
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 28 July 2020
Sec. Chemical Physics and Physical Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00633
This article is part of the Research TopicVibrationally-Mediated Chemical DynamicsView all 9 articles
 Michael D. Horbury1*
Michael D. Horbury1* Matthew A. P. Turner2
Matthew A. P. Turner2 Jack S. Peters3
Jack S. Peters3 Matthieu Mention4
Matthieu Mention4 Amandine L. Flourat4
Amandine L. Flourat4 Nicholas D. M. Hine2
Nicholas D. M. Hine2 Florent Allais4
Florent Allais4 Vasilios G. Stavros2*
Vasilios G. Stavros2*The photochemistry and photostability of a potential ultraviolet (UV) radiation filter, dehydrodiethylsinapate, with a broad absorption in the UVA region, is explored utilizing a combination of femtosecond time-resolved spectroscopy and steady-state irradiation studies. The time-resolved measurements show that this UV filter candidate undergoes excited state relaxation after UV absorption on a timescale of ~10 picoseconds, suggesting efficient relaxation. However, steady-state irradiation measurements show degradation under prolonged UV exposure. From a photochemical standpoint, this highlights the importance of considering both the ultrafast and “ultraslow” timescales when designing new potential UV filters.
In recent years, several artificial ultraviolet (UV) filters, serving the purpose of providing a front-line defense to UV radiation exposure, used in commercial sunscreen formulations, have come under scrutiny due to concerns about their safety (Saija et al., 2000; Matsui et al., 2009; Burnett and Wang, 2011; Loden et al., 2011; Afonso et al., 2014; Skotarczak et al., 2015; Sharma et al., 2017). Alongside this “sunscreen controversy,” incidences of skin cancer are on the rise (Stavros, 2014), even with increasing use of sunscreen formulations. This highlights the need for, not only improved education on general sun exposure and how to apply sunscreen formulas, but also a requisite for improved formulations containing safer UV filters. These new UV filters need to provide enhanced photoprotection along with being non-toxic, particularly when exposed to UV radiation.
One of the approaches to tackling these issues has been to study the intrinsic properties of photoprotective molecules found throughout nature (Saewan and Jimtaisong, 2015), which has had a few billion years head start in UV photoprotection. To this end, nature-based UV filters found in plants have garnered considerable interest as a starting point. One such molecule is the sinapate ester, sinapoyl malate. Indeed, a structurally related cinnamate, ethylhexyl methoxycinnamate, has already been employed as an artificial UV filter. However this has been recently shown to be genotoxic (Sharma et al., 2017), highlighting the urgent need for new non-toxic UV filters. The photochemistry responsible for sinapoyl malate's photoprotective capabilities has been proposed, along with that for related sinapate esters (Dean et al., 2014; Baker et al., 2016; Horbury et al., 2017a, 2018; Luo et al., 2017; Zhao et al., 2020). These studies have shown that, upon absorption of UV, these sinapate esters undergo an effective and ultrafast (femto- to picosecond, 10−15 and 10−12 s, respectively) trans-to-cis and cis-to-trans photoisomerization, which is responsible for their apparent long-term photostability and photoprotective nature. Therefore, gaining an understanding of the initial light/matter interaction which drives the overall photochemistry of the molecule can provide powerful insight in the development of new UV filters and there long-term photostability. Additionally, these sinapate esters possess strong absorptions in the UVA (400–315 nm) and in the UVB region (315–280 nm) of the solar spectrum.
While these sinapate esters strongly absorb in the UVA, they do not cover the entirety of the UVA spectrum, i.e., sinapoyl malate's UVA absorption cuts off around 360 nm (Baker et al., 2016), which is particularly pertinent as this spectral region is linked to premature skin aging (Berneburg et al., 2000). Therefore, and ideally, if their absorption can be broadened across the entire UVA spectrum, it provides the means to a superior UV filter, spanning both the UVB and UVA. However, it is worth noting that increasing the UVA absorption does not imply a better UV filter; the molecule still needs to display a high level of photostability. One simple method of broadening, as well as spectrally red-shifting the absorption of these sinapate esters, is to increase the extent of conjugation in the π-system of the chromophore unit. An intuitive starting point is to add functionalization to the acrylic double bond. This was recently shown to be a promising approach by the addition of an identical ester group being added to the double bond of ethyl sinapate (shown in blue in Figure 1), which resulted in a redshift of the ground state absorption spectrum along with an increase in photostability (Horbury et al., 2019). We therefore utilize this approach, again, this time by dimerizing ethyl sinapate (Neudörffer et al., 2004). Ethyl sinapate has been shown to display very similar intrinsic properties to the plant UV filter, sinapoyl malate (Horbury et al., 2018). The resultant dimer is dehydrodiethylsinapate (DHDES, see Figure 1). As anticipated, DHDES possess a strong absorption across the (almost) entirety of the UVA region (Figure 1).
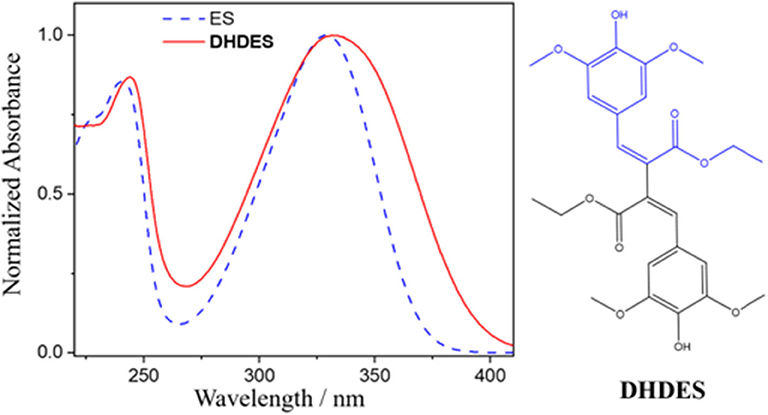
Figure 1. Normalized UV/Vis spectrum of DHDES (red) in ethanol. Additionally, the UV/Vis spectrum of ethyl-sinapate (ES) is overlaid (blue dashed line). The chemical structures of DHDES is shown right; chemical structure of ES is shown as blue in the structure of DHDES.
