 Xixi Cui
Xixi Cui Qingtian Meng
Qingtian Meng- Shandong Province Key Laboratory of Medical Physics and Image Processing Technology, School of Physics and Electronics, Shandong Normal University, Jinan, China
Recently, the successful incorporation of artificial base pairs in genetics has made a significant progress in synthetic biology. The present work reports the proton transfer and photoisomerization of unnatural base pair ZP, which is synthesized from the pyrimidine analog 6-amino-5-nitro-3-(1-β-D-2′-deoxyribo-furanosyl)-2 (1H)-pyridone (Z) and paired with its Watson-Crick complement, the purine analog 2-amino-8-(1′-β-D-2′- deoxyribofuranosyl)-imidazo[1,2-a]-1,3,5-triazin-4(8H)-one (P). To explain the mechanism of proton transfer process, we constructed the relaxed potential energy surfaces (PESs) linking the different tautomers in both gas phase and solution. Our results show that the double proton transfer in the gas phase occurs in a concerted way both in S0 and S1 states, while the stepwise mechanism becomes more favorable in solution. The solvent effect can promote the single proton transfer, which undergoes a lower energy barrier in S1 state due to the strengthened hydrogen bond. In contrast to the excited state ultrafast deactivation process of the natural bases, there is no conical intersection between S0 and S1 states along the proton transfer coordinate to activate the decay mechanism in ZP. Of particular relevance to the photophysical properties, charge-transfer character is obviously related to the nitro rotation in S1 state. We characterized the molecular vibration effect on the electronic properties, which reveals the electronic excitation can be tuned by the rotation-induced structural distortion accompanied with the electron localization on nitro group.
Introduction
In the field of biochemistry, the four natural nucleotide letters [guanine (G), cytosine (C), adenine (A), and thymine (T)] can encode virtually all genetic information. Their selective pairings to form two base pairs (i.e., AT and CG) through complementary hydrogen-bond formation underlie the storage and retrieval of all biological information. Expanding the two-base-pair genetic alphabet has been a quest in the design of artificial life since the DNA structures were discovered, followed by the mechanism of genetic material transfer being understood during cell replication (Watson and Crick, 1953a,b). In the past few years, Romesberg et al. reported a class of unnatural base pairs formed between nucleotides containing hydrophobic nucleobases, successfully replicating artificial base pairs in vivo (McMinn et al., 1999; Tae et al., 2001; Malyshev et al., 2014). Moreover, they have optimized different components of semisynthetic organisms by using genetic and chemical approaches, eventually making them grow robustly and be capable of storing the increased information unrestrictedly in practice (Zhang et al., 2017). In 2019, Benner and collaborators doubled the number of life's building blocks, creating a synthetic, eight-letter language (G, C, A, T, and Z, P, B, S) (Hoshika et al., 2019), which forms the base pairs through hydrogen bonding interactions and seems to store and transcribe information (Geyer et al., 2003; Yang et al., 2006; Kim et al., 2014; Zhang et al., 2015; Biondi and Benner, 2018). Previous studies demonstrated that the artificial base pairs can not only mimic the natural base pairs in terms of both structure and stability (Chawla et al., 2016), but also present novel characters in contrast to the natural bases. For example, it has been revealed that DNA strands containing artificial ZP base pairs could better combine with breast cancer cells by exponential enrichment experiment and thus can be transformed into “cancer cell hunters” (Sefah et al., 2014).
Current advances in the synthesis of artificial bases require further insight into the stability and photochemistry properties of the additional genetic code. As is known, the proton and electron transfer play an important role in regulating the properties of the system, and therefore attract widespread attention both in experimental and theoretical studies (Florián and Leszczyński, 1996; Guallar et al., 1999; Sauri et al., 2013; Bull and Thompson, 2018; Zhao et al., 2018a,b; Gonzalez-Garcia et al., 2019; Liu et al., 2019; Cheng et al., 2020; Liu S. S. et al., 2020). Especially, the proton transfer between two pairing bases acts as a key part in many biological and chemical phenomena and processes, like genetic mutation, radiation-induced DNA damage and dynamics of charge transfer in DNA. For example, Jośe Ortega et al. studied the double proton transfer of GC base pair in B-DNA and illustrated the influence of DNA biological environment on the stability of the genetic code (Soler-Polo et al., 2019). In addition, another work reports the DNA damage due to hydrogen-bonded proton transfer in the protonated GC base pairs (Lin et al., 2011). Recently, one steered molecular dynamic simulation presents that the tautomerization of the T*-A* mispair via double proton transfer is an effective pathway of the T-A to C-G transition (Tolosa et al., 2020).
Although great efforts have been made to study the proton transfer reaction of natural base pairs, to the best of our knowledge, the tautomerism of artificial bases are still poorly understood. On the basis of the mentioned above, we presented a detailed theoretical study on the proton transfer process and photoisomerization of the artificial bases ZP, paired by the pyrimidine analog 6-amino-5-nitro-3-(1-β-D-2'-deoxyribofuranosyl)-2(1H)-pyridone (Z) and its Watson-Crick complement, the purine analog 2-amino-8-(1'-β-D-2'-deoxyribofuranosyl)-imidazo[1,2-a]-1,3,5-triazin-4(8H)-one (P) (Yang et al., 2007, 2011; Chen et al., 2011), as shown in Scheme 1. It is found that the double proton transfer in the gas phase is a concerted mechanism both in ground (S0) and the first excited (S1) states, while the process in solution turns to be stepwise along the S0-PES and only single proton transfer is available in the S1 state. The solvent effect is not only conducive to the single proton transfer but also beneficial to the stabilization of related ZP products in both S0 and S1 states. Compared with the case in S0 state, the proton transfer undergoes a low energy barrier reaction pathway in S1 state due to the hydrogen bond enhancement.
Scheme 1. View of ZP with corresponding atom labels involved in three possible hydrogen-bonds.
Previous studies revealed that the existence of conical intersection of natural bases leads to highly efficient radiationless deactivation pathways, which returns the molecules to their ground states before chemical reactions in the excited states can lead to profound damage, and thus endow additional photostability of the natural base pairs (Sobolewski and Domcke, 2004; Sobolewski et al., 2005; Groenhof et al., 2007; Schwalb et al., 2009). However, there is no conical intersection between S0 and S1 states along the proton transfer coordinate to activate this decay mechanism in ZP. Clearly, the artificial base pair ZP has a longer lifetime in the S1 state and possesses weaker photostability than that of natural base pair GC.
