 Seefat Farzin
Seefat Farzin Tyler J. Johnson
Tyler J. Johnson Shyambo Chatterjee
Shyambo Chatterjee Ehsan Zamani
Ehsan Zamani Shudipto K. Dishari
Shudipto K. Dishari
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 26 August 2020
Sec. Green and Sustainable Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00690
This article is part of the Research TopicWomen in Science: ChemistryView all 43 articles
Converting industrial/agricultural lignin-rich wastes to efficient, cost-effective materials for electrochemical devices (e.g., fuel cells) can aid in both bio- and energy economy. A major limitation of fuel cells is the weak ion conductivity within the ~2–30-nm thick, ion-conducting polymer (ionomer)-based catalyst-binder layer over electrodes. Here, we strategically sulfonated kraft lignin (a by-product of pulp and paper industries) to design ionomers with varied ion exchange capacities (IECs) (LS x; x = IEC) that can potentially overcome this interfacial ion conduction limitation. We measured the ion conductivity, water uptake, ionic domain characteristics, density, and predicted the water mobility/stiffness of Nafion, LS 1.6, and LS 3.1 in submicron-thick hydrated films. LS 1.6 showed ion conductivity an order of magnitude higher than Nafion and LS 3.1 in films with similar thickness. The ion conductivity of these films was not correlated to their water uptake and IECs. Within the three-dimensional, less dense, branched architecture of LS 1.6 macromolecules, the –SO3H and –OH groups are in close proximity, which likely facilitated the formation of larger ionic domains having highly mobile water molecules. As compared to LS 1.6, LS 3.1 showed a higher glass transition temperature and film stiffness at dry state, which sustained during humidification. On the contrary, Nafion stiffened significantly upon humidification. The smaller ionic cluster within stiff LS 3.1 and Nafion films thus led to ion conductivity lower than LS 1.6. Since LS x ionomers (unlike commercial lignosulfonate) are not water soluble, they are suitable for low-temperature, water-mediated ion conduction in submicron-thick films.
For environmental, economic, and societal growth, we need to attain both sustainable energy- and bioeconomy. The Department of Energy's Biotechnologies Office states that to achieve sustainable bioeconomy, we need to strategically design and utilize bio-based products from non-food waste sources (Strategic Plan for a Thriving and Sustainable Bioeconomy, 2016). Lignin is the second most abundant polymer in nature (next to cellulose) and accounts for 15–40 mass% of the plant cell walls (Aro and Fatehi, 2017). Besides, lignocellulosic biorefineries, and pulp and paper industries produce lignin-rich waste (>70 million ton/year) (Aro and Fatehi, 2017), only 1–2% of which is utilized to produce value-added chemicals (Aro and Fatehi, 2017), and the rest is usually combusted to generate heat (Aro and Fatehi, 2017). Some of the lignin valorization efforts have yielded products, like, concrete additives (Danner et al., 2015), plasticizers (Kalliola et al., 2015; Naseem et al., 2016), stabilizing agents (Cerrutti et al., 2012), dispersing agents (Konduri et al., 2015; Kai et al., 2016), corrosion inhibitors (Abu-dalo et al., 2013), thermoplastics (Lange et al., 2013), fuel (Che et al., 2018), aromatic diols (Che et al., 2018) (after delignification), block copolymers (Holmberg et al., 2014a,b) (from monolignols after breaking lignin), and more (Lange et al., 2013; Norgren and Edlund, 2014). To best utilize the abundant natural resources and make bioprocessing industries more economically viable, we critically need more unique lignin valorization efforts to design products with commercial and technological importance for sectors, like energy.
On the other hand, to achieve energy sustainability, we need low-cost, eco-friendly materials that can also overcome the technical barriers of energy conversion and storage devices (fuel cells, electrolyzers, batteries). A major technical challenge of the hydrogen fuel cell, one of the most promising energy conversion devices, is the ion conduction limitations (Astill et al., 2009; Peron et al., 2011; Holdcroft, 2013; Modestino et al., 2013; Kusoglu and Weber, 2017) at the interface of ion-conducting polymer (ionomer) and catalyst particles on electrodes. The current state-of-the-art ionomer Nafion conducts protons very efficiently in bulk membrane separator [~25–50-μm thick (Choi et al., 2005; Xu et al., 2011), used in between two electrodes to conduct protons from anode to cathode]. But when the same ionomer is coated over a substrate as a submicron-thick film (Dishari and Hickner, 2013; Holdcroft, 2013; Modestino et al., 2013; Dishari, 2014; Kusoglu and Weber, 2017; Farzin et al., 2019) or over catalyst particles on cathodes as a ~2–30-nm thick catalyst-binder (Astill et al., 2009; Peron et al., 2011; Holdcroft, 2013), it shows significantly lower proton conductivity. The confinement effect (Richter and Kruteva, 2019) and interfacial interactions (Keddie et al., 1994) between water, polymer, and substrate start to dominate as the film thickness approaches a few multiples of the radius of gyration (Rg) of polymer chains (Innis-samson and Sakurai, 2011), impacting glass transition temperature (Keddie et al., 1994; Forrest and Dalnoki-veress, 2001; Roth and Dutcher, 2005), polymer chain diffusion coefficient/mobility (Frank and Gast, 1996), proton conductivity (Modestino et al., 2013), and many other properties (Campo, 2008; Smith et al., 2009). Moreover, Nafion is very expensive ($500/kg, according to the 2018 cost projection report of the Department of Energy's Fuel Cell Technologies Office) (James, 2018) and not eco-friendly because it is fluorocarbon based.
There have been significant efforts to design low cost, but efficiently proton-conducting, hydrocarbon-based ionomers to substitute fluorocarbon-based ionomers. Many of these ionomers exhibited proton conductivity comparable to or slightly higher than Nafion in bulk membrane format (Kreuer, 2001; Lufrano et al., 2001; Hickner et al., 2004; Miyatake et al., 2012; Chang et al., 2013, 2015; Li and Guiver, 2014; Lee et al., 2015; Li et al., 2015; Miyake et al., 2017) but they could not exceed Nafion considerable in sub-micron thick films. In general, poly(aromatic)-type hydrocarbon-based ionomers designed so far have some key attributes, which are distinctly different from fluorocarbon-based ionomers (Kreuer, 2001; Seung et al., 2004; Peron et al., 2011; Holdcroft, 2013), such as less acidic sulfonic acid groups at the side chain (pKa between −1 and −2) (Kreuer, 2001; Peron et al., 2011) as compared to perfluorosulfonic acid (pKa ~ −6) of Nafion) (Kreuer, 2001; Seung et al., 2004), less flexible backbone, less-pronounced hydrophilic-hydrophobic phase separation (Kreuer, 2001; Seung et al., 2004), low protonic mobility (Kreuer, 2001), and narrow ion-conducting channels (Kreuer, 2001) with dead-ends/pockets and high tortuosity (Kreuer, 2001). None of these features are favorable for proton conduction, especially for submicron-thick ionomer films (Astill et al., 2009; Peron et al., 2011; Holdcroft, 2013; Kusoglu and Weber, 2017; Karan, 2019). Therefore, just like perfluorinated ionomers, the reported polyaromatic ionomers experienced a drastic decrease in proton conductivity (about two orders of magnitude) (Ma et al., 2003; Peron et al., 2011) in catalyst layers from that in corresponding bulk membranes. The limited reports on polyaromatic ionomer-based catalyst layers suggested lower proton conductivity or higher binder-phase diffusion limitations as compared to Nafion (Ma et al., 2003; Sambandam and Ramani, 2010; Peron et al., 2011). Also, proton conductivity (Ma et al., 2003; Peron et al., 2011), and O2 permeability (Sambandam and Ramani, 2010) of a reaction layer (mimicking ionomer-based catalyst layer) made of sulfonated poly(ether sulfone) was significantly lower than a nafion-based one. These transport resistances are detrimental for oxygen reduction reaction kinetics and have hindered the transition from fluorocarbon to hydrocarbon-based catalyst binders. We, therefore, need new formulations of ionomers that are environment-friendly (fluorine-free), cost-effective, and at the same time, can conduct protons efficiently, especially under thin-film confinement.
Lignin has multiple structural features and characteristics, which makes it an ideal precursor of ionomer. Of the plant cell wall polymers, cellulose has been widely explored to make green electron (Zhu et al., 2016) or ion-conducting materials (Seo et al., 2009; Bayer et al., 2016; Zhu et al., 2016; Vilela et al., 2019). However, lignin has been underutilized as an ionomer and majorly used to synthesize porous carbon (graphene, carbon fiber) (Kontturi, 1988; Naseem et al., 2016) or charge storage materials (Kontturi, 1988) [requiring oxidation of phenol groups of lignin to quinone (Furman and Lonsky, 1988; Milczarek and Inganas, 2012; Milczarek and Nowicki, 2013; Thakur et al., 2014; Zhu et al., 2016) or mixing with quinone (Furman and Lonsky, 1988)]. Lignin is a three-dimensional (3D) amorphous, aromatic polymer functionalized with polar ether linkages (-O-) and hydroxyl (–OH) groups. These –OH groups are parts of p-coumaryl, coniferyl, and sinapyl alcohols, the three types of monolignols constituting lignin (Kim et al., 2011). The monolignols are connected with each other via interunit linkages (β-O-4, β-β, β-5, β-1, 5-5, and 4-O-5), giving the ultimate 3D, branched macromolecular structure and mechanical integrity (Kim et al., 2011). If ion-conducting functionalities, such as sulfonic acid (–SO3H) groups are covalently attached to the lignin structure, the neutral lignin polymer can act like an ionomer. The polar –OH groups and ether (-O-) linkages, alongside –SO3H groups, can attract water molecules inside the polymer network, form additional hydrogen-bonded (Grunwald and Puar, 1967; Nagamani et al., 2011, 2012; Ye et al., 2019) proton conduction pathways and facilitate water-mediated proton conduction within lignin sulfonate-based ionomeric materials. To date, a few works have been reported where neutral lignin (Uddin et al., 2017; Ye et al., 2019) or water-soluble, polyelectrolyte-like lignosulfonate (Zhang et al., 2006; Gonggo et al., 2012) were blended with a neutral polymer matrix material (polystyrene or polysulfone) to make composite membranes for methanol fuel cells and redox flow batteries. However, these demonstrations were limited to thick, bulk membranes only (not to submicron-thick films). Also, the lignosulfonates used in these reports (Zhang et al., 2006; Gonggo et al., 2012) and commercial lignosulfonates (Aro and Fatehi, 2017; Inwood et al., 2018) are highly water soluble that may lead to dissolution/degradation of ionomer membranes or thin films. The high water solubility of commercial lignosulfonate may originate from a high degree of sulfonation (Lange et al., 2013) (during the sulfite pulping process) and a low degree of cross-linking. Thus, for practical applicability in hydration-mediated ion conduction, we need to limit the water solubility of lignin sulfonate.
As our first set of efforts along this direction, here we report kraft lignin-based synthesis of ionomers. We explored properties of these ionomers in submicron-thick films to demonstrate the excellent ion conduction properties of lignin-based ionomers under thin-film confinement. Kraft lignin (Figure 1A) is generally precipitated by acidification (using CO2 and/or H2SO4) of black liquor of kraft pulping (Lange et al., 2013; Thakur et al., 2014; Carvajal et al., 2016; Upton and Kasko, 2016; Aro and Fatehi, 2017) and is made commercially available as a water-insoluble (at low to neutral pH) (Thakur et al., 2014), neutral polymer [0.23–3 wt% sulfur, but no sulfonated content (Upton and Kasko, 2016; Aro and Fatehi, 2017)]. We sulfomethylated and subsequently cross-linked this commercial kraft lignin (Aro and Fatehi, 2017) to yield sulfonated lignin ionomers, LS x, where x is the ion exchange capacity of the ionomer (Figures 1B,C). By tuning the ratio of reactants (Aro and Fatehi, 2017), LS x ionomers with controlled ion exchange capacities (IECs) were achieved. Controlled sulfonation and subsequent cross-linking limited the water solubility of the LS x ionomers and fulfilled a critical requirement of practical ionomers for many forms of fuel cells, redox flow batteries, and electrolyzers. In order to understand and explain the route to the higher thin-film proton conductivity of LS x ionomers over Nafion, we measured the water uptake, density, ionic domain characteristics, and water mobility/stiffness (qualitative) of the same films as a function of relative humidity (% RH). While further investigations on mechanical, chemical, and thermal stabilities are needed for this new range of ionomers, this work provides valuable insights into the proton conduction and morphological behavior of LS x ionomers under thin-film confinement. This may guide future designs of lignin-based ionomers.