Whether the structural variant of sinapoyl malate, DHDES, with its enhanced UVA absorption, possesses the photostability (dynamics) displayed by sinapoyl malate and ethyl sinapate is the central question we seek to address in this work. Allied to this is the emerging trends that we may obtain through this work. Therefore, we investigate the photochemistry (dynamics) of DHDES, as a potential starting point for future nature-inspired UV filters with enhanced function; broader UVA coverage and efficient excitation-recovery cycles. To implement our structure-dynamics-function approach, we utilize femtosecond (fs) transient electronic (UV/visible, abbreviated to UV/Vis henceforth) absorption spectroscopy (TEAS) which has already provided valuable insight into the photochemistry of sinapate esters and related cinnamates (Vengris et al., 2005; Baker et al., 2016; Horbury et al., 2016, 2017a,b, 2018; Zhao et al., 2020). Additionally, the long-term photostability of this molecule was assessed via steady-state UV irradiation, monitored by UV/Vis absorption and 1H NMR spectroscopy. The experiments are complemented by computational results based on (time-dependant) density functional theory.
The fs TEAS setup used to explore the photochemistry and photophysics of DHDES has been described in detail previously (Greenough et al., 2014a,b), however, information specific to the present experiments is provided herein. Samples of DHDES were made to a concentration of 1 mM in ethanol (absolute, VWR), ethylene glycol (technical, Arcos Organics), and glycerol (99.93%, Fisher. The fs pump pulses were generated by an optical parametric amplifier (TOPAS-C, Spectra-Physics) with a fluence of 200–800 μJ·cm−2. The pump excitation wavelength used was 332 nm for DHDES in ethanol, ethylene glycol, and glycerol; this wavelength correspond to the associated UV absorption maxima. The probe pulse was a broadband white light supercontinuum generated in a CaF2 window with a thickness of 2 mm, providing a probe spectral window of 330–675 nm. The pump-probe time delay (Δt) was varied by adjusting the optical delay of the probe pulse, the maximum obtainable Δt was 2 nanoseconds (ns). Changes in the optical density (ΔOD) of the samples were calculated from transmitted probe intensities, collected using a spectrometer (Avantes, AvaSpec-ULS1650F). The sample delivery system was a flow-through cell (Demountable Liquid Cell by Harrick Scientific Products Inc.) consisting of two CaF2 windows with a thickness of 1 mm for the front window and 2 mm for the back window, except in the case of DHDES in glycerol, where both windows were 2 mm thick due to the build-up pressure in the flow system (owing to glycerol's viscosity). The windows were spaced 100 μm apart to limit temporal dispersion of the pump and probe pulses. The sample was circulated using a diaphragm pump (SIMDOS, KNF) recirculating sample from a 25 mL reservoir to provide each pulse-pair with fresh sample.
Steady-state UV/Vis absorption spectra of DHDES in ethanol, were collected to determine long-term photostability. The sample was irradiated with an arc lamp (Fluorolog 3, Horiba) for up to 2 h, with the UV/Vis spectra taken at various time points, at the corresponding TEAS excitation wavelength, using an 8 nm bandwidth of the irradiation source. The fluence used during irradiation of DHDES was set to 100–200 μJ·cm−2 to mimic solar incidence conditions. The UV/Vis spectra were measured using a UV/Vis spectrometer (Cary 60, Agilent Technologies).
The structure of DHDES was generated in its cis,cis-isomer and trans,trans-isomer (see Figures 1, 4), as well as a contorted cis,trans-isomer, using VMD (Humphrey et al., 1996) with the molefacture plugin. Each of these structures underwent a density functional theory (DFT) geometry optimization with a cc-pVTZ basis set (Dunning, 1989) and the PBE0 functional (Adamo and Barone, 1999), using the NWChem software (Valiev et al., 2010). The cis,trans-isomer of DHDES was significantly energetically disfavored with respect to the other two isomers, and the geometry optimization did not converge with a reasonable level of convergence criteria; therefore this isomer was discounted from further study. Following relaxation, linear-response time-dependant DFT calculations were conducted at the optimized ground state geometries of the cis,cis-isomer and trans,trans-isomer to obtain their optical absorption spectra. Again, the level of theory was cc-pVTZ/PBE0 and the NWChem software was employed. In all calculations, the conductor-like screening model (COSMO) was used to approximate the effect of the solvent (Klamt and Schüürmann, 1993; York and Karplus, 1999). The default COSMO solvent model for ethanol within NWChem was used, the descriptors of which are based on the Minnesota Solvent Descriptor Database (Winget et al., 1999).
DHDES and Me-DHDES were synthesized using the procedure published by Mention et al. (2020) 1H NMR spectra of DHDES and Me-DHDES (we discuss the reason for studying this system below) in CDCl3 were recorded at 300 MHz on a Bruker Fourier 300, pre- and post-irradiation using a Rayonet RPR-200 after irradiation of 60 min at 302 nm; this wavelength was used due to the limited spectral choices of the Rayonet RPR-200.
To retrieve the dynamical information contained within the transient absorption spectra, a sequential global (across all wavelengths 330–675 nm) fitting technique was performed, using the software package Glotaran (Mullen and Van Stokkum, 2007; Snellenburg et al., 2012). The transient absorption spectra of DHDES were fit using four time-constants (τn, where n = 1–4). Each time-constant is linked to an evolution associated difference spectrum (EADS) that represents the evolving spectral features related to that time-constant. All fits were convoluted with a Gaussian function to model our instrument response function (~80 fs, for glycerol ~100 fs). The final time-constant used in our model (τ4 for DHDES) accounts for the long-lived photoproduct; this time-constant is reported to be >> 2 ns.
First, we consider the resulting transient absorption spectra of DHDES in ethanol shown in Figure 2A (see ESI Figure S1 for transient absorption spectra of DHDES in ethylene glycol and glycerol). Note that the related Me-DHDES, a methylated version of DHDES, is also shown in Figure 2B, but will be discussed later. After initial photoexcitation at 332 nm, the transient absorption spectrum consist of three distinct spectral features, the first being a ground state bleach centered at ~340 nm, the second is a large excited state absorption at ~400 nm and the third is a smaller excited state absorption at ~675 nm. As Δt increases, the excited state absorption at 675 nm begins to decay, leaving a broad flat absorption alongside the large excited state absorption feature at 400 nm. At longer Δt, the ground state bleach and both excited state absorption features tend toward the baseline. Once the excited state absorption features have completely decayed away, a new absorption feature (not immediately evident) spanning 390–600 nm is revealed. Some residual ground state bleach at 340 nm remains. Both features persist beyond the maximum Δt (see ESI Figure S2 for transient absorption spectrum at Δt = 2 ns).
