg); white powder; melting point, 112–114 °C (literature 112–114 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.83 (s, 1H), 7.78 (brs, 2H), 7.37 (dd, JHF = 8.0 Hz, JHH = 8.0 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 164.0 (d, J = 225.3 Hz), 130.2, 128.0 (d, J = 9.0 Hz), 116.5 (d, J = 22.5 Hz), 110.1, 99.6; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C8H4N2O1F1Na2, 209.0098; found, 209.0101.
2-(Hydroxyimino)-2-(3-fluoro)-phenylacetonitrile (2j) (Alam et al., 2020)
Yield, 50% (16.4 mg); white powder; melting point, 120–122 °C (literature 120–122 °C); 1H NMR (400 MHz, DMSO-d6): δ 14.03 (s, 1H), 7.53–7.61 (m, 2H), 7.49 (d, J = 9.6 Hz, 1H), 7.39 (td, JHH = 8.0 Hz, JHF = 3.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 162.7 (d, J = 243.6 Hz), 132.2 (d, J = 8.4 Hz), 132.1 (d, J = 8.4 Hz), 130.7 (d, J = 3.6 Hz), 122.6 (d, J = 2.8 Hz), 118.3 (d, J = 21.3 Hz), 112.3 (d, J = 24.2 Hz), 110.3.
2-(Hydroxyimino)-2-(2-fluoro)-phenylacetonitrile (2k) (Alam et al., 2020)
Yield, 43% (14.1 mg); white powder; melting point, 128–130 °C (literature 132–134 °C); 1H NMR (400 MHz, DMSO-d6): δ 14.12 (s, 1H), 7.69 (dd, JHH = 7.6 Hz, JHF = 6.4 Hz, 1H), 7.59 (td, JHH = 8.0 Hz, JHF = 5.2 Hz, 1H), 7.34–7.43 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.5 (d, J = 250.7 Hz), 133.4 (d, J = 8.6 Hz), 129.5, 127.1, 125.8 (d, J = 3.5 Hz), 118.2 (d, J = 10.4 Hz), 117.2 (d, J = 20.8 Hz), 110.5.
2-(Hydroxyimino)-2-(4-trifluoromethyl)-phenylacetonitrile (2l) (Neel and Zhao, 2018)
Yield, 47% (20.1 mg); white powder; melting point, > 200 °C; 1H NMR (400 MHz, DMSO-d6): δ 14.21 (s, 1H), 7.88–7.95 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 133.5, 130.6 (q, J = 32.0 Hz), 130.3, 126.4, 126.3 (q, J = 3.7 Hz), 123.9 (q, J = 270.8 Hz), 109.9.
2-(Hydroxyimino)-2-(2,5-dimethyl)-phenylacetonitrile (2m)
Yield, 55% (19.2 mg); white powder; melting point, > 200 °C; 1H NMR (400 MHz, DMSO-d6): δ 13.75 (s, 1H), 7.30 (s, 1H), 7.24 (brs, 2H), 2.38 (s, 3H), 2.32 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 136.2, 133.7, 132.0, 131.7, 131.4, 129.9, 128.9, 111.3, 20.8, 20.6.
2-(Hydroxyimino)-2-(3,5-dimethyl)-phenylacetonitrile (2n)
Yield, 56% (19.5 mg); white powder; melting point, 153–155 °C; 1H NMR (400 MHz, DMSO-d6): δ 13.70 (s, 1H), 7.32 (s, 2H), 7.16 (s, 1H), 2.32 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ 139.1, 132.8, 131.7, 129.9, 123.6, 110.7, 21.2; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C10H9N2O1Na2, 219.0505; found, 219.0504.
2-(Hydroxyimino)-2-(3,4-dimethoxyl)-phenylacetonitrile (2o)
Yield, 56% (23.1 mg); white powder; melting point, 183–185 °C (literature 183–191 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.48 (s, 1H), 7.25 (s, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 3.82 (s, 3H), 3.81 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 151.7, 149.6, 131.3, 122.5, 120.1, 112.2, 110.7, 107.6, 56.1, 55.9; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C10H9N2O3Na2, 251.0403; found, 251.0403.
N-Hydroxybenzo[d][1,3]dioxole-5-carbimidoyl Cyanide (2p)
Yield, 57% (21.7 mg); white powder; melting point, 76–78 °C (literature 76–78 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.55 (s, 1H), 7.23 (s, 1H), 7.19 (d, J = 8.1 Hz, 1H), 7.06 (d, J = 8.1 Hz, 1H), 6.12 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 150.2, 148.8, 131.2, 124.1, 121.7, 110.6, 109.2, 104.8, 102.5; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C9H5N2O3Na2, 235.0090; found, 235.0089.
2,4-Difluoro-N-hydroxybenzimidoyl Cyanide (2q)
Yield, 20% (14.6 mg) for 0.4 mmol; pale yellow powder; melting point, > 200 °C; 1H NMR (400 MHz, DMSO-d6): δ 14.14 (s, 1H), 7.72–7.80 (m, 1H), 7.49 (t, J = 8.8 Hz, 1H), 7.26 (t, J = 8.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 164.1 (dd, J = 262.3, 11.7 Hz), 160.0 (dd, J = 253.1, 12.8 Hz), 131.2 (dd, J = 10.3, 3.2 Hz), 126.4, 115.0 (dd, J = 10.6, 3.7 Hz), 113.3 (dd, J = 22.0, 3.6 Hz), 110.4, 105.8 (t, J = 26.0 Hz); HRMS m/z (ESI) [M + H+]: calculated for C8H5N2O1F2, 183.0370; found, 183.0342.
2-(Hydroxyimino)-2-naphthylacetonitrile (2r) (Gao et al., 2018)
Yield, 25% (9.8 mg); white powder; melting point, 143–145 °C (literature 145 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.88 (s, 1H), 8.22 (s, 1H), 8.11 (d, J = 9.2 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.98 (d, J = 9.2 Hz, 1H), 7.89 (d, J = 8.8 Hz, 1H), 7.58–7.65 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 134.2, 133.0, 131.9, 129.6, 129.2, 128.3, 127.73, 127.71, 127.0, 121.8, 110.6; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C12H7N2O1Na2, 241.0348; found, 241.0350.
Hydroxy-1-naphthimidoyl Cyanide (2s)
Yield, 32% (25.1 mg) for 0.4 mmol; pale yellow powder; melting point, > 200 °C; 1H NMR (400 MHz, DMSO-d6): δ 14.05 (s, 1H), 8.45 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 8.04 (d, J = 7.2 Hz, 1H), 7.82 (d, J = 7.2 Hz, 1H), 7.60–7.68 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 134.0, 131.8, 131.5, 129.9, 129.4, 129.0, 128.3, 127.2, 126.7, 125.9, 125.0, 111.6; HRMS m/z (ESI) [M + Na+]: calculated for C12H8N2O1Na, 219.0534; found, 219.0514.
