 Xiaodong Chen
Xiaodong Chen Ya Chen4†
Ya Chen4† Lifeng Cui
Lifeng Cui- 1School of Materials Science and Engineering, Dongguan University of Technology, Dongguan, China
- 2Center for Clean Energy Technology, Faculty of Science, School of Mathematical and Physical Science, University of Technology Sydney, Sydney, NSW, Australia
- 3Department of Applied Chemistry, School of Science, Xi'an Jiaotong University, Xi'an, China
- 4School of Physical Science and Technology, ShanghaiTech University, Shanghai, China
- 5College of Chemistry and Materials Science, Shanghai Normal University, Shanghai, China
The reverse water-gas shift reaction (RWGSR), a crucial stage in the conversion of abundant CO2 into chemicals or hydrocarbon fuels, has attracted extensive attention as a renewable system to synthesize fuels by non-traditional routes. There have been persistent efforts to synthesize catalysts for industrial applications, with attention given to the catalytic activity, CO selectivity, and thermal stability. In this review, we describe the thermodynamics, kinetics, and atomic-level mechanisms of the RWGSR in relation to efficient RWGSR catalysts consisting of supported catalysts and oxide catalysts. In addition, we rationally classify, summarize, and analyze the effects of physicochemical properties, such as the morphologies, compositions, promoting abilities, and presence of strong metal-support interactions (SMSI), on the catalytic performance and CO selectivity in the RWGSR over supported catalysts. Regarding oxide catalysts (i.e., pure oxides, spinel, solid solution, and perovskite-type oxides), we emphasize the relationships among their surface structure, oxygen storage capacity (OSC), and catalytic performance in the RWGSR. Furthermore, the abilities of perovskite-type oxides to enhance the RWGSR with chemical looping cycles (RWGSR-CL) are systematically illustrated. These systematic introductions shed light on development of catalysts with high performance in RWGSR.
Introduction
The increasing emissions of anthropogenic CO2 into our atmosphere through the unrestricted use of fossil fuels to drive industrial processes and human activity, particularly over the past few decades, has resulted in damage to the “carbon neutral” status of the earth and thus caused serious harm to the ecological system and to sustainable human development (Aresta et al., 2014). Therefore, the extensive efforts are needed to develop CO2 utilization technologies to address these issues (Mikkelsen et al., 2010). Benefiting from plentiful low-cost CO2 raw materials as well as the increasingly advanced CO2 capture and separation technologies, CO2 utilization is promising for commercial-scale applications (Aresta et al., 2016; Klankermayer et al., 2016).
The reverse water-gas shift reaction (RWGSR) is an indispensable part of CO2 utilization because it is a non-fossil route for providing feedstock for important chemical processes, such as methanol synthesis (Gao et al., 2016; Huš et al., 2017), Fischer-Tropsch synthesis (Riedel et al., 1999), and Monsanto/Cativa acetic acid synthesis (Maitlis et al., 1996; Jones, 2000). When it is used as an intermediate step in the direct thermochemical transformation of CO2 to hydrocarbons, such as methane (Sahebdelfar and Takht Ravanchi, 2015; Avanesian et al., 2016), ethanol (Sahebdelfar and Takht Ravanchi, 2015), low-carbon olefin (Liu et al., 2008; Zheng et al., 2017), and dimethyl ether (Centi and Perathoner, 2009), the RWGSR renders the process more practical. An important workable application of the RWGSR is associated with scarce H2 reutilization in the Mars Exploration Program, in which it could regenerate H2O more easily for astronauts to utilize (Avanesian et al., 2016). In biomass-based solid oxide fuel cells, the ratio of CO2/CO/H2 in the biomass gas can be considerably dictated by the RWGSR to realize its maximum energy storage efficiency (Chen et al., 2017a). Additionally, the RWGSR can be used to couple CO2 with alkylene oxide or low alkanes to generate valuable chemicals, including ethylene glycol (Arunajatesan et al., 2001), styrene (Burri et al., 2007; Batista et al., 2010), and light olefins (Mukherjee et al., 2016; Kang et al., 2017). In contrast to the direct thermal cracking process, these coupled reactions can effortlessly break the thermodynamic equilibrium constraints and effectively accelerate their utilization (Reddy et al., 2008; Rao et al., 2009).
The chemically inert CO2, with its high C-O bond energy of 806 kJ mol−1, enables the chemical transformation of CO2 to CO via the RWGSR (Wang et al., 2011). According to activation theory, the adsorption of CO2 on the oxygen vacancy sites of certain catalysts initiates the first step of the RWGSR when it involves the cleavage of its own C-O bond under thermal energy-driven conditions (Su et al., 2019). There are two idiographic activation mechanisms proposed for the production of CO from the RWGSR based on experimental observations and theoretical calculations (Goguet et al., 2016). The first pathway is CO2 hydrogenation to CO via the RWGSR, which proceeds via more reactive carboxyl (COOH*) or formate (HCOO*) intermediates, and the other pathway is the decomposition of CO2 to CO* + O*via the direct C-O bond cleavage pathway (Weatherbee and Bartholomew, 1984; Kattel et al., 2016a). Once activated, these adsorbed intermediates will be instantaneously dissociated or desorbed on the constructed active centers of these catalysts to form the CO product (Tang et al., 2009; Roiaz et al., 2016). Based on this objective analysis, it is imperative to develop effective catalysts for CO2 activation in the RWGSR.
In this review, we concentrate on the catalytic performance of the RWGSR with two major categories of heterogeneous catalysts, including supported metal catalysts and oxide catalysts, which is a subject of increasing interest. Utilizing the thermodynamics and kinetics analyses and the atomic-level mechanisms, the principles of RWGSR catalyst design will be comprehensively described. In addition, the physicochemical properties of supported catalysts, such as the morphologies, compositions, promoting abilities, and presence of metal-support interactions, which affect the catalytic activity and CO selectivity of the RWGSR, will be systematically introduced to elucidate the structure-activity relationships. The relationships among the surface structure, oxygen storage capacity (OSC) and catalytic performance of oxide catalysts in the RWGSR are highlighted, especially for the application of perovskite-type oxides to enhance the RWGSR-CL. The present review provides general guidelines for the state-of-the-art architecture of heterogeneous catalysts for the RWGSR and a discussion of their challenges and further prospects.
Thermodynamic Analysis
Since CO is arguably the most important C1-builiding block, the synthetic route of “CO2-to-CO” is considered an economical and valuable strategy (Barnard, 2008; Brennführer et al., 2009; Wu et al., 2011). Based on the thermodynamic standard enthalpy, the transformation of CO2 to CO via the RWGSR is more thermodynamically favorable at elevated temperature because it is reversible and endothermic and because its chemical equilibrium is pressure independent, as shown in Equation (1). However, the RWGSR is always accompanied by undesired CO2 methanation over the catalysts because of its excessive hydrogenation under ambient pressure (Kim et al., 2015; Ishito et al., 2016; Zhou et al., 2017). In addition, methanation is exothermic, favored at lower temperature, and pressure dependent, as shown in Equation (2).
For both the parallel and the cascade reactions over the catalysts, the CO yield is seriously restricted to H2 utilization in additional competitive methanations. From the thermodynamic standpoint, as shown in Figure 1, the equilibrium composition favors the production of CH4 rather than that of CO in the RWGSR at lower temperatures. Therefore, it is challenging to construct heterogeneous catalysts to restrict the production of undesirable CH4 as a lower value-added by-product for applications at lower temperatures.
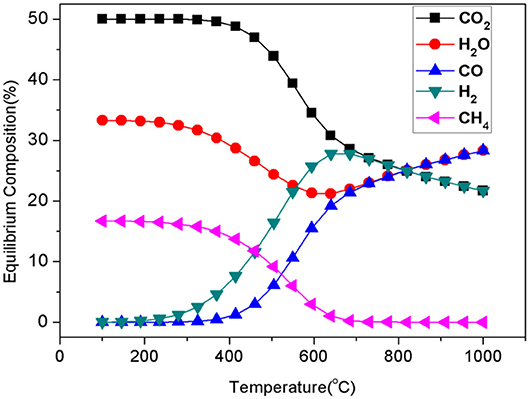
Figure 1. Equilibrium composition of the RWGSR and CO2 methanation (CO2:H2 = 1:1).
Mechanism
The well-known catalytic mechanisms proposed for the RWGSR reaction can be classified into two categories: surface redox mechanisms and associative mechanisms (Su et al., 2017). The major difference between these mechanisms is whether the dissociated H2 species is involved in the formation of the carbon-containing intermediates, namely, formats, and carboxyls (Lin et al., 2017).
Surface Redox Mechanism
The alternative oxido-reduction of active sites on the catalyst surface in the atmosphere of CO2/H2 feedstock is believed to be a prerequisite for the sustainability of the RWGSR. For Cu-based catalysts, e.g., the reaction mechanism can be described as follows (Chen et al., 2000; Xu and Ge, 2016):
In these reactions, Cu0, the active site in the RWGSR, is involved in the rate-controlling step, CO2 reduction. The CO2 oxidizes the Cu0 to generate Cu+ and CO, while the H2 reduces the Cu+ to Cu0 to form H2O; thus, the reaction conforms to a redox mechanism. When the whole catalytic process of the RWGSR is considered in detail, the mechanism of the surface redox reaction can be decomposed into the following basic steps (“*” denotes the vacancy sites) (Gines et al., 1997; Fornero et al., 2017):
The study of kinetics is an important tool for establishing the redox mechanism of the RWGSR. Based on the Monte Carlo method to approximately simulate the RWGSR process over the Cu-based catalysts, CO2 dissociates immediately to give CO and adsorbed oxygen species and is then reduced by H2 with equivalent stoichiometric coefficients (Gines et al., 1997; Xu and Ge, 2016). In this process, the dissociative adsorption of CO2 on the Cu particles is the rate-determining step, and the reduction of the adsorbed oxygen-containing species and surface hydroxyls follows (Fujita et al., 1992; Wang et al., 2013b). Real-time temporal analysis of the products confirms that a surface-reduced Au/CeO2 catalyst can be reoxidized by exposure to CO2 pulses and that the surface oxygen deposited in this way can be reactively removed again, which is a prerequisite for the redox mechanism in the RWGSR. Furthermore, neglecting the changes in the hydroxyls and H2O on the surface imposed by the presence of H2 in the feed, the activity for active oxygen deposition is sufficient to make the redox mechanism the dominant reaction pathway (Fornero et al., 2017). Realistically, the RWGSR proceeds though a redox mechanism over Au/TiO2 catalysts in which the existing surface hydroxyls, surface Ti3+, and oxygen vacancies can jointly participate in the formation of a hydroxycarbonyl intermediate, which quickly decomposes to CO (Bobadilla et al., 2018). According to Density Functional Theory (DFT) calculations, the RWGSR on Cu@Mo2C (001) is preferentially selective for CO via a redox mechanism, and compared to the reaction via a COOH mechanism, the HCOO mechanism is kinetically less favorable due to its higher activation barrier in the rate-determining step, as shown in Figure 2. In the same way as the redox mechanism, the RWGSR occurs first by spontaneous dissociation of H2 to form H*, second by CO* and O* formation from the direct C-O bond cleavage of molecular CO2, third by the reaction of H* and O* to produce OH*, fourth by the reaction of two OH* species to generate H2O*, and finally by the desorption of CO and H2O gas on the Cu@Mo2C (001) catalyst. Notably, the step for OH* formation rather than CO* formation in the redox mechanism, which has a higher activation barrier of 1.4 eV, is the rate-determining step (Jing et al., 2019).
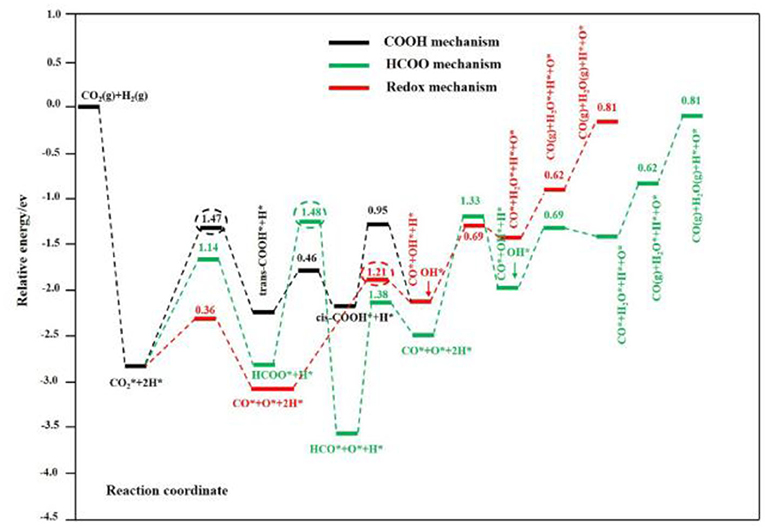
Figure 2. Calculated potential energy profile of the most favorable redox (red line), HCOO (green line), and COOH (black line) mechanisms for the RWGSR on the Cu@Mo2C (001) surface. The numbers in the figure are the activation barriers of the elementary steps. The numbers in red, green, and black circles are the activation barriers of the rate-limiting steps in the redox, HCOO, and COOH pathways, respectively. Reprinted with permission from Jing et al. (2019). Copyright (2019) American Chemical Society.
Associative Mechanism
Formate Species
The RWGSR pathway involves a formate (HCOO*) intermediate that is formed by the initial hydrogenation step and subsequently undergoes an instantaneous dissociation reaction to produce CO (Arunajatesan et al., 2007; Cao et al., 2016; Chen et al., 2016; Wolf et al., 2016). The reaction is described by the following steps (“*” denotes the vacancy sites) (Chen et al., 2017a,b):
As indicated by the temperature-programmed desorption spectra of H2/CO2 co-adsorbed on Cu/SiO2 and Cu/K/SiO2 catalysts, the H atoms either associate with CO2-Cu to form formates or migrate to the surfaces of the interfacial sites, resulting in the formation of K2O and CO by decomposition (Chen and Cheng, 2002; Chen et al., 2003). Regarding the chemical state of Cu in the RWGSR, the Cu0 and Cu+ atoms are proposed to coexist on the Cu-based catalyst surface, and their roles are possibly to dissociate H2 and stabilize the formats, respectively (Chen et al., 2000). Based on transient diffused reflection Fourier transform infrared spectroscopy (DRIFTS), the appearance of interfacial sites may result from an electron transfer from the Pt to the neighboring O in the KOx species over the Pt/K/mutille and Pt/K/L catalysts, which are responsible for the decomposition of the formates to produce CO (Liang et al., 2017; Yang et al., 2017). The in-situ DRIFTS also shows that when strong basic sites, such as those of KOH, are introduced into Ni/Al2O3 catalysts, the formates instead of the carbonates are strongly absorbed on their surface, promoting the hydrogenation of CO2 to CO via the RWGSR (Zhang et al., 2019a). For Cu/CeO2-nanorode catalysts, in-situ DRIFTS points to bidentate formate as the active intermediates for the RWGSR because the preferential formation of a high bidentate formate coverage on their surface may have the surface geometry of a CeO2 (110) termination in which the nearest surface oxygen distance is 2.71Å, which is a more suitable spacing for the formation of bidentate formates and could thus be the main reason for the excellent performance (Lin et al., 2018a,b). The results from in-situ DRIFTS indicate that the CO2 must first react with the surface hydroxyls on Al2O3 to form bicarbonates, which subsequently react with the adsorbed H on Ru or Au to produce adsorbed formates, most likely at the metal/oxide interface, and then react rapidly with the adsorbed H to form CO (Wang et al., 2016a; Bobadilla et al., 2018). Based on a steady-state isotopic transient kinetic analysis, the disappearance trend of the infrared signatures of H12COO* is consistent with the MS signals of 12CO products when the feed gas is switched from 12CO2/H2/Ar to 13CO2/H2/Ar over a Pd/Al2O3 catalyst, suggesting that H12COO* is the reactive intermediate rather than a spectator, and the rate-determining step for the CO formation is related to HCOO* (Wang et al., 2017). DFT calculations demonstrate that a formate mechanism is feasible for the RWGSR catalyzed by M1/W6S8 (M = Fe, Ru, and Os). More concretely, HCOO* formation starts with the most stable adsorption configuration of , in which an H* is placed on the S site, and H* tends to attack the C atom of CO2 to form a C-H bond. When HCOO* is formed, it is hydrogenated to form a HCOOH* intermediate with a relatively low activation energy barrier, and the conversion will proceed to form the final product, CO. In the overall process, the H2 dissociation on M1/W6S8 (M = Fe, Ru, and Os) is the rate-determining step (Zhang et al., 2018).
Carboxyl Species
The carbonyl species, the predominately active intermediate, is selectivity produced through activation of the C-O bond of the CO2 molecule followed by H-assisted formation of COOH* (Tibiletti et al., 2004; Kim et al., 2012a,b). This intermediate immediately decomposes into CO by two different pathways, as shown below (“*” denotes the vacancy sites) (Chen et al., 2017a,b).