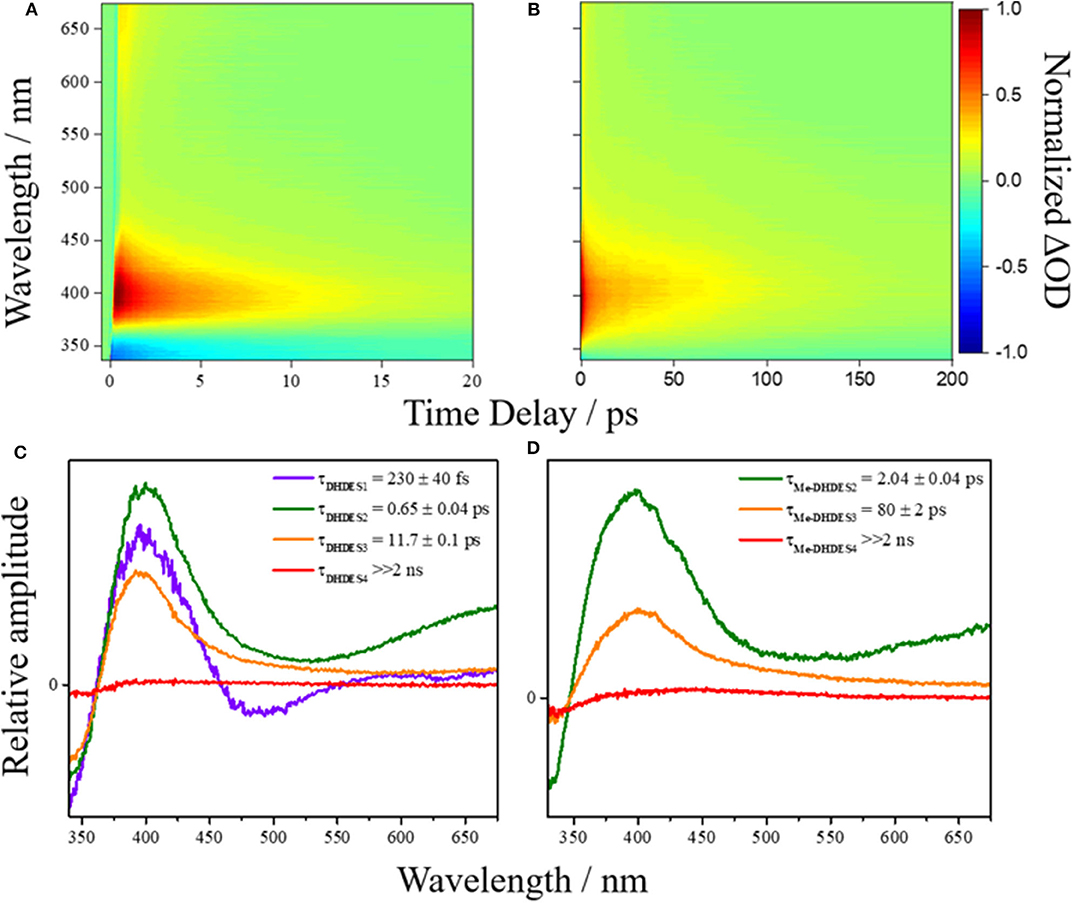
Figure 2. False colormap of the TAS of (A) DHDES and (B) Me-DHDES in ethanol, with intensity representing normalized change in optical density (OD). Additionally, the EADS produced by the sequential global fitting of the TAS are also displayed for (C) DHDES and (D) Me-DHDES.
The time-constants produced from fitting the transient absorption spectra are provided in Table 1, and the corresponding EADS are shown in Figures 2C,D (see ESI Figure S3 for additional EADS for DHDES in ethylene glycol and glycerol, along with residuals Figures S4–S8 for DHDES in ethanol, ethylene glycol, glycerol, and Me-DHDES in ethanol, respectively).
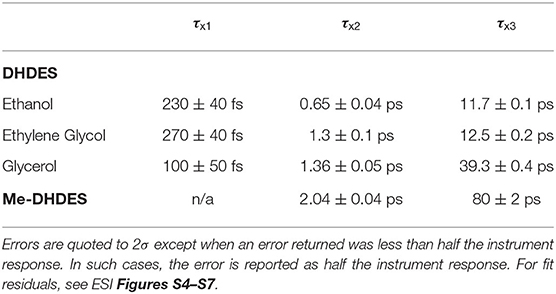
Table 1. The values of the time-constants (τxN, where x represents either DHDES or Me-DHDES) provided from globally fitting the transient absorption spectra of DHDES in ethanol, ethylene glycol, and glycerol, Me-DHDES in ethanol.
Steady-state irradiation of DHDES in ethanol was carried out to determine its long-term photostability under constant UV exposure. To monitor any changes to the sample, both UV/Vis (Figure 3A as well as extracted evolution associated spectra, EAS, Figure 3B) and 1H NMR (Figure 3C and Figure S9) spectra were recorded at various time points of irradiation. Similar experiments were carried out for Me-DHDES (see ESI Figures S10, S11 for further details).
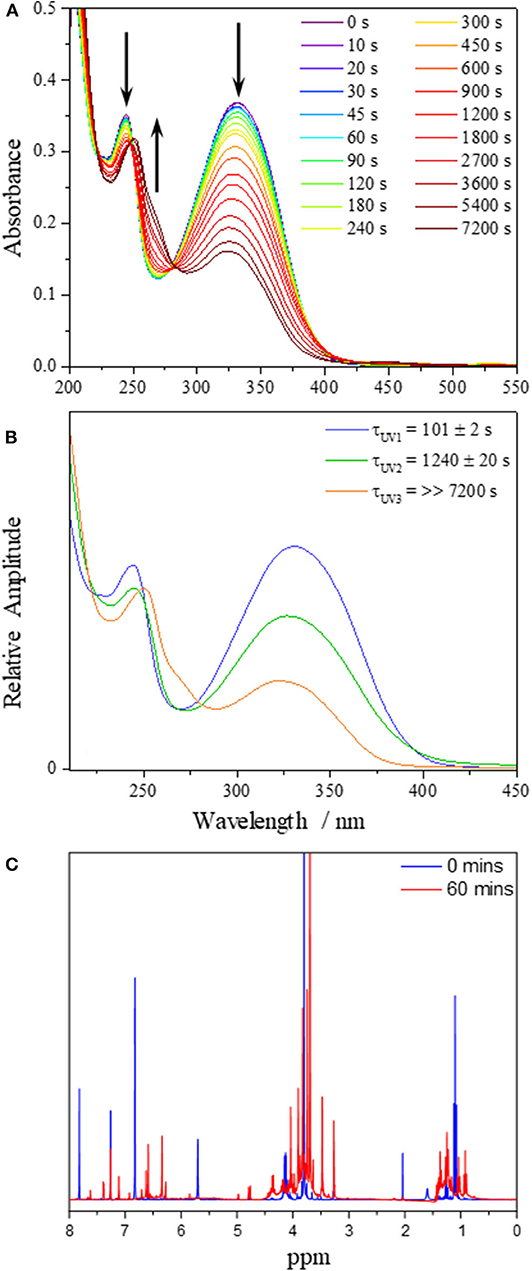
Figure 3. (A) UV/Vis spectra of DHDES in ethanol taken at various time points during irradiation at 332 nm over 7,200 s. (B) EAS from the sequential global fit of the data displayed in (A). (C) 1H NMR spectra of DHDES pre (blue) and post (red) irradiation in ethanol.