N-Hydroxythiophene-2-carbimidoyl Cyanide (2t)
Yield, 37% (22.5 mg) for 0.4 mmol; white powder; melting point, 103–105 °C (literature 103–104 °C); 1H NMR (400 MHz, DMSO-d6): δ 14.22 (s, 1H), 7.97 (dd, J = 5.2, 1.2 Hz, 1H), 7.65 (dd, J = 3.6, 1.2 Hz, 1H), 7.25 (dd, J = 5.2, 4.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 134.1, 131.7, 129.7, 128.6, 127.3, 114.9; HRMS m/z (ESI) [M + H+]: calculated for C6H5N2O1S1, 153.0123; found, 153.0114.
N-Hydroxythiophene-3-carbimidoyl Cyanide (2u)
Yield, 28% (17 mg) for 0.4 mmol; white powder; melting point, 98–100 °C (literature 96–106 °C); 1H NMR (400 MHz, DMSO-d6): δ 13.83 (s, 1H), 8.47 (dd, J = 3.2, 1.2 Hz), 7.73 (dd, J = 5.2, 3.2 Hz), 7.59 (dd, J = 5.2, 1.2 Hz); 13C NMR (100 MHz, DMSO-d6): δ 132.5, 129.7, 128.8, 128.08, 128.06, 116.1; HRMS m/z (ESI) [M + H+]: calculated for C6H5N2O1S1, 153.0123; found, 153.0114.
Phthalimide (4a) (Wang et al., 2019b)
Yield, 79% (23.3 mg); white powder; melting point, > 200 °C (literature 232–234 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.40 (s, 1H), 7.83 (s, 4H); 13C NMR (100 MHz, DMSO-d6): δ 169.7, 134.8, 133.1, 123.4.
3-Methyl-phthalimide (4b)
Yield, 53% (17.1 mg); white powder; melting point, 188–190 °C (literature 193–195 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.22 (s, 1H), 7.66 (t, J = 7.4 Hz, 1H), 7.60 (d, J = 7.0 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 2.58 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 170.6, 169.5, 137.6, 136.8, 134.3, 133.5, 129.6, 121.0, 17.4; HRMS m/z (ESI) [M + NH4+]: calculated for C9H11N2O2, 179.0820; found, 179.0815.
4-Methylphthalimide (4c) (Wang et al., 2019b)
Yield, 75% (24.1 mg) for 6-methyl-indanone, 57% (18.4 mg) for 5-methyl-indanone; white powder; melting point, 196–198 °C (literature 195–197 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.22 (s, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.62 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 2.46 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 169.77, 169.68, 145.6, 135.2, 133.4, 130.5, 123.8, 123.3, 21.8.
4-tert-Butyl-phthalimide (4d)
Yield, 75% (30.5 mg); white powder; melting point, 134–136 °C (literature 132–134 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.24 (s, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.78 (s, 1H), 7.73 (d, J = 7.6 Hz, 1H), 1.32 (s, 9H); 13C NMR (100 MHz, DMSO-d6): δ 169.4, 169.1, 158.0, 132.9, 131.3, 130.1, 122.8, 119.7, 35.4, 30.8; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C12H12N1O2Na2, 248.0658; found, 248.0656.
4-Methoxylphthalimide (4e) (Wang et al., 2019b)
Yield, 80% (28.3 mg) for 6-methoxyl-indanone, 75% (26.6 mg) for 5-methoxyl-indanone; white powder; melting point, > 200 °C (literature 224–225 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.17 (s, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.29 (s, 1H), 7.28 (d, J = 8.0 Hz, 1H), 3.90 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 168.90, 168.85, 164.3, 135.3, 124.8, 124.5, 119.9, 107.9, 56.2.
4-Benzyloxy-phthalimide (4f) (Punchi Hewage et al., 2019)
Yield, 75% (38.0 mg); white powder; melting point, 154–156 °C (literature 159–161 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.20 (s, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.46 (s, 1H), 7.33–7.42 (m, 5H), 5.28 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 168.88, 168.86, 163.3, 136.2, 135.2, 128.6, 128.2, 127.9, 124.80, 124.68, 120.7, 108.6, 70.1.
4-Phenylphthalimide (4g) (Wang et al., 2019b)
Yield, 78% (34.8 mg); white powder; melting point, 198–200 °C (literature 200–202 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.38 (s, 1H), 8.10 (d, J = 7.6, 1H), 8.04 (s, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.80 (d, J = 7.6 Hz, 2H), 7.54 (t, J = 7.6 Hz, 2H), 7.47 (t, J = 7.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 169.01, 168.99, 146.2, 138.4, 133.7, 132.5, 131.3, 129.2, 128.8, 127.3, 123.6, 120.9.
4-Bromophthalimide (4h) (de Nanteuil et al., 2013)
Yield, 75% (35.8 mg); white powder; melting point, > 200 °C (literature 230–233 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.48 (s, 1H), 8.02 (d, J = 7.8 Hz, H), 8.00 (s, 1H), 7.75 (d, J = 7.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 168.5, 167.9, 137.0, 134.7, 131.6, 127.9, 125.9, 124.9.
4-Chlorophthalimide (4i) (Wang et al., 2019b)
Yield, 71% (25.8 mg); white powder; melting point, > 200 °C (literature 228–230 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.50 (s, 1H), 7.88 (s, 1H), 7.87 (d, J = 7.0 Hz, 1H), 7.82 (d, J = 7.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 168.4, 168.0, 139.1, 134.7, 134.1, 131.2, 124.8, 123.1.
4-Fluorophthalimide (4j)
Yield, 65% (21.5 mg) for 6-fluoro-indanone, 71% (23.4 mg) for 5-fluoro-indanone; white powder; melting point, 175–177 °C (literature 178–179 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.46 (s, 1H), 7.89 (dd, JHH = 8.0, JHF = 4.4 Hz, 1H), 7.69 (dd, JHF = 7.6, JHH = 2.4 Hz, 1H), 7.64 (td, JHF/HH = 8.8, JHH = 2.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 168.3, 167.9 (d, J = 1.8 Hz), 165.8 (d, J = 251.3 Hz), 135.6 (d, J = 9.5 Hz), 128.8 (d, J = 2.4 Hz), 125.7 (d, J = 9.7 Hz), 121.2 (d, J = 23.7 Hz), 110.7 (d, J = 24.7 Hz); HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C8H3N1O2F1Na2, 209.9938; found, 209.9959.
4-Cyano-phthalimide (4k)
Yield, 10% (6.9 mg for 0.4 mmol); white powder; melting point, > 200 °C (literature 237–238 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.75 (s, 1H), 8.35 (s, 1H), 8.29 (d, J = 7.6 Hz, 1H), 8.00 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 168.0, 167.8, 138.5, 136.1, 133.3, 126.8, 123.8, 117.6, 116.4; HRMS m/z (ESI) [M + NH4+]: calculated for C9H8N3O2, 190.0617; found, 190.0575.
