 Heesun Yu
Heesun Yu Hyoungsu Kim
Hyoungsu Kim Dongjoo Lee
Dongjoo Lee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 29 July 2020
Sec. Organic Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00629
This article is part of the Research Topic Synthetic Methodologies for Privileged Natural Product Scaffolds View all 8 articles
A highly efficient metal-free oxidative direct C(sp3)–H functionalization of N-acyl/sulfonyl 1,2,3,4-tetrahydroisoquinolines (THIQs) with a wide range of electron-rich nucleophiles was accomplished under mild conditions through oxidation with DDQ and subsequent trapping of the resulting reactive and stable N-acyl/sulfonyl iminium ions. The synthetic utility of this method was illustrated by a concise and efficient total synthesis of (±)-benzo[a]quinolizidine (10) in 3 steps from the known N-Cbz 1,2,3,4-THIQ 4b.
C(1)-Substituted 1,2,3,4-tetrahydroisoquinolines (THIQs) constitute an important family of biologically active alkaloids, and their derivatives are found as major structural motifs in a wide range of natural products as well as medicines such as (–)-ecteinascidin 743 (Yondelis®, 1, anti-tumor activity) (Rinehart, 2000), (–)-emetine (2, treatment of amoebiasis and amebic dysentery) (Akinboye and Bakare, 2011), and (–)-noscapine (3,anti-tussive agent) (Segal et al., 1957) (Figure 1). Not surprisingly, natural and synthetic C(1)-substituted 1,2,3,4-THIQs have attracted much interest from synthetic organic as well as medicinal chemists due to their interesting structural features, in conjunction with a diverse range of biological activities (Bentley, 2001; Scott and Williams, 2002; Chrzanowska and Rozwadowska, 2004), and the development of a new and efficient strategy toward the construction of the C(1)-substituted 1,2,3,4-THIQs still remains imperative.
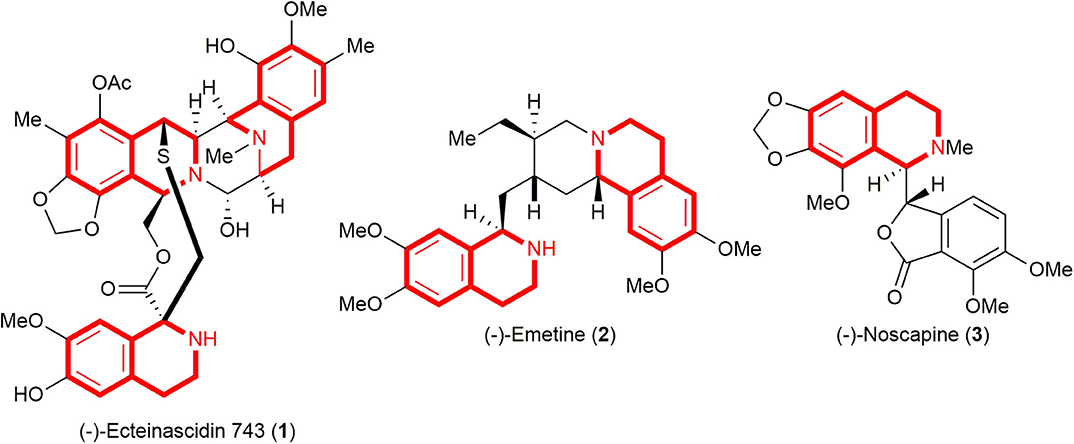
Figure 1. Selected biologically active natural products embodying C(1)-substituted 1,2,3,4-tetrahydroisoquinoline (THIQ) subunit.