The evolution of the UV/Vis spectra during irradiation for DHDES in ethanol, is shown in Figure 3A and clearly indicates that the main absorption feature spanning the UVA and UVB regions of the absorption spectrum undergoes a significant decay (~60% reduction in 2 h). However, this decay appears to consist of two distinct spectral evolutions, as represented by the arrows in Figure 3A; a decay to yield (existing) features at ~330 and ~240 nm, and a growth of new absorption features at ~250 and ~270 nm.
To better distinguish the processes that are occurring, we performed a sequential global (210–450 nm) fit of the UV/Vis spectra. The resulting EAS and time-constants are shown in Figure 3B. Cursory comparison of EAS(τUV1) (101 ± 2 s) with EAS(τUV2) (1240 ± 20 s) suggest that upon irradiation of DHDES, the absorption intensity drops. Under prolonged irradiation the spectra continue to show a decrease in absorption; however, additional spectral features begin to grow in. These new absorption features consist of a peak at ~270 nm and a marked red-shift of the peak at ~240 nm to ~250 nm as seen in EAS(τUV3) (>>7,200 s). This is very likely indicative of a new species being generated.
To identify any newly generated species after UV radiation exposure, a 1H NMR spectrum of DHDES was taken before and after 60 min irradiation (see Figure 3C). One can observe that the peaks corresponding to the benzene ring protons of DHDES (6.78 and 7.83 ppm) have disappeared and peaks appear at ~6.4 and ~6.6 ppm, along with many new peaks in the 3.5–5.0 ppm as well as the 0.7–1.5 ppm regions, demonstrating the transformation of the latter into multiple new compounds under radiation (Figure 3C). The appearance of broad peaks in the 1H NMR spectrum after irradiation hints at the formation of oligomers. Additionally it is likely that DHDES is undergoing an esterification with ethanol when exposed to UV; this has previously been seen with 1,4-diphenyl-1,3-dibutene (Saltiel and Redwood, 2016). We will return to discuss another potential product, a tricyclic compound, in the discussion (vide infra).
The calculated ground state isomers of two stable isomers of DHDES (cis,cis- and trans,trans-isomers), are shown in Figure 4 (NB the trans,trans-isomer of DHDES is also show in in Figure 1). The cis,cis-isomer has the lower energy of the two conformers with the trans,trans-isomer being ~0.3 eV higher in energy. A molecule of this size likely has a complicated ground-state energy surface with multiple local minima corresponding to different stable geometries. As such, it is challenging to determine that the structure chosen for both conformers is the global minimum energy structures and, indeed, it is likely multiple structures exist in solution. Owing to this, the predicted energies for these species, as well as predicted vertical excitation energies, are presented as an approximation of each form rather than as quantitative data. Furthermore, the effect of explicit solvent interactions are not captured by our technique, these likely also have an effect on the relative energies of the species (Zuehlsdorff et al., 2017; Turner et al., 2019). Additionally, the two isomers display several significant differences in geometry, beyond just isomerization around the double bonds. In the case of the cis, cis-isomer, the two ethyl sinapate subunits are at ~90° to each other, which hinders conjugation between the two ethyl sinapate subunits. It should be noted that a planar cis,cis-DHDES is sterically unfavorable, as several atoms would need to occupy the same space. However, the trans,trans-isomer is almost planar (~24° twist between the two benzene rings), but the two ethyl sinapate subunits are sufficiently twisted relative to one another to weaken the extended conjugation across both subunits. This lack of conjugation is further supported by the UV/Vis spectra of DHDES (Figure 1); if there was extended conjugation across the entire molecule, one would anticipate a starker red-shift in the absorption.
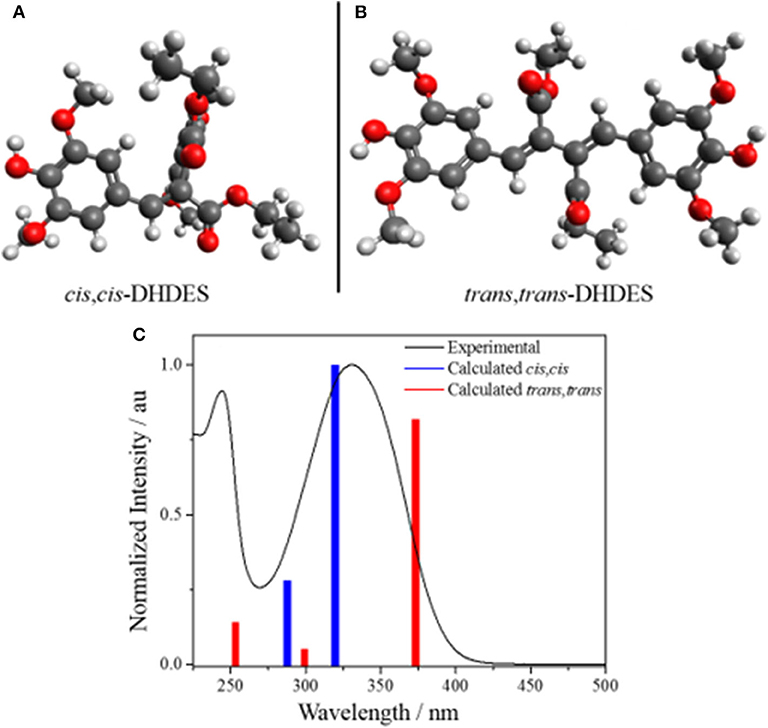
Figure 4. Geometry calculations of the stable ground state isomers (A) cis,cis-DHDES and (B) trans,trans-DHDES. (C) Predicted UV/Vis stick spectra (cis,cis-DHDES blue bars, trans,trans-DHDES red bars) overlaid with experimental UV/Vis spectrum of DHDES in ethanol (black line).
In addition to the calculated geometries, UV/Vis spectra for the cis,cis-DHDES and trans,trans-DHDES were calculated, and also shown in Figure 4. The spectra show the main absorption peak for cis,cis-DHDES is at 322 nm, while for trans,trans-DHDES it is at 371 nm; the cis,cis-isomer has a higher oscillator strength. Due to the calculated peaks for both isomers being within the experimental absorption peak, it possible that our sample of DHDES contains both cis,cis- and trans,trans-isomers, which in turn gives rise to the increased broadness in absorption compared to ethyl sinapate (see Figure 1). While a calculated energy difference of 0.3 eV would suggest that the cis,cis-isomer would be heavily favored, it is possible that the calculated energy difference would be reduced if an explicit solvent model was used. Furthermore, we believe that during the synthesis of DHDES both isomers are generated.