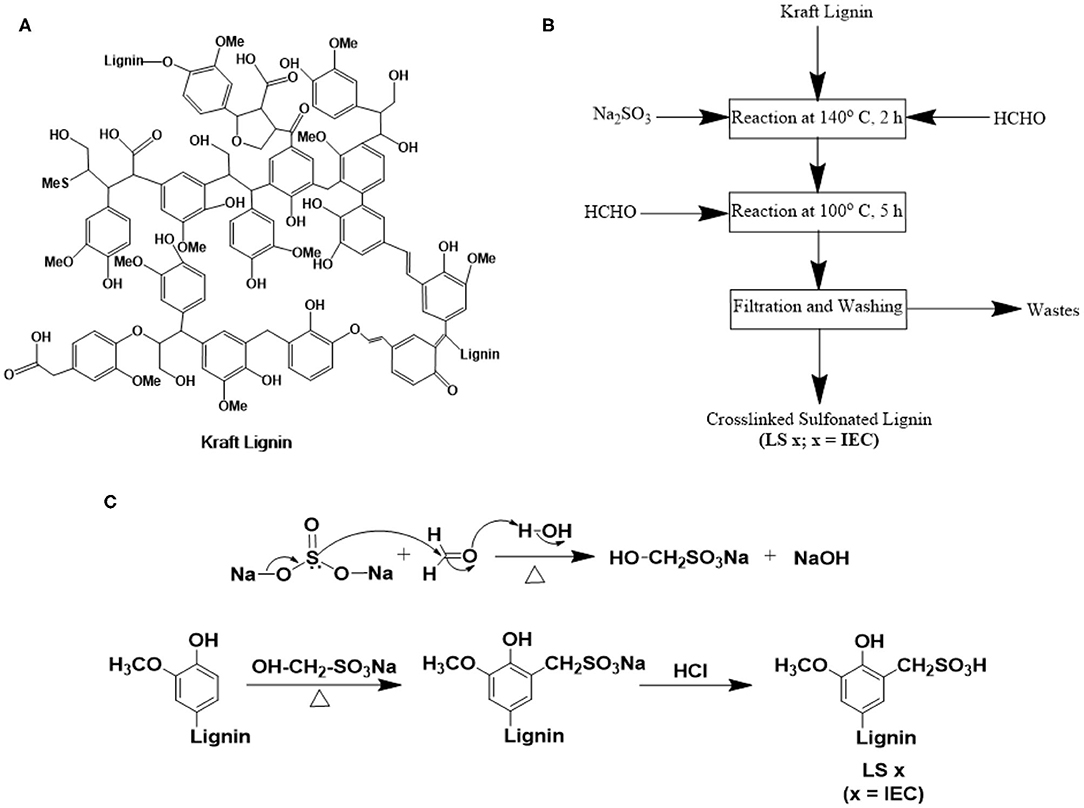
Figure 1. (A) Chemical structure of kraft lignin (Lange et al., 2013); (B) step-by-step procedure; and (C) reaction mechanism of sulfonation of kraft lignin to yield lignin sulfonate ionomers (LS x, where x = IEC).
Kraft lignin [isolated from Norway Spruce, Mn ~ 8,564 g/mol, Mw ~ 21,795 g/mol, polydispersity index (PDI) ~ 2.54], 20 wt% Nafion solution (EW ~ 1,100, IEC ~ 0.909 meq/g), and rotor probe [9-(2-carboxy-2-cyanovinyl) julolidine] (CCVJ) were purchased from MilliporeSigma (St. Louis, MO). Sulfonation was done using the same lot of kraft lignin to maintain the original composition of lignin consistent for all LS x ionomers. To compare with the LS x ionomers (water insoluble) prepared in this work, water-soluble sodium lignosulfonate (LScom) was also purchased (Santa Cruz Biotechnology, Dallas, TX) and studied. Silicon wafers coated with native oxide of silicon (n-SiO2 wafers) were purchased from Wafer Pro (San Jose, CA). n-SiO2-supported gold interdigitated electrode (IDE) arrays (Revtek, Inc., Torrance, CA) and n-SiO2 coated 5 MHz Au crystals (Inficon, Syracuse, NY) were purchased for impedance and water uptake measurements of ionomer films, respectively. All the solvents, reagents, and filter papers (Whatman, grade 1) were purchased from Fisher Scientific (Hampton, NH).
The sulfonation of kraft lignin was carried out as described previously (Aro and Fatehi, 2017). Briefly, 1 g of kraft lignin was dissolved in 3 mL of formaldehyde (HCHO) to which anhydrous sodium sulfite (Na2SO3) was added and stirred for 2 h at 140°C to produce sulfomethylated kraft lignin. The ratio of lignin to Na2SO3 was maintained as 1:0.5, and 1:0.8 to obtain LS 1.6 and LS 3.1 ionomers, respectively. After 2 h, 2 mL of HCHO was added further to initiate the cross-linking reaction. The cross-linking took place for another 5 h at 100°C. In the end, a few drops (~200–300 μL) of 1 M HCl were added to the reaction mixture to convert –CH2SO3Na to –CH2SO3H groups. Once the reaction was complete, the reaction mixture was washed thrice with water (to remove water-soluble fractions of LS x), filtered using filter paper, and finally dried to yield LS x ionomers as brown solids. The yields of dried LS x were calculated to be ~69% for both IECs (1.6, 3.1). The experimentally measured molecular weights and PDIs were: LS 1.6: Mn ~ 11,000, Mw ~ 28,000, PDI ~ 2.54; and LS 3.1: Mn ~ 13,000, Mw ~ 33,427, PDI ~ 2.57. This PDI was significantly lower than that of commercial lignosulfonate from Norway Spruce (PDI ~ 4–9) (Lange et al., 2013).
The water-insoluble, dry powder of LS x was then mixed with an acetone-water (3:1) mixture, vortexed for 5 min, and ultrasonicated for 30 min. Even after ultrasonication, some of the LS x (~ <30%) did not dissolve in the acetone-water solvent system, which could be due to the higher degree of cross-linking. By centrifugation (8,500 rpm), the acetone-water soluble fraction of LS x was thus separated from undissolved fraction. This protocol was consistently followed for any further sample preparation.
Typically, IECs of ionomers are calculated by soaking the dry ionomer in a 1 M NaCl solution to exchange the protons (H+) with Na+ ions. The H+ ions, transferred into the aqueous solution (Smitha et al., 2003; He and Frank, 2014) was then titrated with 0.01 N NaOH. Phenolphthalein was used during the titration as a pH indicator. Since LS x ionomers were brown powder, it was difficult to monitor the pH change during the titration. Therefore, as an alternate approach, we mixed LS x (the fraction soluble in 3:1 acetone: water) with Nafion (LS x:Nafion = 1:9 w/w) and made composite membranes, soaked in NaCl solution (for ion exchange and transfer of H+ from membrane to aqueous solution), and then titrated as mentioned earlier. Equation (1) was used to calculate the IECs of the composite membranes:
Equation (2) (Lavorgna, 2009) was then used to calculate the IECs of LS x present in the composite membranes:
We repeated this process in three replicates for each batch of LS x to confirm the IEC, and the values varied within ±2% of the one reported here. The sulfonation of kraft lignin through sulfomethylation reactions was also confirmed by performing the elemental analysis (described next).
Elemental analysis of kraft lignin and LS x were carried out to determine the elemental composition by sending the samples to Galbraith Laboratories (Knoxville, TN). The samples were dried at 80°C for 6 h before the measurements. The wt% of carbon (C), hydrogen (H), and nitrogen (N) were measured using PerkinElmer 2400 Series II CHNS/O analyzer; while the Thermo Finnigan FlashEATM 1112 elemental analyzer and LECO SC-632 carbon/sulfur determinator were used to determine the wt% of oxygen (O), and sulfur (S), respectively.
TGA was carried out to determine the thermal stability and decomposition temperature of kraft lignin and LS x using TGA 209 F1 Libra (resolution: 0.1 μg) operating under a nitrogen environment. The scans were done from room temperature (25°C) to 600°C with a heating rate of 5°C/min.
To determine the Tg of powder LS x ionomers (dried under vacuum at 100°C, overnight), differential scanning calorimetry measurements were carried out using DSC 204F1 Phoenix under nitrogen environment from room temperature 22°C to 250°C, with 5 and 10°C/min as heating and cooling rates, respectively.
We started with the 1–10 wt% dry LS x powder and followed the protocol mentioned in the earlier section to prepare LS x ionomer solution dissolved in a 3:1 acetone-water mixture. This solution was then used to make LS x ionomer films with thickness ~20–200 nm. Also, 20 wt% Nafion stock solution was diluted with ethanol to yield 0.5–5 wt% dispersion for making Nafion films with similar thickness. n-SiO2 wafers were used as substrates for making ionomer films unless otherwise stated. The wafers were cut into small pieces (2 × 2.5 cm) and cleaned using the following steps: (1) dust particles were removed from substrate surface using compressed air, (2) the substrate was then rinsed with acetone and ethanol, dried with compressed air, and finally (3) treated using a UV-ozone cleaner for 20 min. The ionomer films were then spin-coated using EC 101 spin coater (Headway Research, Inc., Garland, TX) for 1.5 min. The spinning speed was varied in the range of 1,000–7,000 rpm to obtain films with different thicknesses. The spun films were placed inside a vacuum oven [VWR (Model # 1415 M), Radnor, PA], dried at 42°C for 3 h, annealed at 100°C for 7 h, and cooled down to room temperature for 12 h under vacuum. The films were then placed inside an appropriate humidity chamber and exposed to air with varied RHs for different measurements.
The thickness of annealed films was measured at ambient condition (~20–30% RH) using variable angle (65–75°) spectroscopic ellipsometry (α-SETM, J.A. Woollam Co., Inc., Lincoln, NE) with a spectral wavelength range of 381–893 nm. The thickness of the n-SiO2 on a bare silicon wafer (~1.77 nm) was used as a reference for ellipsometric modeling. The Cauchy model was used to obtain the thickness of all the ionomer films.
The impedance of ionomer films was measured using a Solartron 1260a Impedance/Gain-Phase analyzer (Solartron Analytical, Leicester, England), which was connected to a probe station with four gold probes, a Peltier temperature stage, a temperature controller, and an environmental chamber (Nextron, Busan, South Korea). The thin-film impedance and ion conductivity were measured using a 2-microprobe-based technique as used by others (Modestino et al., 2013). Here, gold IDEs, fabricated on a silicon wafer (110) with a thermally grown SiO2 as an electrically insulating layer, were used as substrate. Each IDE had 110 gold teeth. Each tooth was 10-μm wide. The teeth were spaced 100 μm apart, with an overlapping length of 8 mm. Ionomer films (~20–250 nm thick) were spin-coated on these IDEs and annealed following the procedure mentioned in the “Thin-Film Preparation” section.
After annealing, the parts of the films covering the contact pads were removed using a small brush wetted with the same solvent as used to spin-coat the selected ionomers. This process was repeated as needed to ensure the contact pads were as clean as possible. The contact pads were allowed to dry before the impedance measurement. Then the film-coated IDEs were quickly placed on a temperature stage (set at 22°C) inside the environmental chamber, and the gold tip probes were carefully positioned onto the IDE contact pads. The environmental chamber was maintained at 90% RH by bubbling a stream of dry supply air [0.5 SCFH (~235 ccm)] through a bottle containing saturated Na2SO4 solution (placed in a water bath at 23°C) and passing the humid air through the environmental chamber. A humidity sensor was placed at the outlet of the environmental chamber to monitor and ensure that the target RH was attained and maintained inside the chamber.
After the sample was placed inside the chamber, its impedance was measured periodically in the range of 1–10 kHz to monitor the equilibration of the sample. Once equilibrated at 90% RH, the impedance response of the humidified ionomer film was collected in the range of 10 MHz−1 Hz at 100 mV AC potential. The impedance data were then fitted to an appropriate equivalent circuit model (Figure S3) using ZView software (Scribner Associates, Southern Pines, NC), which gave the value of film resistance (Rf). The details of the rationale of choosing the appropriate components for the equivalent circuit model and the corresponding fits are presented in the supporting information (Figure S4 and Table S2). The film resistance (Rf), film thickness (t), and the specifications of the IDEs [spacing between teeth of IDE electrodes (d), length of each teeth (l), number of teeth (N)] were then used in the Equation (3) to calculate the values of ion conductivity (kf):
Impedance measurements were done using two different IDE designs where IDE teeth spacing (100 and 40 μm) and teeth width (10 and 8 μm) were varied. In both cases, the obtained film resistance values were similar (within 10–20% variation between measurements on two IDEs). Therefore, for all the measurements reported here, IDEs with teeth spacing of 100 μm and width of 10 μm were used.
Water uptake of ionomers films was measured using quartz crystal microbalance (QCM) (Stanford Research Systems, Sunnyvale, CA). Nafion, LS 1.6, and LS 3.1 films were spin-coated on n-SiO2/Au coated 5 MHz crystals followed by the annealing procedure stated in the “Thin-Film Preparation” section. Ionomer film was placed on the crystal holder inside a custom-built plastic humidity chamber connected to an ibidi humidifying system (ibidi USA, Inc., Fitchburg, WI). The total mass of water sorbed by ionomer films (Δm) at each RH was calculated from the corresponding frequency change (Δf ) of the films using the Sauerbrey equation (Equation 4):
where fo is the resonant frequency (Hz) of the fundamental mode of the crystal (5 MHz here), A is active crystal area (1.27 cm2), ρq is the density of quartz crystal (2.648 g/cm3), and μq is the shear modulus of quartz (2.947 × 1011 g/cm.sec2) (Kushner and Hickner, 2017)1. The dry mass of each ionomer film was obtained from Δf of the QCM crystal before and after depositing the film (measured at <10% RH). Furthermore, the mass of water uptake for each ionomer film was corrected by subtracting the water uptake of bare QCM crystals at the same %RH since water accumulates within the porous structure of the SiO2 layer (Kobayashi et al., 2002; Shim et al., 2015; Kushner and Hickner, 2017). Hydration numbers (λw), moles of water per mole of sulfonic acid, at different RH values were calculated using Equation (5):
where m0 and mRH were the sample masses at the dry state and a certain RH, respectively. MH2O was the molecular mass of water, and IEC was the ion-exchange capacity of polymer.
The density of the films was measured by dividing the mass/area (from films on n-SiO2-coated QCM crystals) by volume/area (from the thickness measurement of a film with a similar thickness on n-SiO2 wafers using SE). Here, it was assumed that the mass of the ionomer /area of films made on n-SiO2 crystal and n-SiO2 wafer were similar.