Furthermore, our results show that the optimized configuration of ZP in S1 state is a non-planar structure induced by the nitro rotation accompanied with electron localization on nitro group, which indicates the lowest excited singlet state of ZP possesses charge-transfer character. It is well-known that the molecular distortion induced by the vibration is an inherent property of a molecule, which can also be activated by external energy pulse and lead to the changes of the geometric as well as the electronic properties (Huynh and Meyer, 2007; Sánchez-Carrera et al., 2010; Zimmerman et al., 2013; Feng et al., 2016; Zhang et al., 2016). Accordingly, it is worth illustrating the influence of nitro rotation on the absorption spectra and electron distribution of the system. It can be seen that the absorption maximum of ZP presents continuous red-shift associated with the rotation-induced electron localization on nitro group, and thus broadens the absorption spectrum compared with the case of natural base pairs. Our work demonstrates the proton transfer mechanism and the photophysical behaviors of artificial bases to evaluate the stability of artificial bases as the genetic code. The corresponding results can also serve as an impetus for the design of target drug based on unnatural bases.
Computational Details
Here, the structures of ZP base pair in S0 and S1 states were fully optimized by using the DFT and TD-DFT methods at the B3LYP-D3(BJ)/ 6-311++G(d,p) level (Lee et al., 1988; Becke, 1993; Theilacker et al., 2011), which have been successful applied in other similar systems (Mazurkiewicz et al., 2006; Jiao et al., 2009). Corresponding calculations were also confirmed by utilizing the wB97-XD (Chai and Head-Gordon, 2008) and M06-2X (Zhao and Truhlar, 2008) functionals and the wave function methods (DLPNO-STEOM-CCSD and CASSCF) (Andersson et al., 1990, 1992; Hald et al., 2000; Schreiber et al., 2008; Triandafillou and Matsika, 2013). In this work, the bulk solvent (water) has been modeled with the integral equation formalism polarizable continuum model (IEFPCM) (Mennucci et al., 1997; Tomasi et al., 2005). All molecular configurations were verified to be local minima with no imaginary frequency and no constrain of bond lengths and angles were adopted except for additional remarks. To illustrate the proton transfer mechanism, the relaxed PESs linking the different tautomers in both gas and solution were constructed by scanning the bond length of N1-H1 and H2-N3 with a step of 0.05 Å. Subsequently, to adequately investigate the intermolecular hydrogen bonding interactions of ZP system, the infrared (IR) spectra, reduced density gradient (RDG), topological properties and electrostatic potential (ESP) were calculated based on optimized structures and visualized by Multiwfn program (Lu and Chen, 2012). Moreover, the variation tendencies of the electron distribution, orbital energies and absorption spectrum with the nitro rotation were also presented by utilizing the single-point calculation based on rotation-distorted configurations. The electronic excitation types were confirmed from the hole-electron distributions drawn by the Multiwfn program (Liu Z. Y. et al., 2020). The Chole-Cele diagram is employed to smooth out the complex isosurfaces of hole and electron distribution. All calculations were carried out by the Gaussian 16 suite of programs (Frisch et al., 2016), except that the DLPNO-STEOM-CCSD method was completed by the ORCA package (Neese, 2012, 2017).
Results and Discussion
Three possible hydrogen-bonded proton-transfer pathways of ZP base pair are considered as shown in Scheme 1. H1 proton transfer from the N1 site of Z base to the N2 site of P base is defined as the SPT1. The H2 proton transfer from the N3 site of P base to the O1 site of Z base is viewed as SPT2. Meanwhile, the H3 proton transfer from the N4 site of Z base to the O2 site of P base is regarded as SPT3. Our calculated results reveal that the SPT1 is the most common route followed by SPT2, however, SPT3 is unavailable in both S0 and S1 states.
Proton Transfer in the Gas Phase
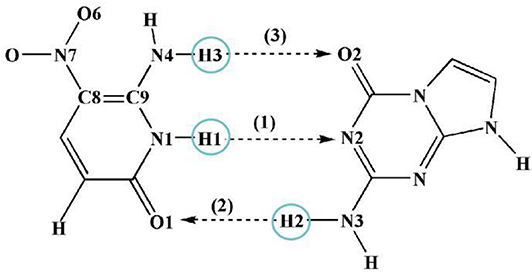
Since the main purpose of the present work is to investigate the proton transfer reaction and photophysical properties of ZP in solution, it is helpful to first know relevant basic information about ZP in the gas phase. To this end, the gaseous mechanism of proton transfer is firstly demonstrated in detail by constructing the PESs of the S0 and S1 states as functions of N1-H1 and H2-N3 bonds shown in Figure 1. The structures of normal ZP (A0, A1), dual-proton transfer ZP-DPT (C0, C1), and single-proton transfer ZP-SPT2 (D1) are obtained (Supplementary Figure 1 and Supplementary Table 1), with a subsequent vibrational frequency analysis to ensure the validity of these structures. As shown in Figure 1, the energies of structure A0 and A1 are the minima in the S0 and S1 states, respectively, which indicates the canonical Watson-Crick configuration is the most stable whether in S0 or S1 state. Subsequently, it is realized that the double-proton-transfer process can take place in both S0 and S1 states. As shown in Table 1, the concerted mechanism is recognized in S0 state with a reaction barrier of 16.8 kcal/mol, and the reverse barrier is 4.6 kcal/mol. However, there are two possible paths in the S1 state: the stepwise reaction undergoes and the concerted path involves the transfer of . Above discussion reveals that the double-proton-transfer in both S0 and S1 states is a concerted process, however, the product ZP-DPT is unstable due to the low reverse reaction barrier. On the other hand, the single-proton-transfer can occur in S1 state and the 9.3 kcal/mol reverse barrier allows the product ZP-SPT2 sufficiently long-lived.