We now consider the ultrafast photochemistry and photophysics of DHDES. As discussed supra, due to the lack of conjugation, a consequence of the twisted geometries between the two ethyl sinapate substructures (see Figures 4A,B), one would anticipate that initial photoexcitation would yield a transient absorption spectrum analogous to the transient absorption spectrum of ethyl sinapate in ethanol. Whilst this is not immediately apparent in the measured transient absorption spectra of DHDES, the EADS(τDHDES1) (230 ± 40 fs) in ethanol is mildly similar to the first EADS of ethyl sinapate in ethanol (see ESI Figures S12, S13 for more details). We therefore assign the first time-constant τDHDES1, and its corresponding EADS(τDHDES1), to the DHDES undergoing a molecular motion that allows for increased coupling between the two ethyl sinapate substructures, thus the decay of the ethyl sinapate-like features; this is substantiated by the significant change between EADS(τDHDES1) and EADS(τDHDES2) (0.65 ± 0.04 ps), see Figure 2C. With the decay of the ethyl sinapate-like features, the comparison of DHDES to ethyl sinapate now ends.
Next, we consider the time-constant τDHDES2 and its corresponding EADS(τDHDES2). The EADS(τDHDES2) is dominated by two large excited state absorption features at ~400 and ~675 nm, which we attribute to a specific DHDES isomer (see Figure 4). We draw confidence here, by noting that the excited state absorption feature at ~675 nm is like the excited state absorption seen in TEAS recorded for 1,4-disyringol-1,3-butadiene (DSB, see ESI Figures S14, S15). DSB is the backbone structure of DHDES, which remains planar, unlike DHDES, due to the absence of the ester functionalities (see ESI Figure S16). Consequently, it is plausible that the absorption feature at 675 nm in DHDES is attributed to a planar form with increased conjugation, akin to DSB. Since trans,trans-DHDES, is near-planar in the ground state, this isomer serves as the leading candidate; the excess energy imparted by the absorption of a UV photon may allow trans,trans-DHDES to approach planarity, as excited state population samples the excited state potential energy landscape. While we cannot rule out that this is also happening in cis,cis-DHDES, one notes that this would require severe nuclear rearrangement, including cis,cis-to-trans,trans isomerization (vide supra), to even approach planarity. With this in mind, we suggest that the excited state absorption seen at 400 nm is likely attributed to the absorption from the cis, cis-DHDES electronic excited state. When comparing the EADS(τDHDES2) and EADS(τDHDES3) (11.7 ± 0.1 ps), the major difference is the absence of the excited state absorption at ~675 nm. Whether or not this is due to trans,trans-DHDES, relaxing back to the electronic ground-state on the timescale of τDHDES2, is unknown. Unfortunately, due to the sample potentially consisting of both isomers of DHDES, the transient absorption spectra and EADS are heavily convoluted making it hard to distinguish which process is associated with which DHDES isomer.
Thirdly, the EADS(τDHDES3) resembles the decay of the remaining excited state absorption at 400 nm. As this excited state absorption is attributed to the cis,cis-DHDES isomer (see above) it represents the decay of the electronically excited state of cis,cis-DHDES, but with the caveat that the trans,trans-DHDES could be present and also contributes to the EADS(τDHDES3) and timescale. Finally, EADS(τDHDES4) (>>2 ns), models the long-lived photoproduct (see ESI Figure S2 for transient absorption spectrum at Δt = 2 ns). Whether this is due to a transient species such as a triplet state or a molecular photoproduct is unknown.
To help determine whether the electronic excited state relaxation, for either isomer, is mediated by a photoisomerization motion leading to a conical intersection between the electronic excited state and ground state, akin to ethyl sinapate (Horbury et al., 2018), additional TEAS measurements were performed in more viscous solvents: ethylene glycol (η = 21) (Tsierkezos and Molinou, 1998) and glycerol (η = 1,412) (Segur and Oberstar, 1951) (see ESI for transient absorption spectra, ethanol η = 1.19) (Khattab et al., 2012). From these measurements, we determined the viscosity dependence, α, of the time-constant τDHDES2 and τDHDES3, as these time-constant are linked to the decay of the electronic excited state, which may be mediated by photoisomerization (Espagne et al., 2006; Horbury et al., 2017a). The retrieved α value for τDHDES2 was 0.09 ± 0.06 and for was τDHDES3 0.18 ± 0.07. The value returned for τDHDES2 suggests that large amplitude nuclear motion linked to this time-constant is minor, meaning that either the trans,trans-DHDES undergoes very little (large amplitude) nuclear motion to relax or that this process involves a change in electronic states. While τDHDES3 is a smaller value compared to related sinapate esters (~0.34) (Horbury et al., 2017a), it still suggests that the electronic excited state decay of DHDES involves some substantial nuclear motion, of which a photoisomerization could be a candidate.
The incomplete recovery of the ground state bleach suggests that (at least) a small fraction of DHDES is not returning to its original form. This incomplete recovery of the ground state bleach is not unexpected as steady-state irradiation has demonstrated DHDES degrades over persistent UV exposure. It has previously been reported that a quinone methide species can be formed upon electrochemically-induced oxidation and one could assume that a similar mechanism could also occur upon irradiation, this mechanism is shown in Scheme 1 (Neudörffer et al., 2006).
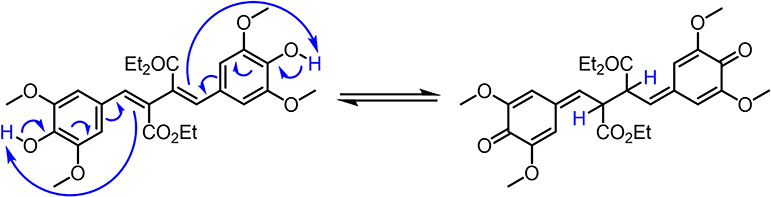
Scheme 1. Generation of the quinone methide from DHDES.
Another potential mechanism for the formation of the quinone methide is that it is initiated by O–H bond fission. While one would anticipate that this pathway would be blocked by the presence of an intramolecular hydrogen bond between the hydroxy and methoxy groups, previous studies on syringol, show it can undergo O–H bond fission. Owing to the non-planar geometry of syringol in the excited electronic state, this serves to weaken the intramolecular hydrogen bond sufficiently enough to allow for O–H bond fission to occur (Young et al., 2014). It is therefore not unreasonable to assume that the strained and twisted nature (see above) of DHDES induces a similar affect, by weakening the intramolecular hydrogen bonding, thus, opening up the O–H fission pathway along, say, a dissociative 1πσ* state, in keeping with previous studies (Roberts and Stavros, 2014).