Ionic domain characteristics within ionomer films were investigated using the RSAXS technique at ~60% RH using a Rigaku Smartlab Diffractometer operating at 40 kV and 44 mA using a sealed Cu anode X-ray tube with an average wavelength (λ*) of X-ray as 1.5418 . RSAXS is a special case of grazing incidence SAXS (GISAXS) technique where the thin film sample is illuminated at grazing incidence angle, but the off-specular reflection signal is captured using a point detector (0D approach). While the 2D detector (used for conventional GISAXS) has the advantage of simultaneously measuring scattering intensities in both in-plane (qp) and out of plane (qz) directions, using the 0D approach, one can resolve one of these (qp or qz) components at a time (Ogi and Inaba, 2011).
In RSAXS measurement, asymmetric ω/2θ geometry (i.e., ω≠θ, where ω and 2θ are incidence and scattering angles, respectively) was used to collect the data. The scan was performed by intentionally offsetting ω by an appropriate angle, in this case, to avoid the specular reflection (in specular reflection, θi = θf; where θi and θf are the incidence and exit angles, respectively), and captured the off-specular scattering (where θi ≠ θf) signal (Ito, 2009). The scattered beams were scanned in the angular range (2θ) of 0–6° (step size = 0.02°). From this off-specular scattering, an in-plane scattering component (qp) was obtained, which provided the in-plane structure of the sample (Muller-Buschbaum, 2009). Thin films of Nafion and LS x, with a thickness of ~250 nm, were deposited on n-SiO2 wafers. The films were then placed on a sample holder disc and optically aligned. The distance between the sample and the detector was 30 cm. The in-plane scattering vector, qp is directly related to its scattering angle, 2θ and x-ray wavelength, λ* by Equation (6):
The spacing between ionic domains (d) within the films was determined based on their primary scattering peak maxima, qp, max in their specified region (~1.0–2.00 nm−1 for LS x; ~1.60–3.50 nm−1 for Nafion films) using Equation (7):
In the cases where the H+-form of ionomer did not provide enough scattering contrast (e.g., LS x), the ionomers were converted to Cs+-form (Tricoli, 1998; Shi et al., 2016). As mentioned earlier, to prepare an LS x solution, the undissolved fraction had to be removed from the dissolved one (in acetone-water) by centrifugation first. The mass of this undissolved fraction was difficult to quantify but was considerably small and neglected thereby. For ion exchange, the number of moles of H+ ions present in 10 wt% LS x solution was thus calculated first [by multiplying the IEC (mmol of –SO3H/g polymer) by total weight of LS x ionomer used to make the solution] based on a 10 wt% LS x solution (before centrifugation) and an equivalent number of moles of Cs+ ions (in the form of CsCl) was added to the solution subsequently. This yielded a 1 wt% Cs+ ion in the final solution. The solutions were spin-coated on prewashed n-SiO2 wafers to prepare ~200–230 nm-thick films followed by the annealing procedure described in the “Thin-Film Preparation” section.
The surface morphology of the annealed Nafion and LS x films on n-SiO2 wafers were investigated using AFM (MFP-3D-BIO AFM; Oxford Instruments Asylum Research, Santa Barbara, CA) at ambient condition. An AC240TS silicon cantilever (Olympus Microcantilevers, Tokyo, Japan) with a nominal spring constant of 10.06 N/m and a nominal tip radius of 7 nm was used for imaging in tapping mode. The measurements were performed in 10 × 10 nm2 and 1 × 1 μm2 scans with a scan rate of 0.2 Hz and 256 scanning lines.
An appropriate volume from a stock solution of CCVJ (in DMSO) was added to LS x solutions to yield 5–10 wt% LS x solutions containing 0.07 wt% CCVJ. n-SiO2 wafers were washed following the protocol described in the “Thin-Film Preparation” section. The dye-ionomer solutions were then spin-coated on these prewashed n-SiO2 wafers at 3,000 rpm to prepare ~100 and ~250 nm-thick films. The films were annealed following the same procedure mentioned earlier in the “Thin-Film Preparation” section. The films were then placed inside a quartz humidity chamber (2.25″ × 2.25″ × 2.875″) and exposed to air with varied %RH to measure fluorescence spectra using steady-state fluorescence spectroscopy (PTI Quantamaster 400, Horiba, NJ). The excitation wavelength (λexc) of CCVJ was 440 nm, while the emission wavelength (λem) ranged between 470 and 560 nm. Same spectroscopic parameters (excitation/emission slit width = 1 mm; step size = 10 under excitation correction and zero bias) were used to measure fluorescence of all samples.
We first converted the neutral kraft lignin to lignin sulfonate ionomers with different IECs (LS x) following literature (Aro and Fatehi, 2017; Zhang et al., 2017) (please see the Experimental section for synthetic details). The major steps of LS x ionomer synthesis were: (1) a sulfomethylation reaction starting with neutral lignin, (2) acidification to convert –CH2SO3Na to –CH2SO3H; and (3) cross-linking the polymer to minimize water solubility (Figures 1B,C). Since this was our first effort to design LS x ionomers, we chose the kraft lignin from Norway Spruce (PDI ~ 2.54) for its commercial availability. All LS x ionomers, reported here were synthesized using the same lot of kraft lignin to ensure the consistent composition of the starting material. Sulfomethylation was done by reacting kraft lignin with Na2SO3 and HCHO. The use of Na2SO3 and HCHO together allowed a sulfonation reaction at mild condition, where Na2SO3 and HCHO reacted together to produce sodium hydroxymethane sulfonate (OH-CH2-SO3Na) first. This molecule attacked the C-5 positions of benzene rings of kraft lignin in the next step to yield sulfonated lignin in Na-form (–CH2SO3Na form) (Zhang et al., 2017). Subsequent reaction with HCl converted Na-form to H-form (i.e., –CH2SO3H form). This sulfomethylation route was more convenient as compared to sulfonation done using Na2SO3 alone where high temperature and pressure were needed (to attack C-α position of kraft lignin structural units) (Zhang et al., 2017). The Na2SO3-HCHO based sulfonation strategy was also more advantageous over the one based on butane sultone-NaOH (Tongiani et al., 2005; Rosu et al., 2013; Zhang et al., 2017) as the former one kept the –OH groups intact in the LS x structure (Zhang et al., 2017). These –OH groups can (1) itself take part in ion conduction as hydrogen bond donor and acceptor (Nagamani et al., 2011, 2012), and (2) facilitate hydrophilic ion channel formation alongside the ether linkages and –SO3H groups of LS x ionomer chains. The ratio of lignin-to-Na2SO3 was the key to control the IEC values of LS x ionomers (described in the “Synthesis of Lignin Sulfonate” subsection under the “Methods” section).
The compositions of kraft lignin and LS x ionomers obtained from combustion-based elemental analysis (Table 1) were in close agreement with what was obtained from XPS (Table S1). Both analyses confirmed increased sulfonation of lignin with the increase in IEC. The wt% of S increased from 1.69 wt% (neutral kraft lignin) to 3.48 wt% for LS 1.6 and 7.29 wt% for LS 3.1 as shown in Table 1. Also, XPS spectra provided evidence of the presence of S in the form of –SO3H in LS x samples (Figure S1). It is to be noted that the ~1.69 wt% of S present in neutral kraft lignin (Table 1) was majorly due to S-containing functional groups [such as thioethers (–SCH3)] other than sulfonic acid (–SO3H) and in agreement with prior literature (Upton and Kasko, 2016; Aro and Fatehi, 2017).
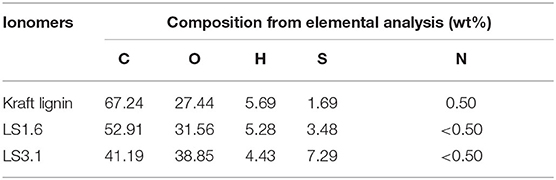
Table 1. Elemental analysis of kraft lignin and LS x ionomers.
Due to the complex structure of lignin, its glass transition temperature (Tg) is often more difficult to detect than the synthetic polymers. In such cases, the Tg was either represented as a discrete value (Thielemans et al., 2002; Ayoub and Venditti, 2013) or as a range of temperature (Sameni et al., 2014; Thakur et al., 2014). The reported value of Tg for kraft lignin is ~142°C (Thielemans et al., 2002) (or 124–174°C) (Thakur et al., 2014). We found the Tg of LS 1.6 and LS 3.1 to be ~140 and ~170°C, respectively from DSC (Figure S2a). Tg was seen to increase with the increase in IEC of LS x, which was typical and could be attributed to the hindered internal rotation owing to increased intermolecular interactions by hydrogen bonding of –SO3H groups (Noshay and Robeson, 1976; Zaidi et al., 2000). Any degradation below 120°C was considered as moisture loss which was prominent for LS 3.1, based on TGA (Figure S2b). A clear degradation started from ~130°C for LS 3.1 and ~150°C for LS 1.6. Within the temperature range of ≥100–400°C, degradation of –SO3H groups in many hydrocarbons (Marani et al., 2006; Koziara et al., 2016; Takenaka et al., 2018) and formation of CO, CO2, and SO2 (Lemes et al., 2010) were reported for sodium lignosulfonate. Thus, the decomposition of LS x above 100°C could be a combined effect of the degradation of macromolecular structure (Hulin et al., 2015) as well the –SO3H groups present in LS x ionomers. Since –SO3H groups may degrade just above 100°C, we heat-treated (rather than annealed) our LS x samples at 100°C for all our measurements (unless otherwise stated). This was done to ensure that there was no moisture left in LS x films and the dry films can be subjected to gradually increased %RHs.
Figure 2 represents the ion conductivity of ~20–200 nm thick Nafion, LS 1.6, LS 3.1, and LScom films at 90% RH. The impedance curves were fitted into an equivalent circuit model (Figure S3a), and the fits are shown as solid lines in Figures 2A,B. The details on the choice of fitting parameters in the equivalent circuit model (Figure S3a), and the sample fits are shown in Figures S3b, S4. The diameter of the semicircular region of an impedance curve (Figures 2A,B) roughly indicates the resistance of that ionomer film on the gold IDE electrode. A visual inspection of the impedance curves of ~25-nm thick (Figure 2A) and ~85-nm thick (Figure 2B) Nafion, LS 1.6 and LS 3.1 films thus clearly indicated that Nafion has the lowest and LS 1.6 has the highest ion conductivity of all three ionomers at 90% RH for the whole film thickness range studied. The quantitative values of ion conductivity (obtained by fitting the impedance curves in equivalent circuit model), when plotted as a function of film thickness (Figure 2C), showed the trend: LS 1.6 > LS 3.1 ≥ Nafion; where LS 1.6 exhibited proton conductivity about an order of magnitude higher than Nafion over the whole film thickness range. Nafion had an IEC lower than LS 1.6, but even after IEC normalization (i.e., ion conductivity/IEC), the ion conductivity of LS 1.6 (8.12 mS/cm, ~170 nm-thick film at 90% RH) exceeded that of Nafion (1.20 mS/cm, ~170 nm-thick film at 90% RH).
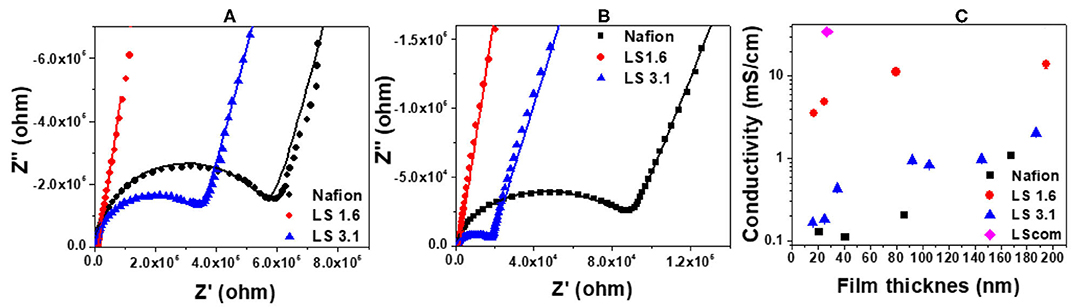
Figure 2. Comparison of electrochemical impedance response of Nafion, LS 1.6, and LS 3.1 films with the thickness (A) ~25 nm, and (B) ~85 nm. The dotted and straight lines represent the original impedance data and the fits to the data (using an equivalent circuit model), respectively; and (C) conductivity values (with error bars) of Nafion, LS 1.6, LS 3.1, and LScom films are plotted as a function of film thickness. All the films were heat-treated at 100°C, and the conductivity values (with fit errors) were measured at ~90% RH and ~22°C.
A significant decrease in ion conductivity was observed for LS 1.6 and LS 3.1 when the film thickness went below ~80 nm (Figure 2C) suggesting a stronger confinement effect in films thinner than this value. The confinement effect and interfacial attractive interactions of –OH and –SO3H groups of LS x ionomers with the substrate (–SiOH groups of the n-SiO2 substrate) may also have been in effect as expected (Dishari and Hickner, 2013). Even at this highly confined state, LS 1.6 retained consistently high proton conductivity (Figure 2C). At a film thickness (~20 nm thick) comparable to the ionomer layer at catalyst interfaces of hydrogen fuel cell electrodes, the conductivity values were ~3.52, ~0.17, and ~0.13 mS/cm for LS 1.6, LS 3.1, and Nafion, respectively (Figure 2C). The data shown above are for films heat-treated at 100°C. When Nafion and LS 1.6 films (~20-nm thick) were annealed at their corresponding Tgs [100°C (Nafion); 140°C (LS 1.6)], the conductivity values were ~0.13 and ~0.15 mS/cm, respectively (data not shown in Figure 2). All of these suggested that LS x ionomers with controlled IEC can be potential candidates for cost-effective catalyst binders. Commercial lignosulfonate (LScom), on the other hand, showed ion conductivity (~34.72 mS/cm in ~25 nm-thick films, Figure 2C) higher than LS x ionomers at similar film thickness. However, the high conductivity of LScom could be due to its high water solubility and thus limited the practical application of LScom as an ionomer for energy technologies. Despite high IEC, LS 3.1 showed proton conductivity lower than LS 1.6 (Figure 2C). This is not unusual and in some cases it is to excessive water uptake leading to dilution of ions in bulk membranes (Xu et al., 2014; Lin et al., 2017; Li et al., 2018; Long et al., 2019). However, for thin ionomer films, the connection between IEC and ion conductivity may not be that straight forward as many other factors (such as ion mobility, organization of ion-conducting groups/phase segregation, interfacial interactions among water-ionomer and substrate, and more) (Farzin et al., 2019) may come into play in a complex manner to give rise to a certain value of ion conductivity.