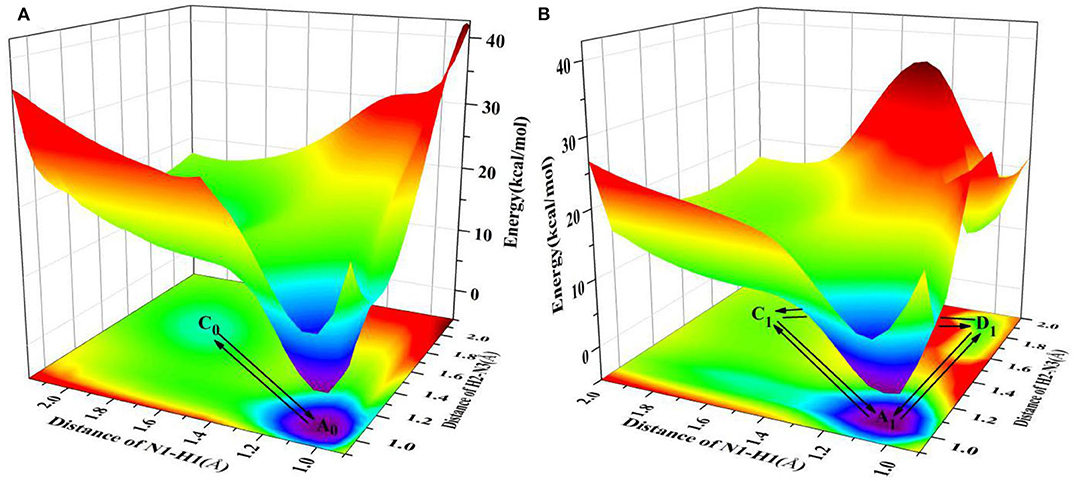
Figure 1. PESs on both S0 and S1 states of ZP as the functions of N1-H1 and H2-N3 length in the gas phase. (A) S0 state PES; (B) S1 state PES.
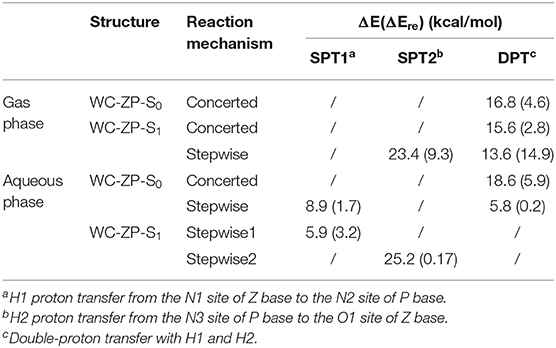
Table 1. The potential barrier of proton transfer process (ΔE) and reverse reaction (ΔEre) in the gas phase and solution.
Proton Transfer in Solution
It is well-known that the biological micro-surrounding or aqueous solution plays an important role in the proton transfer process, which has been investigated both experimentally and theoretically (Adhikary et al., 2009; Kumar and Sevilla, 2009; Ceron-Carrasco et al., 2011). Therefore, it is necessary to consider the solvent effect in the extended calculations to explore the mechanism of proton transfer for the ZP base pairs in biological environment. Here, the SPT3 is unavailable in both S0 and S1 states (Supplementary Figure 3), and thus we constructed the 2-dimension relaxed PESs with respect to the N-H bond length of ZP in S0 and S1 states. As shown in Figure 2, there are six local minimum points in the PESs, namely A0, B0, C0 (S0), and A1, B1, D1 (S1), and the related geometric configurations are displayed in Supplementary Figure 2 and Supplementary Table 2. Similar to the case in gas phase, the configurations A0 and A1 are the most stable in S0 and S1 respectively, revealed by the corresponding energies of reactants and products listed in Supplementary Table 3. As shown in Figure 2A, there are two paths for the double proton transfer in the S0 state: and . Obviously, the stepwise process is more likely to happen than the concerted mechanism in S0 state. It can also be seen that the product ZP-DPT is unstable due to the shallow potential energy well. In addition, there are two single proton transfer pathways in S1 state (Figure 2B), which are and . For the latter one, it takes place through a high energy barrier of 25.2 kcal/mol, and the whole process is endothermic by 25.0 kcal/mol. It is important to note that the barrier for the reverse process is only 0.17 kcal/mol, which indicates the energy well of the product is quite unlikely to support any bound states, so we speculate that the ZP→ZP-SPT2 process will not occur in the S1 state. On the other hand, the energy of the product ZP-SPT1 is just 2.7 kcal/mol higher than that of the reactant (Table 1 and Supplementary Table 3), and the obvious potential well in Figure 2A shows that the ZP-SPT1 is relatively stable in the S1 state. Parenthetically, the H1 atom of the N-H···N fragment is firstly transferred in both S0 and S1 states. Also, the reverse barrier of the ZP→ZP-SPT1 process is a non-negligible obstacle, so once the ZP-SPT1 configuration is formed, it will probably survive for a relatively long period. It should be noticed that the potential barrier in S1 state is 3.0 kcal/mol lower than that of S0 state, which indicates an easier transfer of proton H1 in S1 state. Above discussions are reproduced with considering the explicit solvent molecules (5 H2O molecules) by using the DFT and TD-DFT methods at the B3LYP-D3(BJ)/ 6-311++G(d,p) level as shown in Supplementary Figure 4 and Supplementary Table 4.
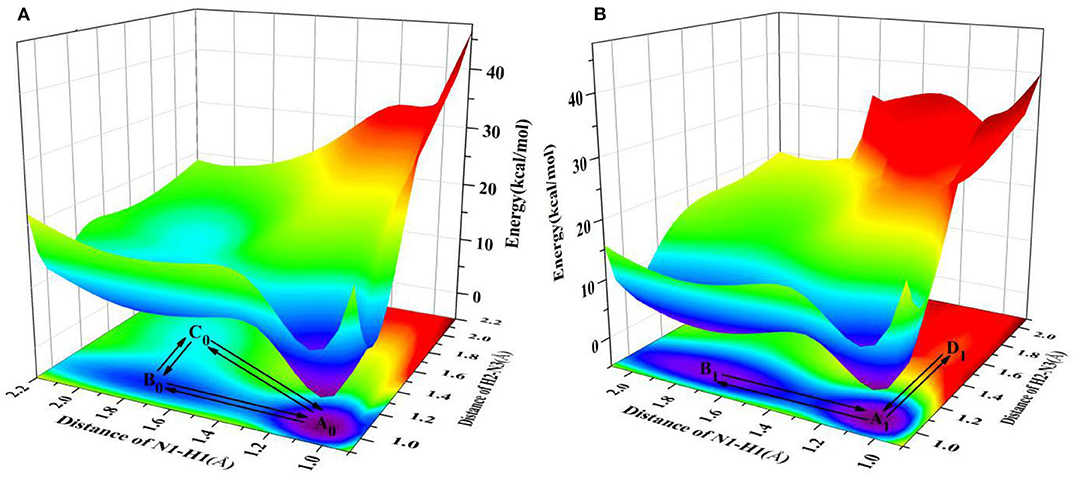
Figure 2. PESs on both S0 and S1 states of ZP as the functions of N1-H1 and H2-N3 length in the aqueous phase. (A) S0 state PES; (B) S1 state PES.