To test such a hypothesis for O–H bond fission shown in Scheme 1, we have replaced O–H with O–CH3; previous studies in similar systems have shown that O–H fission is more facile over O–C fission. (Hadden et al., 2012) Therefore, methylation could lead to stabilization of DHDES under prolonged UV exposure. The methylated form of DHDES, termed Me-DHDES, in ethanol (1 mM), was then studied using TEAS (photoexcited at 310 nm) and steady-state absorption to see if the formation of the proposed quinonic methide species is prevented. The resulting transient absorption spectra and EADS are shown in Figures 2B,D. While the spectral features that are present in the transient absorption spectra match the ones seen in DHDES, the time-constants (see Table 1) clearly differ in two ways: (1) τDHDES1 is absent; and (2) τMe−DHDES3 is substantially longer than τDHDES3 (80 ± 2 ps versus 12.5 ± 0.2 ps for DHDES). However, the EADS(τMe−DHDES2) (2.04 ± 0.04 ps) and EADS(τMe−DHDES3) are remarkably similar spectrally to those of DHDES. Therefore, we are confident that Me-DHDES undergoes the same relaxation mechanism as DHDES. Additionally, the steady-state UV/Vis spectra (see ESI Figure S10) taken during prolonged UV irradiation and the appearance of new proton peaks in the 1H NMR spectra taken pre- and post-irradiation (see ESI Figure S11), demonstrate Me-DHDES is also undergoing degradation. Since, the addition of the methoxy group is likely to supress bond-rearrangement proposed in Scheme 1, the revised degradation pathway is the formation of the tricyclic compound X, formed through a photo cyclo-addition followed by a re-aromatization via an 1,5-H shift (Scheme 2); this photoproduct would be expected to exhibit 1H NMR peaks at ~6.3 and ~6.6 ppm in Figure 3C. Consideration of the above and given the same proton peaks are present in the post-irradiation 1H-NMR spectra of DHDES and Me-DHDES (see ESI Figures S9, S11), the degradation pathway between the two is likely the same, this pathway is likely occurring alongside the photoinduced esterification with ethanol (vide supra).
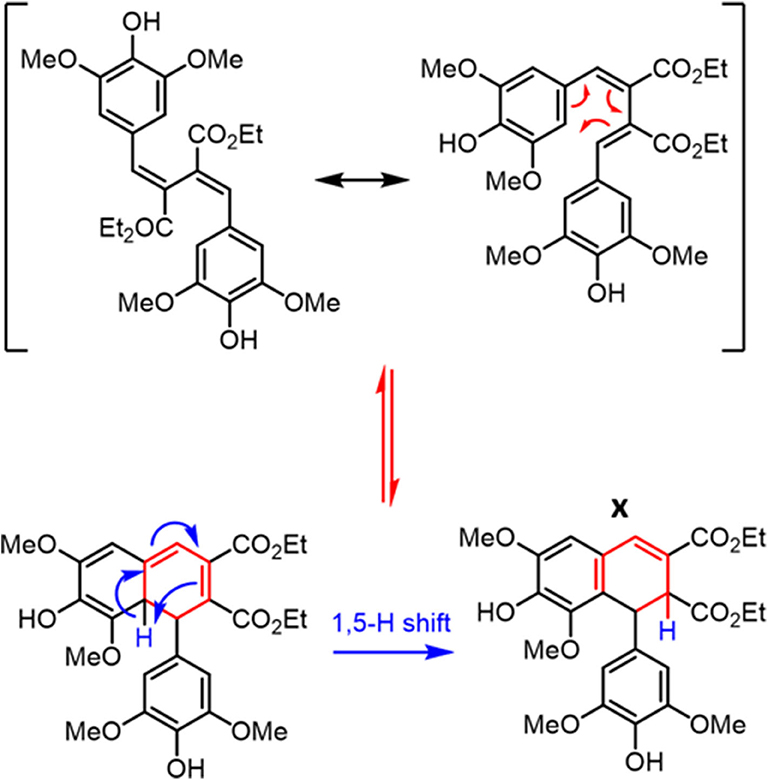
Scheme 2. Generation of the tricyclic species X from DHDES.
We have explored the photochemistry and photostability of a potential UV filter, dehydrodiethylsinapate (DHDES), which provides almost full absorption coverage of the UVA region of the electromagnetic spectrum. This was achieved using a combination of femtosecond transient electronic (UV/Vis) absorption spectroscopy (TEAS) and steady-state UV irradiation, monitored by both UV/Vis and 1H NMR spectroscopy.
The photodynamics observed in the TEAS measurements suggest DHDES undergoes an ultrafast electronic excited state relaxation, potentially mediated via a photoisomerization pathway. However, the steady-state irradiation studies demonstrate that DHDES is not photostable under prolonged UV exposure. We attempted to thwart degradation by protecting the O–H with O–Me without success. Through the result of this methylation in combination with 1H NMR, we have identified a potential cause of degradation, due to the formation of a tricyclic species. It is likely that the tricyclic species further degrades under UV exposure, leading to the formation of the myriad of unidentified molecular species seen in the post irradiation 1H NMR spectrum. Therefore, if one can by-pass the formation of this tricyclic species, this could expand the molecular diversity based around a core DHDES structure, potentially leading to promising next generation, broad-spectrum and nature-inspired, UV filter molecules.
Importantly, this work demonstrates that manipulating a molecules' structure, whilst trying to preserve the ultrafast dynamics, can have both positive and negative implications on its function. Indeed, we have shown that DHDES has enhanced UVA absorption, whilst still possessing a short-lived excited state. However, this is at the cost of long term photostability. Undeniably, what pervades the present study is that chemical intuition, increased conjugation and thus broader UVA absorption, does not necessarily result in a better (nature-inspired) UV filter. It is clear that in order to progress this structure-dynamics-function approach, we will need to seek increasing levels of guidance from theory, a current strategy underway in our laboratory, in our search for next generation, nature inspired UV filters which could be included in commercial sunscreen formulations.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://zenodo.org/record/3741408, doi: 10.5281/zenodo.3741408.