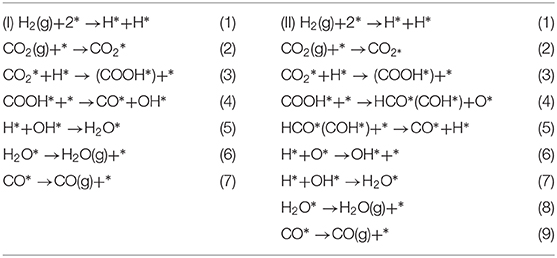
On the basis of in-situ Fourier transform infrared spectroscopy (FT-IR) analyses, a Pt/TiO2 catalyst treated at a high temperature and possessing reducible TiO2 sites but no Pt sites is exclusively active for CO product, and thus the carboxyl species formed on the reducible TiO2 sites are the intermediates in the formation of CO in the RWGSR (Kim et al., 2013). H/D isotopic substitution and kinetics and the results of the in-situ DRIFTS experiments illustrate that the CO formation proceeds via a mechanism in which H assists the dissociation of the C-O bond and that a carboxyl is a more plausible intermediate than is a formate. In addition, the formates is still present on the Cu surface under the reaction conditions, but a fraction of them can be considered spectators of the reaction mechanism (Karelovic et al., 2019). The results from transient DRIFT-MS steady-state isotopic transient kinetic analysis analyses indicate that the characteristic exchange time (defined here as the time at which the DRIFTS signal of the intermediate decreases by 50% following the isotopic switch) of the carboxyl species agrees with that of the CO product (defined here as the time needed to achieve 50% exchange between the two isotopes, e.g., 12CO(g) and 13CO(g) of the main reaction product from MS measurements) when the feed gas is switched from 13CO2/H2 to 12CO2/H2 over the Pt/CeO2 catalyst. These data quantitatively demonstrated that, for the present catalyst and conditions, the main reaction pathway is the formation of CO from the carboxyl species at the oxygen vacancies over the Pt-CeOx interface (Goguet et al., 2004a). DFT calculations indicate that the formation of COOH* over Mo6S8-TM (TM = Pd, Pt, Ag) nanoclusters by the binding of the H* atom to the O atom of followed by its decomposition to CO is very favorable. Note that the COOH* dissociation over Mo6S8-Ag is the rate-determining step in the overall process, whereas the rate-determining step of Mo6S8-Pd and Mo6S8-Pt in the carboxyl pathway is the transition step of the H2 dissociation (Zheng et al., 2017). Moreover, DFT calculations show that the RWGSR complies with a carboxyl mechanism over a Ni5/YSZ (111) catalyst through the identification of the structures and calculation of the energies of the intermediate state and two transition states, as shown in Figure 3. It has been suggested that one of the H* atoms migrates toward the nearest O atom of the to form the COOH* intermediate and subsequently involves its protonation, allowing the formation of H2O* adsorbed on the surface and the CO* adsorbed on the Ni cluster. This calculation also shows that the second transition state for the dissociation of the COOH* intermediate is the rate-determining step of the overall pathway and has an energy barrier of 1.51 eV (Cadi-Essadek et al., 2018).
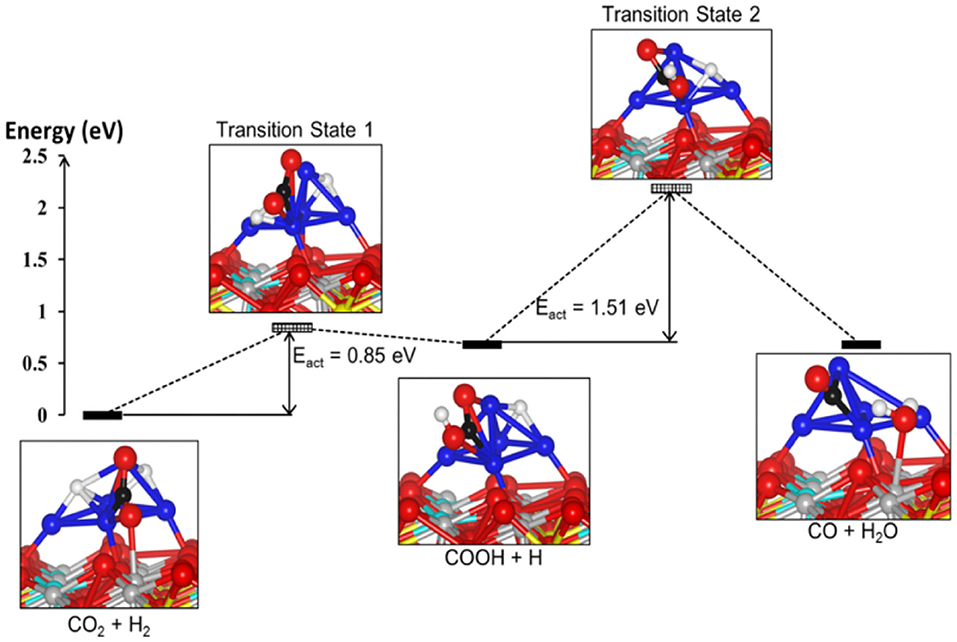
Figure 3. Energy profile showing the reactants, intermediates, transition states, and final products for the RWGSR on the Ni5/YSZ(111) interface (Color key: O, Zr, Y, Ni, C, and H are represented by red, gray, cyan, blue, black, and white spheres, respectively. The yellow spheres represent the oxygen vacancies). Reprinted with permission from Cadi-Essadek et al. (2018). Copyright (2018) American Chemical Society.
Catalytic System
In recent decades, heterogeneous catalysts that promote the RWGSR have been extensively studied because of the gradual realization of their widespread application prospects for CO2 utilization. At the early stage, much research has focused on the oxide catalysts due to their effluent oxygen vacancies sites, such as CeO2, CuO, ZnO, Al2O3, Fe2O3, Cr2O3, In2O3, and MnO2 (Saeidi et al., 2017; Su et al., 2017; He et al., 2019). Although the CO selectivities of these oxide catalysts are desirable in RWGSR, their disadvantages of lower CO2 activation and feasible poisons and sintering are hindering their extended application. In order to address these issues, persistent studies have concentrated on the fabrication of composite oxides (i.e., CuO/ZnO/Al2O3, NiO/CeO2, ZnO/Al2O3, ZnO/Cr2O3, CuOx/CeO2, CuO–CeO2/SBA-15, In2O3-CeO2, FeOx, etc.) (Liu et al., 2015; Dai et al., 2018; Ronda-Lloret et al., 2018; Panarities et al., 2020), spinel oxides (i.e., ZnAl2O4, ZnCr2O4, CuAl2O4, CoAl2O4, etc.) (Joo and Jung, 2003; Bahmanpour et al., 2019, 2020), solid solution oxides (i.e., ZnxZr1−xO2−y, Ce0.5Zr0.5O2, NixCe0.75Zr0.25−xO2, etc.) (Zonetti et al., 2014), and perovskite-type oxides (i.e., BaZr0.8Y0.16Zn0.04O3, La0.75Sr0.25CoO3−δ, La0.75Sr0.25FeO3, La0.75Sr0.25Fe1−YCuYO3, LaNiO3, La0.9Sr0.1NiO3+δ, La0.9Sr0.1FeO3−δ, La0.9Sr0.1Ni0.5Fe0.5O3−δ, La0.75Sr0.25Cr0.5Mn0.5O3−δ, SrCe0.9Y0.1O3−δ, etc.) (Yamazoe et al., 1982; Ten Elshof et al., 1996; Klvana et al., 1999; Yang and Lin, 2006; Radovic et al., 2008; Zhuang et al., 2019; Bogolowski et al., 2020; Liu et al., 2020), which have the approvable characteristics of both stable structure and reverse oxygen storage capacity to increase RWGSR performance.
Recently, considerable efforts have been devoted to the design of metal-based catalysts (i.e., Pt, Pd, Au, Rh, Ru, Cu, Ni, Re, Co, Fe, Mo, etc.) immobilized onto the metal oxide support material (i.e., CeO2, TiO2, Al2O3, ZnO, ZrO2, SiO2, etc.) (Goguet et al., 2004b; Wang et al., 2011; Liu et al., 2015; Álvarez Galván et al., 2016; Ro et al., 2016; Nielsen et al., 2018) because they have metal/oxide interfaces with high reducibility to facilitate CO2 activation in RWGSR. However, the CH4 may be easily produced on these catalysts in the case of excessive hydrogenation of the C-O bond of the CO2 molecule, which can be detrimental to CO selectivity (Yeung et al., 2005; Sun et al., 2015; Wang et al., 2016b; Kattel et al., 2017). In reality, an effective supported metal catalyst must be capable of both the C-O bond scission of CO2 and the appropriate hydrogenation. Thus, the supported metal catalysts must not only dissociate hydrogen relatively easily but also allow it to migrate onto the adjacent oxygen vacancies, where the adsorbed CO2 is further hydrogenated (Chen and Cheng, 2002; Wang et al., 2017). Furthermore, the details of the activity and selectivity of some representative RWGSR catalysts under reaction conditions are presented in Table 1.
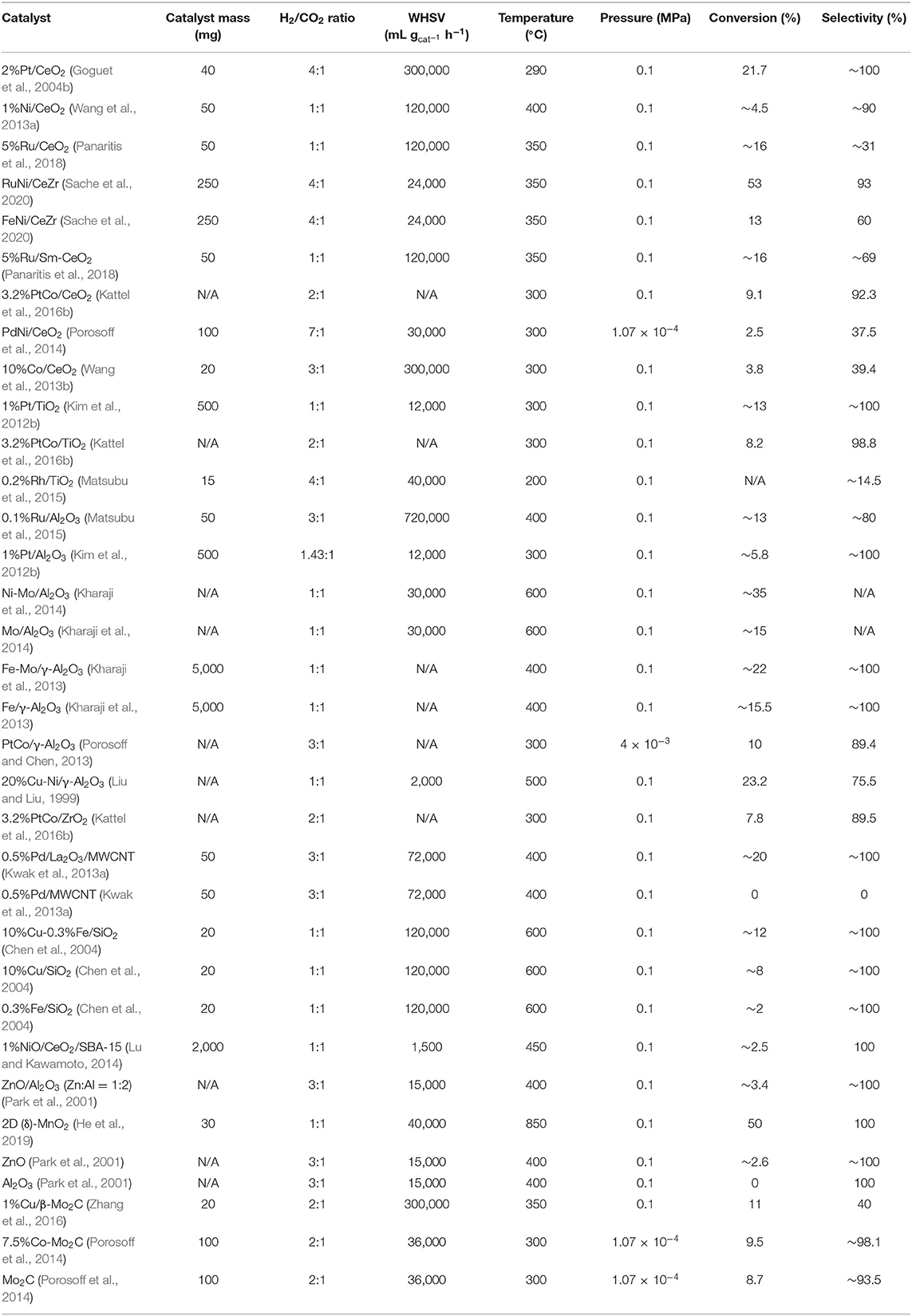
Table 1. Summary of the reaction conditions with conversion to and selectivity for CO, when available, for selected RWGSR catalysts.
Supported Metal Catalysts
The supported catalysts that can be seriously considered in the RWGSR due to their bifunctional catalytic roles for CO2 activation and appropriate hydrogenation (Porosoff et al., 2016). However, RWGSR is demonstrated to be structure sensitive reaction; thus, the CO selectivity of which can be dictated by tailoring the structure functionality of supported catalysts through the SMSI effect, metal size effect, shape and crystal face effect, bimetallic effect, and alkali promoter effect to boost their concentrated activity.
Strong Metal-Support Interaction (SMSI) Effect
The importance of support has been increasingly recognized in the decades following the discovery of SMSIs (Garin, 2001; Diebold, 2003; Neophytides et al., 2005; Fu and Wagner, 2007; Liu et al., 2013). In addition to dispersing metallic particles, the support also functions to influence the catalytic properties of the supported metal catalysts through geometric or electronic effects (Naito et al., 2006; Krstajić et al., 2008; Delgado et al., 2011; Li et al., 2015). In this section, the mechanisms by which SMSI effects provide catalytic characteristics of supported catalysts for the RWGSR are further elucidated.
The high electron donating property of metallic Pt in contact with a Ti3+ ion site is caused by the SMSI effect, which generates new Pt-Ov-Ti3+ sites for CO production over the Pt/TiO2 catalyst (Kim et al., 2012a,b, 2013). Additionally, by replacing the ZrO2 support by TiO2, the SMSI effect selectively weakens the binding of the C-O bond and O-bond intermediates at the PtCo-oxide interface, thus leading to the high selectivity toward CO in the RWGSR (Kattel et al., 2016b). For TiO2-supported Rh catalysts, an adsorbate-mediated SMSI (A-SMSI) encapsulation state can be formed as a result of its treatment in a 20CO2:2H2 environment at 250°C. The high coverage of the adsorbates (HCOx) on the support induces oxygen vacancy formation, driving the migration of the HCOx-functionalized support onto the metal. This A-SMSI encapsulation state is more stable against reoxidation by H2O in the RWGSR process compared with the SMSI encapsulation state formed as a result of only H2 treatment, which modifies the reactivity of all the remaining exposed Rh sites and appears to be comprehensive in covering the Rh but permeable to reactants, due to its amorphous properties. Consequently, formation of the A-SMSI state induces a selectivity switch in the CO2-reduction reaction from the CH4 production on the bare Rh particles to the CO product in the A-SMSI state, thus effectively rendering Rh less active for C-H bond formation (Matsubu et al., 2017). For Ir/CeO2 catalysts, the SMSIs can enable more oxygen atoms to be incorporated into the metal surface, resulting in a weaker CO adsorption strength over the partially oxidized Ir nanoparticles and giving a near 100% selectivity toward CO compared with that over the corresponding metallic Ir. Therefore, modulation of the chemical state of the metal species by the SMSI is more important for the regulation of the observed CO selectivity in the RWGSR (Li et al., 2017). For the Cu/CeO2 catalyst, the Ce3+-Ov-Cu0 and Cu0-CeO2−δ interface structures can be generated by the electron transfer from Cu to Ce on its surface through SMSI effect, which can boost the adsorption and activation performance of reactant CO2 and H2 molecules for RWGSR (Zhou et al., 2020). In the simulation of catalytic CO2 reduction by Pd-decorated silicon-hydride nanosheets (Pd@SiNS), the direct SMSI between the Pd nanoparticle and the Si nanosheet causes H transfer from the Pd to the oxidized SiNS surface, which may occur repeatedly by two mechanisms. First, an H atom adsorbed on the Pd nanoparticle interacts with a surface Si-O-Si and creates a Si-OH; second, another H from the Pd nanoparticle forms a bond with the Si-OH, which leads to desorption of the H2O, creating a surface radical, thereby enabling a catalytic cycle. Furthermore, the strain induced in the SiNS by the Si-O-Si bonds enhances the reactivity of the oxidized SiNS surface toward the transformation of CO2 to CO under mild conditions (Qian et al., 2019). In the context of Mo2C-supported Co catalysts, the SMSI effect facilitates the formation of the amorphous CoMoCyOz phase formed during the CO2 hydrogenation, in which the Co with a positive charge is identified as the critical active site that dissociates CH4 to CO. Therefore, the addition of 7.5% Co to Mo2C leads to an increase in conversion from 8.7 to 9.5%, while the CO:CH4 ratio increases from 15 to 51 (Porosoff et al., 2014). When Cu is added to the β-Mo2C support during the preparation process, the SMSI effect not only promotes the dispersion of supported copper and prevents the aggregation of Cu particles but also enables a portion of the electrons to transfer from Cu to Mo2C so that the Cu+ and Cu0 species coexist in the Cu/β-Mo2C catalyst. Its modulated electronic structure makes the highly dispersed Cu species more active in the CO2 activation and accelerates the CO* desorption in the following transformation reactions, which accounts for its excellent activity in the RWGSR, as depicted in Figure 4 (Zhang et al., 2016).