The water uptake of Nafion and LS x films were measured and corresponding hydration numbers (λw) were plotted as a function of film thickness and %RH (Figure 3). While Nafion (Figure 3A) and LS 3.1 (Figure 3C) showed thickness-dependent water uptake behavior, the water uptake by LS 1.6 films (Figure 3B) was relatively less sensitive to film thickness. In the thicker films (e.g., ~250 nm-thick films), water uptake was the highest for LS 1.6 with a trend: LS 1.6 (λw ~ 12.22) > LS 3.1 (λw ~ 6.237) > Nafion (λw ~ 2.76). But in thinner films (~25 nm thick), λw of all three ionomers were almost similar [15.38 (Nafion); 16.04 (LS 1.6); 19.59 (LS 3.1) at 90% RH] and very high. The higher water uptake in thinner films has been reported many times (Dishari and Hickner, 2012, 2013; Modestino et al., 2013; Farzin et al., 2019). The consistently high-water uptake of LS 1.6 at all film thickness could be attributed to its relatively low Tg (140°C) and low dry-state stiffness. On the contrary, the high Tg (170°C) and corresponding higher extent of hydrogen bonding between –SO3H and –OH groups in the dry polymer may have made LS 3.1 films stiff enough at dry state impeding the water uptake. Having said that, the higher water uptake did not necessarily lead to higher proton conductivity. This observation was consistent with multiple reports on Nafion films with a thickness range similar to ours (Dishari and Hickner, 2012, 2013; Farzin et al., 2019). Despite having similar water uptake, different proton conductivity values of ~25 nm-thick LS 1.6 film (3.6 mS/cm) from LS 3.1 (0.18 mS/cm) and Nafion (0.13 mS/cm) films thus suggested the importance of other factors. Some of the already-identified factors influencing confined ionomeric systems are state (Kim et al., 2003; Seung et al., 2004) (frozen vs. loosely bound vs. free water), mobility (Dishari and Hickner, 2012; Dishari et al., 2018), and distribution (interfacial vs. bulk) (Dishari and Hickner, 2012; Decaluwe et al., 2018; Dishari et al., 2018; Shrivastava et al., 2018) of water, film nanostructure, and ionic domain characteristics (Dishari and Hickner, 2013; Farzin et al., 2019) within the films. The role of water-polymer mobility (or stiffness) and ionic domain characteristics on water uptake and proton conductivity are discussed later in detail with experimental evidence.
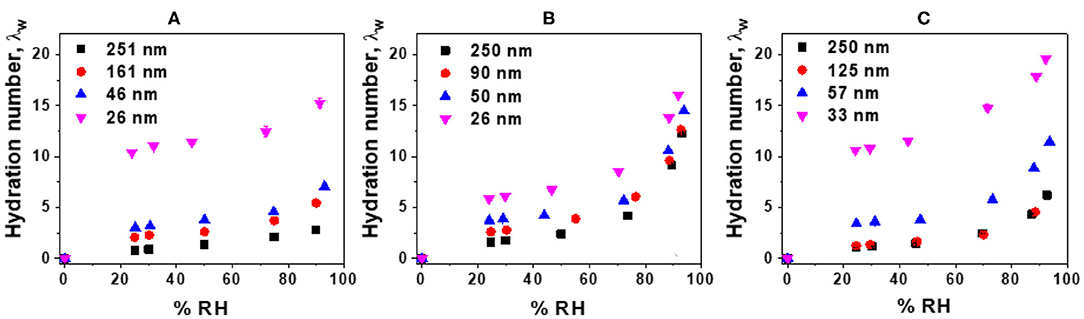
Figure 3. Corrected hydration numbers (λw) (with error bars) as a function of %RH: (A) Nafion, (B) LS 1.6, and (C) LS 3.1 films deposited on SiO2/Au crystal.
Table 2 represents the density of Nafion, LS 1.6, and LS 3.1 at two different thicknesses (25 and 250 nm). LS x ionomer films were less dense than Nafion films at comparable thickness. Lignin macromolecules are branched by nature. Moreover, the acetone-soluble fractions of LS x (used in this work) can possess a higher degree of branching as compared to water soluble fractions (Crestini et al., 2017). This branched architecture may have worked in favor of creating less dense ionomeric materials, unlike other hydrocarbon-based ionomers. For example, a ~250 nm-thick sulfonated polysulfone (S-Radel) film had a density of ~1.45 g/cm3 (Dishari et al., 2018), which was higher than LS 1.6 film with a similar thickness (~1.29 g/cm3, Table 2). The less compact, branched structure may potentially have facilitated 3D ion channel formation and led to higher ion conductivity of LS 1.6 over Nafion and S-Radel films. On the contrary, LS 3.1 showed lower proton conductivity despite having the lowest film density. While the density was the lowest, the Tg was the highest in LS 3.1 without a significant increase in molecular weight (from LS 1.6), suggesting high intramolecular cross-linking during the chemical reaction. Such chemical cross-linking is likely to involve the –CH2OH groups as a result of which the cross-linked material may not be able to leverage the polar –OH groups to create ion conduction pathways.
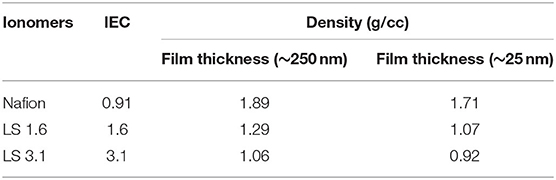
Table 2. Density of Nafion and LS x ionomers in thin films.
It was also noted that the thinner films (~25 nm thick) showed lower density than thicker (~250 nm thick) films (Table 2) and was in agreement with prior literature (Shi et al., 2000; Petrina, 2013; Dishari et al., 2018). As compared to a thicker film, a thinner film is made with more dilute (i.e., lower wt% of polymer) and less viscous solution of a polymer with less chain entanglement (Wu et al., 1994). Moreover, we made ionomer solutions with highly volatile solvents [such as acetone (for LS x) or ethanol (for Nafion)]. During the spin-coating of a thinner film, the solvent from the dilute polymer solution evaporates so fast that the polymer chains get locked (non-equilibrium conformation) without getting any chance to reorganize, leaving voids within the thinner films (Shi et al., 2000; Petrina, 2013; Dishari et al., 2018). Chains in such non-equilibrium conformations get an opportunity to equilibrate if the films are annealed above the glass transition temperature (Wu et al., 1994). However, we could not do so for LS x as around Tg (~140–170°C for LS x), the –SO3H groups were likely to degrade (discussed earlier). This is why thinner films of LS x ionomers retained lower density than thicker films. Nafion, despite annealing above its Tg, showed a decrease in density in thinner films at ambient condition (Table 2). This could be attributed to stronger surface interactions of Nafion with SiO2 (an effect similar to polymer chain locking), which retained after annealing as evident from the film stiffening at ~20% RH and beyond (Figure 4, discussed in the next section) and prior works (Dishari and Hickner, 2012, 2013).
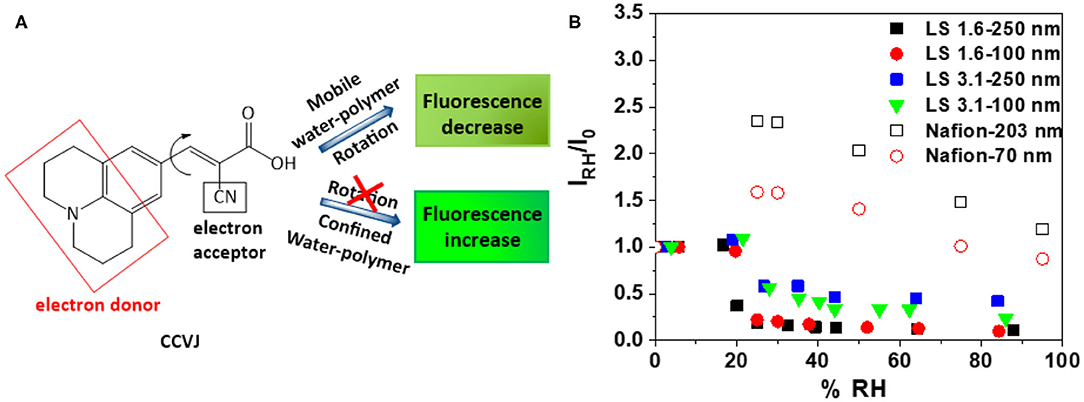
Figure 4. (A) Chemical structure and working principle of fluorescent rotor probe CCVJ; and (B) relative fluorescence intensity (IRH/I0) of CCVJ in ~100 nm and ~250 nm-thick LS 1.6, LS 3.1, and Nafion films as a function of %RH [(λexc)CCVJ = 440 nm, (λem)CCVJ = 470–560 nm].
While ionomers can be stiff at dry state, hydration can induce a different degree of stiffening or antiplasticization of ionomer films and increase/decrease the water-polymer mobility within films. Since (1) ion conductivity is a function of both ion concentration and ion mobility; and (2) ion mobility is impacted by water-polymer mobility, it is important to explore the mobility within the ionomer films. A qualitative, but conventional way (Goodelle et al., 2002; Ellison et al., 2004; Dishari and Hickner, 2012, 2013; Dishari et al., 2018) to predict the mobility inside thin ionomer films is to incorporate fluorescent rotor probes inside films and monitor the RH induced changes in its fluorescence response (Figure 4). Here, we used CCVJ (Figure 4A) as the rotor probe. In general, the fluorescence of this dye increases when it experiences a constricted environment (Goodelle et al., 2002; Ellison et al., 2004; Dishari and Hickner, 2012, 2013; Dishari et al., 2018). On the other hand, the fluorescence of the dye decreases when polymeric matrix around it plasticizes or softens. The fluorescence intensity of CCVJ in ionomer films was recorded at dry reference state (I0) and different elevated RH (IRH). Thus, an increase in the ratio (IRH/I0) will indicate RH-induced film stiffening or reduced mobility of water molecules and ionomer chains within the film.
As the dry (0% RH) films were hydrated to ~20% RH (Figure 4B), the water-polymer mobility in LS 1.6 and LS 3.1 films changed negligibly (i.e., IRH/I0 changed negligibly). On the contrary, the Nafion films showed a drastic decrease in mobility (or high antiplasticization) when RH increased from 0 to 20% RH. Above 20% RH, the Nafion films plasticized, but not to the extent like LS x films. The low mobility of water molecules may have contributed to the lower ion conductivity of Nafion films since water molecules have to continuously rotate, break, and form hydrogen bonds for efficient proton conduction.
Another thing to note here is the dry state mobility/stiffness of the films. By normalizing the dry state fluorescence of CCVJ in ionomers films (I0) with corresponding ionomer film thickness (L), we were able to roughly approximate the relative differences in stiffness of the films of the three ionomers at similar thickness. I0/L values for ~250 nm-thick films were 119, 222, and 504 nm−1 for Nafion, LS 1.6, and LS 3.1, respectively. This indicated that at dry state, LS 3.1 was already stiffer than LS 1.6 (consistent with Tg of LS 3.1 higher than LS 1.6 shown earlier); while the mobility did not decrease much further upon humidification (Figure 4B). This also indicated that the stiffening of Nafion was more humidity dependent than LS x. Based on IRH/I0 (Figure 4B) and I0/L values, it can also be inferred that the stiffening of LS 1.6 films (both dry and hydrated state) happened in moderation, favoring its ion conductivity (Figure 2). The higher RH-induced antiplasticization of Nafion (over LS x) could be attributed to the flexible perfluorosulfonic acid chain, which may reach out to the water and –SiOH groups for hydrogen bonding.
Figure 5 represents in-plane RSAXS patterns of Nafion, LS 1.6, and LS 3.1 films at 60% RH. A common practice to (1) enhance the electron density contrast between hydrocarbon matrix and the ionic cluster, and (2) see the ionic domain peaks using RSAXS, is to convert hydrocarbon-based ionomers from H+ to Cs+ form (Sivashinsky and Tanny, 1983; Li et al., 2010). We did so for LS x ionomers as we could not see any ionic domain peak in LS x films in H+ form (LS x-H+); while scattering peaks were prominent in Cs+ form (LS x-Cs+) (Figure 5). On the contrary, Nafion did not show any ionic domain peak in Cs+ form in our thin films, while it showed peaks in H+ form. A similar observation for Nafion has been reported by others (Tricoli, 1998; Shi et al., 2016). It was suggested that Cs+ ions may have strongly interacted with –SO3H groups of Nafion causing disruption (Shi et al., 2016) of ionic domains, and the disappearance of ionic domain peak in Nafion-Cs+. While it was difficult to be certain whether the evolution of scattering peak in LS x-Cs+ was due to (1) increase in electron density contrast between ionic and non-ionic regions of the ionomer, or (2) alter morphologies/internal structure within ionic domains (Shi et al., 2016), we did a couple of measurements. First of all, the UV/Vis absorbance of LS 3.1 (Figure S5a) at 210 and 280 nm [characteristic of aromatic groups in lignosulfonate (Zhou et al., 2016)] did not show any difference between H+ and Cs+ forms. Second, no significant change in full width at half maxima (FWHM) or shift of ionic domain peak was observed as the extent of Cs+ ion exchange was increased (Figure S5b), suggesting an insignificant alteration of ionic domain spacing and distribution in LS 3.1 due to ion exchange. Finally, the ion conductivity (kf) values of LS 1.6-H+ and LS 1.6-Cs+ were not drastically different [(kf)LS1.6−H+ ≈ 2 (kf)LS1.6−Cs+]; while Nafion-H+ showed conductivity more than an order of magnitude higher than Nafion-Cs+ (Shi et al., 2016). Thus, it can be inferred that Cs+ ion mainly induced electron density contrast in LS x ionomer films.