MH acquired and analyzed the time-resolved and steady-state spectroscopic data and prepared the manuscript. JP provided assistant in the acquisition of the time-resolved and steady-state spectroscopic data. AF, MM, and FA conceived and performed the syntheses, as well as contributing to the preparation of the manuscript. MT and NH performed and analyzed the computational calculations. VS conceived the experiments and provided guidance in data analysis and interpretation and the writing of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. M. Staniforth for useful discussion. They also acknowledge the Warwick Centre for Ultrafast Spectroscopy (WCUS) for the use of the Cary 60 and Fluorolog 3. MH thanks the Leverhulme Trust for postdoctoral funding. MT thanks EPSRC for a doctoral studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science, Grant Number EP/L015307/1. NH and MT acknowledge the support of EPSRC through grant number EP/P0229X/1. Computing facilities were provided by the Scientific Computing Research Technology Platform of the University of Warwick. MM, AF, and FA thank the Grand Reims, the Conseil Départemental de la Marne and the Region Grand Est for financial support, and the ANR for the SINAPUV grant (ANR-17-CE07-0046). Finally, VS thanks the EPSRC for an equipment grant (EP/J007153), the Leverhulme Trust for a research grant (RPG-2016-055), and the Royal Society and Leverhulme Trust for a Royal Society Leverhulme Trust Senior Research Fellowship. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the grant agreement No. 828753.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00633/full#supplementary-material
Adamo, C., and Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170. doi: 10.1063/1.478522
Afonso, S., Horita, K., Silva, J. P. S., Almeida, I. F., Amaral, M. H., Costa, P. C., et al. (2014). Photodegradation of avobenzone: stabilization effect of antioxidants. J. Photochem. Photobiol. B Biol. 140, 36–40. doi: 10.1016/j.jphotobiol.2014.07.004
Baker, L. A., Horbury, M. D., Greenough, S. E., Allais, F., Walsh, P. S., Habershon, S., et al. (2016). Ultrafast photoprotecting sunscreens in natural plants. J. Phys. Chem. Lett. 7, 56–61. doi: 10.1021/acs.jpclett.5b02474
Berneburg, M., Plettenberg, H., and Krutmann, J. (2000). Photoaging of human skin. Photodermatol. Photoimmunol. Photomed. 16, 239–244. doi: 10.1034/j.1600-0781.2000.160601.x
Burnett, M. E., and Wang, S. Q. (2011). Current sunscreen controversies: a critical review. Photodermatol. Photoimmunol. Photomed. 27, 58–67. doi: 10.1111/j.1600-0781.2011.00557.x
Dean, J. C., Kusaka, R., Walsh, P. S., Allais, F., and Zwier, T. S. (2014). Plant sunscreens in the UV-B: ultraviolet spectroscopy of jet-cooled sinapoyl malate, sinapic acid, and sinapate ester derivatives. J. Am. Chem. Soc. 136, 14780–14795. doi: 10.1021/ja5059026
Dunning, T. H. Jr. (1989). Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023. doi: 10.1063/1.456153
Espagne, A., Paik, D. H., Changenet-Barret, P., Martin, M. M., and Zewail, A. H. (2006). Ultrafast photoisomerization of photoactive yellow protein chromophore analogues in solution: influence of the protonation state. ChemPhysChem 7, 1717–1726. doi: 10.1002/cphc.200600137
Greenough, S. E., Horbury, M. D., Thompson, J. O. F., Roberts, G. M., Karsili, T. N. V., Marchetti, B., et al. (2014a). Solvent induced conformer specific photochemistry of guaiacol. Phys. Chem. Chem. Phys. 16, 16187–16195. doi: 10.1039/C4CP02424A
Greenough, S. E., Roberts, G. M., Smith, N. A., Horbury, M. D., Mckinlay, R. G., Zurek, J. M., et al. (2014b). Ultrafast photo-induced ligand solvolysis of cis-[Ru (bipyridine)2(nicotinamide)2]2+: experimental and theoretical insight into its photoactivation mechanism. Phys. Chem. Chem. Phys. 16, 19141–19155. doi: 10.1039/C4CP02359E
Hadden, D. J., Roberts, G. M., Karsili, T. N., Ashfold, M. N., and Stavros, V. G. (2012). Competing 1 πσ* mediated dynamics in mequinol: O–H versus O–CH 3 photodissociation pathways. Phys. Chem. Chem. Phys. 14, 13415–13428. doi: 10.1039/c2cp42289a
Horbury, M., Flourat, A. L., Greenough, S. E., Allais, F., and Stavros, V. (2018). Investigating isomer specific photoprotection in a model plant sunscreen. Chem. Commun. 54, 936–939. doi: 10.1039/C7CC09061G
Horbury, M. D., Baker, L., Quan, W., Greenough, S. E., and Stavros, V. (2016). Photodynamics of potent antioxidants: ferulic and caffeic acids. Phys. Chem. Chem. Phys. 18, 17691–7. doi: 10.1039/C6CP01595F
Horbury, M. D., Baker, L. A., Rodrigues, N. D., Quan, W.-D., and Stavros, V. G. (2017b). Photoisomerization of ethyl ferulate: a solution phase transient absorption study. Chem. Phys. Lett. 673, 62–67. doi: 10.1016/j.cplett.2017.02.004
Horbury, M. D., Holt, E. L., Mouterde, L. M., Balaguer, P., Cebrián, J., Blasco, L., et al. (2019). Towards symmetry driven and nature inspired UV filter design. Nat. Commun. 10:4748. doi: 10.1038/s41467-019-12719-z
Horbury, M. D., Quan, W.-D., Flourat, A., Allais, F., and Stavros, V. (2017a). Elucidating nuclear motions in a plant sunscreen during photoisomerization through solvent viscosity effects. Phys. Chem. Chem. Phys. 19, 21127–21131. doi: 10.1039/C7CP04070A
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Khattab, I. S., Bandarkar, F., Fakhree, M. A. A., and Jouyban, A. (2012). Density, viscosity, and surface tension of water+ ethanol mixtures from 293 to 323K. Korean J. Chem. Eng. 29, 812–817. doi: 10.1007/s11814-011-0239-6
Klamt, A., and Schüürmann, G. (1993). COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2, 799–805. doi: 10.1039/P29930000799
Loden, M., Beitner, H., Gonzalez, H., Edström, D. W., Åkerström, U., Austad, J., et al. (2011). Sunscreen use: controversies, challenges and regulatory aspects. Br. J. Dermatol. 165, 255–262. doi: 10.1111/j.1365-2133.2011.10298.x