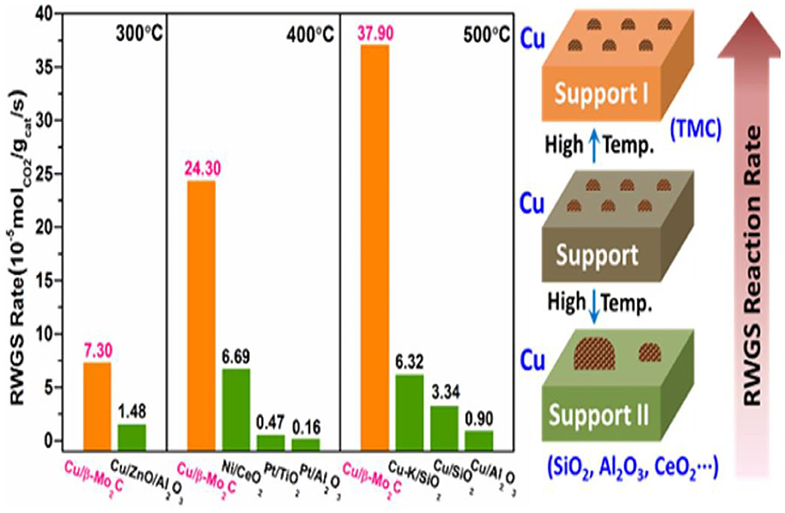
Figure 4. RWGSR rate and SMSI effect of Cu/β-Mo2C and reference catalysts. Reprinted with permission from Zhang et al. (2016). Copyright (2017) American Chemical Society.
Metal Size Effect
The RWGSR is considered structure sensitive for supported catalysts, of which the intermediate dissociation pathway associated with H assistance is substantially dependent on the size of the anchored metal active sties and thus exerts an influence on the CO selectivity of the RWGSR (Chen et al., 2017a).
Multiple studies have been conducted to study the size effect of metal sites relative to CO selectivity in supported noble metal catalysts. The metal active sites (i.e., Pt, Pd, Ru) dispersed at an atomic level contribute more to the CO product compared to metal clusters at a 3D level. This phenomenon is a consequence of the absence of larger metals clusters in which the initially formed COad can be further activated during the continuous reaction (Kwak et al., 2013a,b; Wang et al., 2016a; Chen et al., 2017a). In addition, the Pd sites that slightly retain the CO surface species formed from the formates and other intermediates are more prevalent on the surface of the smaller Pd particles and thus exhibit a higher selectivity toward the CO product. In contrast, the larger Pd particles, due to a higher population of terrace sites in which it is easier to form multi-bound CO and dissociated H2 bound in the vicinity of CO, reveal a stronger interaction with CO. These stable CO species are mainly in multi-bound forms and act as the direct intermediates to CH4 (Wang et al., 2015, 2017). Matsubu et al. have utilized DRIFTS with known site-specific extinction coefficients to quantify the fraction of Rh sites residing as atomically dispersed isolated sites (Rhiso), as well as Rh sites on the surface of Rh nanoparticles (RhNP) for a series of TiO2 supported Rh catalysts. The reaction condition-induced disintegration of RhNP, which form the Rhiso active sites, have been observed to control the CO selectivity of the RWGSR (Matsubu et al., 2015). Furthermore, we have determined that the difference between the desorption energy and dissociation barrier of metal carbonyls is a critical factor for determining the CO selectivity of the RWGSR by combining DFT calculations and experiments, as shown in Figure 5. Specifically, narrowing the size of the Ir active sites by decreasing the Ir loading over Ir/TiO2 catalysts can hinder the carbonyl dissociation but improve the CO desorption, giving rise to CO selectivity (Chen et al., 2017a).
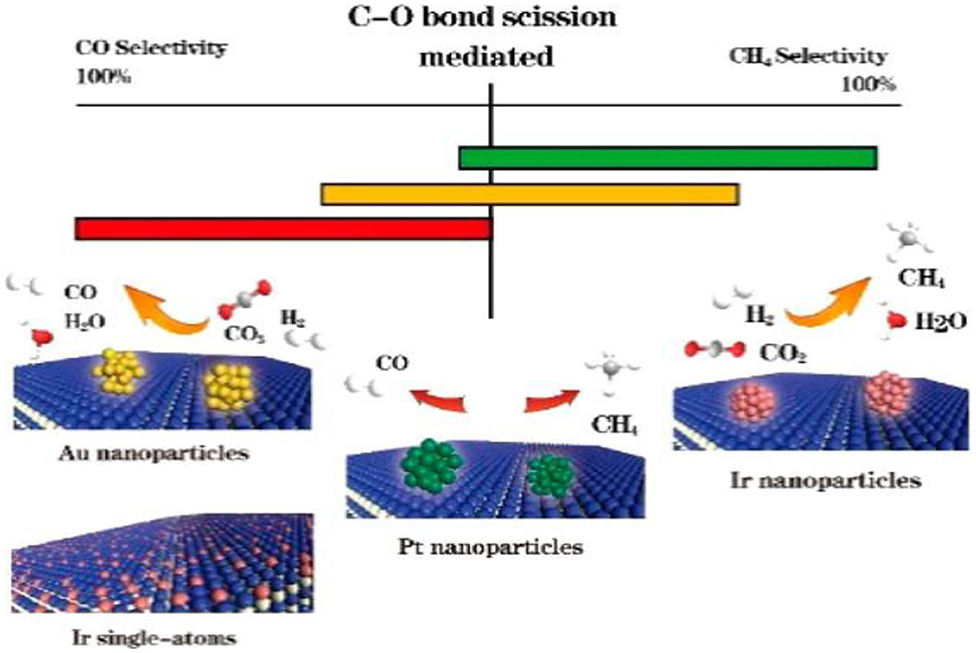
Figure 5. Comparative CO and CH4 selectivities of the Ir1/TiO2, Ir5/TiO2, Pt5/TiO2, and Au5/TiO2 catalysts. Reprinted with permission from Chen et al. (2017a). Copyright (2017) American Chemical Society.
For supported non-noble metal catalysts, significant efforts on Ni-based catalysts have also shown that smaller anchored Ni active sites are beneficial to produce CO in the RWGSR (Wang et al., 2013a,d; Lu and Kawamoto, 2014). The consecutive pathway is favored on small Ni particles, which is attributed to low H2 coverage on the Ni surface, thus leading to the dissociation of the intermediates and high CO selectivity. Whereas the RWGSR on large Ni particles may be controlled by mixed consecutive and parallel pathways, it increases the likelihood that the intermediates will be competitively hydrogenated to CO or CH4 as part of a parallel reaction pathway (Wu et al., 2015). Millet et al. reported on the activation of CO2 on Ni single-atom catalysts that are synthesized using a solid solution approach by controlled substitution of 1–10 atom % of Mg2+ by Ni2+ inside the MgO structure. The Ni atoms are preferentially located on the surface of the MgO and, as predicted by hybrid-functional calculations, favor the low-coordinated sites, where they can reduce the strength of the CO2 binding and promote H2 dissociation. Thus, the Ni atoms are active for CO2 conversion through the RWGSR but are unable to conduct its further hydrogenation to CH4, for which Ni clusters are needed (Millet et al., 2019).
Shape and Crystal Face Effect
RWGSR activities are also significantly depending on the shape and exposed crystal face of catalysts because they can determine the virtual adsorption energy and desorption energy of intermediates in the reaction process (Liu et al., 2019).
Up to now, abundant efforts have been dedicated to study the effect of surface structure of Cu-based catalysts on RWGSR performance. Through the simulation of the adsorption of CO2, H2, H, O, OH, CO, and H2O on the Cu(hkl) surfaces at low coverage, it has been demonstrated that the trend in the calculated activation barriers for the reaction is CO2 dissociative adsorption (namely CO2, g COs + Os) follows the order of Cu(110)<Cu(100)<Cu(111), suggesting that the most efficient crystal surface for catalyzing RWGSR by copper is Cu(110), and the more densely packed Cu(111) surface is the least active among the Cu(hkl) surfaces studied here (Nakamura et al., 1998; Wang et al., 2003; Wang and Nakamura, 2010). When the Cu particles are doped onto the CeO2-Nanaorod and CeO2-Nanosphere surfaces, respectively, which can be marked as Cu/CeO2-NR(111) and Cu/CeO2-NS(110), by comparation, the Cu/CeO2-NR displays the higher RWGSR activity. This is mainly because that the CO2 dissociative activation and the formation of active bidentate carbonate and formate intermediates over CeO2(110) become more feasible (Kovacevic et al., 2016; Lin et al., 2018b). Furthermore, self-assembled CeO2 with 3D hollow nanosphere, nanoparticle, and nanocube morphologies are used to support Cu particles, which can be denoted as Cu/CeO2-hs(111), Cu/CeO2-np(111), and Cu/CeO2-nc(200), respectively. Thereinto, the Cu/CeO2-hs(111) presents the best catalytic RWGSR performance among these as-prepared catalysts due to its high concentration of active oxygen vacancies sites (Zhang et al., 2020). For PtCo/TiO2(110), *HCOO is formed as an intermediate, which may eventually produce CO, whereas for PtCo/CeO2(110), the aside from the route that proceeds via *HCOO, a pathway via a *CH3O intermediate is operating in parallel, which likely leads to the formation of CH4. Moreover, DFT calculation demonstrates that the adsorption of CO2 is stronger at the Nin/YSZ(111) (n = 4–7, 10, and 20) interface than on the clean YSZ(111) between the Ni clusters and the YSZ(111) surface, which facilitates the transformation of CO2 to CO (Cadi-Essadek et al., 2018). For Cu@Mo2C(001) and Cu4@Mo2C(001) surfaces, although the dissociative adsorption of H2 on these two surface is barrier-free and highly exothermic, the activation barrier of carboxyl formation or C-O bond scission as a rate-limiting step on the Cu4@Mo2C(001) surfaces is smaller, and the desorption of CO at the Cu site needs less heat than Mo site, thereby accelerating CO2 conversion in RWGSR (Chen and Cheng, 2002).
Bimetallic Effect
The behavior of a catalyst is modulated by its interaction with other catalyst components, such as a second metal, which influences it through electronic interactions, generates interfacial active sites, or is directly involved in the reaction by bonding to reactants or intermediates (Liu and Liu, 1999; Liu et al., 2015). Therefore, supported bimetallic catalysts have been extensively used for the RWGSR due to their tuning catalytic activity that may be achieved by two metals working synergistically.
The existence of Mo in the structure of the Fe-Mo/Al2O3 catalyst enhances its catalytic performance for the RWGSR due to the electronic effect, which transfers electrons from Fe to Mo and leads to an electron-deficient state of the Fe species, in which it is not helpful for CO adsorption and hence inhibits the continuous hydrogenation of the intermediates (Kharaji et al., 2013; Panaritis et al., 2018). It is reasonable for the Ni species with the electron deficient state to possess high catalytic performance for RWGSR when Ni is added as a second metal component to the Mo/Al2O3 catalyst (Kharaji et al., 2014). As for RuNi/CeZr catalyst, the addition of Ru enhances the Ni reducibility and leads to greater Ni dispersion on the catalyst surface, thus promoting overall activity and CO selectivity for the RWGSR (Sache et al., 2020). Typically, MOF-74 plays a role in helping adsorb and deliver electrons, whereas the low amount of Au@Pd NPs in Au@Pd@MOF-74 results in the poor photon adsorption strength of the Au@Pd active sites. Based on this feature, the core-shell Au@Pd@MOF-74 nanostructure is more propitious to generate CO than MOF-74 in the RWGSR because CO generation is a two-electron reaction, while CH4 generation requires eight electrons; thus, it is more difficult to produce CH4 (Han et al., 2019). Because of the functional characteristics of Au@Pd nanoparticles, the Pt/Au@Pd@UIO-66 catalyst is synthesized to improve its catalytic activity in the RWGSR, as shown in Figure 6. In this system, the core-shell monodispersed Au@Pd nanosphere is encapsulated in the UIO-66 to control its morphology and impart nanoparticle functionality. Additionally, the microporous nature of the UIO-66 assists the adsorption of the Pt nanoparticles, which enhances the interaction between them, favoring the formation of isolated and well-dispersed Pt nanoparticle active sites. This advanced architecture results in excellent catalytic activity and CO selectivity for the RWGSR, and the concept of inserting nanoparticles into microporous MOFs will revolutionize future industrial applications (Zheng et al., 2018). DFT calculations indicate that the catalytic behavior of a Cu12TM (TM = Co, Rh, Ir, Ni, Pd, Ag, Au) bimetallic nanocluster in the RWGSR is dependent on the position of the d-band center. In general, the closer the d-band center is to the Fermi level of these catalysts, the greater is the CO2 adsorption energy, and the smaller is the C-O bond dissociation barrier. Therefore, Cu12Co delivers better catalytic activity for the RWGSR, with a TOF value of 8.96*10−13 s−1, than do the Cu13 and Cu12TM bimetallic systems, due to its d-center value of −0.547 eV, which is higher than that of the other two systems (Zhang and Guo, 2018). Furthermore, for γ-Al2O3- and CeO2-supported Co catalysts, with the addition of Pt as the second component, the values of the d-band center move from the Fermi level toward more negative values, which prevents the excessive hydrogenation of the C-O bond of the CO2 molecule and thereby increases the CO selectivity of the RWGSR (Porosoff and Chen, 2013). In addition, the deposition of Mo onto Au/SiO2 catalyst generates new Au/MoOx interfacial sites since it preferentially occurs on undercoordinated Au sites. The heat of CO adsorption (ΔHads) for the Au/MoOx sites is 33 kJ mol−1, considerably lower than that of the Au0 sites, indicating that these interfacial sites are more selective than the Au0 sites for the RWGSR (Carrasquillo-Flores et al., 2015). Similarly, the Cu/Fe interfacial active sites are generated after introduction of additional Fe into the Cu/SiO2 catalyst, on which the formation of the Fe-Cu bond also prevents Cu from being sintered and oxidized during the RWGSR (Chen et al., 2004). Moreover, the addition of Cu to the Mo/FAU catalyst results in an improvement in the reducibility of MoO3. Therefore, the Mo(0.8)Cu(0.2)/FAU catalyst, which contains co-supported Mo-Cu at an atomic ratio of 4:1, exhibits the higher CO yield of 18.5% and selectivity of 99% compared with the supported Mo catalyst for the RWGSR with the feed gas (H2:CO2 = 1:1) at atmosphere pressure and 773K (Okemoto et al., 2020). The bulk Pt3Ni intermetallic parent compound is formed selectively over the Pt-Ni bimetallic catalysts supported on mesoporous silica, which is related to the thermodynamics of the phase equilibria with a metal silicate that precludes the formation of more Ni-rich intermetallics during the operando conditions of the RWGSR, as shown in Figure 7. This proposed intermetallic structure for these ~1 nm supported clusters, shows a surface/interfacial speciation of the Ni in which only heterometallic Pt-Ni interactions are present in an atomic arrangement within the catalytically active bimetallic sites, which afford exceptionally high activity and CO selectivity in the RWGSR (Liu et al., 2018).