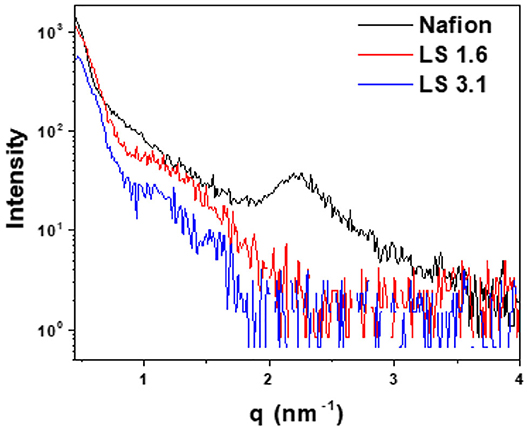
Figure 5. In-plane RSAXS profiles of ~200–250 nm thick Nafion (H+ form), LS 1.6 (Cs+ form), and LS 3.1 (Cs+-form) films on n-SiO2 wafers at 60% RH.
Based on FWHM (Table 3), LS 1.6 and LS 3.1 had slightly broader distributions of ionic domain size and spacing as compared to Nafion (~0.74 nm−1) in films (Eastman et al., 2012; Kusoglu et al., 2016; Kusoglu and Weber, 2017). The average values of d-spacing (=2π/qmax) for LS 1.6 (~4.52 nm) and LS 3.1 (~5.61 nm) were larger than Nafion (~2.79 nm) (Table 3). The closed packing of neighboring ionic domains in Nafion films agreed with a large degree of phase mixing as observed in our prior work (Modestino et al., 2013; Farzin et al., 2019). Also, smaller d-spacing was often accompanied by the smaller size of ionic domains for ionomer films (Kreuer, 2001; Hwang et al., 2011; Farzin et al., 2019). The d-spacing we reported here for submicron-thick LS x films were larger than some well-known hydrocarbon-based ionomers even in several tens of micron thick, bulk membrane format (Sivashinsky and Tanny, 1983; Kreuer, 2001; Chang et al., 2013). This possibly suggested that LS x ionomers may have ionic domains bigger than those hydrocarbon and Nafion ionomers. By fitting the ionic domain peak to different models developed for specific self-assembled geometry [e.g., cylindrical, core-shell (spherical), or oblate spheroid (ellipsoid)], ionic domain shape and size are often predicted (Yarusso and Cooper, 1983; Matsuoka et al., 1988; Choi et al., 2010; Kreuer and Portale, 2013; Farzin et al., 2019). We attempted to do so for all three ionomers using Nanosolver (a built-in model of RSAXS system) to obtain the size of ionic domains within films (Table 4).
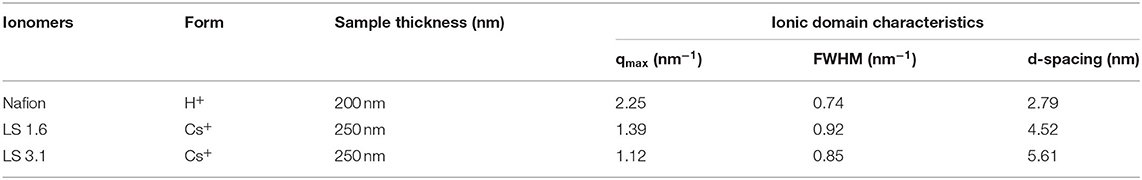
Table 3. qmax and domain spacing (d-spacing) of Nafion, LS 1.6, and LS 3.1 films on n-SiO2 from in-plane RSAXS data at ~60% RH.
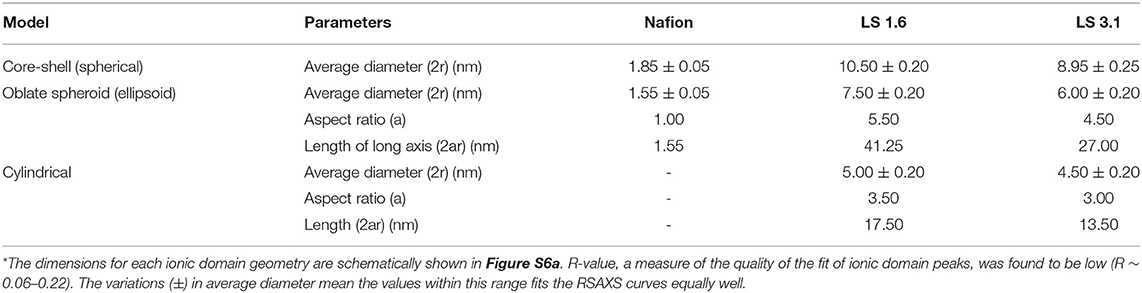
Table 4. Comparison of the fitting parameters of ~200–250 nm-thick Nafion, LS 1.6, and LS 3.1 films at 60% RH obtained by fitting ionic domain peaks (RSAXS) using Nanosolver*.
For Nafion, we used a core-shell-based spherical model only as of the existing literature on Nafion bulk membrane (Gebel, 2000; Haubold et al., 2001) and thin films (Farzin et al., 2019) strongly support that. At 90% RH, the volume fraction of water sorbed by Nafion thin film was <0.25, which did not support the formation of cylindrical, connected morphology (Farzin et al., 2019). Even when we tried to fit ionic domain peaks of Nafion films in a spheroid model (Table 4), we found an aspect ratio of 1.0, which strongly supported the formation of spherical ionic domains in Nafion films.
On the other hand, above critical aggregation concentration (CAC), a broad range of geometries of self-assembled structures has been proposed for water-soluble sodium lignosulfonate [CAC ~ 0.38 g/L (Qiu et al., 2010)], such as oblate (in solutions and films) (Pasquini et al., 2002; Vainio et al., 2008; Yan et al., 2010), irregularly shaped flat disc-like (in films) (Goring et al., 1979; Qiu et al., 2010), and spherical (in solution) (Kontturi, 1988; Qiu et al., 2010). Please note that not all of this water-soluble lignosulfonate is commercial (i.e., some of those were made from different natural sources in the lab). Therefore, we avoided using the term “LScom” onward, whenever appropriate. Also, we should keep in mind that all the literature reporting varied morphology do also vary in the sources of lignin (Goring et al., 1979; Pasquini et al., 2002; Vainio et al., 2004), the process of making lignosulfonates (Rezanowich et al., 1964; Goring et al., 1979; Yan et al., 2010), molecular weight and polydispersity of polymers, and solution/film preparation conditions (Kontturi, 1988; Vainio et al., 2008); and no straightforward trend was found in structural geometry as a function of any of the above-mentioned parameters. This suggested that we should evaluate different possible geometries of LS x particles/domains in films rationally. We, therefore, primarily selected three major ionic domain geometries to fit the ionic domain peaks in LS 1.6 and LS 3.1 films for subsequent evaluation: core-shell, spheroid, and cylindrical (disc-like) (as shown in Figure S6a). The corresponding best-fit parameters for all three ionomers are tabulated (Table 4). Representative fits of the ionic domain peaks using Nanosolver are shown in Figures S6b,c.
By fitting the ionic domain peaks into a core-shell model, the average size of ionic domains was found to be the highest for LS 1.6 (~10.5 nm) and the lowest for Nafion (~1.85 nm) (Table 4). The size of the ionic domain as big as ~9–10.5 nm appeared to be too high [the largest reported ionic domain is ~4 nm of ~25–50 μm-thick Nafion membrane at hydrated state (Gebel, 2000)]. However, the diameter of a single lignosulfonate macromolecule was often roughly approximated to be ~8 nm (based on particle size distribution from dynamic light scattering measurements in solution) (Yan et al., 2010). This suggested that if our LS x films truly have core-shell ionic domain structure, the individual domains are likely to be made of single LS x macromolecules where the core is the ion-conducting region.
While the spherical morphology of lignosulfonate particles in solution is reported, the spherical morphology at the interface (Qiu et al., 2010) or in concentrated solution (Kontturi, 1988; Vainio et al., 2008) of these molecules is debatable (based on the interpretation of the exponent value in Mark-Howink equation correlating diffusion coefficient of a macromolecule to its molar mass) (Vainio et al., 2008). Our AFM topographical (Figure S7) and 3D (Figure S8) images of LS x ionomer films clearly indicated large oblate spheroid-like, elongated, aggregated structures standing perpendicular to the substrate. Therefore, we looked for more evidences based on SAXS to comment on the ionic domain size within LS x films. Vainio et al. (2008) fitted SAXS data of lignosulfonate particles, which yielded oblate-shaped particles with an aspect ratio of 3.5. The presence of such oblate spheroid structures has also been evidenced in solid samples of dry kraft lignin (SAXS, USAXS) (Vainio et al., 2004), Langmuir-Blodgett films of saccharified lignin (AFM) (Pasquini et al., 2002), and drop-cast lignosulfonate films on silicon flakes [environmental scanning electron microscopy (SEM)] (Yan et al., 2010). All of these prior evidences convinced us that the oblate spheroid (or ellipsoid) model can be a rational choice to fit our RSAXS data and obtain ionic domain size on LS x thin films.
The ionic domain size trend (LS1.6 > LS 3.1 > Nafion) was still valid when the dimensions of ionic domains of Nafion using the core-shell model and LS x using the ellipsoid model were compared (Table 4). When we employed the ellipsoid (or oblate spheroid) model to fit RSAXS data of our LS x films, the following dimensions of ionic domains were obtained: LS 1.6: diameter (2r) ~ 7.5 nm; length (2ar) ~ 41.25 nm; LS3.1: diameter (2r) ~ 6 nm; length (2ar) ~ 27 nm. The aspect ratios were found to be 5.5 (LS 1.6) and 4.5 (LS 3.1), which were higher than that for lignosulfonate in solution (~3.5) (Vainio et al., 2008). This could be attributed to the conformational difference between solution (Vainio et al., 2008) and solid-state as well as the nature of the solvent used [LS x (acetone-water) and lignosulfonate (water) used (Vainio et al., 2012)]. All of these factors can impact the solution and solid-state self-assembly and so are the aspect ratios of ionic domains.
If the average dimension of single LS x macromolecules in films was closer to what was reported for single lignosulfonate macromolecules in solution (~8 nm) (Yan et al., 2010), the oblate spheroidal domains in LS x films from RSAXS [length ~ 41 nm (LS 1.6); ~27 nm (LS 3.1); Table 4] suggested the likelihood of aggregation or association of multiple LS x macromolecules to form single oblate spheroidal ionic cluster or particle. Such association of up to 2–10 oblate spheroid-shaped lignosulfonate macromolecules has been suggested in solution (SAXS) (Vainio et al., 2008, 2012) and in the solid-state (SEM) (Yan et al., 2010). However, how the LS x molecules are organizing within these oblate clusters requires further investigation to understand. Based on the common understanding of ionomers in solid films, it can be predicted that when the %RH is not extremely high (i.e., when RH ≤ 90%), the sulfonic acid groups of LS x are located at the interior, and aromatic rings are located at the surface of the macromolecules. Thus, the hydrophobic interaction (Deng et al., 2012; Vainio et al., 2012; Ma et al., 2018) may drive the intermolecular association to give large aggregates. The AFM topography (Figure S7) and 3D images of LS x films (Figure S8) suggested a much stronger aggregation and formation of larger elongated clusters (as compared to the cluster size obtained from RSAXS, Table 4) at the air/film interface. However, as the film thickness decreased to ~20–25 nm, these ellipsoidal perpendicular features disappeared from AFM images (Figure S9). Despite that, the high ion conductivity of ~20 nm-thick LS 1.6 films (Figure 2C) suggested a rearrangement/reorientation of ionic domains still favorable for proton conduction.
On another note, the possibility of having disc-like/cylindrical structures [proposed by others (Goring et al., 1979; Vainio et al., 2012) of lignosulfonate at the interface] is less in LS x films. This is based on a simple hypothesis: if multiple cylinders are stacked to form large-scale structures, it is highly unlikely to obtain such elongated oblate shapes (seen in AFM images, Figure S8) out of cylindrical units, unless cylinders with gradually smaller diameter are systematically stacked from the center to edges. Without applying some precise self-assembly-based film preparation techniques, such hierarchical stacking may not be possible. With limited prior information available about lignin sulfonate similar to what we designed, it may not be ideal to try to make comments with more certainty than this.