4-Nitro-phthalimide (4l) (Song et al., 2018)
Yield, 10% (7.7 mg for 0.4 mmol); pale yellow powder; melting point, 196–198 °C (literature 198–202 °C); 1H NMR (400 MHz, DMSO-d6): δ 11.84 (s, 1H), 8.60 (d, J = 8.0 Hz, 1H), 8.43 (s, 1H), 8.07 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 167.7, 167.4, 151.4, 137.4, 134.1, 129.5, 124.6, 117.9.
4,5-Dimethoxyl-phthalimide (4m)
Yield, 70% (29.0 mg); white powder; melting point, > 200 °C (literature 318–320 °C); 1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, 1H), 7.33 (s, 2H), 3.91 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ 169.3, 153.6, 125.7, 105.4, 56.3; HRMS m/z (ESI) [M + Na+]: calculated for C10H9N1O4Na, 230.0430; found, 230.0474.
7,8-Dihydro-1H-furo[3,2-e]isoindole-1,3(2H)-dione (4n)
Yield, 43% (16.3 mg); white powder; melting point, > 200 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.11 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 4.73 (t, J = 8.8 Hz, 2H), 3.39 (t, J = 8.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 168.8, 166.0, 129.4, 125.7, 124.5, 123.9, 112.9, 73.2, 27.0; HRMS m/z (ESI) [M + 2Na+ - H+]: calculated for C10H6N1O3Na2, 234.0138; found, 234.0141.
Results and Discussion
All the reactions were carried out under a dioxygen atmosphere in solvents (1 mL). We choose phenylacetonitrile (1a) as the model substrate and optimized the reaction by changing ingredients to find out the most efficient conditions (Table 1). Based on the yields obtained, it is found that the reaction of 1a (0.6 mmol), nitromethane (0.2 mmol), CuSO4.5H2O (0.2 mmol), 4-dimethylaminopyridine (DMAP, 0.4 mmol), and sodium hydroxide (0.4 mmol) in THF (1 mL) at 120 °C under dioxygen atmosphere for 24 h gives the best result, yielding the target product 2-hydroxyimino-2-phenylacetonitrile (2a) in 78% (entry 1). Other copper sources such as Cu(ClO4)2·6H2O and Cu(OTf)2 afford the compound 2a in similar yields (entries 2-3). However, the use of copper salt Cu(OAc)2·H2O for reaction would lead to the decline of yield to 54% (entry 4). It is proposed that the coordination interaction between the acetate ligands and the Cu(II) center was strong, which made the replacement of the OAc− groups with the deprotonated nitromethane more difficult. Therefore, the formation of the active intermediate of [CuII(DMAP)2(CH2NO2)]− became difficult accordingly (see the following mechanism section, Scheme 6). No compound 2a could be isolated without the participation of copper salt or with the replacement of copper salts with NiSO4.6H2O and ZnSO4.7H2O (entries 6-7). The base of NaOH is found to be necessary for the processing of reaction. Both the strong base tBuOK and the weak base K2CO3 resulted in the decreasing of yield to trace (entries 8-9). Interestingly, the use of the organic base of Et3N generated the product in a considerable good yield 68% (entry 10), probably due to the perfect solubility of it in THF. The type of solvent is essential for the production of 2a. The coupling reaction proceeds efficiently in THF, but it turns out to be difficult to process in the solvents of DMF, 1,4-dioxane, and toluene with the yields decreasing down to trace (entries 11–13). An excess of 1a (3 equivalents of nitromethane) would help to enhance the synthetic rate and increase the production of 2a (78%, entry 1), but overaddition of 1a (5 equivalents) did not do help to improve the rate of production (77%, entry 16). In contrast, the use of less amount of 1a, such as one equivalent or two equivalents, would decrease the yields down to 54% and 66% (entries 14-15), respectively. It has been shown that the strong base of EtONa and tBuOK et al. would promote an extra transformation of phenylacetonitrile to β-enaminonitrile and/or 4-aminopyrimidine by mean of self-condensation (Zhu et al., 2019; Li et al., 2020), which would lead to the loss of the starting material of 1a unwillingly. In addition, the dioxygen atmosphere is crucial for the activation of nitromethane. The reaction in an atmosphere of air would lead to the decline of yield to 66%, and no product could be isolated from the reaction under N2 atmosphere (entries 17-18). This result indicates the role of dioxygen as an oxidant in the reaction. Finally, the influence of ligand on the production of 2a was examined with pyridine (L2), 4,4′-dimethyl-2,2′-bipyridyl (L3), 1,10-phenanthroline (L4), and N,N,N′,N′-tetramethylethylenediamine (L5). It showed that the production of 2a could be achieved with the employment of those ligands, but their yields were generally low at a level of ca. 30% (entries 19–22).
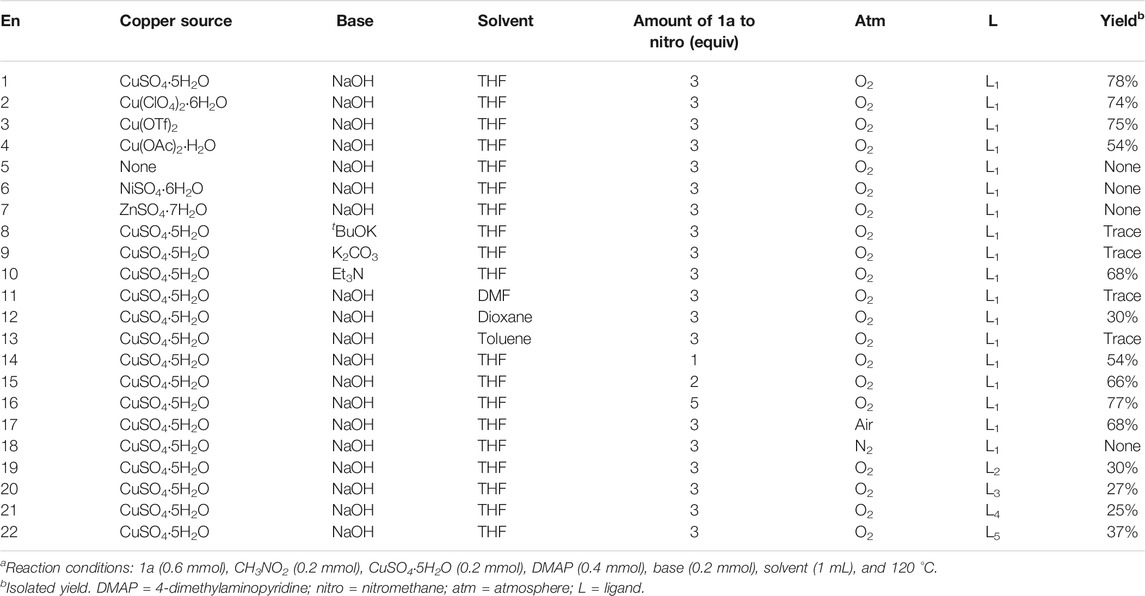
TABLE 1. Optimization of the formation of 2-hydroxyimino-2-phenylacetonitrilea.