To further explain the mechanism of excited-state proton transfer in the solvent, we calculated the hydrogen bond parameters of the ZP system. The corresponding electronic distribution of ZP and its tautomers are shown in Figure 3. In addition, the parameters of bond length and bond angle related to the intermolecular hydrogen bond are listed in Supplementary Table 2. Herein, we just investigated the hydrogen bond N1-H1···N2, because it is the primary reaction path in both S0 and S1 states. Upon photo-excitation, the bond length of N1-H1 is changed from 1.04 Å (S0) to 1.05 Å (S1), while the bond length of the hydrogen bond H1···N2 is changed from 1.86 Å (S0) to 1.78 Å (S1). Meanwhile, the bond angle of N1-H1···N2 is changed from 179.0° (S0) to 179.2° (S1). These phenomena indicate that the intermolecular hydrogen bond N1-H1···N2 is enhanced in the S1 state, which provides the driving force for H1 proton transfer in the excited state.
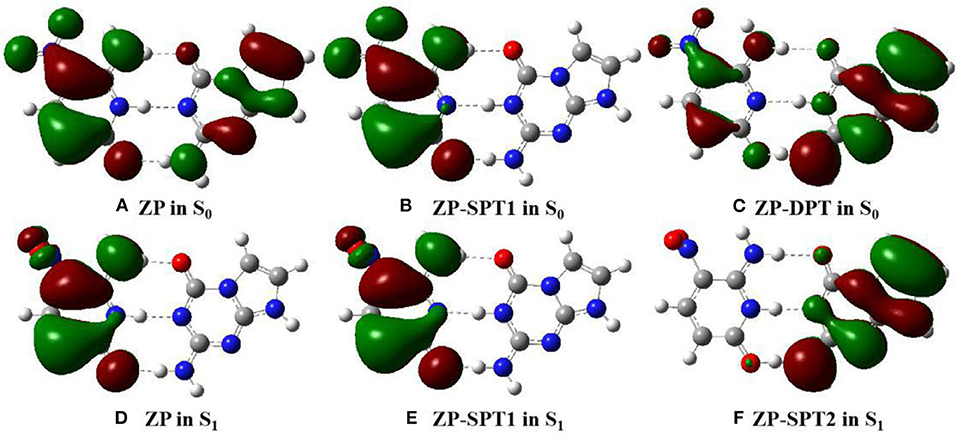
Figure 3. View of HOMO for the normal ZP and tautomers in solution at B3LYP-D3(BJ)/6-311++G(d,p)/IEFPCM level. The green opaque shade and red opaque shade are for positive and negative parts of the wave function (isovalue = 0.02), respectively. (A) ZP in S0. (B) ZP-SPT1 in S0. (C) ZP-DPT in S0. (D) ZP in S1. (E) ZP-SPT1 in S1. (F) ZP-SPT2 in S1.
To further reveal the intermolecular hydrogen bonding interactions of ZP in real space, the RDG versus sign(λ2)ρ is displayed via the Multiwfn and the VMD programs (see Supplementary Figure 5). The relevant formulas can be written as
in which λ2(r) reflects the type of interaction and the electron density ρ(r) is used to give the strength of interaction. Besides, the sign(λ2)ρ is a good indicator to characterize the interaction strength (Johnson et al., 2010). Specifically, if the value of sign(λ2)ρ is negative, the interaction is attractive and is zero meaning that there is a weak interaction, while a positive value of sign(λ2)ρ is an indication of non-bonding repulsive interaction. The spike peak of ZP is located around −0.03 a.u. to −0.04 a.u. in the S0 state, while the S1 state spike peak is between −0.04 a.u. and −0.05 a.u., and the isosurface of the hydrogen bond H1···N2 in S1 state is deeper blue distinctly than that in S0 state. These calculations indicate that the H1···N2 is strengthened upon photo-excitation. Similar trends are considered in the topology structure (Supplementary Figure 6 and Supplementary Table 5) and the IR spectra (Supplementary Figure 7) of ZP. We also calculated the ESP along the N1-H1···N2 proton transfer path (Supplementary Figure 8) to recognize the electrostatic interaction between molecules. The related results show the ESP of the H1 proton in S1 is significantly higher than that in S0, which leads to an easier proton transfer from N1 to N2 in S1 state. However, the ESP of ZP-SPT1 in the S0 state is much higher than that in S1 state, indicating the reverse proton transfer is more likely to take place in S0 state. Similar trends can also be observed in the differences of atom charges in the S0 and S1 states. As shown in Supplementary Table 6, the charge of N1 is changed form −0.268 (S0) to −0.265(S1), while the charge of N2 is changed form −0.424 (S0) to −0.438 (S1), which has contributions to the H1 proton transfer in the excited state.
In summary, the solvent effect can decrease the potential barriers of single proton transfer, and thus promote the reaction in both S0 and S1 states. On the other hand, we presented the different trends of proton transfer between ZP and natural base pair GC, which has similar three intermolecular hydrogen bonds like ZP. Although the most easily transferred site is also the H1 proton, the photophysical properties of ZP are different from the GC. Previous studies have proposed a coupled intermolecular electron-proton transfer mechanism for ultrafast electronic deactivation of the GC base pair (Supplementary Figure 9A calculated at CASSCF/CASSPT2 level), which demonstrates the barrierless minimum energy pathway in S1 state finally leads to a conical intersection with the S0 state at the N1-H1 bond length of about 2.35 Å, and the S1 state population returns to S0 and the initial Watson-Crick structure is restored. However, the PESs of the S0 and S1 states of ZP are almost parallel (Supplementary Figures 9B–11), which supports ZP having a longer lifetime in the S1 state and possessing weaker photostability than that of natural base pair GC.