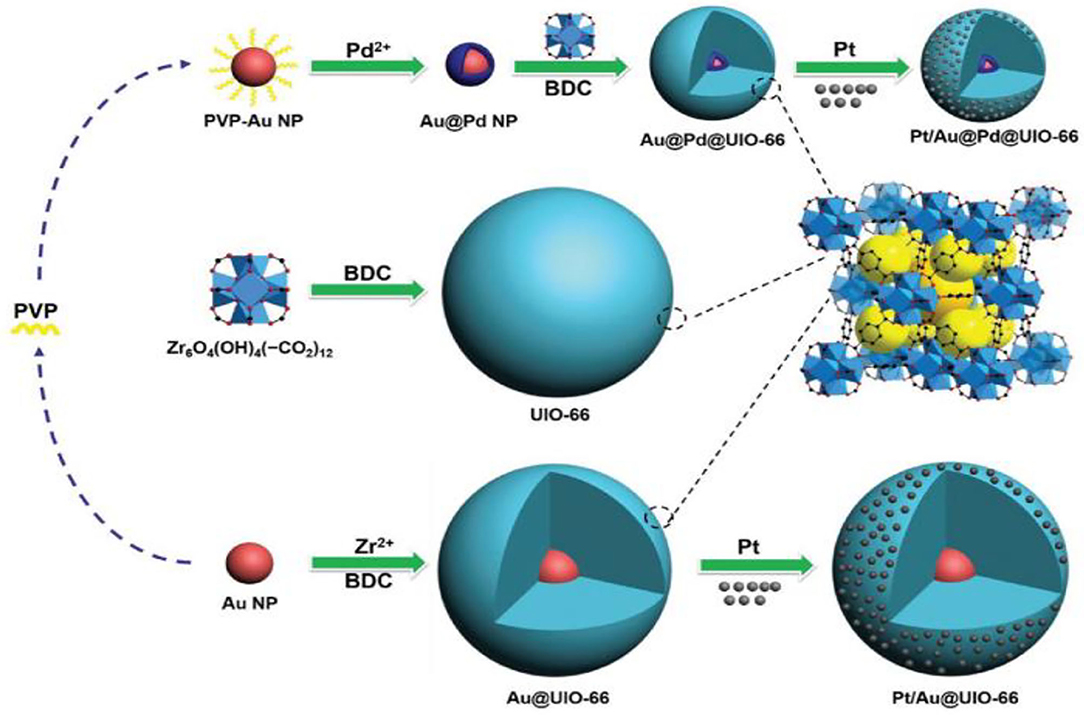
Figure 6. Synthetic route for the production of Au@Pd NPs and other nanocomposites. Reprinted with permission from Zheng et al. (2018). Copyright (2017) Wiley-VCH.
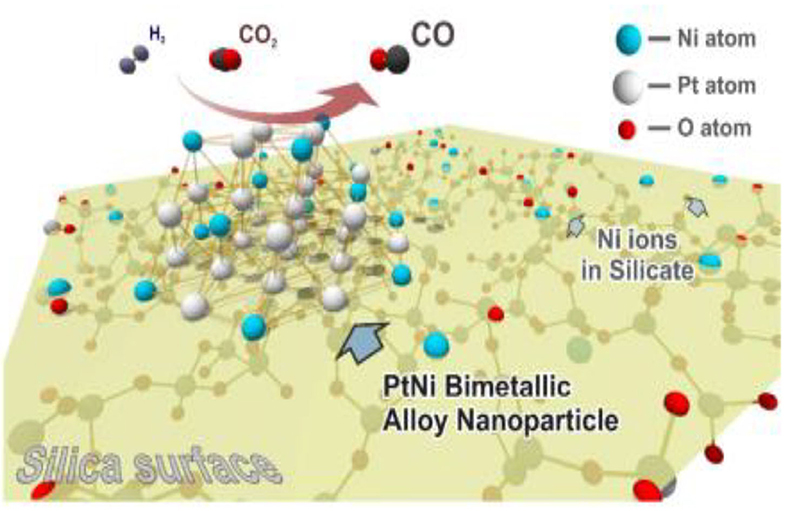
Figure 7. Schematic model of the PtNi/SBA-15 catalyst. Reprinted with permission from Liu et al. (2018). Copyright (2018) American Chemical Society.
Alkali Promoter Effect
For supported heterogeneous catalysts, alkali metal components are typically introduced as promoters to increase the amount of adsorption sites and mediate the adsorption strength of the reactants and intermediates on the “inert support” (Li et al., 1998; Gálvez et al., 2013; Obalová et al., 2013; Connor and Holland, 2017; Pacultová et al., 2017). For instance, the addition of alkali metal promoters is crucial for industrial catalysts substantially applied in the Fisher-Tropsch synthesis (Mirzaei et al., 2006; Okabe et al., 2007; Feyzi et al., 2011; Cosultchi et al., 2012) and ammonia synthesis (Shimoda et al., 2017; Lin et al., 2018a; Jafari et al., 2019; Rogowski, 2019; Zhou et al., 2019). In the field of RWGSRs, abundant studies have shown that K and other promoter additives are essential for some supported catalysts to acquire the expected CO product (Arunajatesan et al., 2007).
The highly K-promoted Fe/Al2O3 and Cu/SiO2 catalysts give much higher CO formation rates than do their counterparts in the RWGSR mainly because the addition of K introduces abundant weak, medium, and strong basic sites, which helps to adsorb/activate CO2 and further converts the CO2 to CO through reaction (Choi et al., 1996; Chen et al., 2003). For the Ni/Al2O3 catalyst, carbonates are the main intermediates, decomposition of which to CO is relatively difficult. However, via modification of this catalyst with a strong base such KOH, the formates rather than the carbonate forms as the main intermediate, which strongly absorbs on the surface of the Ni-KOH/Al2O3 catalyst, contributes to CO formation and hinders further hydrogenation of the CO to CH4 through C-O bond scission (Zhang et al., 2019a). Furthermore, it is demonstrated that the introduction of alkali promoter (K, Mg) by co-impregnation technique enhance the dispersity of Ni active species on the Al2O3, thus increasing the RWGSR performance (Ranjabar et al., 2019). The addition of K promoter leads to an electron transfer from Pt to O in KOx species, resulting in the generation of interfacial active sites over the Pt/mutille catalyst, which is proposed to be more responsible for the production of CO (Liang et al., 2017). Similarly, the K promoter acts as a reducing agent relative to the Fe metal, and the observed increase in the ratio of the Fe2+/Fe3+ ions over the BaFe-hexaaluminates after the K addition reflects the increasing concentration of reduced Fe2+ ions in the hexaaluminate lattice, which is accompanied by the appearance of oxygen vacancies due to the cleavage of one of the neighboring Fe-O-M (M = Al, Ba) bonds in the first coordination sphere of Fe ions. These vacancies play a role in the sites for CO2 adsorption forming monodentate surface carbonates followed by redox transformation evolving CO and leaving the second oxygen bonded to the Fe3+ ion (Wang et al., 2013b,c; Utsis et al., 2018). The results of DFT calculations demonstrate that the K adatom greatly stabilizes the adsorption of all oxygenate intermediates through direct K-O bonding formation on K-modified Cu(111) and Cu(110) surfaces, thus promoting CO2 dissociation in the RWGSR. In general, the different promoting effects of alkali metals on CO2 dissociation are due to their electronegativities, which induce different work function changes and surface dipole moments. Correspondingly, the promoting effects on CO2 dissociation induced by alkali metals increase in the order of Na < K < Rb < Cs, while the electronegativity of various alkali metals decreases in the order of Na > K > Rb > Cs (Wang and Wang, 2019). In accordance with the above effects, the electronegative character facilitates the electronic transfer from Cs to Mo and Fe and leads to an electronically rich surface, which favors the selectivity toward CO over the corresponding catalysts (Pastor-Pérez et al., 2018; Zhang et al., 2019b). For the WC/γ-Al2O3 catalyst, the addition of the K promoter not only has a structural effect to promote the dispersion of the WC species across the high surface area support but also can serve as electronic promotion to strengthen CO and CO2 adsorption while weakening H2 adsorption, which is therefore hypothesized to result in a lower H2/Cox on the catalyst surface, thus inhibiting hydrogenation activity for CH4 and accelerating the generation of CO via the RWGSR (Morse et al., 2020). Specifically, our studies have systematically investigated the effect of K promoter on the activities and selectivities of zeolite L-supported Pt catalysts for the RWGSR, as shown in Figure 8. This study concluded that an additive K promoter not only alters the work function of Pt through their interaction but also forms Pt-O(OH)-K interfacial sites. In addition, the electronic properties of Pt-O(OH)-K sites, with a charge transfer from the Pt surface to the adjacent O in KOx, facilitate the formation of formate intermediates and desorption of the CO. However, with excessive addition of K, the access of the reactants to the Pt surface and interface is tightly blocked. Thus, the activity of the RWGSR is significantly promoted by the controlled addition of K promoter (Yang et al., 2017).
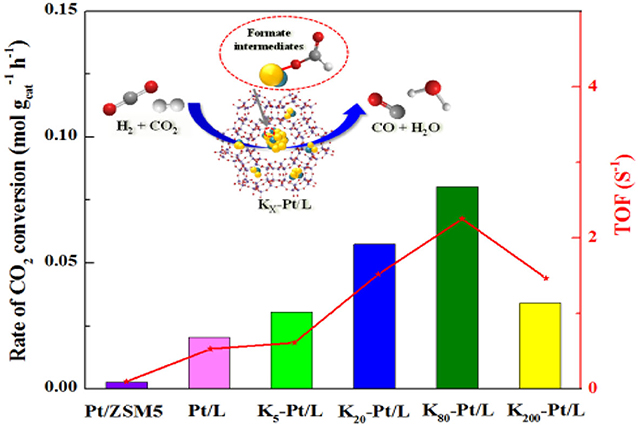
Figure 8. RWGSR over Pt/L catalysts with different K contents. Reprinted with permission from Yang et al. (2017). Copyright (2017) Elsevier.
Oxide Catalysts
Reducible transition oxides are intensively employed in RWGSR catalysts due to their relative abundance and their OSC, which is the ability to reversibly store and release oxygen while formally switching the valence state of the metal ion in its own structure under a CO2/H2 atmosphere (Reddy et al., 2010; Dong et al., 2012; Yao et al., 2013). The O atom can be deprived of H2 in the oxide lattice to generate surface oxygen vacancies, which are much more favorable to the generation of CO rather than CH4, because the oxygen from the C-O bond cleavage of the CO2 molecule can be accommodated, but this leads to unsatisfactory catalytic activity and thermal stability (Katta et al., 2010; Ahn et al., 2012; Graciani et al., 2014). Normally, the additional introduction of heteroatoms into the oxides leads to the formation of spinel, solid solution, and perovskite-type oxides, and their ultra-stable structure is conductive to reversible oxygen donor-acceptor over oxygen vacancies sites, which thus effectively overcome the disadvantages of pure oxides as RWGSR catalysts (Ringuedẽ and Fouletier, 2001; Royer et al., 2005).
ZnO-based oxides are preferentially utilized to catalyze the RWGSR during the CAMARE process due to its ease of formation of metal composite oxides with high stability and specific activity (Li et al., 2002; Schmale et al., 2013). Nonetheless, ZnO-based oxide catalysts are vulnerable to the reduction of ZnO to Zn metal and therefore the loss of ZnO active components when exposed to high thermal reaction conditions, which contributes to their catalytic deactivation (Park et al., 2001). The formation of the spinel structure of the ZnAl2O4 phase by addition of Al2O3 to the ZnO catalyst can cause resistance to its catalytic deactivation in the RWGSR (Joo and Jung, 2003). Similarly, when Fe2O3 is substituted by ZnO over the Fe2O3/Cr2O3 catalyst, the corresponding ZnCr2O4 phase is formed and thus becomes stable (Park et al., 2000). In the synthetic process of the ZnZrOx mixed oxide, the substitution of the Zr in the first layers of the m-ZrO2 lattice with Zn causes the formation of a surface solution (ZnxZr1−xO2−y), which generates oxygen vacancies and improves its stability, reducibility, and oxygen mobility, thus increasing the CO2 conversion in the RWGSR (Silva-Calpa et al., 2016).
The widespread application of CeO2-based oxide in RWGSR catalysts is mainly due to its high OSC, which is inextricably correlated with the catalytic activity (Masui et al., 1997; Wang and Liu, 2018). Both the manipulation of the CeO2 shape with emphasis on tuning its fraction of reactive crystal planes and the doping CeO2 with heterocations to alter its structure and chemical properties are effective strategies to obtain a superior OSC (Sun et al., 2012; Zhou and Li, 2012). Considering their distinct morphologies (particles, rods, and cubes), the higher activity of CeO2 cubes in the RWGSR is due to the superior inherent reactivity of the CeO2 (100) crystal planes enclosing the cubes, contrary to the less inherently reactive CeO2 (111) facets enclosing the rods and particles in the RWGSR (Kovacevic et al., 2016). The CeO2 lattice distortion caused by the incorporation of heterocations such as Zn increases the oxygen vacancy defects and thus accelerates the mobility of the oxygen ions, leading to a higher OSC, thus markedly enhancing its catalytic activity in the RWGSR (Lin et al., 2015; Wenzel et al., 2017). In addition, either Ce0.75Zr0.25O2 or Ce0.75Zr0.5O2 solid solution can be formed by the addition of Zr to the CeO2 lattice, increasing its ability to generate oxygen vacancies and, more importantly, promoting its thermal stability, which is a very promising aspect of catalytic systems employed in reactions in which the RWGSR is one of the steps in the processes that generate hydrocarbons from CO2 (Zonetti et al., 2014; Wenzel et al., 2018).
The adsorption of CO2 on In2O3 has an adsorption energy of −1.25 eV, which is sufficiently exothermic and thus favorable, so the O-C-O angle of the CO2 on adsorbed on the In2O3 is significantly distorted relative to the gas phase structure, significantly increasing the activity of the CO2 in the RWGSR (Ye et al., 2012; Sun et al., 2014). The oxygen vacancies are increasingly created and stabilized on In2O3 with the presence of CeO2 in the In2O3-CeO2 catalyst, on which the dissociated H2 adsorption is enhanced and the amount of bicarbonate species resulting from activated CO2 is increased, which thus exhibits enhanced catalytic activity for the RWGSR (Wang et al., 2016c). Cubic In2O3[denoted as c-In2O3(110)] exhibits a higher RWGSR rate than the hexagonal In2O3[denoted as h-In2O3(110)] at temperature below 350°C due to its enhanced dissociative adsorption of H2, facile formation of the oxygen vacancies, and enhanced ability to adsorb and activate CO2 on the oxygen vacancies (Wang et al., 2020). DFT calculations indicate that the oxygen vacancies sites on the In2O3 (110) surface assist CO2 activation and hydrogenation and stabilize the key intermediates involved in CO formation (Ye et al., 2013). Furthermore, an In2O3−x(OH)y surface containing both Lewis base hydroxide groups and Lewis acid In sites together with oxygen vacancies can heterolytically dissociate H2 to form a hydride bonded to In metal and a proton bonded to a lattice O. This hydrogenated In2O3−x(OH)y surface facilitates CO2 reduction by mediating the charge transfer between the In2O3−x(OH)y surface and adsorbed reactants CO2 and H2 to form CO and H2O (Ghuman et al., 2015). Well-tempered MetaD-biased AIMD simulations have been performed, taking the temperature into account, to probe the mechanism for the RGWS reaction over the In2O3−x(OH)y surface at temperatures of 20 and 180°C, as shown in Figure 9, and the results show that the reduction of gaseous CO2 is the rate-limiting step, with no significant change resulting from increased temperature. However, the energy barrier corresponding to the adsorption of CO2 is slightly reduced at 180°C compared to the that at 20°C, suggesting that the thermal effects may only be relevant to the reaction step characterized by an adsorptive mechanisms and that the increased thermal conditions may enhance the reactivity by enabling the surface frustrated Lewis pairs to become further spatially separated (Ghoussoub et al., 2016).
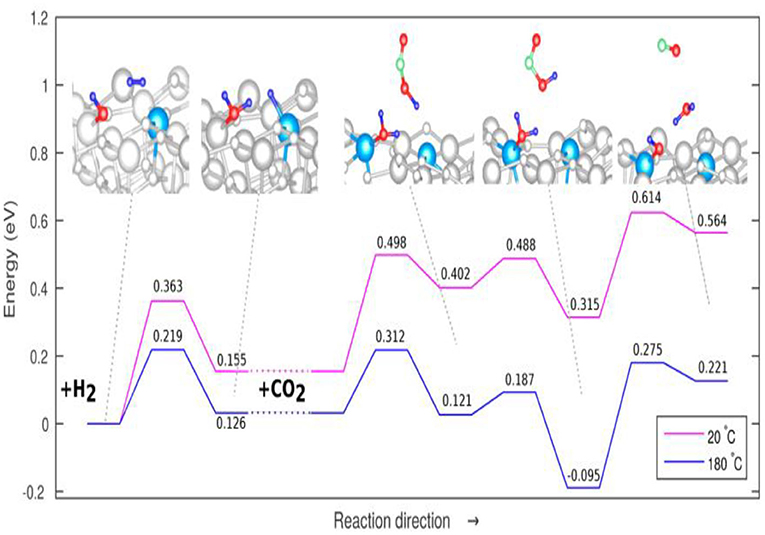
Figure 9. Overall proposed mechanism for the RWGSR on In2O3−x(OH)y at 20°C (pink line) and 180°C (blue line). Reprinted with permission from Ghoussoub et al. (2016). Copyright (2016) American Chemical Society.