High acidity of ion-conducting groups (Chang et al., 2011), and connected, hydrogen-bonded network of water molecules (Ueki and Watanabe, 2008) with sufficient rotational mobility (Laage, 2006) are considered as prerequisites for efficient proton conduction. As opposed to the long, flexible perfluorosulfonic acid side chains of Nafion (pKa ~ −14), the pKa of LS x should be less negative with short methylsulfonic acid side chains (predicted based on pKa reported for –C6H4SO3H as −2.5) (Chang et al., 2011). Therefore, LS x with low acidity was expected to exhibit lower proton conductivity as compared to highly acidic Nafion (Chang et al., 2011). However, it appeared that the significantly larger ionic domains/clusters (comprised of multiple LS x macromolecules) (Table 4) helped LS x films overcome the side-chain acidity effect. The branched, 3D, less-compact (low density, Table 2) structures likely offered ample free spaces (or free volumes) within the hydrophilic interior of individual LS x macromolecules. Based on experimental evidences, we anticipated that within this hydrophilic interior, the close proximity of –SO3H and polar ether and –OH groups attracted water molecules, allowed them to move, and assisted connectivity between individual macromolecules to create ion-conducting cluster networks. The rotor probe experiments (Figure 4) provided evidence of high-water mobility within these networks; while the high aspect ratio of the LS 1.6 clusters (based on the spheroid model) supported the good connectivity among clusters made of individual LS x macromolecules. Together, it created a bulk-like ion-conducting environment [which so far has been defined as ~4-nm size ionic domains in bulk Nafion membrane (Gebel, 2000)] in submicron-thick LS x films. As the film thickness of LS 1.6 approached ~20 nm, the protruded, ellipsoidal, perpendicular features disappeared (Figure S9), but the ion conductivity was still maintained at high value suggesting reorientation/alignment of ionic domains, which was still favorable for ion conduction. As opposed to some of the traditional hydrocarbon-based ionomers (Kreuer, 2001; Seung et al., 2004; Peron et al., 2011; Chang et al., 2013), the phase segregated, large, and connected ionic domains showed the promise of LS 1.6 as an ionomer for thin ion-conducting materials.
LS 3.1, on the other hand, showed ion conductivity lower than LS 1.6 but higher than Nafion in most of the thickness range we studied (Figure 2C). The low ion conductivity of LS 3.1 was in agreement with its higher dry-state Tg (~170°C) as compared to LS 1.6 (Tg ~ 140°C) and could be attributed to the higher extent of hydrogen-bonding between –SO3H and –OH groups in LS 3.1 As a result, LS 3.1 films were already stiff enough at the dry state (further supported by I0/L of CCVJ) and might have impeded the water uptake (Figure 3C). At hydrated state, LS 3.1 films retained this high stiffness (insignificant change in IRH/I0, Figure 4) and low water mobility, which had a negative impact on proton conductivity. On the other hand, both Nafion and LS 1.6 films were less stiff than LS 3.1 films in the dry state. But upon humidification, Nafion films significantly stiffened up, while LS 1.6 films did not. The comparison of IRH/I0 (Figure 4) values suggested that at 60% RH, Nafion was still approximately six times stiffer than LS 1.6 and LS 3.1. Therefore, the low stiffness, high water mobility, larger ionic domains all together led to such a high ion conductivity of LS 1.6 as compared to Nafion and LS 3.1 films.
We have systematically synthesized sulfonated kraft lignin (LS x) (from Norway Spruce) with controlled IECs and limited water solubility and explored the potential of LS x as an ion conductor in submicron-thick films. While commercial lignosulfonate (LScom) is completely water soluble, we successfully limited the water solubility of LS x (through adjustable polymer cross-linking), while making them suitable for hydration-mediated proton conduction. The ion conductivity followed a trend: LS 1.6 > LS 3.1 ≥ Nafion in 20–200 nm-thick films at 90% RH. Using LS 1.6, ion conductivity about an order of magnitude higher than Nafion was achieved in submicron-thick films, which suggested the great potential of LS x-based ionomers in overcoming ion conduction limitations at the ionomer-catalyst interface. Water uptake, density, dimension and d-spacing of ionic clusters, and qualitative estimation of the extent of water-polymer mobility (or film stiffness) were obtained for all three ionomers in films. The higher value of proton conductivity of LS 1.6 was believed to be an individual or synergistic effect of: (1) the presence of –OH groups in close proximity of –SO3H groups in highly branched, 3D structure; (2) low film density, (3) higher water uptake (in thicker films only), (4) lower stiffness (higher water mobility), and (5) significantly larger ionic domains with domain connectivity. On the other hand, denser Nafion films with lower water uptake (thicker films only), smaller ionic domains, and significant hydration-induced antiplasticization led to lower ion conductivity of Nafion films over LS 1.6 films (and LS 3.1 over some thickness range). For LS 3.1 films, higher dry-state Tg and stiffness were observed. This suggested that higher IEC of an ionomer may not be beneficial to ion conductivity if the ionomer chains and water experience stronger confinement. As LS 1.6 ionomer, made from cheap and abundant kraft lignin, exhibited much higher ion conductivity over Nafion in films with a thickness comparable to catalyst binder layer (~2–30 nm), it may have a great potential to address ion conduction limitations. Such understanding of ion conduction behavior of LS x in thin films, along with further consideration of material viability (to be reported in the future) can inform and guide the future design of lignin-based ionomers for energy conversion and storage devices. This also potentially indicates a new way to valorize waste lignin from industrial (biorefinery or pulp/paper industries) and natural resources.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
SF performed sulfonation, water uptake, TGA, DSC, RSAXS, SE, XPS, elemental analysis, density, CCVJ experiments, and fitting of ionic domain peak in RSAXS. TJ performed EIS measurements, and EZ performed the UV/Vis experiments, while TJ and EZ worked together for AFM experiments. SC determined molecular weight and helped SF with the sulfonation of kraft lignin. SD, SF, and TJ were involved in manuscript writing. SD proposed the main concept of the work, supervised the experimental plans, led the manuscript writing, and revisions. All authors contributed to the article and approved the submitted version.
SD acknowledges supports from the Nebraska Center for Energy Science Research (NCESR) Competitive Grant (26-1217-0020-502, Cycle 14), Layman Award (NU Foundation 1024460), Research Council Faculty Seed Grant, University of Nebraska-Lincoln (UNL) core facility grant, and UNL start-up fund. SD acknowledges supports from the NSF CAREER Award (NSF-DMR # 1750040) for purchasing the EIS instrument. SF thanks NSF (NSF-DMR # 1750040) and NCESR Competitive Grant for partial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Nano-Engineering Research Core Facility (NERCF) at UNL for AFM measurements. SD thanks Dr. Kevin G. Yager (Brookhaven National Laboratory) for useful discussion on interpreting the RSAXS data. Part of this research (DSC, TGA, RSAXS, SE, XPS) was performed in the Nebraska Nanoscale Facility: National Nanotechnology Coordinated Infrastructure and the Nebraska Center for Materials and Nanoscience, which are supported by the National Science Foundation under Award NNCI-1542182, and the Nebraska Research Initiative.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00690/full#supplementary-material
1. ^Operation and Service Manual-Research Quartz Crystal Microbalance- Inficon.
Abu-dalo, M. A., Al-rawashdeh, N. A. F., and Ababneh, A. (2013). Evaluating the performance of sulfonated kraft lignin agent as corrosion inhibitor for iron-based materials in water distribution systems. Desalination 313, 105–114. doi: 10.1016/j.desal.2012.12.007
Aro, T., and Fatehi, P. (2017). Production and application of lignosulfonates and sulfonated lignin. ChemSusChem 10, 1861–1877. doi: 10.1002/cssc.201700082
Astill, T., Xie, Z., Shi, Z., Navessin, T., and Holdcroft, S. (2009). Factors influencing electrochemical properties and performance of hydrocarbon-based electrolyte PEMFC catalyst layers. J. Electrochem. Soc. 156, B499–B508. doi: 10.1149/1.3082119
Ayoub, A., and Venditti, R. (2013). “The effect of irradiation on the processability of lignin for carbon fiber applications,” in AIChE Annual Meeting, At Hilton San Francisco Union Square (San Francisco, CA).
Bayer, T., Cunning, B. V., Selyanchyn, R., Nishihara, M., Fujikawa, S., Sasaki, K., et al. (2016). High temperature proton conduction in nanocellulose membranes: paper fuel cells. Chem. Mater. 28, 4805–4814. doi: 10.1021/acs.chemmater.6b01990
Campo, E. A. (2008). “Thermal properties of polymeric materials,” in Selection of Polymeric Materials (Norwich, NY: William Andrew), 103–140. doi: 10.1016/B978-081551551-7.50005-X
Carvajal, J. C., Gómez, Á., and Cardona, C. A. (2016). Comparison of lignin extraction processes: economic and environmental assessment. Bioresour. Technol. 214, 468–476. doi: 10.1016/j.biortech.2016.04.103
Cerrutti, B. M., De Souza, C. S., Castellan, A., Ruggiero, R., and Frollini, E. (2012). Carboxymethyl lignin as stabilizing agent in aqueous ceramic suspensions. Ind. Crops Prod. 36, 108–115. doi: 10.1016/j.indcrop.2011.08.015
Chang, Y., Brunello, G. F., Fuller, J., Disabb-Miller, M. L., Hawley, M. E., Kim, Y. S., et al. (2013). Polymer electrolyte membranes based on poly(arylene ether sulfone) with pendant perfluorosulfonic acid. Polym. Chem. 4, 272–281. doi: 10.1039/C2PY20666H
Chang, Y., Brunello, G. F., Hawley, M., Kim, Y. S., Disabb-miller, M., Hickner, M. A., et al. (2011). Aromatic ionomers with highly acidic sulfonate groups: acidity, hydration, and proton conductivity. Macromolecules 44, 8458–8469. doi: 10.1021/ma201759z
Chang, Y., Mohanty, A. D., Smedley, S. B., Abu-Hakmeh, K., Lee, Y. H., Morgan, J. E., et al. (2015). Effect of superacidic side chain structures on high conductivity aromatic polymer fuel cell membranes. Macromolecules 48, 7117–7126. doi: 10.1021/acs.macromol.5b01739
Che, C., Vagin, M., Wijeratne, K., Zhao, D., Warczak, M., Jonsson, M. P., et al. (2018). Conducting polymer electrocatalysts for proton-coupled electron transfer reactions: toward organic fuel cells with forest fuels. Adv. Sustain. Syst. 2:1800021. doi: 10.1002/adsu.201800021
Choi, J. H., Kota, A., and Winey, K. I. (2010). Micellar morphology in sulfonated pentablock copolymer solutions. Ind. Eng. Chem. Res. 49, 12093–12097. doi: 10.1021/ie1002476
Choi, P., Jalani, N. H., and Datta, R. (2005). Thermodynamics and proton transport in nafion. J. Electrochem. Soc. 152, E123–E130. doi: 10.1149/1.1859814
Crestini, C., Lange, H., Sette, M., and Arguropoulos, D. S. (2017). On the structure of softwood kraft lignin. Green Chem. 19, 4104–4121. doi: 10.1039/C7GC01812F
Danner, T., Justnes, H., Geiker, M., and Lauten, R. A. (2015). Phase changes during the early hydration of Portland cement with Ca-lignosulfonates. Cem. Concr. Res. 69, 50–60. doi: 10.1016/j.cemconres.2014.12.004
Decaluwe, S. C., Baker, A. M., Bhargava, P., Fischer, J. E., and Dura, J. A. (2018). Structure-property relationships at nafion thin-film interfaces: thickness effects on hydration and anisotropic ion transport. Nano Energy 46, 91–100. doi: 10.1016/j.nanoen.2018.01.008
Deng, Y., Feng, X., Yang, D., Yi, C., and Qiu, X. (2012). π-π stacking of the aromatic groups in lignosulfonates. Bioresouces 7, 1145–1156. doi: 10.15376/biores.7.1.1145-1156
Dishari, S. K. (2014). Current understanding of proton conduction in confined ionomeric systems. PostDoc J. 2, 30–39. doi: 10.14304/SURYA.JPR.V2N4.3
Dishari, S. K., and Hickner, M. A. (2012). Antiplasticization and water uptake of nafion® thin films. ACS Macro Lett. 1, 291–295. doi: 10.1021/mz200169a
Dishari, S. K., and Hickner, M. A. (2013). Confinement and proton transfer in nafion thin films. Macromolecules 46, 413–421. doi: 10.1021/ma3011137
Dishari, S. K., Rumble, C. A., Maroncelli, M., Dura, J. A., and Hickner, M. A. (2018). Unraveling the complex hydration behavior of ionomers under thin film confinement. J. Phys. Chem. C 122, 3471–3481. doi: 10.1021/acs.jpcc.7b11888
Eastman, S. A., Kim, S., Page, K. A., Rowe, B. W., Kang, S., Soles, C. L., et al. (2012). Effect of confinement on structure, water solubility, and water transport in nafion thin films. Macromollecules 45, 7920–7930. doi: 10.1021/ma301289v
Ellison, C. J., Miller, K. E., and Torkelson, J. M. (2004). In situ monitoring of sorption and drying of polymer films and coatings: self-referencing, nearly temperature-independent fluorescence sensors. Polymer 45, 2623–2632. doi: 10.1016/j.polymer.2004.02.034
Farzin, S., Sarella, A., Yandrasits, M. A., and Dishari, S. K. (2019). Fluorocarbon-based ionomers with single acid and multiacid side chains at nanothin interfaces. J. Phys. Chem. C 123, 30871–30884. doi: 10.1021/acs.jpcc.9b10015
Forrest, J. A., and Dalnoki-veress, K. (2001). The glass transition in thin polymer films. Adv. Colloid Interface Sci. 94, 167–196. doi: 10.1016/S0001-8686(01)00060-4
Frank, B., and Gast, A. P. (1996). Polymer mobility in thin films. Macromolecules 29, 6531–6534. doi: 10.1021/ma960749n
Furman, G. S., and Lonsky, W. (1988). Charge-transfer complexes in kraft lignin part 1: occurance. J. Wood Chem. Technol. 8, 165–189. doi: 10.1080/02773818808070678
Gebel, G. (2000). Structural evolution of water swollen perfluorosulfonated ionomers from dry membrane to solution. Polymer 41, 5829–5838. doi: 10.1016/S0032-3861(99)00770-3
Gonggo, S. T., Radiman, C. L., Bundjali, B., and Arcana, I. M. (2012). Properties of polymer electrolyte membranes prepared by blending sulfonated polystyrene with lignosulfonate. ITB J. Sci. 44, 285–295. doi: 10.5614/itbj.sci.2012.44.3.8
Goodelle, J. P., Pearson, R. A., and Santore, M. M. (2002). Water-uptake kinetics in poly(methyl methacrylate) films with a fluorescent rotor probe. J. Appl. Polym. Sci. 86, 2463–2471. doi: 10.1002/app.10964
Goring, D. A. I., Vuong, R., Gancet, C., and Chanzy, H. (1979). The flatness of lignosulfonate macromolecules as demonstrated by electron microscopy. J. Appl. Polym. Sci. 24, 931–936. doi: 10.1002/app.1979.070240406
Grunwald, E., and Puar, M. S. (1967). Proton exchange of phenol in aqueous acid. J. Phys. Chem. 71, 1842–1845. doi: 10.1021/j100865a047
Haubold, H., Vad, T., Jungbluth, H., and Hiller, P. (2001). Nano structure of NAFION: a SAXS study. Electrochim. Acta 46, 1559–1563. doi: 10.1016/S0013-4686(00)00753-2
He, S. S., and Frank, C. W. (2014). Facilitating hydroxide transport in anion exchange membranes via hydrophilic grafts. J. Mater. Chem. A 2, 16489–16497. doi: 10.1039/C4TA02942A
Hickner, M. A., Ghassemi, H., Kim, Y. S., Einsla, B. R., and Mcgrath, J. E. (2004). Alternative polymer systems for proton exchange membranes (PEMs). Chem. Rev. 104, 4587–4612. doi: 10.1021/cr020711a
Holdcroft, S. (2013). Fuel cell catalyst layers: a polymer science perspective. Chem. Mater. 26, 381–393. doi: 10.1021/cm401445h
Holmberg, A. L., Reno, K. H., Wool, R. P., and Epps, T. H. (2014a). Biobased building blocks for the rational design of renewable block polymers. Soft Matter 10, 7405–7424. doi: 10.1039/C4SM01220H
Holmberg, A. L., Stanzione, J. F., Wool, R. P., and Epps, T. H. (2014b). A facile method for generating designer block copolymers from functionalized lignin model compounds. ACS Sustain. Chem. Eng. 2, 569–573. doi: 10.1021/sc400497a
Hulin, L., Husson, E., Bonnet, J., Stevanovic, T., and Sarazin, C. (2015). Enzymatic transesterification of kraft lignin with long acyl chains in ionic liquids. Molecules 20, 16334–16353. doi: 10.3390/molecules200916334
Hwang, G. S., Kaviany, M., Gostick, J. T., Kientiz, B., Weber, A. Z., and Kim, M. H. (2011). Role of water states on water uptake and proton transport in nafion using molecular simulations and bimodal network. Polymer 52, 2584–2593. doi: 10.1016/j.polymer.2011.03.056
Innis-samson, V. A., and Sakurai, K. (2011). Hydrophobic switching nature of methylcellulose ultra-thin films: thickness and annealing effects. J. Phys. Condens. Matter. 23:435010. doi: 10.1088/0953-8984/23/43/435010
Inwood, J. P. W., Pakzad, L., and Fatehi, P. (2018). Production of sulfur containing kraft lignin products. Bioresour. Technol. 13, 53–70. doi: 10.15376/biores.13.1.53-70
Ito, Y. (2009). Grazing-incidence small-angle x-ray scattering technique for nanostructure determination of surfaces and interfaces of thin films. Rigaku J. 25, 1–6.