During our investigation of the scope and limitations of using a variety of nucleophiles in the oxidative direct C(sp3)–H functionalization of N-acyl/sulfonyl 1,2,3,4-THIQs (Kim et al., 2018), we recognized that a variety of structurally and electronically different nucleophiles were employed in the majority of reported examples such as styrenes (Richter et al., 2012), terminal alkynes (Su et al., 2011; Freeman et al., 2012; Yu et al., 2013; Sun et al., 2015), nitroalkanes (Tsang and Todd, 2009; Hari and König, 2011; Su et al., 2011; Dhineshkumar et al., 2013; Nobuta et al., 2013), dialkyl malonate (Dubs et al., 2008; Hari and König, 2011), malonitrile (Su et al., 2011), nitrile (Murahashi et al., 2003, 2005; Yan et al., 2014), aldehydes (Xie et al., 2016), α,β-unsaturated aldehydes (Zhang et al., 2012), ketones (Shen et al., 2009; Sud et al., 2009; Alagiri et al., 2012a,b; Chen et al., 2014), α,β-unsaturated γ-butyrolactam (Ma et al., 2014), coumarins (Alagiri et al., 2012a,b; Dhineshkumar et al., 2013), aryl boronic acids (Baslé and Li, 2008), aryl boronates (Liu et al., 2015), organotrifluoroboronates (Xie et al., 2014), phosphonates (Hari and König, 2011; Alagiri et al., 2012a,b; Wang et al., 2012), or difluoramide (Chen et al., 2015). Although sporadic examples were reported on the use of electron-rich aromatic nucleophiles such as indole (Alagiri et al., 2011; Ghobrial et al., 2011; Su et al., 2011; Dhineshkumar et al., 2013) and phenols (Dhineshkumar et al., 2013) in this research area, there has been no practical and general method for oxidative direct C(sp3)–H functionalization of 1,2,3,4-THIQs with electron-rich nucleophiles which are labile to oxidation such as organostannes, silyl enol ethers, or other aromatic rings bearing electron-donating substituents. We postulated that the use of such electron-rich nucleophiles was limited in this area, presumably since they are rapidly oxidized and lose their nucleophilicity under oxidative conditions. In addition, silyl enol ethers or ketene silyl acetals are very unstable under harsh reaction conditions such as a high temperature and long reaction time that most transition metal-catalyzed oxidative direct C(sp3)–H functionalization of 1,2,3,4-THIQs required to proceed to completion. We envisaged that this problem could be circumvented through the direct oxidation of N-protected 1,2,3,4-THIQs with a proper oxidant in the absence of moisture first, thereby leading to a high-yielding in situ reactive and stable iminium ion along with the consumption of the oxidant, then subsequent trapping of the resultant iminium ion with electron-rich nucleophiles, which will afford the corresponding N-protected C(1)-substituted 1,2,3,4-THIQs avoiding the oxidation of electron-rich nucleophiles (Scheme 1).
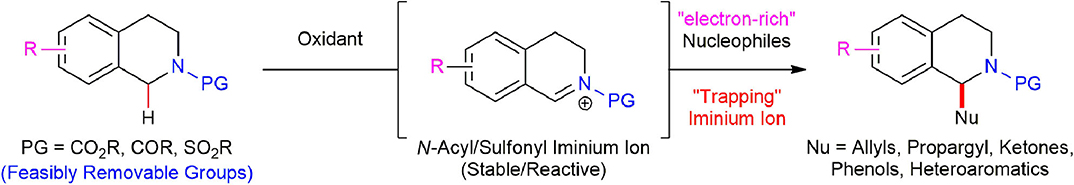
Scheme 1. Proposed strategy for oxidative C(sp3)–H functionalization of N-acyl/sulfonyl 1,2,3,4-THIQs with electron-rich nucleophiles.
The majority of oxidative functionalization reactions widely employed an aryl group as the activating and protecting group for 1,2,3,4-THIQs (Li, 2009; Scheuermann, 2010; Yoo and Li, 2010; Klussmann and Sureshkumar, 2011; Yeung and Dong, 2011; Rohlmann and Mancheño, 2013), since the aryl group on the nitrogen atom activates the C(sp3)–H bond at the C(1)-position of 1,2,3,4-THIQs and stabilizes the resulting iminium ion intermediate. Although Todd and co-workers recently identified that 4-methoxyphenyl (PMP) group is a removable protecting group in the oxidative direct C(sp3)–H functionalization (Tsang et al., 2013), it still proves to be problematic to remove the aryl protecting group from the nitrogen atom in the presence of other functional groups, which significantly limits the synthetic utility of oxidative functionalization of N-aryl 1,2,3,4-tetrahydroisoquinolines. For instance, the phenyl protecting group from amines was removed under harsh reaction conditions where only a small set of organic compounds could be tolerated (Girard et al., 2004, 2005; Girard and Hurvois, 2007). Therefore, use of easily removable N-acyl or N-sulfonyl groups on the nitrogen atom of 1,2,3,4-THIQs in place of the aryl ones would provide an attractive solution for enhancing the scope and synthetic utility of the direct C(sp3)–H functionalization of 1,2,3,4-THIQs through generating a more reactive N-acyl/sulfonyl iminium ion intermediate that can react with a broader range of nucleophiles.
Considering that 1,2,3,4-THIQ motifs are core units found in a multitude of pharmacologically active natural products and medicines, the development of an operationally convenient and practical method to introduce a wide range of nucleophiles is still a worthwhile project to pursue. Herein we wish to report a new direct metal-free direct C(sp3)–H functionalization of N-acyl/sulfonyl 1,2,3,4-THIQs with a variety of electron-rich nucleophiles via 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) oxidation under ambient conditions.