The perovskite-type oxides are represented by an ABO3 formula, where the A-site is typically occupied by lanthanides or alkaline earth metals, and the B-site is usually filled with transition metals (Yamazoe et al., 1981; Peña and Fierro, 2001; de Lima et al., 2009). With multiple cation combinations possible on each site, perovskite-type oxides can be easily customized to achieve desirable properties, such as high oxygen mobility and tunability, together with thermal stability at high temperatures without aggregation (Royer et al., 2005; Kae et al., 2014). Therefore, these materials are attractive for application to the RWGSR with chemical looping cycles (RWGSR-CL) that can convert CO2 and H2 to separate streams of CO and H2O, as depicted in Figure 10 (Ringuedẽ and Fouletier, 2001). The combination of La and Sr in the A-site and metal in the B-site enhances the formation of oxygen vacancies due to the generation of a charge imbalance in the ABO3 structure caused by the difference in their oxidation states (Daza et al., 2014). Regarding Co-based perovskite type oxides (La0.75Sr0.25CoO3−δ), under H2 flow conditions, their phases can change to metallic cobalt and base oxides (Co/SrCO3/La2O3), which are then reoxidized to a layered perovskite (CoO/LaSrCoO4−δ) with a K2NiF4-type structure when exposed to CO2, thus producing CO during this cycle. Additionally, the optimal isothermal reduction and conversion temperatures for maximizing the CO product rates of 113.9 μmole CO/g/min are 500°C (of 400, 500, and 600°C) and 850°C (of 650, 750, and 850°C), presumably due to the formation of mixed oxides and metallic cobalt crystalline phases (observed via X-ray diffraction) in close contact under these conditions (Royer et al., 2005). Fe-based perovskite type oxides [La0.75Sr0.25FeO3 (LSF)] have shown the greatest promise in the RWGSR-CL process due to the low energy barrier for oxidation-state transitions (Fe3+-Fe2+) during the redox cycles (Peña and Fierro, 2001). Enhanced oxygen self-diffusion, material recyclability, and therefore the viability of LSF have been demonstrated for chemical looping when supported by redox materials with more abundant alternatives, such as CeO2, ZrO2, Al2O3, SiO2, and TiO2 (Li et al., 2011; Chen et al., 2014). In comparison, supports such as TiO2 and Al2O3 demonstrate SMSIs, which often result in some degree of LSF particle encapsulation, even at low temperatures, thus hindering the CO2 adsorption on the surface oxygen vacancies, whereas SiO2 demonstrates more moderate interactions that are strong enough and suitable for particle segregation yet weak enough to avoid deactivation (Min et al., 2003; Hare et al., 2019). These behaviors occur because the utilization of SiO2 as a support significantly reduces the average LSF crystallite size and the extent of oxygen self-diffusion retardation, and the CO generation yields of LSF/SiO2 surpass those of LSF alone by ~200%, producing 2.6 mmol of at a peak rate of 0.8 mmol min−1 (Hare et al., 2018). In addition, further modification of Fe-based perovskite type oxides with transition metals helps to increase the strength of the interaction of the active species and support and thus stabilizes the unusual cationic oxidation state in the RWGSR process (Nitarori et al., 1988). The incorporation of Cu in La0.75Sr0.25Fe1−YCuYO3 perovskites [Cu100*Y (with Y = 0, 0.10, 0.25, 0.50, 0.75, and 1)] facilitates the formation of oxygen vacancies at lower temperatures. CO production is promoted in the Cu10 sample vs. Cu0 and Cu25, likely due to a combined effect of better CO2 dissociative chemisorption energies on metallic Cu and decreased thermodynamic stability of the oxygen-deficient perovskites (Daza et al., 2016). The enhanced crystalline structure stability is aroused by the incorporation of Co in the La0.75Sr0.25Co(1−Y) FeYO3 perovskite. Additionally, a computational investigation using DFT calculations correlates CO2 adsorption strength, generally a strong barrier in CO2 conversion, on the (100) crystal facets on La0.75Sr0.25FeO(3−δ) to increasing the surface oxygen vacancies (δ). Therefore, δ in the perovskite is the driving force to break the CO-O bond and reoxidize the La0.75Sr0.25FeO3−δ (Daza et al., 2015; Maiti et al., 2016).
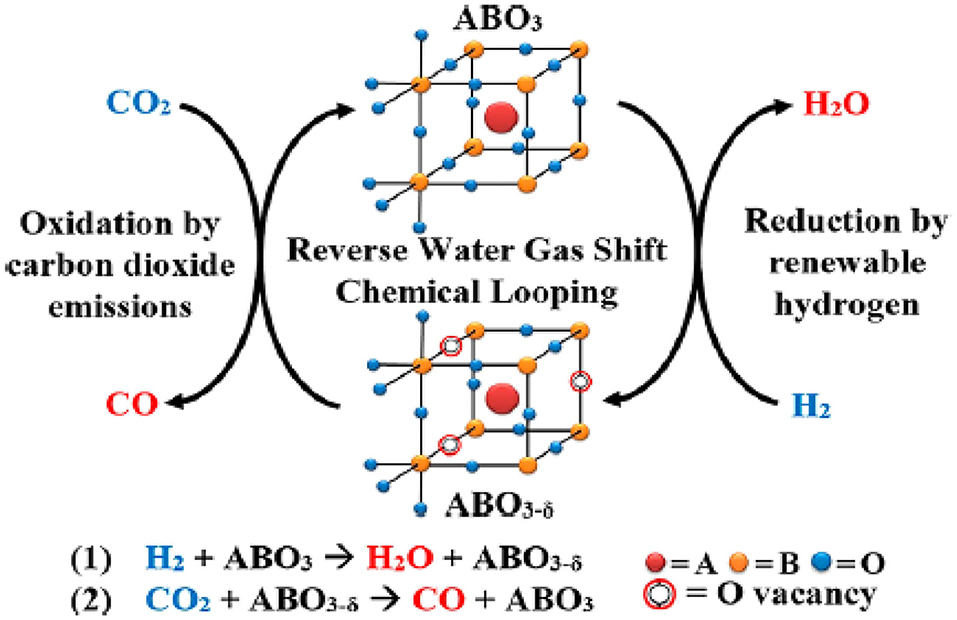
Figure 10. Reverse water gas shift–chemical looping process. Reprinted with permission from Ringuedẽ and Fouletier (2001). Copyright (2018) American Chemical Society.
Conclusion and Outlook
The large-scale conversion of CO2 to CO via the RWGSR is a promising route with great potential for use in the near future, provided a mature technology for commercial production of renewable H2 is also available. The RWGSR also achieves higher CO2 conversion than other relevant technologies that meet the global CO2 emissions standards. Because it is a slightly endothermic and pressure-independent reaction, the current challenge for RWGSR employed in fuel synthesis is the design of thermally stable materials that can achieve high CO selectivity and high production rates. Preferential strategies have recently been enacted to address the existing problems either by modulating the SMSI, size of the active metal, second metal composition, and addition of alkali promoter for supported catalysts or by dipping with additional heteroatoms or tuning their crystal planes for oxide catalysts. Furthermore, the perovskite-type oxides can act as the oxygen donor-acceptor for the RWGSR-CL to not only circumvent thermodynamic and kinetic limitations but also eliminate the possibility of methanation as a side reaction because there is no direct interaction between two feed gases and between two product streams.
From this systematic introduction, the relationships between the nature of the active sites and the main intermediates of RWGSR catalysts are understood through the insights gained from the molecular dynamic simulations and mechanistic work under the operando reaction conditions, which is beneficial to the development of state-of-the-art architecture of RWGSR catalysts. However, even though several materials have been studied, improvements are still possible, especially for commercial development of RWGSR catalysts for laboratory and market applications. If the RWGSR plays a major role in the reduction of the atmospheric CO2 concentration, then designing catalysts with earth-abundant materials will be necessary and desirable. To develop supported metals, both Fe oxides and Ni oxides are chosen to be investigated as representative substitutes for the most commonly used reducible supports, such as CeO2, ZnO, and In2O3, largely due to their oxygen vacancies with high oxygen mobility and stability, which can activate CO2 more easily by accommodating oxygen due to the C-O bond cleavage in the RWGSR. Additionally, transition metal carbides are attractive and convenient alternatives for industrial use in the RWGSR because of their properties, which are similar to those of precious metals, as well as their dual functionality for H2 dissociation and C-O bond scission and their potential to behave similarly to reducible oxides.
Author Contributions
XC, CS, and YC drafted the manuscript, conceived the concept of the review, conducted literature survey, and arranged the figures. NW provided the suggestions. WW and LC revised the manuscript and provided comments. All authors contributed to the article and approved the submitted version.
Funding
This project is financially supported by the Startup Research Fund of Dongguan University of Technology (KCYKYQD2017015), the Postdoctoral Startup Research Fund of Dongguan University of Technology (No.196100040019), and the China Postdoctoral Science Foundation (No. 2019M653608).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, K. Y., Yoo, D. S., Prasad, D. H., Lee, H. W., Chung, Y. C., and Lee, J. H. (2012). Role of multivalent Pr in the formation and migration of oxygen vacancy in Pr-Doped ceria: experimental and first-principles investigations. Chem. Mater. 24, 4261–4267. doi: 10.1021/cm3022424
Álvarez Galván, C., Schumann, J., Behrens, M., Fieero, J. L. G., Schlogl, R., and Frei, E. (2016). Reverse water-gas shift reaction at the Cu/ZnO interface: influence of the Cu/Zn ratio on structure-activity correlations. Appl. Catal. B-Environ. 195, 104–111. doi: 10.1016/j.apcatb.2016.05.007
Aresta, M., Dibenedetto, A., and Angelini, A. (2014). Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels technological use of CO2. Chem. Rev. 114, 1709–1742. doi: 10.1021/cr4002758
Aresta, M., Dibenedetto, A., and Quaranta, E. (2016). State of the art and perspectives in catalytic processes for CO2 conversion into chemicals and fuels: the distinctive contribution of chemical catalysis and biotechnology. J. Catal. 343, 2–45. doi: 10.1016/j.jcat.2016.04.003
Arunajatesan, V., Subramaniam, B., Hutchenson, K. W., and Herkes, F. E. (2001). Fixed-bed hydrogenation of organic compounds in supercritical carbon dioxide. Chem. Eng. Sci. 56, 1363–1369. doi: 10.1016/S0009-2509(00)00359-6
Arunajatesan, V., Subramaniam, B., Hutchenson, K. W., and Herkes, F. E. (2007). In situ FTIR investigations of reverse water gas shift reaction activity at supercritical conditions. Chem. Eng. Sci. 62, 5062–5069. doi: 10.1016/j.ces.2007.01.010
Avanesian, T., Gusmão, G. S., and Christopher, P. (2016). Mechanism of CO2 reduction by H2 on Ru(0001) and general selectivity descriptors for late-transition metal catalysts. J. Catal. 343, 86–96. doi: 10.1016/j.jcat.2016.03.016
Bahmanpour, A. M., Héroguel, F., Kiliç, M., Baranowski, C. J., Artiglia, L., Rothlisberger, U., et al. (2019). Cu-Al spinel as a highly active and stable catalyst for the reverse water gas shift reaction. ACS Catal. 9, 6243–6251. doi: 10.1021/acscatal.9b01822
Bahmanpour, A. M., Héroguel, F., Kiliç, M., Baranowski, C. J., Schouwink, P., and Röthlisberger, U. (2020). Essential role of oxygen vacancies of Cu-Al and Co-Al spinel oxides in their activity for the reverse water gas shift reaction. Appl. Catal. B-Environ. 266:118669. doi: 10.1016/j.apcatb.2020.118669
Barnard, C. F. J. (2008). Palladium-catalyzed carbonylation -a reaction come of age. Organometallics 27, 5402–5422. doi: 10.1021/om800549q
Batista, A. H. M., Ramos, F. S. O., Braga, T. P., Lima, C. L., de Sousa, F. F., de Barros, E. B., et al. (2010). Mesoporous MAl2O4 (M=Cu, Ni, Fe or Mg) spinels: characterisation and application in the catalytic dehydrogenation of ethylbenzene in the presence of CO2. Appl. Catal. A-Gen. 382, 148–157. doi: 10.1016/j.apcata.2010.04.027
Bobadilla, L. F., Santos, J. L., Ivanova, S., Odriozola, J. A., and Urakawa, A. (2018). Unravelling the role of oxygen vacancies in the mechanism of the reverse water-gas shift reaction by operando drifts and ultraviolet-visible spectroscopy. ACS Catal. 8, 7455–7467. doi: 10.1021/acscatal.8b02121
Bogolowski, N., Batalla, B. S., Shin, B., and Drillet, J. F. (2020). Activity of La0.75Sr0.25Cr0.5Mn0.5O3−δ, Ni3Sn2 and Gd-doped CeO2 towards the reverse water-gas shift reaction and carburisation for a high temperature H2O/CO2 co-electrolysis. RSC Adv. 10, 10285–10296. doi: 10.1039/D0RA00362J
Brennführer, A., Neuman, H., and Beller, M. (2009). Palladium-catalyzed carbonylation reactions of aryl halides and related compounds. Angew. Chem. Int. Ed. 48, 4114–4133. doi: 10.1002/anie.200900013
Burri, D. R., Choi, K. M., Han, S. C., Burri, A., and Park, S. E. (2007). Selective conversion of ethylbenzene into styrene over K2O/TiO2-ZrO2 catalysts: unified effects of K2O and CO2. J. Mol. Catal. A-Chem. 269, 58–63. doi: 10.1016/j.molcata.2006.12.021
Cadi-Essadek, A., Roldan, A., Aparicio-Anglès, X., and de Leeuw, N. H. (2018). CO2 and H2 adsorption and reaction at Nin/YSZ(111) interfaces: a density functional theory study. J. Phys. Chem. C 122, 19463–19472. doi: 10.1021/acs.jpcc.8b03488
Cao, Z., Guo, L., Liu, N. Y., Zheng, X. L., Li, W. L., Shi, Y. Y., et al. (2016). Theoretical study on the reaction mechanism of reverse water–gas shift reaction using a Rh–Mo6S8 cluster. RSC Adv. 6, 108270–108279. doi: 10.1039/C6RA23855F
Carrasquillo-Flores, R., Ro, I., Kumbhalkar, M. D., Burt, S., Carrero, C. A., Alba-Rubio, A. C., et al. (2015). Reverse water-gas shift on interfacial sites formed by deposition of oxidized molybdenum moieties onto gold nanoparticles. J. Am. Chem. Soc. 137, 10317–10325. doi: 10.1021/jacs.5b05945
Centi, G., and Perathoner, S. (2009). Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 148, 191–205. doi: 10.1016/j.cattod.2009.07.075
Chen, C. S., and Cheng, W. H. (2002). Study on the mechanism of CO formation in reverse water gas shift reaction over Cu/SiO2 catalyst by pulse reaction, TPD and TPR. Catal. Lett. 83, 121–126. doi: 10.1023/A:1021006718974
Chen, C. S., Cheng, W. H., and Lin, S. S. (2000). Mechanism of CO formation in reverse water-gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 68, 45–48. doi: 10.1023/A:1019071117449
Chen, C. S., Cheng, W. H., and Lin, S. S. (2003). Study of reverse water gas shift reaction by TPD, TPR and CO2 hydrogenation over potassium-promoted Cu/SiO2 catalyst. Appl. Catal. A-Gen. 238, 55–67. doi: 10.1016/S0926-860X(02)00221-1
Chen, C. S., Cheng, W. H., and Lin, S. S. (2004). Study of iron-promoted Cu/SiO2 catalyst on high temperature reverse water gas shift reaction. Appl. Catal. A Gen. 257, 97–106. doi: 10.1016/S0926-860X(03)00637-9
Chen, X. D., Su, X., Duan, H. M., Liang, B. L., Huang, Y. Q., and Zhang, T. (2017a). Catalytic performance of the Pt/TiO2 catalysts in reverse water gas shift reaction: Controlled product selectivity and a mechanism study. Catal. Today 281, 312–318. doi: 10.1016/j.cattod.2016.03.020
Chen, X. D., Su, X., Liang, B. L., Yang, X. L., Ren, X. Y., Duan, H. M., et al. (2016). Identification of relevant active sites and a mechanism study for reverse water gas shift reaction over Pt/CeO2 catalysts. J. Energy Chem. 25, 1051–1057. doi: 10.1016/j.jechem.2016.11.011