Rotation Modulation of the Electronic Transition
The optimized geometries and related electron distributions for the normal ZP and tautomers are depicted in Figure 3. It can be seen that the canonical ZP Watson-Crick configuration in the S0 state is a co-planar structure, in which electrons are delocalized on two bases. However, the rotation of the NO2 group on the C5 position of the Z base makes the system non-planar in the S1 state, and the electrons concentrated on the Z-base. Obviously, the nitro rotation dominates the transient changes of the electron distributions. It is well-known that as one of the molecular inherent properties, the structural vibrations are persistent, which has been proved to be an important role in the geometries as well as the electronic properties. Accordingly, it is essential to monitor the change of electronic properties along with the geometrical distortions. Here, we considered the configuration of nitro rotation mode, which is quantified by dihedral angle δ(O6-N7-C8-C9).
As a spectral indicator of electronic properties of ZP, the variation tendency of the absorption maximum with the nitro rotation is recorded (Supplementary Figure 12). The calculated results shows a continuous red-shift with the enlargement of the dihedral angles δ(O6-N7-C8-C9) and thus indicates a broad absorption spectrum (Supplementary Figure 13). To examine the electron excitation characteristics, we plot the HOMO and LUMO of the ZP base pair. As seen from Figure 4, the electron in both the HOMO and LUMO transfers from the aromatic ring to the nitro group with the enlargement of the dihedral angles. Besides, the hole-electron analysis is also utilized to reveal the nature of electron excitation (i.e., the departure and arrival of the excited electrons). As shown in Figure 5, the ZP undergoes the charge transfer excitation at the most stable geometry in S0 with δ = 0°, where the electrons are transferred from P base to Z base. However, the vertical electronic excitation induced charge transfer characteristics become increasingly weak with the enlargement of the dihedral angles. It can be seen that both electrons and holes convergence to the nitro group accompanied with the increase of the rotational displacements, and thus lead to an easier electronic transition from S0 to S1.
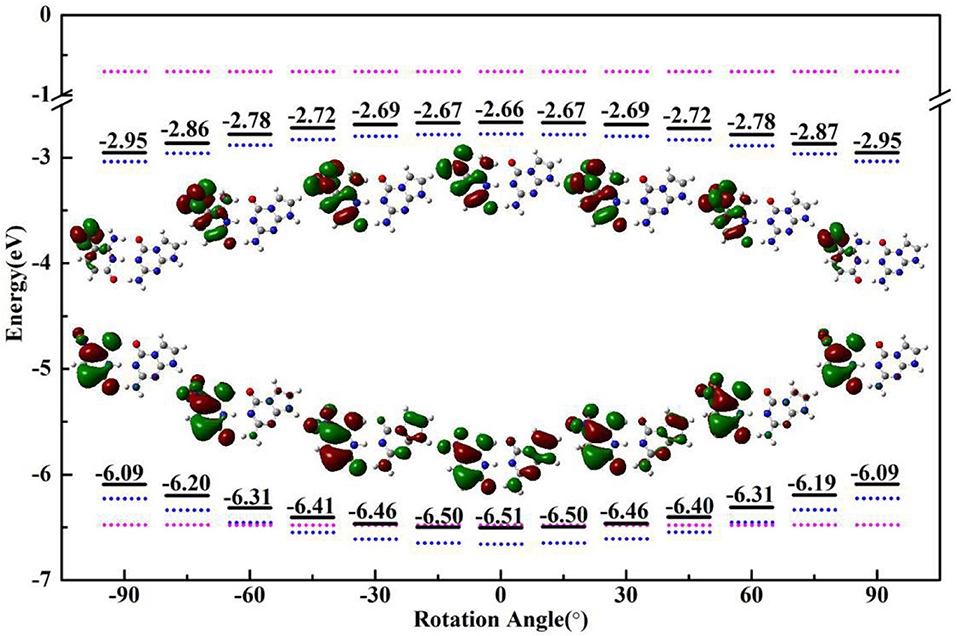
Figure 4. The HOMO/LUMO energy and electron distribution of ZP (black solid line), single Z (blue dotted line) and P (pink dotted line) with the rotational displacements of −90°, −60°, −30°, 0°, 30°, 60°, 90°.
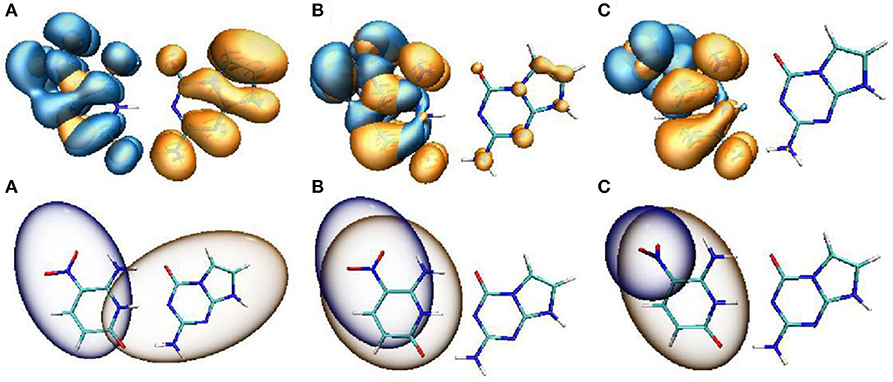
Figure 5. Hole-electron distribution (upper panel) and the Chole-Cele diagrams (lower panel) of ZP with the rotational displacements of 0° (A), 45° (B), 90° (C). Orange and blue regions (isovalue = 0.0002) denote the electron distributions and the holes, respectively.