Table 1. Optimization of the formation of 2-hydroxyimino-2-phenylacetonitrile.
Under the optimal conditions, we set out to examine the scope of phenylacetonitriles for the generation of benzyl-cyano-oxime compounds (Scheme 2). A total of 26 reactions were carried out for the analysis of the implication of reaction, including twenty-one phenylacetonitrile substrates (2a–2u) and five nitro compounds (I–V). The yield distribution and production efficiency of 2-hydroxyimino-2-phenylacetonitriles (2) are discussed based on the electronic effect, steric effect, and the synergic effect of functional groups on the nitriles. Firstly, the experimental results show that the electron-donating groups would help to improve the yields of compounds (2a–2e) compared to the electron-withdrawing groups (2g–2i, 2l), with the yield difference of ca. 25% between them. The phenyl-group-substituted phenylacetonitrile afforded the product in a moderate yield of 60% (2f). Secondly, the steric hindrance to the reaction was studied by employing methyl group (2b-2d) and fluoro group (2i–2k) in the para-, meta-, and orthopositions of phenyl group, respectively. It is found that the size of methyl group or fluoro group is not important for the production of compounds (2b-2d, 2i–2k), but the difunctional groups would lead to a drop of yields about 20% correspondingly (2m–2q) (Scheme 5A). Thirdly, the coupling reaction was examined with varied aromatic-group-substituted acetonitriles as substrates, for example, naphthyl acetonitriles and thienyl acetonitriles. All the reactions could succeed in the production of cyano-oxime compounds, but the efficiency of transformation was relatively low, and the products were obtained in yields about 30% (2r–2u). It is clear that the deprotonation of aromatic acetonitrile is a key step for the formation of 2-hydroxyimino-2-phenylacetonitriles in our system. The reaction of the methyl-substituted phenylacetonitrile in the position of carbon proximal to the cyanide group did not generate the corresponding compound at all. In addition, the scope of nitroalkanes was also examined for comparison. Five nitro compounds, e.g., nitroethane, 1-nitropropane, 2-nitropropane, 1-nitrobutane, and 2-methyl-2-nitropropane, were selected to examine the percent of the transformation efficiency from nitro to oxime. It is shown that the nitro compounds with α-hydrogen on the carbon atom proximal to the cyanide group provided the product 2a in moderate yields (Schemes 2I–V), while no product could be isolated with the use of 2-methyl-2-nitropropane as a starting material (Scheme 2VI). It is proposed that the initial deprotonation of the α-H to the nitro group is critical for the activation of nitro compounds, by which the nitro group would transfer to nitroso group via oxidation and nitrification reactions (see below).
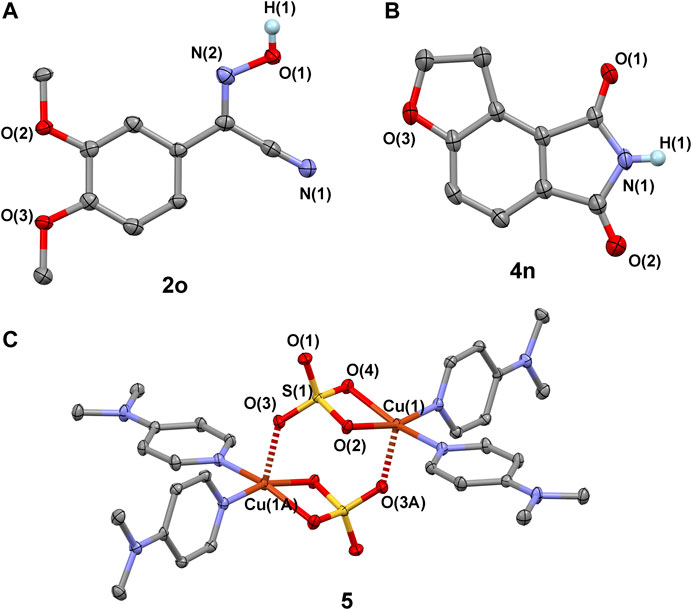
SCHEME 5. Crystal structures of compounds 2o (CCDC 2007798), 4n (CCDC 2007797), and 5 (CCDC 2007804), showing the thermal ellipsoids of 50% probability surfaces. The weak interactions between molecules with the Cu(1)-O(3A) and Cu(1A)-O(3) bond lengths of 2.193(1) Å are shown.
Interestingly, our reaction can also be extended to the formation of functional phthalimides (Scheme 3). Twenty-seven reactions were carried out to examine the transformation of 1-indanones to phthalimides, by using nitromethane as nitrogen donor. The experimental results show that the electronic effect of substituted groups on the phenyl ring of 1-indanones has a significant impact on the production of phthalimides (4). The reaction of 1-indanones, with electron-donating groups (H-, Me-, Me3C-, MeO-, PhCH2O-) and weak electron-withdrawing groups (Ph-, Br-) on the 6′-position of phenyl ring, afforded the phthalimides in moderate to high yields (75–80%; 4a and 4c-4h). The 4′-methyl group substituted indanone provided lower production efficiency (53%), probably due to the steric effect on the activation of the 3-carbon of indanone (4b). In contrast, the production of four would be influenced by the introduction of medium electron-withdrawing groups to the phenyl ring. The reaction of 1-indanones with the substituted chloro and fluoro groups in the 6′-position of phenyl ring afforded the product in lower yields 71% and 65% (4i and 4j), respectively. With the strong electron-withdrawing groups, such as cyano and nitro groups, the substituted 1-indanones would only be able to offer the products in yields about 10% (4k and 4l). In addition, the disubstitution of the phenyl ring of 1-indanones would have a negative influence on the yield of product. The dimethoxyl-group-substituted 1-indanone generated the compound 4m in a lower yield (70%) compared to the single methoxyl group substituted 1-indanone (4e, 80%). The disubstituted groups in the 6′,7′-positions of 1-indanone led to the decline of yield dramatically down to 43% (4n) (Scheme 5B). This point of view is consistent with the result presented in the formation of 4b that the substitution on the carbon atoms adjacent to the five-member ring of 1-indanones would significantly decrease the yields of phthalimides in our system.