Chen, X. D., Su, X. H., Su, Y., Liu, X. Y., Miao, S., Zhao, Y. H., et al. (2017b). Theoretical insights and the corresponding construction of supported metal catalysts for highly selective CO2 to CO conversion. ACS Catal. 7, 4613–4620. doi: 10.1021/acscatal.7b00903
Chen, Y., Galinsky, N., Wang, Z., and Li, F. (2014). Investigation of perovskite supported composite oxide oxides for chemical looping conversion of syngas. Fuel 134, 521–530. doi: 10.1016/j.fuel.2014.06.017
Choi, P. H., Jun, W. K., Lee, J. S., Choi, J. M., and Lee, W. K. (1996). Hydrogenation of carbon dioxide over alumina supported Fe-K catalysts. Catal. Lett. 40 115–118. doi: 10.1007/BF00807467
Connor, C. P., and Holland, P. L. (2017). Coordination chemistry insights into the role of alkali metal promoters in dinitrogen reduction. Catal. Today 286 21–40. doi: 10.1016/j.cattod.2016.08.014
Cosultchi, A., Pérez-Luna, M., Morales-Serna, J. A., and Salmón, M. (2012). Characterization of modified Fischer-Tropsch catalysts promoted with alkaline metals for higher alcohol synthesis. Catal. Lett. 142, 368–377. doi: 10.1007/s10562-012-0765-9
Dai, B., Cao, S. Q., Xie, H. M., Zhou, G. L., and Chen, S. M. (2018). Reduction of CO2 to CO via reverse water-gas shift reaction over CeO2 catalyst. Korean J. Chem. Eng. 35, 421–427. doi: 10.1007/s11814-017-0267-y
Daza, Y. A., Kent, R. A., Yung, M. M., and Kuhn, J. N. (2014). Carbon dioxide conversion by reverse water- gas shift chemical looping on perovskite-type oxides. Int. Eng. Chem. Res. 53, 5828–5837. doi: 10.1021/ie5002185
Daza, Y. A., Maiti, D., Hare, B. J., Bhethanabotla, V. R., Kuhn, J. N., and More, CU, (2016). More problems: decreased CO2 conversion ability by Cu-doped La0.75Sr0.25FeO3 perovskite oxides. Surf. Sci. 648, 92–99. doi: 10.1016/j.susc.2015.11.017
Daza, Y. A., Maiti, D., Kent, R. A., Bhethanabotla, V. R., and Kuhn, J. N. (2015). Isothermal reverse water gas shift chemical looping on La0.75Sr0.25Co1−YFeYO3 perovskite-type oxides. Catal. Today 258, 691–698. doi: 10.1016/j.cattod.2014.12.037
de Lima, R. K. C., Batista, M. S., Wallau, M., Sanches, E. A., Mascarenhas, Y. P., and Urquieta-González, E. A. (2009). High specific surface area LaFeCo perovskite synthesis by nanocasting and catalytic behavior in the reduction of NO with CO. Appl. Catal. B-Environ. 9, 441–450. doi: 10.1016/j.apcatb.2009.04.004
Delgado, J. J., José, M. C., López-Haro, M., del, R. E., Calvino, J. J., Serafín, B, et al. (2011). Recent process in chemical characterization of supported gold catalysts: CO adsorption on Au/ceira-zirconia. Chem. Lett. 40, 1210–1216. doi: 10.1246/cl.2011.1210
Diebold, U. (2003). The surface science of titanium dioxide. Sur. Sci. Rep. 48, 53–229. doi: 10.1016/S0167-5729(02)00100-0
Dong, Q., Yin, S., Guo, C. S., and Sato, T. (2012). Ce0.5Zr0.4Sn0.1O2/Al2O3 catalysts with enhanced oxygen storage capacity and high CO oxidation activity. Catal. Sci. Technol. 2, 2521–2524. doi: 10.1039/c2cy20425h
Feyzi, M., Irandoust, M., and Mirzaei, A. A. (2011). Effects of promoters and calcination conditions on the catalytic performance of iron-manganese catalysts for fisher-tropsch synthesis. Fuel Process. Technol. 92, 1136–1143. doi: 10.1016/j.fuproc.2011.01.010
Fornero, E. L., Chiavassa, D. L., Bonivardi, A. L., and Baltanás, M. A. (2017). Transient analysis of the reverse water gas shift reaction on Cu/ZrO2 and Ga2O3/Cu/ZrO2 catalysts. J. CO2 Util. 22, 289–298. doi: 10.1016/j.jcou.2017.06.002
Fu, Q., and Wagner, T. (2007). Interaction of nanostructured metal overlayers with oxide surfaces. Sur. Sci. Rep. 62, 431–498. doi: 10.1016/j.surfrep.2007.07.001
Fujita, S. L., Usui, M., and Takezawa, N. (1992). Mechanism of the reverse water gas shift reaction over Cu/ZnO catalyst. J. Catal. 134, 220–225. doi: 10.1016/0021-9517(92)90223-5
Gálvez, M. E., Ascaso, S., Moliner, R., and Lázaro, MJ (2013). Influence of the alkali promoter on the activity and stability of transition metal (Cu, Co, Fe) based structured catalysts for the simutaneous removal of soot and NOx. Top Catal. 56, 493–498. doi: 10.1007/s11244-013-0004-7
Gao, S., Lin, Y., Jiao, X. C., Sun, Y. F., Luo, Q. Q., Zhang, W. H., et al. (2016). Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 529, 68–71. doi: 10.1038/nature16455
Garin, F. (2001). Mechanism of NOx decompositon. Appl. Catal. A- Gen. 222, 183–219. doi: 10.1016/S0926-860X(01)00827-4
Ghoussoub, M., Yadav, S., Ghuman, K. K., Ozin, G. A., and Singh, C. V. (2016). Metadynamics-biased ab initio molecular dynamics study of heterogeneous CO2 reduction via surface frustrated lewis pairs. ACS Catal. 6, 7109–7117. doi: 10.1021/acscatal.6b01545
Ghuman, K. K., Wood, T. E., Hoch, L. B., Mims, C. A., Ozin, G. A., and Singh, C. V. (2015). Illuminating CO2 reduction on frustrated lewis pair surfaces: investigating the role of surface hydroxides and oxygen vacancies on nanocrystalline In2O3−x(OH)y. Phys. Chem. Chem. Phys. 17, 14623–14635. doi: 10.1039/C5CP02613J
Gines, M. J. L., March, A. J., and Apesteguia, C. R. (1997). Kinetic study of the reverse water-gas shift reaction over CuO/ZnO/Al2O3 catalysts. Appl. Catal. A-Gen. 154, 155–171. doi: 10.1016/S0926-860X(96)00369-9
Goguet, A., Meunier, F., Breen, J. P., Burch, R., Petch, M. I., and Ghenciu, A. F. (2004b). Study of the origin of the deactivation of a Pt/CeO2 catalyst during reverse water gas shift (RWGSR) reaction. J. Catal. 226, 382–392. doi: 10.1016/j.jcat.2004.06.011
Goguet, A., Meunier, F. C., Tibiletti, D., Breen, J. P., and Burch, T. (2004a). Spectrokinetic investigation of reverse water-gas-shift reaction intermediates over a Pt/CeO2 catalyst. J. Phys. Chem. B 108, 20240–20246. doi: 10.1021/jp047242w
Goguet, A., Shekhtman, S. O., Burch, R., Hardacre, C., Meunier, F. C., and Yablonsky, G. S. (2016). Pulse-response TAP studies of the reverse water-gas shift reaction over a Pt/CeO2 catalyst. J. Catal. 237, 102–110. doi: 10.1016/j.jcat.2005.10.020
Graciani, J., Mudiyanselage, K., Xu, F., Baber, A. E., Evans, J., Senanayake, S. D., et al. (2014). Highly active copper-ceria and copper-ceira-titania catalysts for methanol synthesis from CO2. Science 345 546–550. doi: 10.1126/science.1253057
Han, Y. Q., Xu, H. T., Su, Y. Q., Xu, Z. L., Wang, K. F., and Wang, W. Z. (2019). Noble metal (Pt, Au@Pd) nanoparticles supported on metal organic framework (MOF-74) nanoshuttles as high-selectivity CO2 conversion catalysts. J. Catal. 370, 70–78. doi: 10.1016/j.jcat.2018.12.005
Hare, B. J., Maiti, D., Daza, Y. A., Bhethanabotla, V. R., and Kuhn, J. N. (2018). Enhanced CO2 conversion to CO by silica-supported perovskite oxide at low temperatures. ACS Catal. 8, 3021–3029. doi: 10.1021/acscatal.7b03941
Hare, B. J., Maiti, D., Ramani, S., Ramos, A. E., and Bhethanabotla, V. R. (2019). Thermochemical conversion of carbon dioxide by reverse water-gas shift chemical looping using supported pervoskite oxides. Catal. Today 323, 225–232. doi: 10.1016/j.cattod.2018.06.002
He, Y. L., Yang, K. R., Yu, Z. W., Fishman, Z. S., Achola, L. A., and Tobin, Z. M. (2019). Catalytic manganese oxide nanostructures for the reverse water gas shift reaction. Nanoscale 11, 16677–16688. doi: 10.1039/C9NR06078B
Huš, M., Dasireddy, V. D. B. C., Stefancic, N. S., and Likozar, B. (2017). Mechanism, kinetics and thermodynamics of carbon dioxide hydrogenation to methanol on Cu/ZnAl2O4 spinel-type heterogeneous catalysts. Appl. Catal. B-Environ. 207, 267–278. doi: 10.1016/j.apcatb.2017.01.077
Ishito, N., Hara, K. J., Nakajima, K., and Fukuoka, A. (2016). Selective synthesis of carbon monoxide via formates in reverse water–gas shift reaction over alumina-supported gold catalyst. J. Energy Chem. 25, 306–310. doi: 10.1016/j.jechem.2015.12.005
Jafari, A., Ebadi, A., and Sahedbedlfar, S. (2019). Effect of iron oxide precursor on the properties and ammonia synthesis activity of fused iron catalysts. React. Kinet. Mech. Cat. 126, 307–325. doi: 10.1007/s11144-018-1498-6
Jing, H. J., Li, Q. H., Wu, J., Liu, D. W., and Wu, K. C. (2019). Theoretical study of the reverse water gas shift reaction on copper modified β-Mo2C (001) surfaces. J. Phys. Chem. C 123, 1235–1251. doi: 10.1021/acs.jpcc.8b09884
Jones, J. H. (2000). Te cativa process for the manufacture of acetic acid. Iridium catalyst improves productivity in an established industrial process. Platin. Met. Rev. 44, 94–105.
Joo, O. S., and Jung, K. D. (2003). Stability of ZnAl2O4 catalyst for reverse-water-gas-shift reaction (RWGSR). B Korean Chem. Soc. 24:1. doi: 10.5012/bkcs.2003.24.1.086
Kae, H. K., Park, J. L., Park, E. J., Dim, Y. D., and SUhm, H. (2014). Dopant effect of barium zirconate-based perovskite-type catalysts for the intermediate-temperature reverse water gas shift reaction. ACS Catal. 4, 3117–3122. doi: 10.1021/cs500476e
Kang, J., Czaja, A. D., and Guliants, V. V. (2017). Carbon dioxide as feedstock in selective oxidation of propane. Eur. J. Inorg. Chem. 2017, 4757–4762. doi: 10.1002/ejic.201701049
Karelovic, A., Galdames, G., Median, J. C., Yévenes, C., Barra, Y., and Jiménez, R. (2019). Mechanism and structure sensitivity of methanol synthesis from CO2 over SiO2-supported Cu nanoparticles. J. Catal. 369, 419–426. doi: 10.1016/j.jcat.2018.11.012
Katta, L., Sudarsamam, P., Thrimurthulu, G., and Reddy, B. M. (2010). Doped nanosized ceria solid solutions for low temperature soot oxidation: zirconium versus lanthanum promoters. Appl. Catal. B-Environ. 101, 101–108. doi: 10.1016/j.apcatb.2010.09.012
Kattel, S., Liu, P., and Chen, J. G. (2017). Tuning selectivity of RWGSR reactions at the metal/oxide interface. J. Am. Chem. Soc. 139, 9739–9754. doi: 10.1021/jacs.7b05362
Kattel, S., Yan, B. H., Chen, J. G. G., and Liu, P. (2016a). RWGSR on Pt, Pt/SiO2 and Pt/TiO2: importance of synergy between Pt and oxide support. J. Catal. 343, 115–126. doi: 10.1016/j.jcat.2015.12.019
Kattel, S., Yu, W. T., Yang, X. F., Yan, B. H., Huang, Y. Q., Wan, W. M., et al. (2016b). CO2 hydrogenation over oxide-supported PtCo catalysts the role of the oxide support in determining the product selectivity. Angew. Chem. Int. Ed. 55, 7968–7973. doi: 10.1002/anie.201601661
Kharaji, A. G., Shariati, A., and Ostadi, M. (2014). Development of Ni-Mo/Al2O3 catalyst for reverse water gas shift (RWGSR) reaction. J. Nanosci. Nanotechno. 14, 6841–6847. doi: 10.1166/jnn.2014.8962
Kharaji, A. G., Shariati, A., and Takassi, M. A. (2013). A novel γ-alumina supported Fe-Mo bimetallic catalyst for reverse water gas shift reaction. Chinese J. Chem. Eng. 21, 1007–1014. doi: 10.1016/S1004-9541(13)60573-X
Kim, D. H., Han, S. W., Yoon, H. S., and Kim, Y. D. (2015). Reverse water gas shift reaction catalyzed by Fe nanoparticles with high catalytic activity and stability. J. Ind. Eng. Chem. 23, 67–71. doi: 10.1016/j.jiec.2014.07.043
Kim, S. S., Lee, H. H., and Hong, S. C. (2012a). The effect of the morphological characteristics of TiO2 supports on the reverse water–gas shift reaction over Pt/TiO2 catalysts. Appl. Catal. B-Environ. 119–120, 100–108. doi: 10.1016/j.apcatb.2012.02.023
Kim, S. S., Lee, H. H., and Hong, S. C. (2012b). A study on the effect of support's reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A-Gen. 423–424, 100–107. doi: 10.1016/j.apcata.2012.02.021
Kim, S. S., Park, K. H., and Hong, S. C. (2013). A study of the selectivity of the reverse water–gas-shift reaction over Pt/TiO2 catalysts. Fuel Process. Technol. 108 47–54. doi: 10.1016/j.fuproc.2012.04.003
Klankermayer, J., Wesselbaum, S., Beydoun, K., and Leitner, W. (2016). Selective catalytic synthesis using the combination of carbon dioxide and hydrogen: catalytic chess at the interface of energy and chemistry. Angew. Chem. Int. Ed. 55, 7296–7343. doi: 10.1002/anie.201507458
Klvana, D., Kirchnerová, J, and Tofan, C. (1999). Catalytic decomposition of nitric oxide by perovskites. Korean J. Chem. Eng. 16, 470–477. doi: 10.1007/BF02698270
Kovacevic, M., Mojet, B. L., van Ommen, J. G., and Lefferts, L. (2016). Effects of morphology of cerium oxide catalysts for reverse water gas shift reaction. Catal. Lett. 146, 770–777. doi: 10.1007/s10562-016-1697-6
Krstajić, V. N., Lačnjevac, C. M., Jakšić, J. M., and Jakšić, M. M. (2008). Interactive supported electrocatalysts and spillover effect in electrocatalysis for hydrogen and oxygen electrode reactions. Chem. Ind. Chem. Q. 14, 119–136. doi: 10.2298/CICEQ0802119J
Kwak, J. H., Kovarik, L., and Szanyi, J. (2013a). Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 3, 2094–2100. doi: 10.1021/cs4001392
Kwak, J. H., Kovarik, L., and Szanyi, J. (2013b). CO2 reduction on supported Ru/Al2O3 catalysts: cluster size dependence of product selectivity. ACS Catal. 3, 2449–2455. doi: 10.1021/cs400381f
Li, D., Ichikuni, N., Shimazu, S., and Uematsu, T. (1998). Catalytic properties of sprayed Ru/Al2O3 and promoter effects of alkali metals in RWGSR. Appl. Catal. A-Gen. 172, 351–358. doi: 10.1016/S0926-860X(98)00139-2
Li, F., Luo, S., Sun, Z., Bao, X., and Fan, L. (2011). Role of metal oxide support in redox reactions of iron oxide for chemical looping applications; experiments and density functional theory calculations. Energy Environ. Sci. 4, 3661–3667. doi: 10.1039/c1ee01325d
Li, L., Hu, L. P., Li, J., and Wei, Z. D. (2015). Enhanced stability of Pt nanoparticle electrocatalysts for fuel cells. Nano Res. 8, 418–440. doi: 10.1007/s12274-014-0695-5
Li, R. X., Yabe, S., Yamashita, M., Momose, S., Yoshida, S., Yin, S., et al. (2002). UV-shielding properties of zinc oxide-doped ceria fine powders derived via soft solution chemical routes. Mater. Chem. Phys. 75, 39–44. doi: 10.1016/S0254-0584(02)00027-5
Li, S. W., Xu, Y., Chen, Y. F., Li, W. Z., Lin, L. L., Li, M. Z., et al. (2017). Tuning the selectivity of catalytic carbon dioxide hydrogenation over iridium/cerium oxide catalysts with a strong metal-support interaction. Angew. Chem. Int. Ed. 56, 10761–10765. doi: 10.1002/anie.201705002.