An analysis of HOMO-LUMO gap is also particularly associated with the absorption characters. In general, a small energy gap is advantageous to the electronic transition from HOMO to LUMO. As shown in Figure 4, the HOMO-LUMO gap calculated at the optimized geometries with different dihedral angles in S0 state gradually decreases with the increase of the nitro rotation, which is consistent with the red-shift in the absorption spectra. Specifically, the HOMO energies increase with the augment of the dihedral angles, while the LUMO energies decrease with the increase of the dihedral angles, which leads to the narrowing of the HOMO-LUMO energy gap (consistent with the energy difference between the S0 and vertical S1 states as shown in Supplementary Figure 14) and thus gives rise to the significant red-shift in the absorption spectra. Furthermore, the variation trends of the orbital energies of the separated Z-base and P-base were presented. The structures of single Z-base and P-base are extracted from the optimized geometries of ZP corresponding to different dihedral angles. It can be seen that the HOMO and LUMO energies of Z-base gradually increase and decrease along the positive and negative displacement directions, respectively. However, the orbital energy of P-base is insensitive to the nitro rotation, which remains relatively a constant. These observations indicate the variation of the orbital energies of Z-base induced by the nitro rotation is responsible for the spectral red-shift of ZP base pair. In fact, it is understandable because the orbital energy is an effective parameter to measure the electron-binding ability of a molecule. When the orbital energy is low, the ability of this molecule in binding electron is relatively large, which indicates an electron is prone to be captured. Obviously, the nitro rotation enhances binding ability of LUMO and recedes electron-binding ability of HOMO, respectively, which is conducive to the vertical electronic excitation. In a word, ZP possesses charge-transfer character due to the rotation-induced electron localization on the nitro group, and the transition of ZP from S0 to S1 is also regulated by the molecular structural fluctuations accompanied with the nitro rotation. The above conclusions have been examined at the wB97-XD and M06-2X functionals (Supplementary Figures 15, 16).
Conclusion
In this paper, we investigated the intermolecular proton-transfer mechanism and photoisomerization of artificial base pair-ZP. Our results demonstrate that the double proton transfer takes place in a concerted way both in the S0 and S1 states in the gas phase. Under the water environment, the stepwise mechanism is more favorable along the S0-PES and only single proton transfer is available in the S1 state. The solvent effect promotes the single proton transfer in both S0 and S1 states by decreasing the activation energy and stabilizing the products. In addition, the proton transfer reaction is more likely to occur in the S1 state because of the excited-state hydrogen bond enhancement. Unlike the excited-state ultrafast deactivation process of the natural bases, there is no conical intersection along the proton transfer coordinate of ZP, which suggests ZP having a longer lifetime in the S1 state and possessing weaker photostability than that of natural base pair GC. Corresponding electron distribution reveals that the lowest excited singlet state of ZP possesses charge transfer characters, where the electron transfers from the aromatic ring to the nitro group induced by the photoexcitation. Moreover, we characterized the molecular vibration effect on the electronic excitation. It can be seen that the absorption maximum shows a continuous red-shift with the increase of the dihedral angles δ(O6-N7-C8-C9) due to the narrowing HOMO-LUMO energy gap with the nitro rotation, which indicates a broadened absorption spectrum in contrast to the case of natural base pairs. It can be concluded that the electronic transition of ZP from S0 to S1 state can be tuned by the rotation-induced structural distortion accompanied with the electron localization on nitro group. This work provides an in-depth understanding on the biological process involved with artificial bases, which may also trigger more promising application prospects on the design of biological drug based on unnatural bases base pairs.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
XC, YZ, and ZL carried out the ab initio calculation. XC analyzed the results and wrote the manuscript. QM and CZ supervised this project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 11674198, 11847224, and 11804195), China Postdoctoral Science Foundation (No. 2018M630796), and Natural Science Foundation of Shandong Province (No. ZR2018BA034).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.605117/full#supplementary-material
References
Adhikary, A., Khanduri, D., and Sevilla, M. D. (2009). Direct observation of the hole protonation state and hole localization site in DNA-oligomers. J. Am. Chem. Soc. 131, 8614–8619. doi: 10.1021/ja9014869
Andersson, K., Malmqvist, P., and Roos, B. O. (1992). Second-order perturbation theory with a complete active space self-consistent field reference function. J. Phys. Chem. 96, 1218–1226. doi: 10.1063/1.462209
Andersson, K., Malmqvist, P. A., Roos, B. O., Sadlej, A. J., and Wolinski, K. (1990). Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 94, 5483–5488. doi: 10.1021/j100377a012
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Biondi, E., and Benner, S. A. (2018). Artificially expanded genetic information systems for new aptamer technologies. Biomedicine 6:53. doi: 10.3390/biomedicines6020053
Bull, G. D., and Thompson, K. C. (2018). Proton transfer and tautomerism in 2-Aminopurine-Thymine and Pyrrolocytosine-Guanine base pairs. Biochemistry-us 57, 4547–4561. doi: 10.1021/acs.biochem.8b00521
Ceron-Carrasco, J. P., Zuniga, J., Requena, A., Perpete, E. A., Michaux, C., and Jacquemin, D. (2011). Combined effect of stacking and solvation on the spontaneous mutation in DNA. Phys. Chem. Chem. Phys. 13, 14584–14589. doi: 10.1039/c1cp20946a
Chai, J. D., and Head-Gordon, J. D. (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. doi: 10.1039/b810189b
Chawla, M., Credendino, R., Chermak, E., Oliva, R., and Cavallo, L. (2016). Theoretical characterization of the H-bonding and stacking potential of two nonstandard nucleobases expanding the genetic alphabet. J. Phys. Chem. B 120, 2216–2224. doi: 10.1021/acs.jpcb.6b00125
Chen, F., Yang, Z. Y., Yan, M. C., Alvarado, B., Wang, G. G., and Benner, S. A. (2011). Recognition of an expanded genetic alphabet by type-II restriction endonucleases and their application to analyze polymerase fidelity. Nucl. Acids Res. 39, 3949–3961. doi: 10.1093/nar/gkq1274
Cheng, R. D., Martens, J., and Fridgen, T. D. (2020). A vibrational spectroscopic and computational study of gaseous protonated and alkali metal cationized G-C base pairs. Phys. Chem. Chem. Phys. 22, 11546–11557. doi: 10.1039/D0CP00069H
Feng, Y. W., Zhang, F. Y., Song, X. Y., and Bu, Y. X. (2016). Molecular vibrations induced potential diradical character in hexazapentacene. J. Phys. Chem. C 120, 10215–10226. doi: 10.1021/acs.jpcc.6b03891
Florián, J., and Leszczyński, J. (1996). Spontaneous DNA mutations induced by proton transfer in the Guanine·Cytosine base pairs: an energetic perspective. J. Am. Chem. Soc. 118, 3010–3017. doi: 10.1021/ja951983g
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16. Version A.03. Inc. Wallingford, CT.