Furthermore, the reactions of 5′-substituted 1-indanones with methyl, methoxyl, and fluoro groups also generated the phthalimides in good yields (Scheme 3, Part b, I–III), even though they have the same structures to compounds 4c, 4e, and 4j. Apart from the 1-indanone with a 5-member ring on the side of ketone, the 1-tetralone with a 6-member ring and the 1-benzosuberone with a 7-member ring are also able to produce the compound 4a under the standard reaction conditions (Scheme 3, Part b, IV-V). It implies that the methylene groups next to the phenyl ring of 1-indanone and its derivatives (3′-carbon in 1-indanone; 4′-carbon in 1-tetralone; 5′-carbon in 1-benzosuberone) would be oxidized in the processing of reaction. The cleavage of C-C bond between the ketone groups and the unsaturated carbon atoms adjacent to them (1′- and 2′-carbon and 2′- and 3′-carbon in 1-indanone; 1′- and 2′-carbon and 3′- and 4′-carbon in 1-tetralone; 1′- and 2′-carbon and 4′- and 5′-carbon in 1-benzosuberone) would occur with the insertion of nitrogen atom. This inference is consistent with our experimental results that the reactions of indan-1,2-dione, indan-1,3-dione, and 1,2,3-indantrione (ninhydrin) in our system are also able to provide the product 4a (Scheme 3, Part b, VI–VIII). Finally, other organic nitro compounds, such as nitroethane, 1-nitropropane, 2-nitropropane, and 1-nitrobutane, are also suitable for the formation of compound 4a, with the yields decreasing gradually from 65% to 30% (Scheme 3, Part c, I–V). No reaction occurred with the use of 2-methyl-2-nitropropane as a nitrogen donor (Scheme 3, Part c, VI).
The above experiment results aroused our great interest in exploring the reaction mechanism. A number of reactions were carried out for a better understanding of how the reactions occurred (Scheme 4). Firstly, the species of 1-indanone was reduced to form an aromatic hydrocarbon compound, indanone, which was found not able to generate the compound 4a under our conditions. It implies that the presence of carbonyl group in 1′or 3′-position of indanone is required for the processing of reaction (Scheme 4A). Secondly, the oxidation of aromatic ketone in our system was examined in the solution of 2-methyl-2-nitropropane (Me3C-NO2). The solvent of 2-methyl-2-nitropropane was used with the aim of maintaining the condition of the solvent close to nitromethane without the presence of α-H. The reaction of 1-indanone with Me3C-NO2 under our conditions afforded phthalic acid (3′) rather than phthalimide (4a) after acidification (Scheme 4B). No phthalic acid could be obtained without the addition of CuSO4.5H2O and DMAP under dinitrogen atmosphere (Scheme 4C). This result indicates that the oxidation of 1-indanone to phthalic acid could be achieved by the activate oxygen, probably coordinating on the Cu(II) intermediate (Wang et al., 2015). The aromatic ketone species underwent a C-C bond cleavage to form an intermediate species of o-phthalic anhydride during the reaction, and the insertion of nitrogen donor occurred with the species of o-phthalic anhydride (Scheme 6). Thirdly, the activation of nitromethane was investigated by using phthalic acid and/or o-phthalic anhydride as the starting materials. The experimental results showed that the reaction of phthalic acid or o-phthalic anhydride under the standard conditions afforded the phthalimide in yield 68% and 75%, respectively (Schemes 4D,E). No product could be isolated in the absence of CuSO4.5H2O and DMAP under dinitrogen atmosphere (Scheme 4F). That is to say, the combination of Cu(II) salt, DMAP, and dioxygen facilitated the activation of nitromethane. Meanwhile, the reaction of o-phthalic anhydride with butyl nitrite under the same conditions provided the target product 2a in yield 70% (Scheme 4G). The reaction of phenylacetonitrile with butyl nitrite afforded the compound 4a in yield 65% (Scheme 4H). It is noted that the butyl nitrite was used instead of methyl nitrite for the reasons of commercial availability, easy handling, and same performance. Thus, the methyl nitrite is thought to be an intermediate of the reaction in the process of the oxidation of nitromethane, which resembles the research of the oxygen consumption in the oxidation of nitromethane by visible light irradiation of Rose Bengal (Bilski et al., 1994). Fourthly, the stirring of CuSO4.5H2O and DMAP in nitromethane at room temperature formed a dark green solution, from which a dinuclear copper(II) species of [CuII(DMAP)2(SO4)] 5) (Scheme 5C) was isolated by diffusion of Et2O into the reaction solution (Scheme 4I). The reactions of 1-indanone or phenylacetonitrile with nitromethane in the presence of compound 5 generated the products 4a and 2a in similar yields (Schemes 4J,K). It implies that compound 5 would be an intermediate of activated copper(II) species for the transformation of nitromethane to methyl nitrite.
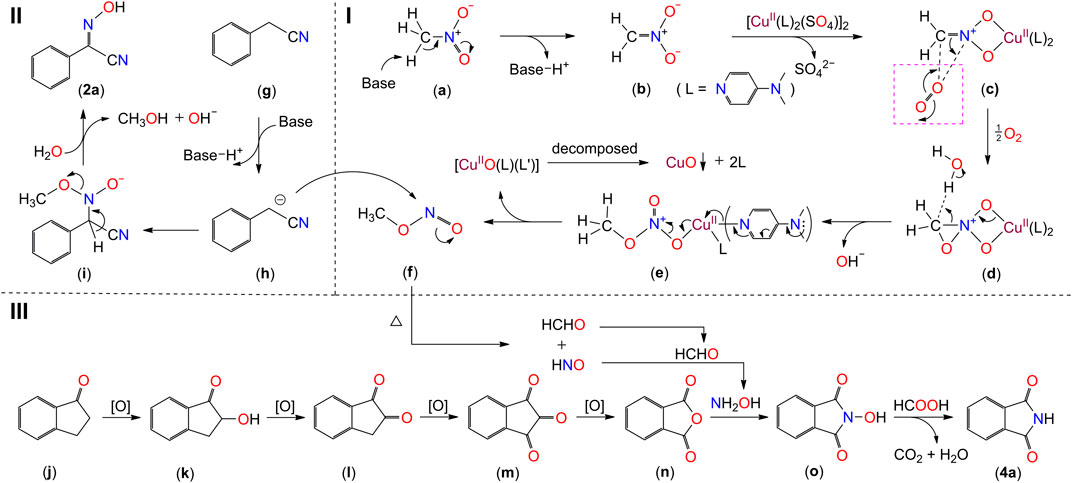
SCHEME 6. The plausible mechanism of the transformation of nitromethane to 2-hydroxyimino-2-phenylacetonitrile (2a) and phthalimide (4a). The image in the dashed-line-box of (c) is shown for illustrative purposes, displaying how the electron transfer occurs.
Based on the above results, a plausible mechanism is depicted in Scheme 6. The model of the reaction is divided into three parts, the transformation of nitromethane to methyl nitrite (Scheme 6-(I)), the formation of 2-hydroxyimino-2-phenylacetonitrile (Scheme 6-(II)), and the generation of phthalimide (Scheme 6-(III)). The deprotonation of CH3NO2 molecule (a) leads to the generation of intermediate (b), by which intermediate (c) is formed with the participation of [CuII(DMAP)2(SO4)]. The latter is produced simultaneously by the reaction of CuSO4.5H2O with two equivalents of DMAP (Scheme 5C). Two molecules of (c) react with one equivalent of dioxygen to generate the intermediate (e), through the transition state of (d). Methyl nitrite (f) is generated with the release of [CuIIO(L) (L′)]2+ (L = DMAP) into the solution (Scriven, 1983; Ragnarsson and Grehn, 1998). The [CuIIO(L) (L’)]2+ species undergoes a decomposition to deposit the metal moiety as a black precipitate CuO. The decomposition process is not clear to us.