James, B. (2018). 2018 Cost Projections of PEM Fuel Cell Systems for Automobiles and Medium-Duty Vehicles (DOE-Fuel Cell Technologies Office). Available online at: https://www.energy.gov/sites/prod/files/2018/04/f51/fcto_webinarslides_2018_costs_pem_fc_autos_trucks_042518.pdf.
Kai, D., Tan, J., Chee, L., Chua, K., and Yap, L. (2016). Towards lignin-based functional materials in a sustainable world. Green Chem. 18, 1175–1200. doi: 10.1039/C5GC02616D
Kalliola, A., Vehmas, T., Liitiä, T., and Tamminen, T. (2015). Alkali-O2 oxidized lignin-a bio-based concrete plasticizer. Ind. Crops Prod. 74, 150–157. doi: 10.1016/j.indcrop.2015.04.056
Karan, K. (2019). Interesting facets of surface, interfacial, and bulk characteristics of perfluorinated ionomer films. Langmuir 35, 13489–13520. doi: 10.1021/acs.langmuir.8b03721
Keddie, J. L., Jones, R. A. L., and Cory, R. A. (1994). Interfacial and surface effect on the glass-transition temperature in thin polymer films. Faraday Discuss. 98, 219–230. doi: 10.1039/fd9949800219
Kim, J., Shin, E., Eom, I., Won, K., Kim, Y. H., Choi, D., et al. (2011). Structural features of lignin macromolecules extracted with ionic liquid from poplar wood. Bioresour. Technol. 102, 9020–9025. doi: 10.1016/j.biortech.2011.07.081
Kim, Y. S., Dong, L., Hickner, M. A., Glass, T. E., Webb, V., and Mcgrath, J. E. (2003). State of water in disulfonated poly(arylene ether sulfone) copolymers and a perfluorosulfonic acid copolymer (nafion) electrochemical properties. Macromolecules 36, 6281–6285. doi: 10.1021/ma0301451
Kobayashi, Y., Zheng, W., Chang, T. B., Hirata, K., Suzuki, R., Ohdaira, T., et al. (2002). Nanoporous structure of sputter-deposited silicon oxide films characterized by positronium annihilation spectroscopy. J. Appl. Phys. 91, 1704–1706. doi: 10.1063/1.1428787
Konduri, M. K., Kong, F., and Fatehi, P. (2015). Production of carboxymethylated lignin and its application as a dispersant. Eur. Polym. J. 70, 371–383. doi: 10.1016/j.eurpolymj.2015.07.028
Kontturi, A. (1988). Determination of diffusion coefficients and effective charge numbers of lignosulphonate. J. Chem. Soc. Faraday Trans. 84, 4033–4041. doi: 10.1039/f19888404033
Koziara, B. T., Kappert, E. J., Ogieglo, W., and Nijmeijer, K. (2016). Thermal stability of sulfonated Poly (ether ether ketone) films thermal stability of sulfonated poly (ether ether ketone) films: on the role of protodesulfonation. Macromol. Mater. Eng. 301, 71–80. doi: 10.1002/mame.201500075
Kreuer, K., and Portale, G. (2013). A critical revision of the nano-morphology of proton conducting ionomers and polyelectrolytes for fuel cell applications. Adv. Funct. Mater. 23, 5390–5397. doi: 10.1002/adfm.201300376
Kreuer, K. D. (2001). On the development of proton conducting polymer membranes for hydrogen and methanol fuel cells. J. Mem. Sci. 185, 29–39. doi: 10.1016/S0376-7388(00)00632-3
Kushner, D. I., and Hickner, M. A. (2017). Water sorption in electron-beam evaporated SiO2 on QCM crystals and its influence on polymer thin film hydration measurements. Langmuir 33, 5261–5268. doi: 10.1021/acs.langmuir.7b00759
Kusoglu, A., Dursch, T. J., and Weber, A. Z. (2016). Nanostructure/swelling relationships of bulk and thin-film PFSA ionomers. Adv. Funct. Mater. 26, 4961–4975. doi: 10.1002/adfm.201600861
Kusoglu, A., and Weber, A. Z. (2017). New insights into perfluorinated sulfonic-acid ionomers. Chem. Rev. 117, 987–1104. doi: 10.1021/acs.chemrev.6b00159
Laage, D. (2006). A molecular jump mechanism of water reorientation. Science 311, 832–835. doi: 10.1126/science.1122154
Lange, H., Decina, S., and Crestini, C. (2013). Oxidative upgrade of lignin–recent routes reviewed. Eur. Polym. J. 49, 1151–1173. doi: 10.1016/j.eurpolymj.2013.03.002
Lavorgna, M. (2009). Enhanced sol gel hybridization of nafion membrane for fuel cell applications (Ph.D. thesis). Loughborough University, Loughborough, United Kingdom.
Lee, S., Ann, J., Lee, H., Kim, J.-H., Kim, C.-S., Yang, T.-H., et al. (2015). Synthesis and characterization of crosslink-free highly sulfonated multi-block poly(arylene ether sulfone) multi-block membranes for fuel cells. J. Mater. Chem. A 3, 1833–1836. doi: 10.1039/C4TA05887A
Lemes, A. P., Waldman, W. R., and Dura, N. (2010). Effect of lignosulfonate on the thermal and morphological behavior of Poly (3-hydroxybutyrate-co-3-hydroxyvalerate). J. Polym. Environ. 18, 250–259. doi: 10.1007/s10924-010-0170-7
Li, G., Zhao, C., Li, X., Qi, D., Liu, C., Bu, F., et al. (2015). Novel side-chain type sulfonated diphenyl-based poly(arylene ether sulfone)s with hydrogen-bonded network as proton exchange membranes. Polym. Chem. 6, 5911–5920. doi: 10.1039/C5PY00700C
Li, L., Lin, C. X., Wang, X. Q., Yang, Q., Zhang, Q. G., Zhu, A. M., et al. (2018). Highly conductive anion exchange membranes with long flexible multication spacer. J. Mem. Sci. 553, 209–217. doi: 10.1016/j.memsci.2018.02.048
Li, N., and Guiver, M. D. (2014). Ion transport by nanochannels in ion-containing aromatic copolymers. Macromolecules 47, 2175–2198. doi: 10.1021/ma402254h
Li, W., Manthiram, A., and Guiver, M. D. (2010). Acid-base blend membranes consisting of sulfonated poly(ether ether ketone) and 5-amino-benzotriazole tethered polysulfone for DMFC. J. Mem. Sci. 362, 289–297. doi: 10.1016/j.memsci.2010.06.059
Lin, C. X., Zhuo, Y. Z., Hu, E. N., Zhang, Q. G., Zhu, A. M., and Liu, Q. L. (2017). Crosslinked side-chain-type anion exchange membranes with enhanced conductivity and dimensional stability. J. Mem. Sci. 539, 24–33. doi: 10.1016/j.memsci.2017.05.063
Long, Z., Miyake, J., and Miyatake, K. (2019). Partially fluorinated polyphenylene ionomers as proton exchange membranes for fuel cells: effect of pendant multi-sulfophenylene groups. ACS Appl. Energy Mater. 2, 7527–7534. doi: 10.1021/acsaem.9b01513
Lufrano, F., Gatto, I., Staiti, P., Antonucci, V., and Passalacqua, E. (2001). Sulfonated polysulfone ionomer membranes for fuel cells. Solid State Ionics 145, 47–51. doi: 10.1016/S0167-2738(01)00912-2
Ma, C., Zhang, L., Mukerjee, S., Ofer, D., and Nair, B. (2003). An investigation of proton conduction in select PEM's and reaction layer interfaces-designed for elevated temperature operation. J. Mem. Sci. 219, 123–136. doi: 10.1016/S0376-7388(03)00194-7
Ma, R., Guo, M., and Zhang, X. (2018). Recent advances in oxidative valorization of lignin. Catal. Today 302, 50–60. doi: 10.1016/j.cattod.2017.05.101
Marani, D., Di Vona, M. L., Traversa, E., Licoccia, S., Beurroies, I., Llewellyn, P. L., et al. (2006). Thermal stability and thermodynamic properties of hybrid proton-conducting polyaryl etherketones. J. Phys. Chem. B 110, 15817–15823. doi: 10.1021/jp062085h
Matsuoka, H., Tsurumi, M., and Ise, N. (1988). Small-angle x-ray scattering study of aqueous solutions of sulfonated calix[6]arene derivatives. Phys. Rev. B 38, 6729–6826. doi: 10.1103/PhysRevB.38.6279
Milczarek, G., and Inganas, O. (2012). Renewable cathode materials from biopolymer/conjugated polymer interpenetrating networks. Science 335, 1468–1472. doi: 10.1126/science.1215159
Milczarek, G., and Nowicki, M. (2013). Carbon nanotubes/kraft lignin composite: characterization and charge storage properties. Mater. Res. Bull. 48, 4032–4038. doi: 10.1016/j.materresbull.2013.06.022
Miyake, J., Taki, R., Mochizuki, T., Shimizu, R., Akiyama, R., Uchida, M., et al. (2017). Design of flexible polyphenylene proton-conducting membrane for next-generation fuel cells. Sci. Adv. 3, 1–8. doi: 10.1126/sciadv.aao0476
Miyatake, K., Hirayama, D., Bae, B., and Watanabe, M. (2012). Block poly(arylene ether sulfone ketone)s containing densely sulfonated linear hydrophilic segments as proton conductive membranes. Polym. Chem. 3, 2517–2522. doi: 10.1039/c2py20318a
Modestino, M. A., Paul, D. K., Dishari, S., Petrina, S. A., Allen, F. I., Hickner, M. A., et al. (2013). Self-assembly and transport limitations in confined nafion films. Macromolecules 46, 867–873. doi: 10.1021/ma301999a
Muller-Buschbaum, P. (2009). “Chapter 3: a basic introduction to grazing incidence small-angle x-ray scattering,” in Applications of Synchroton Light to Scattering and Diffraction in Materials and Life Sciences, eds T. A. Ezquerra, M. Garcia-Gutierrez, A. Nogales, and M. Gomez (Heidelberg: Springer), 61–90. doi: 10.1007/978-3-540-95968-7_3
Nagamani, C., Guo, J., and Thayumanavan, S. (2012). Synthesis and characterization of phenol-based biaryl proton conducting polymers. J. Polym. Sci. A Polym. Chem. 50, 1187–1196. doi: 10.1002/pola.25880
Nagamani, C., Viswanathan, U., Versek, C., Tuominen, M. T., Auerbach, S. M., and Thayumanvan, S. (2011). Importance of dynamic hydrogen bonds and reorientation barriers in proton transport. Chem. Commun. 47, 6638–6640. doi: 10.1039/c1cc11207d
Naseem, A., Tabasum, S., Zia, K. M., Zuber, M., Ali, M., and Noreen, A. (2016). Lignin-derivative based polymer, blends and composites: a review. Int. J. Biol. Macromol. 93, 296–313. doi: 10.1016/j.ijbiomac.2016.08.030
Norgren, M., and Edlund, H. (2014). Lignin: recent advances and emerging applications. Curr. Opin. Colloid Interface Sci. 19, 409–416. doi: 10.1016/j.cocis.2014.08.004
Noshay, A., and Robeson, L. M. (1976). Sulfonated polysulfone. J. Appl. Polym. Sci. 20, 1885–1903. doi: 10.1002/app.1976.070200717
Ogi, A., and Inaba, K. (2011). X-ray thin-film measurement technique VI. Small angle X-ray scattering. Rigaku J. 27, 1–5.