Geyer, C. R., Battersby, T. R., and Benner, S. A. (2003). Nucleobase pairing in expanded Watson-Crick-like genetic information systems. Structure 11, 1485–1498. doi: 10.1016/j.str.2003.11.008
Gonzalez-Garcia, M. C., Herrero-Foncubierta, P., Castro, S., Resa, S., Alvarez-Pez, J. M., Miguel, D., et al. (2019). Coupled excited-state dynamics in N-substituted 2-methoxy-9-acridones. Front. Chem. 7:129. doi: 10.3389/fchem.2019.00129
Groenhof, G., Schafer, L. V., Boggio-Pasqua, M., Goette, M., Grubmuller, H., and Robb, M. A. (2007). Ultrafast deactivation of an excited cytosine-guanine base pair in DNA. J. Am. Chem. Soc. 129, 6812–6819. doi: 10.1021/ja069176c
Guallar, V., Douhal, A., Moreno, M., and Lluch, J. M. (1999). DNA mutations induced by proton and charge transfer in the low-lying excited singlet electronic states of the DNA base pairs: a theoretical insight. J. Phys. Chem. A 103, 6251–6256. doi: 10.1021/jp9908496
Hald, K., Hattig, C., and Jorgensen, P. (2000). Triplet excitation energies in the coupled cluster singles and doubles model using an explicit triplet spin coupled excitation space. J. Chem. Phys. 113, 7765–7772. doi: 10.1063/1.1316033
Hoshika, S., Leal, N. A., Kim, M. J., Kim, M. S., Karalkar, N. B., Kim, H. J., et al. (2019). Hachimoji DNA and RNA: a genetic system with eight building blocks. Science 363, 884–887. doi: 10.1126/science.aat0971
Huynh, M. H. V., and Meyer, T. J. (2007). Proton-coupled electron transfer. Chem. Rev. 107, 5004–5064. doi: 10.1021/cr0500030
Jiao, D. S., Wang, H. Y., Zhang, Y. L., and Tang, Y. (2009). A DFT study of thymine and its tautomers. Can. J. Chemistry 87, 406–415. doi: 10.1139/v08-168
Johnson, E. R., Keinan, S., Mori-Sanchez, P., Contreras-Garcia, J., Cohen, A. J., and Yang, W. T. (2010). Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506. doi: 10.1021/ja100936w
Kim, H. J., Leal, N. A., Hoshika, S., and Benner, S. A. (2014). Ribonucleosides for an artificially expanded genetic information system. J. Org. Chem. 79, 3194–3199. doi: 10.1021/jo402665d
Kumar, A., and Sevilla, M. D. (2009). Influence of hydration on proton transfer in the guanine-cytosine radical cation (G•+-C) base pair: a density functional theory study. J. Phys. Chem. B 113, 11359–11361. doi: 10.1021/jp903403d
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi: 10.1103/PhysRevB.37.785
Lin, Y. X., Wang, H. Y., Gao, S. M., and Schaefer, H. F. (2011). Hydrogen-bonded proton transfer in the protonated guanine-cytosine (GC+H)+ base pair. J. Phys. Chem. B 115, 11746–11756. doi: 10.1021/jp205403f
Liu, H., Cao, J. W., and Bian, W. S. (2019). Double proton transfer in the dimer of formic acid: an efficient quantum mechanical scheme. Front. Chem. 7:676. doi: 10.3389/fchem.2019.00676
Liu, S. S., Lu, J. J., Lu, Q., Fan, J. Z., Lin, L. L., Wang, C. K., et al. (2020). Theoretical study on the sensing mechanism of novel hydrazine sensot TAPHP and its SEIPT and ICT processes. Front. Chem. 7:932. doi: 10.3389/fchem.2019.00932
Liu, Z. Y., Lu, T., and Chen, Q. X. (2020). An sp-hybridized all-carboatomic ring, cyclo[18]carbon: electronic structure, electronic spectrum, and optical nonlinearity. Carbon 165, 461–467. doi: 10.1016/j.carbon.2020.05.023
Lu, T., and Chen, F. W. (2012). Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592. doi: 10.1002/jcc.22885
Malyshev, D. A., Dhami, K., Lavergne, T., Chen, T. J., Dai, N., Foster, J. M., et al. (2014). A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388. doi: 10.1038/nature13314
Mazurkiewicz, K., Bachorz, R. A., Gutowski, M., and Rak, J. (2006). On the unusual stability of valence anions of thymine based on very rare tautomers: a computational study. J. Phys. Chem. B 110, 24696–24707. doi: 10.1021/jp065666f
McMinn, D. L., Ogawa, A. K., Wu, Y. Q., Liu, J. Q., Schultz, P. G., and Romesberg, F. E. (1999). Efforts toward expansion of the genetic alphabet: recognition of a highly stable, self-pairing hydrophobic base. J. Am. Chem. Soc. 121, 11585–11586. doi: 10.1021/ja9925150
Mennucci, B., Cancès, E., and Tomasi, J. (1997). Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 101, 10506–10517. doi: 10.1021/jp971959k
Neese, F. (2012). The ORCA program system, Wiley Interdiscip. Comput. Mol. Sci. 2, 73–78. doi: 10.1002/wcms.81
Neese, F. (2017). Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Comput. Mol. Sci. 8:e1327. doi: 10.1002/wcms.1327
Sánchez-Carrera, R. S., Paramonov, P., Day, G. M., Coropceanu, V., and Brédas, J. L. (2010). Interaction of charge carriers with lattice vibrations in oligoacene crystals from naphthalene to pentacene. J. Am. Chem. Soc. 132, 14437–14446. doi: 10.1021/ja1040732
Sauri, V., Gobbo, J. P., Serrano-Perez, J. J., Lundberg, M., Coto, P. B., Serrano-Andres, L., et al. (2013). Proton/hydrogen transfer mechanisms in the guanine-cytosine base pair: photostability and tautomerism. J. Chem. Theory. Comput. 9, 481–496. doi: 10.1021/ct3006166
Schreiber, M., Silva, M. R., Sauer, S. P. A., and Thiel, W. (2008). Benchmarks for electronically excited states: CASSPT2, CC2, CCSD, and CC3. J. Chem. Phys. 128:134110. doi: 10.1063/1.2889385