On one hand, one of hydrogen atoms on the methylene group of phenyl acetonitrile molecule (g) is deprotonated to form a cyanoalkylide anion (h). The anion attacks an adjacent CH3ONO molecule on the nitrogen atom to form the intermediate species (i). The deprotonation of the second hydrogen atoms of the methylene group of (i) with the transfer of the pair electrons to the C-N bond generates the compound 2a (Scheme 6-(II)). On the other hand, the continuous oxidation of 1-indanone (j) in the presence of [CuII(DMAP)2(SO4)] and dioxygen generates the intermediate species o-phthalic anhydride (n) (Scheme 6-(III)) (Wang et al., 2015). Meanwhile, the methyl nitrite (f) undergoes a thermal decomposition reaction to generate the species HCHO and HNO (Brower, 1988; Zhu et al., 2013). The HNO is reduced to NH2OH in the presence of formaldehyde, and the generated NH2OH is inserted into the intermediate o-phthalic anhydride (n) to form the species (o). The species (o) is reduced by the generated formic acid from the oxidation of formaldehyde (Liu et al., 2019), forming the final product phthalimide (4a).
Conclusion
In summary, we have developed a simple and efficient method for the synthesis of 2-(hydroxyimino)-2-phenylacetonitriles and phthalimides in a total of 51 samples, by using nitromethane as the nitrogen donor. The production of two types of compounds indicates the flexible and diversified characteristics of our system in the activation of nitromethane for the synthesis of N-containing compounds. The extra diversity and stability of compounds are discussed in terms of electronic effect, steric effect, position of substituted groups, and intramolecular charge transfer. The mechanism study shows that the methyl nitrite is generated as an intermediate in the reaction, and the transformation of nitromethane to methyl nitrite is promoted by the Cu(II) intermediate with the participation of dioxygen as oxidant. The proposed nitromethane transformation process is supported by the experimental results. The employment of easily available reagents and mild reaction condition makes this method more interesting for chemists. This work might provide a clue to apply nitromethane as a nitrogen donor in the development of organic synthetic chemistry, biological chemistry, and pharmaceutical chemistry.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author Contributions
SX contributed to the full experimental part and initial draft. YL contributed to the results discussion of Scheme 2. WF contributed to the results discussion of Scheme 3. JJ and WZ contributed to the result discussion of Scheme 4. DH contributed to full organization, supervision, writing of final version of the manuscript, and submission.
Funding
This research was supported by the National Natural Science Foundation of China (Grant no. 21371171) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant no. XDB20000000).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the facilities of the State Key Laboratory of Structural Chemistry at Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. They also thank Yingzu Zhu for helpful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.622867/full#supplementary-material.
References
Aakeröy, C. B., Smith, M. M., and Desper, J. (2012). α,α′,α′′-Tris(hydroxyimino)-1,3,5-benzenetriacetonitrile: a three-fold symmetric, versatile and practical supramolecular building block. Cryst. Eng. Comm. 14 (1), 71–74. doi:10.1039/c1ce05919j
Akagawa, K., and Kudo, K. (2012). Construction of an all-carbon quaternary stereocenter by the peptide-catalyzed asymmetric michael addition of nitromethane to β-disubstituted α, β-unsaturated aldehydes. Angew. Chem. Int. Ed. 51 (51), 12786–12789. doi:10.1002/anie.201206916
Aksenov, A. V., Smirnov, A. N., Aksenov, N. A., Bijieva, A. S., Aksenova, I. V., and Rubin, M. (2015). Benzimidazoles and benzoxazoles via the nucleophilic addition of anilines to nitroalkanes. Org. Biomol. Chem. 13 (14), 4289–4295. doi:10.1039/c5ob00131e
Aksenov, N. A., Aksenov, A. V., Ovcharov, S. N., Aksenov, D. A., and Rubin, M. (2020). Electrophilically activated nitroalkanes in reactions with carbon based nucleophiles. Front. Chem. 8, 77. doi:10.3389/fchem.2020.00077
Alam, T., Rakshit, A., Begum, P., Dahiya, A., and Patel, B. K. (2020). Visible-light-induced difunctionalization of styrenes: synthesis of N-hydroxybenzimidoyl cyanides. Org. Lett. 22 (9), 3728–3733. doi:10.1021/acs.orglett.0c01235
Allen, S. E., Walvoord, R. R., Padilla-Salinas, R., and Kozlowski, M. C. (2013). Aerobic copper-catalyzed organic reactions. Chem. Rev. 113 (8), 6234–6458. doi:10.1021/cr300527g
Basle, O., and Li, C.-J. (2007). Copper catalyzed oxidative alkylation of sp3 C–H bond adjacent to a nitrogen atom using molecular oxygen in water. Green Chem. 9 (10), 1047–1050. doi:10.1039/b707745a
Bilski, P., Reszka, K., and Chignell, C. F. (1994). Oxidation of aci-nitromethane by singlet oxygen in aqueous solution. J. Am. Chem. Soc. 116 (22), 9883–9889. doi:10.1021/ja00101a008
Bohle, D. S., Chua, Z., Perepichka, I., and Rosadiuk, K. (2013). E/Z oxime isomerism in PhC(NOH)CN. Chem. Eur J. 19 (13), 4223–4229. doi:10.1002/chem.201203357
Bohle, D. S., and Perepichka, I. (2009). A new synthetic route to 3-Oxo-4-amino-1,2,3-oxadiazole from the diazeniumdiolation of benzyl cyanide: stable sydnone iminium N-oxides. J. Org. Chem. 74 (4), 1621–1626. doi:10.1021/jo802343k
Brower, K. R. (1988). Thermolysis of nitromethane in pressurized supercritical media. J. Org. Chem. 53, 3776–3779. doi:10.1021/jo00251a021
Chaudhary, P., Gupta, S., Muniyappan, N., Sabiah, S., and Kandasamy, J. (2016). An efficient synthesis of N-nitrosamines under solvent, metal and acid free conditions using tert-butyl nitrite. Green Chem. 18 (8), 2323–2330. doi:10.1039/c5gc02880a
De Nanteuil, F., Loup, J., and Waser, J. (2013). Catalytic Friedel_Crafts reaction of aminocyclopropanes. Org. Lett. 15 (14), 3738–3741. doi:10.1021/ol401616a
Dubery, I. A., Louw, A. E., and van Heerden, F. R. (1999). Synthesis and evaluation of 4-(3-methyl-2-butenoxy) isonitrosoacetophenone, a radiation-induced stress metabolite in Citrus. Phytochemistry 50 (6), 983–989. doi:10.1016/S0031-9422(98)00247-7
El-Faham, A., Elnakdy, Y. A., El Gazzar, S. A. M., Abd El-Rahman, M. M., and Khattab, S. N. (2014). Synthesis, characterization and anti-proliferation activities of novel cyano oximino sulfonate esters. Chem. Pharm. Bull. 62 (4), 373–378. doi:10.1248/cpb.c13-01005
Elwell, C. E., Gagnon, N. L., Neisen, B. D., Dhar, D., Spaeth, A. D., Yee, G. M., et al. (2017). Copper–oxygen complexes revisited: structures, spectroscopy, and reactivity. Chem. Rev. 117 (3), 2059–2107. doi:10.1021/acs.chemrev.6b00636
Fu, M., Li, H., Su, M., Cao, Z., Liu, Y., Liu, Q., et al. (2019). Synthesis of 3-nitroisoxazoles via copper acetate-mediated reaction of benzaldehydes with nitromethane. Adv. Synth. Catal. 361 (14), 3420–3429. doi:10.1002/adsc.201900323
Gao, Y., Liu, J., Li, Z., Guo, T., Xu, S., Zhu, H., et al. (2018). Dichloroimidazolidinedione-activated beckmann rearrangement of ketoximes for accessing amides and lactams. J. Org. Chem. 83 (4), 2040–2049. doi:10.1021/acs.joc.7b02983
Gerasimchuk, N., Goeden, L., Durham, P., Barnes, C., Cannon, J. F., Silchenko, S., et al. (2008). Synthesis and characterization of disubstituted arylcyanoximes and their several metal complexes. Inorg. Chim. Acta. 361 (7), 1983–2001. doi:10.1016/j.ica.2007.10.019
Hennessy, E. T., and Betley, T. A. (2013). Complex N-heterocycle synthesis via iron-catalyzed, direct C-H bond amination. Science 340 (6132), 591–595. doi:10.1126/science.1233701
Itoh, M., Hagiwara, D., and Kamiya, T. (1975). A New tert-butyloxycarbonylating reagent, 2-tert-butyloxycarbonyloxyimino-2-phenylacetonitrile. Tetrahedron Lett. 49, 4393–4394. doi:10.1016/S0040-4039(00)91133-X
Jacobsen, E. (2001). “The nitro-aldol (henry) reaction,” in The nitro Group in organic synthesis. New York: Wiley, 30–69.