Liang, B., Duan, H. M., Su, X., Chen, X. D., Huang, Y. Q., Chen, X. W., et al. (2017). Promoting role of potassium in the reverse water gas shift reaction on Pt/mullite catalyst. Catal. Today 281, 319–326. doi: 10.1016/j.cattod.2016.02.051
Lin, B., Liu, Y., Heng, L., Ni, J., Lin, J. X., and Jiang, L. L. (2018a). Effect of barium and potassium promoter on Co/CeO2 catalysts in ammonia synthesis. J. Rare Earth. 36, 703–707. doi: 10.1016/j.jre.2018.01.017
Lin, F. J., Delmelle, R., Vinodkumar, T., Reddy, B. M., Wokaun, A., and Alxneit, I. (2015). Correlation between the structural characteristics, oxygen storage capacities and catalystic activities of dual-phase Zn-modified ceria nanocrystals. Catal. Sci. Technol. 5, 3556–3567. doi: 10.1039/C5CY00351B
Lin, L. L., Yao, S. Y., Liu, Z. Y., Zhang, F., Li, N., Vovchok, D., et al. (2018b). In situ characterization of Cu/CeO2 nanocatalysts for RWGSR: morphological effects of nanostructured ceria on the cataystic activity. J. Phys. Chem. C 122, 12934–12943. doi: 10.1021/acs.jpcc.8b03596
Lin, W., Stocker, K. M., and Schatz, G. C. (2017). Mechanisms of hydrogen-assisted CO2 reduction on nickel. J. Am. Chem. Soc. 139, 4663–4666. doi: 10.1021/jacs.7b01538
Liu, B. W., Ju, Y. W., Abe, T., and Kawamoto, K. (2015). Dispersion and distribution of bimetallic oxides in SBA-15, and their enhanced activity for reverse water gas shift reaction. Irong. Chem. Front. 2, 741–748. doi: 10.1039/C5QI00062A
Liu, D. Y., Li, Y. Y., Kottwitz, M., Yan, B. H., Yao, S. Y., Gamalski, A., et al. (2018). Identifying dynamic structural changes of active sites in Pt-Ni bimetallic catalysts using multimodal approaches. ACS Catal. 5, 4120–4131. doi: 10.1021/acscatal.8b00706
Liu, L., Das, S. L., Chen, T. J., Dewangan, N., Ashok, J., Xi, S. B., et al. (2020). Low temperature catalytic reverse water-gas shift reaction over perovskite catalysts in DBD plasma. Appl. Catal. B-Environ. 265:118573. doi: 10.1016/j.apcatb.2019.118573
Liu, L. N., Fan, F., Bai, M. M., Xue, F., Ma, X. R., Jiang, Z., et al. (2019). Mechanistic study of methanol synthesis from CO2 hydrogenation on Rh-doped Cu(111) surfaces. Mol. Catal. 466, 26–36. doi: 10.1016/j.mcat.2019.01.009
Liu, X. Y., Wang, A. Q., Zhang, T., and Mou, C. Y. (2013). Catalysis by gold: new insights into the support effect. Nano Today 8, 403–416. doi: 10.1016/j.nantod.2013.07.005
Liu, Y., and Liu, D. Z. (1999). Study of bimetallic Cu-Ni/γ-Al2O3 catalysts for carbon dioxide hydrogenation. Int. J. Hydrogen. Energy 24, 351–354. doi: 10.1016/S0360-3199(98)00038-X
Liu, Y., Zhang, C. H., Wang, Y., Li, Y., Hao, X., Bai, L., et al. (2008). Effect of co-feeding carbon dioxide on fischer–tropsch synthesis over an iron–manganese catalyst in a spinning basket reactor. Fuel Process. Technol. 89, 234–241. doi: 10.1016/j.fuproc.2007.11.002
Lu, B. W., and Kawamoto, K. (2014). Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction. Mater. Res. Bull. 53, 70–78. doi: 10.1016/j.materresbull.2014.01.043
Maiti, D., Daza, Y. A., Yung, M. M., Kuhn, J. N., and Bhethanabotla, V. R. (2016). Oxygen vacancy formation characteristics in the bulk and across different surface terminations of La(1−x)Fe(1−y)CoyO(3−δ) perovskite oxides for CO2 conversion. J. Mater. Chem. A 4, 5137–5148. doi: 10.1039/C5TA10284G
Maitlis, P. M., Haynes Maitlis, A., Sunley, G. J., and Howard, M. J. (1996). Methanol carbonylation revisited: thirty years on. J. Chem. Soc. Dalton. 112, 2187–2196. doi: 10.1039/dt9960002187
Masui, T., Fujiwara, K., Machida, K., and Adachi, G. (1997). Characterization of Cerium(IV) oxide ultrafine particles prepared using reversed micelles. Chem. Mater. 9, 2197–2204. doi: 10.1021/cm970359v
Matsubu, J. C., Yang, V. N., and Christopher, P. (2015). Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity. J. Am. Chem. Soc. 137, 3076–3084. doi: 10.1021/ja5128133
Matsubu, J. C., Zhang, Y., DeRita, L., Marinkovic, N. S., Chen, J. G., Graham, G. W., et al. (2017). Adsorbate-mediated strong metal-support interactions in oxide-supported Rh catalysts. Nat. Chem. 9, 120–127. doi: 10.1038/nchem.2607
Mikkelsen, M., Jørgensen, M., and Krebs, F. C. (2010). The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 3, 43–81. doi: 10.1039/B912904A
Millet, M. M., Algara-Siller, G., Wrabetz, S., Mazheika, A., Girgsdies, F., and Teschner, D. (2019). Ni single atom catalysts for CO2 activation. J. Chem. Soc. 141, 2451–2461. doi: 10.1021/jacs.8b11729
Min, B. K., Santra, A. K., and Goodman, D. W. (2003). Understanding silicasupported metal catalysts: Pd/silica as a case study. Catal. Today 85, 113–124. doi: 10.1016/S0920-5861(03)00380-8
Mirzaei, A. A. R., Habibpour Faizi, M., and Kashi, E. (2006). Characterization of iron-cobalt oxide catalysts: effect of different supports and promoters upon the structure and morphology of precursors and catalysts. Appl. Catal. A-Gen. 301, 272–283. doi: 10.1016/j.apcata.2005.12.022
Morse, J. R., Juneau, M., Baldwin, J. W., Porosoff, M. D., and Willauer, H. D. (2020). Alkali promoted tungsten carbide as a selective catalysts for the reverse water gas shift reaction. J. CO2 Util. 35, 38–46. doi: 10.1016/j.jcou.2019.08.024
Mukherjee, D., Park, S. E., and Reddy, B. M. (2016). CO2 as a soft oxidant for oxidative dehydrogenation reaction: an eco-benign process for industry. J. CO2 Util. 16, 301–312. doi: 10.1016/j.jcou.2016.08.005
Naito, S., Aida, S. G., Kasahara, T., and Miyao, T. H. (2006). Infrared spectroscopic study on the reaction mechanism of CO hydrogenation over Pd/CeO2. Res. Chem. Intermediat. 32, 279–290. doi: 10.1163/156856706777346444
Nakamura, I., Fujitani, T., Uchijima, T., and Nakamura, J. (1998). The synthesis of methanol and the reverse water-gas shift reaction over Zn-deposited Cu(100) and Cu(110) surfaces: comparison with Zn/Cu(111). Surf. Sci. 400, 387–400. doi: 10.1016/S0039-6028(97)00899-6
Neophytides, S. G., Zafeiratos, S., Papakonstantinou, G. D., Jaksic, J. M., Paloukis, F. E., and Jaksic, M. M. (2005). Extended brewer hypo-hyper-d-interionic bonding theory II. strong metal-support interaction grafting of composite electrocatalysts. Int. J. Hydrog. Energy 30, 393–410. doi: 10.1016/j.ijhydene.2004.07.005
Nielsen, D. U., Hu, X. M., Daasbjerg, K., and Skrydstrup, T. (2018). Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicals. Nat. Catal. 1, 244–254. doi: 10.1038/s41929-018-0051-3
Nitarori, T., Ichiki, T., and Misono, M. (1988). Catalytic properties of perovskite-type mixed oxides (ABO3) consisting of rare earth and 3d transition metals. The roles of the A- and B-site ions. B Chem. Soc. Jpn 61, 621–626. doi: 10.1246/bcsj.61.621
Obalová, L., Karásková, K., Wach, A., Kustrowski, P., Mamulová-Kutláková, K., Michalik, S., et al. (2013). Alkali metals as promoters in Co-Mn-Al mixed oxide for N2O decomposition. Appl. Catall. A-Gen. 462–463, 227–235. doi: 10.1016/j.apcata.2013.05.011
Okabe, K., Takahara, I., Inaba, M., Murata, K., and Yoshimura, Y. (2007). Effects of Ru precursors on activity of Ru-SiO2 catalysts prepared by alkoxide method in Fisher-Tropsch synthesis. J. Jpn. Petrol. Inst. 50, 65–68. doi: 10.1627/jpi.50.65
Okemoto, A., Harada, M. R., Ishizaka, T., Hiyoshi, N., and Sato, K. (2020). Catalytic performance of Mo3/FAU zeolite catalysts modified by Cu for reverse water gas shift reaction. Appl. Catal. A-Gen. 592:117415. doi: 10.1016/j.apcata.2020.117415
Pacultová, K., Draštíková, V., Chromčáková, Ž., Bílková, T., Kutlákovác, K. M., Kotarba, A., et al. (2017). On the stability of alkail metal promoters in Co mixed oxides during direct NO catalytic decomposition. J. Mol. Catal. A Chem. 428, 33–40. doi: 10.1016/j.molcata.2016.11.038
Panarities, C., Zgheib, J., Ebrahim, S. A. H., Couillard, M., and Baranova, E. A. (2020). Electrochemical in-situ activation of Fe-oxide nanowires for the reverse water gas shift reaction. Appl. Catal. B-Environ. 269:118826. doi: 10.1016/j.apcatb.2020.118826
Panaritis, C., Edake, M., Couillard, M., Einakchi, R., and Baranova, E. A. (2018). Insight towards the role of ceria-based supports for reverse water gas shift reaction over RuFe nanopartilces. J. CO2 Util. 26, 350–358. doi: 10.1016/j.jcou.2018.05.024
Park, S. W., Joo, O. S., Jung, K. D., Kim, H., and Han, S. H. (2000). ZnO/Cr2O3 catalyst for reverse-water-gas shift reaction of CAMERE process. Korean J. Chem. Eng. 17, 719–722. doi: 10.1007/BF02699123
Park, S. W., Joo, O. S., Jung, K. D., Kim, H., and Han, S. H. (2001). Development of ZnO/Al2O3 catalyst for reverse-water-gas-shift reaction of CAMERE (carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction) process. Appl. Catal. A-Gen. 211, 81–90. doi: 10.1016/S0926-860X(00)00840-1
Pastor-Pérez, L., Shah, M., Saché, L. E., and Reina, T. R. (2018). Improving Fe/Al2O3 catalysts for the reverse water-gas shift reaction: on the effect of Cs as activity/selectivity promoter. Catalysts 8:608. doi: 10.3390/catal8120608
Peña, M. A., and Fierro, J. L. G. (2001). Chemical structures and performance of perovskite oxides. Chem. Rev. 101, 1981–2017. doi: 10.1021/cr980129f
Porosoff, M. D., and Chen, J. G. (2013). Trends in the catalytic reduction of CO2 by hydrogen over supported monometallic and bimetallic catalysts. J. Catal. 301, 30–37. doi: 10.1016/j.jcat.2013.01.022
Porosoff, M. D., Yan, B. H., and Chen, J. G. (2016). Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities. Energ. Environ. Sci. 9:62–73. doi: 10.1039/C5EE02657A
Porosoff, M. D., Yang, X. F., Boscoboinik, J. A., and Chen, J. G. (2014). Molybdenum carbide as alternative catalysts to precious metals for highly selective reduction of CO2 to CO. Angew. Chem. Int. Ed. 53, 6705–6709. doi: 10.1002/anie.201404109
Qian, C. X., Sun, W., Hung, D. L. H., Qiu, C. Y., Makaremi, M., Kumar, S. G. H., et al. (2019). Catalytic CO2 reduction by palladium-decorated silicon-hydride nanosheets. Nat. Catal. 2, 46–54. doi: 10.1038/s41929-018-0199-x
Radovic, M., Speakman, S. A., Allard, L. F., Payzant, E. A., Lara-Curzio, E., Kriven, W. M., et al. (2008). Thermal, mechanical and phase stability of LaCoO3 in reducing and oxidizing environments. J. Power Sour. 184, 77–83. doi: 10.1016/j.jpowsour.2008.05.063
Ranjabar, A., Irankhah, A., and Aghamiri, S. F. (2019). Catalytic activity of rare earth and alkali metal promoted (Ce, La, Mg, K) Ni/Al2O3 nanocatalysts in reverse water gas shift reaction. Res. Chem. Intermediat. 45, 5125–5141. doi: 10.1007/s11164-019-03905-1
Rao, K. N., Reddy, B. M., Abhishek, B., Seo, Y. H., Jiang, N. Z., and Park, S. E. (2009). Effect of ceria on the structure and catalytic activity of V2O5/TiO2-ZrO2 for oxidehydrogenation of ethylbenzene to styrene utilizing CO2 as soft oxidant. Appl. Catal. B-Environ. 91, 649–656. doi: 10.1016/j.apcatb.2009.07.003
Reddy, B. M., Han, D. S., Jiang, N. Z., and Park, S. E. (2008). Dehydrogenation of ethylbenzene to styrene with carbon dioxide over ZrO2-based composite oxide catalysts. Catal. Surv. Asia 12, 56–69. doi: 10.1007/s10563-007-9039-8
Reddy, B. M., Katta, L., and Thrimurthulu, G. (2010). Novel nanocrystalline Ce1−xLaxO2−δ (x = 0.2) solid solutions: structural characteristics and catalytic performance. Chem. Mater. 22, 467–475. doi: 10.1021/cm903282w
Riedel, T., Claeys, M., Schulz, H., Schaub, G., Nam, S. S., Jun, K. W., et al. (1999). Comparative study of fischer-tropsch synthesisi with H2/CO and H2/CO2 syngas using Fe- and Co-based catalysts. Appl. Catal. A-Gen. 186, 201–203. doi: 10.1016/S0926-860X(99)00173-8
Ringuedẽ, A., and Fouletier, J. (2001). Oxygen reaction on strontium-doped lanthanum cobaltite dense electrodes at intermediate temperatures. Solid State Ionics 139, 167–177. doi: 10.1016/S0167-2738(01)00692-0
Ro, I., Sener, C., Stadelman, T. M., Ball, M. R., Venegas, J. M., Burt, S. P., et al. (2016). Measurement of intrinsic catalytic activity of Pt monometallic and Pt-MoOx interfacial sites over visible light enhanced PtMoOx/SiO2 catalyst in reverse water gas shift reaction. J. Catal. 344, 784–794. doi: 10.1016/j.jcat.2016.08.011
Rogowski, J. (2019). TOF-SIMS study of morphology and chemical composition of wustite-based precursor and iron catalyst for ammonia synthesis. Appl. Sur. Sci. 469, 82–89. doi: 10.1016/j.apsusc.2018.10.271
Roiaz, M., Monachino, E., Dri, C., Greiner, M., Knop-Gericke, A., Schlögl, R., et al. (2016). Reverse water-gas shift or Sabatier methanation on Ni(110)? Stable surface species at near ambient pressure. J. Am. Chem. Soc. 138, 4146–4154. doi: 10.1021/jacs.5b13366
Ronda-Lloret, M., Rico-Francés, S., Sepúlveda-Escribano, A., and Ramos-Fernandez, E. V. (2018). CuOx/CeO2 catalyst derived from metal organic framework for reverse water-gas shift reaction. Appl. Catal. A-Gen. 562, 28–36. doi: 10.1016/j.apcata.2018.05.024
Royer, S., Duprez, D., and Kaliaguine, S. (2005). Role of bulk and grainboundary oxygen mobility in the catalytic oxidation activity of LaCo1−xFexO3. J. Catal. 234, 364–375. doi: 10.1016/j.jcat.2004.11.041
Sache, E. L., Pastor-Pérez, L., Haycock, B. J., Villora-Pic,ó, J. J., Sepúlveda-Escribano, A., and Ramirez Reina, T. (2020). Switchable catalysts for chemical CO2 recycling: A step forward in the methanation and reverse water-gas shift reactions. ACS Sustain. Chem. Eng. 11, 4614–4622. doi: 10.1021/acssuschemeng.0c00551
Saeidi, S., Najari, S., Fazlollahi, F., Nikoo, M. F., Sefidkon, F., Klemes, J. J., et al. (2017). Mechanisms and kinetics of CO2 hydrogenation to value-added products: A detailed review on current status and future trends. Renew. Sust. Energ. Rev. 80, 1292–1311. doi: 10.1016/j.rser.2017.05.204
Sahebdelfar, S., and Takht Ravanchi, M. (2015). Carbon dioxide utilization for methane production: A thermodynamic analysis, J. Petrol. Sci. Eng. 134 14–22. doi: 10.1016/j.petrol.2015.07.015