Schwalb, N. K., Michalak, T., and Temps, F. (2009). Ultrashort fluorescence lifetimes of hydrogen-bonded base pairs of guanosine and cytidine in solution. J. Phys. Chem. B 113, 16365–16376. doi: 10.1021/jp904883n
Sefah, K., Yang, Z. Y., Bradley, K. M., Hoshika, S., Jimenez, E., Zhang, L. Q., et al. (2014). In vitro selection with artificial expanded genetic information systems. Proc. Natl. Acad. Sci. U. S. A. 111, 1449–1454. doi: 10.1073/pnas.1311778111
Sobolewski, A. L., and Domcke, W. (2004). Ab initio studies on the photophysics of the guanine–cytosine base pair. Phys. Chem. Chem. Phys. 6, 2763–2771. doi: 10.1039/B314419D
Sobolewski, A. L., Domcke, W., and Hattig, C. (2005). Tautomeric selectivity of the excited-state lifetime of guanine/cytosine base pairs: the role of electron-driven proton-transfer processes. Proc. Natl. Acad. Sci. U. S. A. 102, 17903–17906. doi: 10.1073/pnas.0504087102
Soler-Polo, D., Mendieta-Moreno, J. I., Trabada, D. G., Mendieta, J., and Ortega, J. (2019). Proton transfer in guanine-cytosine base pairs in B-DNA. J. Chem. Theory. Comput. 15, 6984–6991. doi: 10.1021/acs.jctc.9b00757
Tae, E. J. L., Wu, Y. Q., Xia, G., Schultz, P. G., and Romesberg, F. E. (2001). Efforts toward expansion of the genetic alphabet: replication of DNA with three base pairs. J. Am. Chem. Soc. 123, 7439–7440. doi: 10.1021/ja010731e
Theilacker, K., Arbuznikov, A. V., Bahmann, H., and Kaupp, M. (2011). Evaluation of a combination of local hybrid functionals with DFT-D3 corrections for the calculation of thermochemical and kinetic data. J. Phys. Chem. A 115, 8990–8996.doi: 10.1021/jp202770c
Tolosa, S., Sansón, J. A., and Hidalgo, A. (2020). Mechanisms of the T-A to C-G transition studied by SMD simulations: deamination vs. tautomerisation. J. Mol. Liq. 308:113036. doi: 10.1016/j.molliq.2020.113036
Tomasi, J., Mennucci, B., and Cammi, R. (2005). Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3093. doi: 10.1021/cr9904009
Triandafillou, C. G., and Matsika, S. (2013). Excited-state tautomerization of gas-phase cytosine. J. Phys. Chem. A 117, 12165–12174. doi: 10.1021/jp407758w
Watson, J. D., and Crick, F. H. (1953a). Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature 171, 737–738. doi: 10.1038/171737a0
Watson, J. D., and Crick, F. H. (1953b). General implications of the structure of deoxyribonucleic acid. Nature 171, 964–967. doi: 10.1038/171964b0
Yang, Z. Y., Chen, F., Alvarado, J. B., and Benner, S. A. (2011). Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 133, 15105–15112. doi: 10.1021/ja204910n
Yang, Z. Y., Hutter, D., Sheng, P. P., Sismour, A. M., and Benner, S. A. (2006). Artificially expanded genetic information system: a new base pair with an alternative hydrogen bonding pattern. Nucl. Acids Res. 34, 6095–6101. doi: 10.1093/nar/gkl633
Yang, Z. Y., Sismour, A. M., Sheng, P. P., Puskar, N. L., and Benner, S. A. (2007). Enzymatic incorporation of a third nucleobase pair. Nucl. Acids Res. 35, 4238–4249. doi: 10.1093/nar/gkm395
Zhang, F. Y., Feng, Y. W., Song, X. Y., and Bu, Y. X. (2016). Computational insights into intriguing vibration-induced pulsing diradical character in perfluoropentacene and the perfluorination effect. Phys. Chem. Chem. Phys. 18, 16179–16187. doi: 10.1039/C6CP01706A
Zhang, L. Q., Yang, Z. Y., Sefah, K., Bradley, K. M., Hoshika, S., Kim, M. J., et al. (2015). Evolution of functional six-nucleotide DNA. J. Am. Chem. Soc. 137, 6734–6737. doi: 10.1021/jacs.5b02251
Zhang, Y. K., Lamb, B. M., Feldman, A. W., Zhou, A. X., Lavergne, T., Li, L. J., et al. (2017). A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl. Acad. Sci. U. S. A. 114, 1317–1322. doi: 10.1073/pnas.1616443114
Zhao, J. F., Dong, H., and Zheng, Y. J. (2018a). Theoretical insights into the excited state double proton transfer mechanism of deep red pigment alkannin. J. Phys. Chem. A 122, 1200–1208. doi: 10.1021/acs.jpca.7b10492
Zhao, J. F., Dong, H., and Zheng, Y. J. (2018b). Elaborating the excited state multiple proton transfer mechanism for 9H-pyrido[3,4-b]indole. J. Lumin. 195, 228–233. doi: 10.1016/j.jlumin.2017.11.026
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi: 10.1007/s00214-007-0310-x
Keywords: proton transfer, potential energy surface, photoisomerization, artificial bases, ab initio
Citation: Cui X, Zhao Y, Li Z, Meng Q and Zhang C (2020) Proton Transfer and Nitro Rotation Tuned Photoisomerization of Artificial Base Pair-ZP. Front. Chem. 8:605117. doi: 10.3389/fchem.2020.605117
Received: 11 September 2020; Accepted: 09 November 2020;
Published: 30 November 2020.
Edited by:
Michael Staniforth, University of Warwick, United KingdomReviewed by:
Josene Toldo, Aix-Marseille Université, FranceJiri Pittner, J. Heyrovsky Institute of Physical Chemistry (ASCR), Czechia
Copyright © 2020 Cui, Zhao, Li, Meng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingtian Meng, cXRtZW5nQHNkbnUuZWR1LmNu; Changzhe Zhang, emhlODUyNDU2QDEyNi5jb20=