Pasquini, D., Balogh, D. T., Antunes, P. A., Constantino, C. J. L., Curvelo, A. A. S., Aroca, R. F., et al. (2002). Surface morphology and molecular organization of lignins in langmuir-blodgett films. Langmuir 18, 6593–6596. doi: 10.1021/la025729v
Peron, J., Shi, Z., and Holdcroft, S. (2011). Hydrocarbon proton conducting polymers for fuel cell catalyst layers. Energy Environ. Sci. 4, 1575–1591. doi: 10.1039/c0ee00638f
Petrina, S. A. (2013). Water sorption, viscoelastic, and optical properties of thin nafion® films (Ph.D. thesis). State College, PA: Department of Materials Science and Engineering, The Pennsylvania State University.
Qiu, X., Kong, Q., Zhou, M., and Yang, D. (2010). Aggregation behavior of sodium lignosulfonate in water solution. J. Phys. Chem. B 114, 15857–15861. doi: 10.1021/jp107036m
Rezanowich, A., Yean, W. Q., and Goring, D. A. I. (1964). High resolution electron microscopy of sodium lignin sulfonate. J. Appl. Polym. Sci. 8, 1801–1802. doi: 10.1002/app.1964.070080429
Richter, D., and Kruteva, M. (2019). Polymer dynamics under confinement. Soft Matter 15, 7316–7349. doi: 10.1039/C9SM01141B
Rosu, A., Rafin, C., Surpateanu, G., Brabie, G., Miron, D. N., and Veignie, E. (2013). Synthesis of alkylated potato starch derivatives and their potential in the aqueous solubilization of benzo[a]pyrene. Carbohydr. Polym. 93, 184–190. doi: 10.1016/j.carbpol.2012.06.062
Roth, C. B., and Dutcher, J. R. (2005). Glass transition and chain mobility in thin polymer films. J. Electroanal. Chem. 584, 13–22. doi: 10.1016/j.jelechem.2004.03.003
Sambandam, S., and Ramani, V. (2010). Influence of binder properties on kinetic and transport processes in polymer electrolyte fuel cell electrodes. Phys. Chem. Chem. Phys. 12, 6140–6149. doi: 10.1039/b921916a
Sameni, J., Krigstin, S., Rosa, D. D. S., Leao, A., and Sain, M. (2014). Thermal characteristics of lignin residue from industrial processes. Bioresources 9, 725–737. doi: 10.15376/biores.9.1.725-737
Seo, J. A., Kim, J. C., Koh, J. K., Ahn, S. H., and Kim, J. H. (2009). Preparation and characterization of crosslinked cellulose/sulfosuccinic acid membranes as proton conducting electrolytes. Ionics 15, 555–560. doi: 10.1007/s11581-009-0314-8
Seung, Y., Hickner, M. A., Dong, L., Pivovar, B. S., and Mcgrath, J. E. (2004). Sulfonated poly (arylene ether sulfone) copolymer proton exchange membranes: composition and morphology effects on the methanol permeability. J. Mem. Sci. 243, 317–326. doi: 10.1016/j.memsci.2004.06.035
Shi, S., Weber, A. Z., and Kusoglu, A. (2016). Structure-transport relationship of perfluorosulfonic-acid membranes in different cationic forms. Electrochim. Acta 220, 517–528. doi: 10.1016/j.electacta.2016.10.096
Shi, Y., Liu, J., and Yang, Y. (2000). Device performance and polymer morphology in polymer light emitting diodes: the control of thin film morphology and device quantum efficiency. J. Appl. Phys. 87, 4254–4263. doi: 10.1063/1.373062
Shim, H. K. (Key), Paul, D. K., and Karan, K. (2015). Resolving the contradiction between anomalously high water uptake and low conductivity of nanothin nafion films on SiO2 substrate. Macromolecules 48, 8394–8397. doi: 10.1021/acs.macromol.5b02093
Shrivastava, U. N., Fritzsche, H., and Karan, K. (2018). Interfacial and bulk water in ultrathin films of nafion, 3M PFSA, and 3M PFIA ionomers on a polycrystalline platinum surface. Macromolecules 51, 9839–9849. doi: 10.1021/acs.macromol.8b01240
Sivashinsky, N., and Tanny, G. B. (1983). Ionic heterogeneities in sulfonated polysulfone films. J. Appl. Polym. Sci. 28, 3235–3245. doi: 10.1002/app.1983.070281018
Smith, K. E., Sawicki, S., Hyjek, M. A., Downey, S., and Gall, K. (2009). The effect of glass transition temperature on the toughness of photopolymerizable (meth)acrylate networks under physiological conditions. Polymer 50, 5112–5123. doi: 10.1016/j.polymer.2009.08.040
Smitha, B., Sridhar, S., and Khan, A. A. (2003). Synthesis and characterization of proton conducting polymer membranes for fuel cells. J. Mem. Sci. 225, 63–76. doi: 10.1016/S0376-7388(03)00343-0
Strategic Plan for a Thriving Sustainable Bioeconomy U.S. Department of Energy-Bioenergy Technologies Office. (2016). Available online at: https://www.energy.gov/sites/prod/files/2017/09/f36/beto_strategic_plan_december_2016.pdf.
Takenaka, M., Kimura, Y., and Ohara, H. (2018). Influence of decomposition temperature of aromatic sulfonic acid catalysts on the molecular weight and thermal stability of poly(l-lactic acid) prepared by melt/solid state polycondenstaion. Polymer 155, 218–224. doi: 10.1016/j.polymer.2018.09.046
Thakur, V. K., Thakur, M. K., Raghavan, P., and Kessler, M. R. (2014). Progress in green polymer composites from lignin for multifunctional applications: a review. ACS Sustain. Chem. Eng. 2, 1072–1092. doi: 10.1021/sc500087z
Thielemans, W., Can, E., Morye, S. S., and Wool, R. P. (2002). Novel applications of lignin in composite materials. J. Appl. Polym. Sci. 83, 323–331. doi: 10.1002/app.2247
Tongiani, S., Velde, D. V., Ozeki, T., and Stella, V. J. (2005). Sulfoalkyl ether-alkyl ether cyclodextrin derivatives, their synthesis, nmr characterization, and binding of 6α-methylprednisolone. J. Pharm. Sci. 94, 2380–2392. doi: 10.1002/jps.20367
Tricoli, V. (1998). Proton and methanol transport in poly (perfluorosulfonate) membranes containing Cs+ and H+ cations. J. Electrochem. Soc. 145, 3798–3801. doi: 10.1149/1.1838876
Uddin, M., Alaboina, P. K., Zhang, L., and Cho, S. (2017). A low-cost, environment-friendly lignin-polyvinyl alcohol nanofiber separator using a water-based method for safer and faster lithium-ion batteries. Mater. Sci. Eng. B 223, 84–90. doi: 10.1016/j.mseb.2017.05.004
Ueki, T., and Watanabe, M. (2008). Macromolecules in ionic liquids: progress, challenges, and opportunities. Macromolecules 41, 3739–3749. doi: 10.1021/ma800171k
Upton, B. M., and Kasko, A. M. (2016). Strategies for the conversion of lignin to high-value polymeric materials: review and perspective. Chem. Rev. 116, 2275–2306. doi: 10.1021/acs.chemrev.5b00345
Vainio, U., Lauten, R. A., Haas, S., Svedstro, K., Veiga, L. S. I., and Hoell, A. (2012). Distribution of counterions around lignosulfonate macromolecules in different polar solvent mixtures. Langmuir 28, 2465–2475. doi: 10.1021/la204479d
Vainio, U., Lauten, R. A., and Serimaa, R. (2008). Small-angle x-ray scattering and rheological characterization of aqueous lignosulfonate solutions. Langmuir 24, 7735–7743. doi: 10.1021/la800479k
Vainio, U., Maximova, N., Hortling, B., Laine, J., Stenius, P., Simola, L. K., et al. (2004). Morphology of dry lignins and size and shape of dissolved kraft lignin particles by x-ray scattering. Langmuir 20, 9736–9744. doi: 10.1021/la048407v
Vilela, C., Silvestre, A. J. D., Figueiredo, F. M., and Freire, C. S. R. (2019). Nanocellulose-based materials as components of polymer electrolyte fuel cells. J. Mater. Chem. A 7, 20045–20074. doi: 10.1039/C9TA07466J
Wu, W., Orts, W. J., Van Zanten, J. H., and Fanconi, B. M. (1994). Density profile of spin cast polymethylmethacrylate thin films. J. Polym. Sci. B Polym. Phys. 32, 2475–2480. doi: 10.1002/polb.1994.090321504
Xu, K., Oh, H., Hickner, M. A., and Wang, Q. (2011). Highly conductive aromatic ionomers with perfluorosulfonic acid side chains for elevated temperature fuel cells. Macromolecules 44, 4605–4609. doi: 10.1021/ma201188e
Xu, P. Y., Zhou, K., Han, G. L., Zhang, Q. G., Zhu, A. M., and Lin, Q. (2014). Fluorene-containing poly (arylene ether sulfone) s as anion exchange membranes for alkaline fuel cells. J. Mem. Sci. 457, 29–38. doi: 10.1016/j.memsci.2014.01.042
Yan, M., Yang, D., Deng, Y., Chen, P., Zhou, H., and Qiu, X. (2010). Influence of pH on the behavior of lignosulfonate macromolecules in aqueous solution. Colloid Surf. A Physicochem. Eng. Aspects 371, 50–58. doi: 10.1016/j.colsurfa.2010.08.062
Yarusso, D. J., and Cooper, S. L. (1983). Microstructure of ionomers: interpretation of small-angle x-ray scattering data. Macromolecules 16, 1871–1880. doi: 10.1021/ma00246a013
Ye, J., Cheng, Y., Sun, L., Ding, M., Wu, C., Yuan, D., et al. (2019). A green SPEEK/lignin composite membrane with high ion selectivity for vanadium redox flow battery. J. Mem. Sci. 572, 110–118. doi: 10.1016/j.memsci.2018.11.009
Zaidi, S. M. J., Mikhailenko, S. D., Robertson, G. P., Guiver, M. D., and Kaliaguine, S. (2000). Proton conducting composite membranes from polyether ether ketone and heteropolyacids for fuel cell applications. J. Mem. Sci. 173, 17–34. doi: 10.1016/S0376-7388(00)00345-8
Zhang, H., Bai, Y., Zhou, W., and Chen, F. (2017). Color reduction of sulfonated eucalyptus kraft lignin. Int. J. Biol. Macromol. 97, 201–208. doi: 10.1016/j.ijbiomac.2017.01.031
Zhang, X., Glusen, A., and Gracial-Valls, R. (2006). Porous lignosulfonate membranes for direct methanol fuel cells. J. Mem. Sci. 276, 301–307. doi: 10.1016/j.memsci.2005.10.018
Zhou, H., Yang, D., and Zhu, J. Y. (2016). Molecular structure of sodium lignosulfonate from different sources and their properties as dispersant of TiO2 slurry. J. Dispersion Sci. Technol. 37, 296–303. doi: 10.1080/01932691.2014.989572
Keywords: ionomer, thin films, Nafion, kraft lignin, proton conduction, energy
Citation: Farzin S, Johnson TJ, Chatterjee S, Zamani E and Dishari SK (2020) Ionomers From Kraft Lignin for Renewable Energy Applications. Front. Chem. 8:690. doi: 10.3389/fchem.2020.00690
Received: 14 May 2020; Accepted: 03 July 2020;
Published: 26 August 2020.
Edited by:
Jennifer L. Schaefer, University of Notre Dame, United StatesReviewed by:
Geoffrey M. Geise, University of Virginia, United StatesCopyright © 2020 Farzin, Johnson, Chatterjee, Zamani and Dishari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shudipto K. Dishari, c2Rpc2hhcmkyQHVubC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.