Luo, J., Liu, Y., Yang, S., Flourat, A. L., Allais, F., and Han, K. (2017). Ultrafast barrierless photoisomerization and strong ultraviolet absorption of photoproducts in plant sunscreens. J. Phys. Chem. Lett. 8, 1025–1030. doi: 10.1021/acs.jpclett.7b00083
Matsui, M. S., Hsia, A., Miller, J. D., Hanneman, K., Scull, H., Cooper, K. D., et al. (2009). Non-sunscreen photoprotection: antioxidants add value to a sunscreen. J. Invest. Dermatol. Symp. Proc. 14, 56–59. doi: 10.1038/jidsymp.2009.14
Mention, M., Flourat, A. L., Peyrot, C., and Allais, F. (2020). Biomimetic regioselective and high-yielding Cu (I)-catalyzed dimerization of sinapate esters in green solvent CyreneTM: towards sustainable antioxidant and anti-UV ingredients. Green Chem 22, 2077–2085. doi: 10.1039/D0GC00122H
Mullen, K. M., and Van Stokkum, I. H. (2007). TIMP: an R package for modeling multi-way spectroscopic measurements. J. Stat. Softw 18. doi: 10.18637/jss.v018.i03
Neudörffer, A., Bonnefont-Rousselot, D., Legrand, A., Fleury, M.-B., and Largeron, M. (2004). 4-Hydroxycinnamic ethyl ester derivatives and related dehydrodimers: relationship between oxidation potential and protective effects against oxidation of low-density lipoproteins. J. Agric. Food Chem. 52, 2084–2091. doi: 10.1021/jf035068n
Neudörffer, A., Fleury, M.-B., Desvergne, J.-P., and Largeron, M. (2006). Effects of lithium ion-pairing on the electrochemical oxidation of 4-hydroxycinnamate derivatives. Electrochim. Acta 52, 715–722. doi: 10.1016/j.electacta.2006.06.001
Roberts, G. M., and Stavros, V. G. (2014). The role of πσ* states in the photochemistry of heteroaromatic biomolecules and their subunits: insights from gas-phase femtosecond spectroscopy. Chem. Sci. 5, 1698–1722. doi: 10.1039/c3sc53175a
Saewan, N., and Jimtaisong, A. (2015). Natural products as photoprotection. J. Cosmet. Dermatol. 14, 47–63. doi: 10.1111/jocd.12123
Saija, A., Tomaino, A., Trombetta, D., De Pasquale, A., Uccella, N., Barbuzzi, T., et al. (2000). In vitro and in vivo evaluation of caffeic and ferulic acids as topical photoprotective agents. Int. J. Pharm. 199, 39–47. doi: 10.1016/S0378-5173(00)00358-6
Saltiel, J., and Redwood, C. E. (2016). Photochemistry of the 1, 4-Diphenyl-1, 3-butadienes in Ethanol. Trapping conical intersections. J. Phys. Chem. A 120, 2832–2840. doi: 10.1021/acs.jpca.6b02330
Segur, J. B., and Oberstar, H. E. (1951). Viscosity of glycerol and its aqueous solutions. Ind. Eng. Chem. 43, 2117–2120. doi: 10.1021/ie50501a040
Sharma, A., Bányiová, K., Babica, P., El Yamani, N., Collins, A. R., and Cupr, P. (2017). Different DNA damage response of cis and trans isomers of commonly used UV filter after the exposure on adult human liver stem cells and human lymphoblastoid cells. Sci. Tot. Environ. 593, 18–26. doi: 10.1016/j.scitotenv.2017.03.043
Skotarczak, K., Osmola-Mankowska, A., Lodyga, M., Polanska, A., Mazur, M., and Adamski, Z. (2015). Photoprotection: facts and controversies. Eur. Rev. Med. Pharmacol. Sci. 19, 98–112.
Snellenburg, J. J., Laptenok, S., Seger, R., Mullen, K. M., and Van Stokkum, I. H. M. (2012). Glotaran: a java-based graphical user interface for the R package TIMP. J. Stat. Softw. 49, 1–22. doi: 10.18637/jss.v049.i03
Stavros, V. G. (2014). A bright future for sunscreens. Nat. Chem. 6, 955–956. doi: 10.1038/nchem.2084
Tsierkezos, N. G., and Molinou, I. E. (1998). Thermodynamic properties of water+ ethylene glycol at 283.15, 293.15, 303.15, and 313.15 K. J. Chem. Eng. Data 43, 989–993. doi: 10.1021/je9800914
Turner, M., Horbury, M., Stavros, V., and Hine, N. (2019). Determination of secondary species in solution through pump-selective transient absorption spectroscopy and explicit-solvent TDDFT. J. Phys. Chem. A 123, 873–880. doi: 10.1021/acs.jpca.8b11013
Valiev, M., Bylaska, E. J., Govind, N., Kowalski, K., Straatsma, T. P., Van Dam, H. J., et al. (2010). NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477–1489. doi: 10.1016/j.cpc.2010.04.018
Vengris, M., Larsen, D. S., Van Der Horst, M. A., Larsen, O. F. A., Hellingwerf, K. J., and Van Grondelle, R. (2005). Ultrafast dynamics of isolated model photoactive yellow protein chromophores: “chemical perturbation theory” in the laboratory. J. Phys. Chem. B 109, 4197–4208. doi: 10.1021/jp045763d
Winget, P., Dolney, D. M., Giesen, D. J., Cramer, C. J., and Truhlar, D. G. (1999). Minnesota Solvent Descriptor Database. Minneapolis, MN: Department of Chemistry and Supercomputer Institute; University of Minnesota.
York, D. M., and Karplus, M. (1999). A smooth solvation potential based on the conductor-like screening model. J. Phys. Chem. A 103, 11060–11079. doi: 10.1021/jp992097l
Young, J. D., Staniforth, M., Dean, J. C., Roberts, G. M., Mazzoni, F., Karsili, T. N., et al. (2014). Towards understanding photodegradation pathways in lignins: the role of intramolecular hydrogen bonding in excited states. J. Phys. Chem. Lett. 5, 2138–2143. doi: 10.1021/jz500895w
Zhao, X., Ji, F., Liang, Y., Li, P., Jia, Y., Feng, X., et al. (2020). Theoretical and spectroscopic investigation on ultrafast nonadiabatic photoprotective mechanism of novel ultraviolet protective compounds inspired by natural sunscreens. J. Lumin. 223:117228. doi: 10.1016/j.jlumin.2020.117228
Keywords: photoprotection, femtosecond, sinapates, photochemistry, spectroscopy
Citation: Horbury MD, Turner MAP, Peters JS, Mention M, Flourat AL, Hine NDM, Allais F and Stavros VG (2020) Exploring the Photochemistry of an Ethyl Sinapate Dimer: An Attempt Toward a Better Ultraviolet Filter. Front. Chem. 8:633. doi: 10.3389/fchem.2020.00633
Received: 22 April 2020; Accepted: 17 June 2020;
Published: 28 July 2020.
Edited by:
Jacob Dean, Southern Utah University, United StatesReviewed by:
Ryoji Kusaka, Japan Atomic Energy Agency, JapanCopyright © 2020 Horbury, Turner, Peters, Mention, Flourat, Hine, Allais and Stavros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael D. Horbury, TS5ELkhvcmJ1cnlAbGVlZHMuYWMudWs=; Vasilios G. Stavros, Vi5TdGF2cm9zQHdhcndpY2suYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.