Schmale, K., Daniels, M., Buchheit, A., Grünebaum, M., Haase, L., Koops, S., et al. (2013). Influence of zinc oxide on the conductivity of ceria. J. Electrochem. Soc. 160, 1081–1087. doi: 10.1149/2.119309jes
Shimoda, N., Kimura, Y., Kobayashi, Y., Kubota, J., and Satokawa, S. (2017). Ammonia synthesis over yttrium-doped barium zirconate and cerate-based perovskite-type oxide supported ruthenium catalysts. Int. J. Hydrog. Energ. 42, 29745–29755. doi: 10.1016/j.ijhydene.2017.10.108
Silva-Calpa, L. D. R., Zonetti, P. C., Rodrigues, C. P., Alves, O. C., Appel, L. G., and de Avillez, R. R. (2016). The ZnxZr1−xO2−y solid solution on m-ZrO2: creating O vacancies and improving the m-ZrO2 redox properties. J. Mol. Catal. A-Chem. 425, 166–173. doi: 10.1016/j.molcata.2016.10.008
Su, X., Yang, X. F., Huang, Y. Q., Liu, B., and Zhang, T. (2019). Single-atom catalysis toward efficient CO2 conversion to CO and formate products. Acc. Chem. Res. 52, 656–664. doi: 10.1021/acs.accounts.8b00478
Su, X., Yang, X. L., Zhao, B., and Huang, Y. Q. (2017). Designing of highly selective and high-temperature endurable RWGSR heterogeneous catalysts: recent advances and the future directions. J. Energy Chem. 26, 854–867. doi: 10.1016/j.jechem.2017.07.006
Sun, C. W., Li, H., and Chen, L. Q. (2012). Nanostructured ceria-based materials: synthesis, properties, and applications. Energy Environ. Sci. 5, 8475–8505. doi: 10.1039/c2ee22310d
Sun, F. M., Yan, C. F., Wang, Z. D., and Guo, C. Q. (2015). Ni/Ce-Zr-O catalyst for high CO2 conversion during reverse water gas shift reaction (RWGSR). Int. J. Hydrog. Energ. 40, 15985–15993. doi: 10.1016/j.ijhydene.2015.10.004
Sun, Q. D., Ye, J. Y., and Liu, C. J. (2014). In2O3 as a promising catalyst for CO2 utilization: a case study with reverse water gas shift over In2O3. Greenhouse Gases 4, 140–144. doi: 10.1002/ghg.1401
Tang, Q. L., Hong, Q. J., and Liu, Z. P. (2009). CO2 fixation into methanol at Cu/ZrO2 interface from first principles kinetic Monte Carlo. J. Catal. 263, 114–122. doi: 10.1016/j.jcat.2009.01.017
Ten Elshof, J. E., Bouwmeester, H. J. M., and Verweij, H. (1996). Oxygen transport through La1−XSrXFeO3−δ membranes II. Permeation in air/CO, CO2 gradients. Solid State Ionics 89, 81–92. doi: 10.1016/0167-2738(96)00255-X
Tibiletti, D., Goguet, A., Meunier, F. C., Breen, J. P., and Burch, R. (2004). On the importance of steady-state isotopic techniques for the investigation of the mechanism of the reverse water-gas-shift reaction. Chem. Commun. 14, 1636–1637. doi: 10.1039/b403438d
Utsis, N., Landau, M. V., Erenburg, A., Nehemya, R. V., and Hersowitz, M. (2018). Performance of reverse water gas shift on co-precipitated and carbon-templated BaFe-hexaaluminate effect of iron loading, texture, and promotion with Postassium. ChemCatChem 10, 3795–3805. doi: 10.1002/cctc.201800709
Wang, C. C., and Nakamura, J. (2010). Structure sensitivity for forward and reverse water-gas shift reactions on copper structure surfaces: a DFT study. J. Phys. Chem. Lett. 1, 3053–3057. doi: 10.1021/jz101150w
Wang, F., He, S., Chen, H., Wang, B., Zheng, L. R., Wei, M., et al. (2016b). Active site dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J. Am. Chem. Soc. 138, 6298–6305. doi: 10.1021/jacs.6b02762
Wang, G. C., Jiang, L., Pang, X. Y., Cai, Z. S., Pan, Y. M., Zhao, X. Z., et al. (2003). A theoretical study of surface-structural sensitivity of the reverse water-gas shift reaction over Cu(hkl) surfaces. Surf. Sci. 543, 118–130. doi: 10.1016/S0039-6028(03)00876-8
Wang, J. Y., Liu, C. Y., Senfile, T. P., Zhu, J., Zhang, G. H., Guo, X. W., et al. (2020). Variation in In2O3 crystal phase alters catalytic performance toward the reverse water gas shift reaction. ACS Catal. 10, 3264–3273. doi: 10.1021/acscatal.9b04239
Wang, L., Liu, H., Liu, Y., Chen, Y., and Yang, S. Q. (2013a). Effect of precipitants on Ni-CeO2 catalysts prepared by a co-precipitation method for the reverse water-gas shift reaction. J. Rare Earths 31, 969–974. doi: 10.1016/S1002-0721(13)60014-9
Wang, L. C., Khazaneh, M. T., Widmann, D., and Behm, R. J. (2013b). TAP reactor studies of the oxidizing capability of CO2 on a Au/CeO2 catalyst – A first step toward identifying a redox mechanism in the reverse water–gas shift reaction. J. Catal. 302, 20–30. doi: 10.1016/j.jcat.2013.02.021
Wang, L. H., and Liu, H. (2018). Mesoporous Co-CeO2 catalyst prepared by colloidal solution combustion method for reverse water-gas shift reaction. Catal. Today 316, 155–161. doi: 10.1016/j.cattod.2018.04.015
Wang, L. H., Liu, H., Chen, Y., Zhang, R. K., and Yang, S. Q. (2013c). K-promoted Co-CeO2 catalyst for reverse water–gas shift reaction. Chem. Lett. 42, 682–683. doi: 10.1246/cl.130137
Wang, L. H., Liu, H., Liu, Y., Chen, Y., and Yang, S. Q. (2013d). Influence of preparation method on performance of Ni-CeO2 catalysts for reverse water-gas shift reaction. J. Rare Earths 31, 559–564. doi: 10.1016/S1002-0721(12)60320-2
Wang, W., Wang, S. P., Ma, X. B., and Gong, J. L. (2011). Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 40, 3703–3727. doi: 10.1039/c1cs15008a
Wang, W., Zhang, Y., Wang, Z. Y., Yan, J. M., Ge, Q. F., and Liu, C. J. (2016c). Reverse water gas shift over In2O3-CeO2 catalysts. Catal. Today 259, 402–408. doi: 10.1016/j.cattod.2015.04.032
Wang, X., Hong, Y. C., Shi, H., and Szanyi, J. (2016a). Kinetic modeling and transient DRIFTS–MS studies of CO2 methanation over Ru/Al2O3 catalysts. J. Catal. 343, 185–195. doi: 10.1016/j.jcat.2016.02.001
Wang, X., Shi, H., Kwak, J. H., and Szanyi, J. (2015). Mechanism of CO2 hydrogenation on Pd/Al2O3 catalysts: Kinetics and transient DRIFTS-MS studies. ACS Catal. 5, 6337–6349. doi: 10.1021/acscatal.5b01464
Wang, X., Shi, H., and Szanyi, J. (2017). Controlling selectivities in CO2 reduction through mechanistic understanding. Nat. Commun. 8, 513–518. doi: 10.1038/s41467-017-00558-9
Wang, Y. X., and Wang, G. C. (2019). A systematic theoretical study of water gas shift reaction on Cu(111) and Cu(110): potassium effect. ACS Catal. 9, 2261–2274. doi: 10.1021/acscatal.8b04427
Weatherbee, G. D., and Bartholomew, C. H. (1984). Hydrogenation of CO2 on group-VIII metals.4. Specific activities and selectivities of silica-supported Co, Fe, and Ru. J. Catal. 87, 352–362. doi: 10.1016/0021-9517(84)90196-9
Wenzel, M., Dharanipragada, V. R. A., Galvita, V. V., Poelman, H., Marin, G. B., Rihko-Struckmann, L., et al. (2017). CO product from CO2 via reverse water-gas shift reaction performed in a chemical looping mode: Kinetics on modified iron oxide. J. CO2 Util. 17, 60–68. doi: 10.1016/j.jcou.2016.10.015
Wenzel, M., Rihko-struckmann, L., and Sundmacher, K. (2018). Continuous production of CO from CO2 by RWGSR chemical looping in fixed and fluidized bed reactors. Chem. Eng. J. 336, 278–296. doi: 10.1016/j.cej.2017.12.031
Wolf, A., Jess, A., and Kern, C. (2016). Syngas production via reverse water-gas shift reaction over a Ni/Al2O3 catalyst: catalyst stability, reaction kinetics, and modelling. Chem. Eng. Technol. 39, 1040–1048. doi: 10.1002/ceat.201500548
Wu, H. C., Chang, Y. C., Wu, J. H., Lin, J. H., Lin, I. K., and Chen, C. S. (2015). Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: the influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 5, 4154–4163. doi: 10.1039/C5CY00667H
Wu, X.-F., Neumann, H., and Beller, M. (2011). Palladium-catalyzed carbonylative coupling reactions between Ar-X and carbon nucleophiles. Chem. Soc. Rev. 40, 4986–5009. doi: 10.1039/c1cs15109f
Xu, H. C., and Ge, L. (2016). Progress on the catalytic hydrogenation of CO2 via reverse water gas shift reaction. Ind. Eng. Pro. 35, 3180–3189. doi: 10.16085/j.issn.1000-6613.2016.10.023
Yamazoe, N., Furukawa, S., Teraoka, Y., and Seiyama, T. (1982). The effect of oxygen sorption on the crystal structure of La1−XSrXCoO3−δ. Chem. Lett. 10, 2019–2022. doi: 10.1246/cl.1982.2019
Yamazoe, N., Teraoka, Y., and Seiyama, T. (1981). TPD and XPS study on thermal behavior of absorbed oxygen in La1−XSrXCoO3. Chem. Lett. 11, 1767–1770. doi: 10.1246/cl.1981.1767
Yang, Q., and Lin, J. Y. S. (2006). Fixed-bed performance for production of oxygen-enriched carbon dioxide stream by perovskite-type ceramic sorbent. Separation and purification technology. Sep. Purif. Technol. 49, 27–35. doi: 10.1016/j.seppur.2005.08.004
Yang, X. L., Su, X., Chen, X. D., Duan, H. M., Liang, B. L., Liu, Q. G., et al. (2017). Promotion effects of potassium on the activity and selectivity of Pt/zeolite catalysts for reverse water gas shift reaction. Appl. Catal. B-Environ. 216, 95–105. doi: 10.1016/j.apcatb.2017.05.067
Yao, X. J., Tang, C. J., Ji, Z. Y., Dai, Y., Cao, Y., Gao, F., et al. (2013). Investigation of the physicochemical properties and catalytic activities of Ce0.67M0.33O2 (M = Zr4+, Ti4+, Sn4+) solid solutions for NO removal by CO. Catal. Sci. Technol. 3, 688–698. doi: 10.1039/C2CY20610B
Ye, J. Y., Liu, C. J., and Ge, Q. F. (2012). DFT study of CO2 adsorption and hydrogenation on the In2O3 surface. J. Phys. Chem. C 116, 7817–7825. doi: 10.1021/jp3004773
Ye, J. Y., Liu, C. J., Mei, D. H., and Ge, Q. F. (2013). Active oxygen vacancy site for methanol synthesis from RWGSR on In2O3 (110): a DFT study. ACS Catal. 3, 1293–1306. doi: 10.1021/cs400132a
Yeung, C. M. Y., Yu, K. M. K., Fu, Q. J., Thompsett, D., Petch, M. I., and Tsang, S. C. (2005). Engineering Pt in ceria for a maximum metal-support interaction in catalysis. J. Am. Chem. Soc. 127, 18010–18011. doi: 10.1021/ja056102c
Zhang, Q., and Guo, L. (2018). Mechanism of the reverse water-gas shift reaction catalyzed by Cu12TM bimetallic nanocluster: a density functional theory study. J. Clust. Sci. 29, 867–877. doi: 10.1007/s10876-018-1346-x
Zhang, Q., Guo, L., and Hao, Z. J. (2018). Computational investigation of M1/W6S8(M=Fe, Ru, and Os) single-atom catalysts for CO2 hydrogenation. Catal. Surv. Asia 22, 195–207. doi: 10.1007/s10563-018-9252-7
Zhang, Q., Pastor-Pérez, L., Jin, W., Gu, S., and Reina, T. R. (2019b). Understanding the promoter effect of Cu and Cs over highly effective β-Mo2C catalysts for the reverse water-gas shift reaction. Appl. Catal. B-Environ. 244, 889–898. doi: 10.1016/j.apcatb.2018.12.023
Zhang, X., Zhu, X. B., Lin, L. L., Yan, S. Y., Zhang, M. T., Liu, X., et al. (2016). Highly dispersed copper over β-Mo2C as an efficient and stable catalyst for the reverse water gas shift (RWGSR) reaction. ACS Catal. 7, 912–918. doi: 10.1021/acscatal.6b02991
Zhang, Y., Liang, L., Chen, Z. Y., Wen, J. J., Zhong, W., Zou, S. B., et al. (2020). Highly efficient Cu/CeO2-hollow nanospheres catalyst for the reverse water-gas shift reaction: Investigation on the role of oxygen vacancies through in situ UV-Raman and DRIFTS. Appl. Sur. Sci. 516:146035. doi: 10.1016/j.apsusc.2020.146035.
Zhang, Z. M., Xun, H., Wang, Y., Hu, S., Xiang, J., Li, C. C., et al. (2019a). Regulation the reaction intermediates in methanation reactions vis modification of nickel catalysts with strong base. Fuel 237, 566–579. doi: 10.1016/j.fuel.2018.10.052
Zheng, X. L., Guo, L., Li, W. L., Cao, Z. R., Liu, N. Y., Zhang, Q., et al. (2017). Insight into the mechanism of reverse water-gas shift reaction and ethanol formation catalyzed by Mo6S8-TM clusters. Mol. Catal. 439, 155–162. doi: 10.1016/j.mcat.2017.06.030
Zheng, Z. Z., Xu, H. T., Xu, Z. L., and Ge, J. P. (2018). A monodispersed spherical Zr-based metal-organic framework catalyst, Pt/Au@Pd@UIO-66, comprising an Au@Pd core-shell encapsulated in a UIO-66 center and its highly selective RWGSR to produce CO. Small 14:1702812. doi: 10.1002/smll.201702812
Zhou, G., Dai, B., Xie, H. M., Zhang, G. Z., Xiong, K., and Zheng, X. X. J. (2017). CeCu composite catalyst for CO synthesis by reverse water–gas shift reaction: effect of Ce/Cu mole ratio. CO2 Util. 21, 292–301. doi: 10.1016/j.jcou.2017.07.004
Zhou, G. L., Xie, F. Q., Deng, L. D., Zhang, G. Z., and Xie, H. M. (2020). Supported mesoporous Cu/CeO2−δ catalyst for CO2 reverse water-gas shift reaction to syngas. Int. J. Hydrog. Energy 45, 11380–11393. doi: 10.1016/j.ijhydene.2020.02.058
Zhou, K., and Li, Y. D. (2012). Catalysis based on nanocrystals with well-defined facets. Angew. Chem. Int. Ed. 51, 602–613. doi: 10.1002/anie.201102619
Zhou, P., Ma, Y. C., Lan, G. J., Tang, H. D., Han, W. F., Liu, H. Z., et al. (2019). A highly stable and active mesoporous rethenium catalyst for ammonia synthesis prepared by a RhCl3/SiO2-templated approach. Chinese J. Catal. 40, 114–123. doi: 10.1016/S1872-2067(18)63192-4
Zhuang, S. J., Han, N., Wang, T. T., Meng, X. X., Meng, B., Li, Y., et al. (2019). Enhanced CO selectivity for reverse water-gas shift reaction using Ti4O7-doped SrCe0.9Y0.1O3−δ hollow fibre membrane reaction. Canad. J. Chem. Eng. 97, 1619–1626. doi: 10.1002/cjce.23384
Keywords: RWGSR, catalytic mechanism, catalytic performance, supported metal catalysts, oxide catalysts, chemical looping cycles
Citation: Chen X, Chen Y, Song C, Ji P, Wang N, Wang W and Cui L (2020) Recent Advances in Supported Metal Catalysts and Oxide Catalysts for the Reverse Water-Gas Shift Reaction. Front. Chem. 8:709. doi: 10.3389/fchem.2020.00709
Received: 15 May 2020; Accepted: 09 July 2020;
Published: 31 August 2020.
Edited by:
Leilei Xu, Nanjing University of Information Science and Technology, ChinaReviewed by:
Miao Zhichao, Shandong University of Technology, ChinaBo Yang, Nanjing University of Information Science and Technology, China
Mei Wu, Huaiyin Institute of Technology, China
Qinghui Shou, Qingdao Institute of Bioenergy and Bioprocess Technology (CAS), China
Copyright © 2020 Chen, Chen, Song, Ji, Wang, Wang and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nannan Wang, d2FuZ25uQGRndXQuZWR1LmNu; Wenlong Wang, MjAxNjgzM0BkZ3V0LmVkdS5jbg==; Lifeng Cui, bGN1aUBkZ3V0LmVkdS5jbg==
†These authors have contributed equally to this work