Kim, S., and Hong, S. H. (2015). Copper-Catalyzed N-Aryl-β-enaminonitrile synthesis utilizing isocyanides as the nitrogen source. Adv. Synth. Catal. 357 (5), 1004–1012. doi:10.1002/adsc.201400973
Kryshenko, F. I., Knyazeva, E. A., Konstantinova, L. S., and Rakitin, O. A. (2016). Synthesis of 4-substituted 3-chloro-1,2,5-thiadiazoles from monosubstituted glyoximes. Russ. Chem. Bull. Int. Ed. 65 (11), 2678–2681. doi:10.1007/s11172-016-1635-1
Kunai, A., Doi, T., Nagaoka, T., Yagi, H., and Sasaki, K. (1990). Stereoselective synthesis of (E)-2-Hydroxyimino-2-pheylacetonitrile by photolysis of 4-Azido-3-pheynlfurazan 2-oxide. Bull. Chem. Soc. Jpn. 63 (6), 1843–1844. doi:10.1246/bcsj.63.1843
Kwiatkowski, P., Cholewiak, A., and Kasztelan, A. (2014). Efficient and highly enantioselective construction of trifluoromethylated quaternary stereogenic centers via high-pressure mediated organocatalytic conjugate addition of nitromethane to β, β-disubstituted enones. Org. Lett. 16 (22), 5930–5933. doi:10.1021/ol502941d
Lemercier, B. C., and Pierce, J. G. (2014). Synthesis of thiohydroxamic acids and thiohydroximic acid derivatives. J. Org. Chem. 79 (5), 2321–2330. doi:10.1021/jo500080x
Li, Y., Zhu, Y., Xiang, S., Fan, W., Jin, J., and Huang, D. (2020). Temperature controlled condensation of nitriles: efficient and convenient synthesis of β-enaminonitriles, 4-aminopyrimidines and 4-amidinopyrimidines in one system. RSC Adv. 10 (11), 6576–6583. doi:10.1039/c9ra10866a
Li, Z., Bohle, D. S., and Li, C.-J. (2006). Cu-catalyzed cross-dehydrogenative coupling: a versatile strategy for C–C bond formations via the oxidative activation of sp3 C–H bonds. Proc. Natl. Acad. Sci. Unit. States Am. 103 (24), 8928–8933. doi:10.1073/pnas.0601687103
Liu, J., Zhang, C., Zhang, Z., Wen, X., Dou, X., Wei, J., et al. (2019). Nitromethane as a nitrogen donor in Schmidt-type formation of amides and nitriles. Science 367, 6475–6479. doi:10.1126/science.aay9501
Ma, J.-A., Ma, Z.-H., Ma, H.-M., Huang, R.-Q., and Shao, R.-L. (1999). A convenient synthesis of α-hydroxyiminoacetonitriles from aldoximes. Synth. Commun. 29 (22), 3863–3868. doi:10.1080/00397919908085906
Mudithanapelli, C., Dhorma, L. P., and Kim, M.-H. (2019). PIFA-promoted, solvent-controlled selective functionalization of C(sp2)–H or C(sp3)–H: nitration via C–N bond cleavage of CH3NO2, cyanation, or oxygenation in water. Org. Lett. 21 (9), 3098–3102. doi:10.1021/acs.orglett.9b00751
Mudshinge, S. R., Potnis, C. S., Xu, B., and Hammond, G. B. (2020). HCl·DMPU-assisted one-pot and metal-free conversion of aldehydes to nitriles. Green Chem. 22 (13), 4161–4164. doi:10.1039/d0gc00757a
Nagase, Y., Sugiyama, T., Nomiyama, S., Yonekura, K., and Tsuchimoto, T. (2014). Zinc-Catalyzed direct cyanation of indoles and pyrroles: nitromethane as a source of a cyano group. Adv. Synth. Catal. 356 (2–3), 347–352. doi:10.1002/adsc.201301073
Neel, A. J., and Zhao, R. (2018). Mild synthesis of substituted 1,2,5-oxadiazoles using 1,1′-carbonyldiimidazole as a dehydrating agent. Org. Lett. 20 (7), 2024–2027. doi:10.1021/acs.orglett.8b00568
Noble, A., and Anderson, J. C. (2013). Nitro-mannich reaction. Chem. Rev. 113 (5), 2887–2939. doi:10.1021/cr300272t
Ogiwara, Y., Morishita, H., Sasaki, M., Imai, H., and Sakai, N. (2017). Copper-catalyzed cyanation of aryl iodides using nitromethane. Chem. Lett. 46 (12), 1736–1739. doi:10.1246/cl.170798
Punchi Hewage, A. N. D., Yao, H., Nammalwar, B., Gnanasekaran, K. K., Lovell, S., Bunce, R. A., et al. (2019). Small molecule inhibitors of the BfrB–bfd interaction decrease Pseudomonas aeruginosa fitness and potentiate fluoroquinolone activity. J. Am. Chem. Soc. 141 (20), 8171–8184. doi:10.1021/jacs.9b00394
Qiu, S., Guo, C., Wang, M., Sun, Z., Li, H., Qian, X., et al. (2018). Mild dealkylative N-nitrosation of N,N-dialkylaniline derivatives for convenient preparation of photo-triggered and photo-calibrated NO donors. Org. Chem. Front. 5 (22), 3206–3209. doi:10.1039/c8qo00818c
Ragnarsson, U., and Grehn, L. (1998). Novel amine chemistry based on DMAP-catalyzed acylation. Acc. Chem. Res. 31 (8), 494–501. doi:10.1021/ar980001k
Robertson, D., Cannon, J. F., and Gerasimchuk, N. (2005). Double-stranded metal-organic networks for one-dimensional mixed valence coordination polymers. Inorg. Chem. 44 (23), 8326–8342. doi:10.1021/ic050465w
Sakai, N., Sasaki, M., and Ogiwara, Y. (2015). Copper(ii)-catalyzed oxidative N-nitrosation of secondary and tertiary amines with nitromethane under an oxygen atmosphere. Chem. Commun. (J. Chem. Soc. Sect. D) 51 (58), 11638–11641. doi:10.1039/c5cc03675e
Scriven, E. F. V. (1983). 4-Dialkylaminopyridines: super acylation and alkylation catalysts. Chem. Soc. Rev. 12 (2), 129–161. doi:10.1039/cs9831200129
Shimokawa, S., Kawagoe, Y., Moriyama, K., and Togo, H. (2016). Direct transformation of ethylarenes into primary aromatic amides with N-bromosuccinimide and I2–aqueous NH3. Org. Lett. 18 (4), 784–787. doi:10.1021/acs.orglett.6b00048
Soliman, S. M., Ghabbour, H. A., Khattab, S. N., Siddiqui, M. R. H., and El-Faham, A. (2017). Synthesis, crystallographic characterization, DFT and TD-DFT studies of Oxyma-sulfonate esters. J. Chem. Sci. 129 (9), 1469–1481. doi:10.1007/s12039-017-1354-7
Solomon, E. I., Heppner, D. E., Johnston, E. M., Ginsbach, J. W., Cirera, J., Qayyum, M., et al. (2014). Copper active sites in biology. Chem. Rev. 114 (7), 3659–3853. doi:10.1021/cr400327t
Song, X., Zhao, J., Zhang, W., and Chen, L. (2018). Novel n-channel organic semiconductor based on pyrene-phenazine fused monoimide and bisimides. Chin. Chem. Lett. 29 (2), 331–335. doi:10.1016/j.cclet.2017.09.015
Thorseth, M. A., Tornow, C. E., Tse, E. C. M., and Gewirth, A. A. (2013). Cu complexes that catalyze the oxygen reduction reaction. Coord. Chem. Rev. 257 (1), 130–139. doi:10.1016/j.ccr.2012.03.033
Ushakov, P. Y., Tabolin, A. A., Ioffe, S. L., and Sukhorukov, A. Y. (2019). In Situ generated magnesium cyanide as an efficient reagent for nucleophilic cyanation of nitrosoalkenes and parent nitronates. Eur. J. Org Chem. 2019 (9), 1888–1892. doi:10.1002/ejoc.201801761
Uysal, Ş. (2010). Synthesis and characterization of a new dioxime: thermal and magnetic behavior of its heterotrinuclear -capped complexes. J. Coord. Chem. 63 (13), 2370–2378. doi:10.1080/00958972.2010.501106
Wang, M., Lu, J., Ma, J., Zhang, Z., and Wang, F. (2015). Cuprous oxide catalyzed oxidative C-C bond cleavage for C-N bond formation: synthesis of cyclic imides from ketones and amines. Angew. Chem. Int. Ed. 54 (47), 14061–14065. doi:10.1002/anie.201508071
Wang, N., Liu, L., Xu, W., Zhang, M., Huang, Z., Shi, D., et al. (2019a). Rhodium(III)-Catalyzed oxidative annulation of ketoximes with sulfonamide: a direct approach to indazoles. Org. Lett. 21 (2), 365–368. doi:10.1021/acs.orglett.8b03488
Wang, Y., Zhou, Y., Lei, M., Hou, J., Jin, Q., Guo, D., et al. (2019b). PPh3/I2/HCOOH: an efficient CO source for the synthesis of phthalimides. Tetrahedron 75 (9), 1180–1185. doi:10.1016/j.tet.2019.01.023
Wu, Q., Luo, Y., Lei, A., and You, J. (2016a). Aerobic copper-promoted radical-type cleavage of coordinated cyanide anion: nitrogen transfer to aldehydes to form nitriles. J. Am. Chem. Soc. 138 (9), 2885–2888. doi:10.1021/jacs.5b10945
Wu, X., Miao, J., Li, Y., Li, G., and Ge, H. (2016b). Copper-promoted site-selective carbonylation of sp3 and sp2 C–H bonds with nitromethane. Chem. Sci. 7 (8), 5260–5264. doi:10.1039/c6sc01087c
Xiang, S., Zhang, X., Chen, H., Li, Y., Fan, W., and Huang, D. (2019). Copper(ii) facilitated decarboxylation for the construction of pyridyl–pyrazole skeletons. Inorg. Chem. Front. 6 (9), 2359–2364. doi:10.1039/c9qi00599d
Xu, K., Zhang, Z., Qian, P., Zha, Z., and Wang, Z. (2015). Electrosynthesis of enaminones directly from methyl ketones and amines with nitromethane as a carbon source. Chem. Commun. (J. Chem. Soc. Sect. D) 51 (55), 11108–11111. doi:10.1039/c5cc02730f
Zhu, R. S., Raghunath, P., and Lin, M. C. (2013). Effect of roaming transition states upon product branching in the thermal decomposition of CH3NO2. J. Phys. Chem. 117 (32), 7308–7313. doi:10.1021/jp401148q
Keywords: copper-dioxygen, nitrogen source, nitromethane transformation, methyl nitrite, phthalimide, 2-hydroxyimino-2-phenylacetonitrile
Citation: Xiang S, Li Y, Fan W, Jin J, Zhang W and Huang D (2021) Copper(II)-Dioxygen Facilitated Activation of Nitromethane: Nitrogen Donors for the Synthesis of Substituted 2-Hydroxyimino-2-phenylacetonitriles and Phthalimides. Front. Chem. 8:622867. doi: 10.3389/fchem.2020.622867
Received: 29 October 2020; Accepted: 18 December 2020;
Published: 29 January 2021.
Edited by:
Ming-Yu Ngai, Stony Brook University, United StatesReviewed by:
Alexey Sukhorukov, N. D. Zelinsky Institute of Organic Chemistry (RAS), RussiaAndrey A. Tabolin, N.D. Zelinsky Institute of Organic Chemistry (RAS), Russia
Copyright © 2021 Xiang, Li, Fan, Jin, Zhang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deguang Huang, ZGh1YW5nQGZqaXJzbS5hYy5jbg==