At the outset of our studies, we examined the C(1)-allylation of N-Boc 1,2,3,4-THIQ 4a (Hickin et al., 2014), which is ubiquitous structural frameworks in numerous pharmacologically active THIQ natural products, as a model substrate to test the viability of the envisioned direct metal-free C(sp3)–H functionalization. The allyl moiety is exceptionally versatile and synthetically useful in that this functional group offers a wealth of opportunities to further functionalization (Denmark and Fu, 2003). Although Wang and co-workers (Yan et al., 2015) recently reported the use of allyltrimethylsilane (Me3SiCH2CH=CH2) as the nucleophile in direct oxidative C(1)-allylation of N-acyl/N-sulfonyl 1,2,3,4-THIQs employing 2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (T+), success of such a direct oxidative transformation with an electron-rich allyltrialkylstannane was not yet to be proven, presumably, due to their high propensity of oxidation in the presence of oxidizing agents. It is difficult to generate N-acyl or N-sulfonyl iminium ion intermediates with commonly used transition metal catalysts or non-metal organic oxidants (Luo et al., 2020). Therefore, a judicious selection of oxidant is critical. We selected 1,2-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (Walker and Hiebert, 1967; Fu and Harvey, 1978; Wendlandt and Stahl, 2015) since it is inexpensive and stable organic solid that is conveniently handled under ambient conditions, and permits mild and more practical reaction conditions. To test the compatibility of allyltributylstannane ((n-Bu)3SnCH2CH=CH2) in the presence of DDQ, DDQ (1.1 equiv) was added to a mixture of 4a (1.0 equiv) and (n-Bu)3SnCH2CH=CH2 (2.5 equiv) in the presence of 4Å MS in DCM, and the reaction mixture was stirred for 1 h at ambient temperature (Scheme 2). However, the desired C(1)-allylated N-Boc 1,2,3,4-THIQ (±)-5a was not obtained, but most of 4a was recovered, presumably due to faster oxidation of electron-rich nucleophile (n-Bu)3SnCH2CH=CH2 than 4a. Pleasingly, treatment of 4a with DDQ (1.1 equiv) as an oxidant in the presence of 4Å MS in DCM at room temperature for 30 min, thereby leading in situ high yield of the reactive N-Boc iminium ion along with consumption of the oxidant. The subsequent addition of (n-Bu)3SnCH2CH=CH2 (2.5 equiv) afforded the desired (±)–5a in excellent yield (98%). Molecular sieves (4Å) was added to eliminate moisture that might be present in the reaction mixture and the reactivity of the N-Boc iminium ion lasted for several hours at room temperature under argon atmosphere. To the best of our knowledge, such a DDQ-mediated direct functionalization of C(sp3)–H functionalization of N-Boc 1,2,3,4-THIQ with electron-rich (n-Bu)3SnCH2CH=CH2 as a nucleophile has not been reported yet.
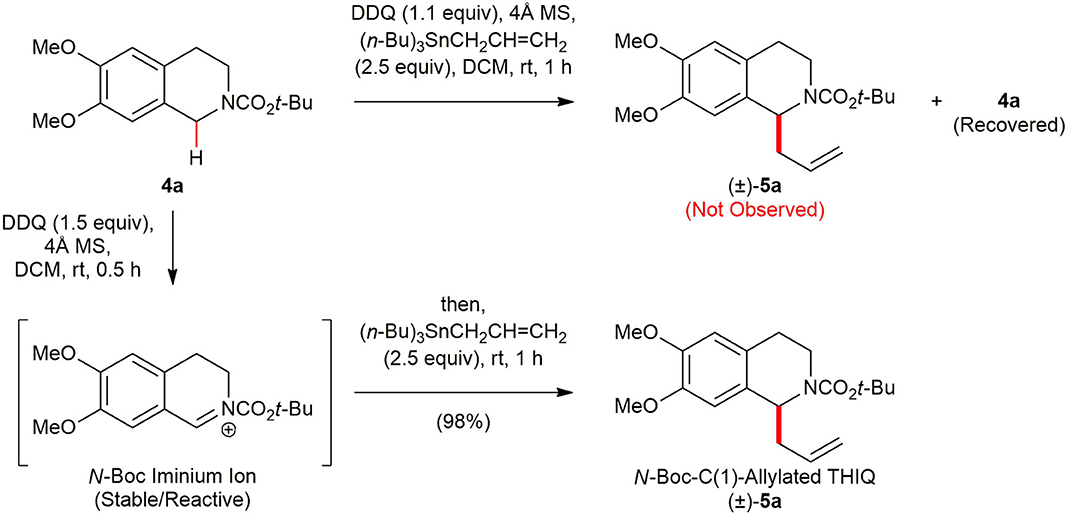
Scheme 2. DDQ-promoted C(1)-allylation of N-Boc 1,2,3,4-THIQ 4a with an electron-rich allyltributylstannane.
Among oxidants tested in this study, ceric ammonium nitrate (CAN) proved to be an effective oxidant albeit with lower yield (62%) (entry 5, Table 1) compared with DDQ. However, other oxidants including (diacetoxyiodo)benzene (PhI(OAc)2), 1,4-benzoquinone (1,4-BQ), TBHP (tert-butyl hydroperoxide), and silver acetate (AgOAc) did not promote the oxidative allylation reaction, and only unreacted starting material 4a was recovered (entries 1–4, Table 1). Solvent screening studies revealed that most organic solvents tested were effective (entries 6–14, Table 1). When EtOAc or THF was used, the desired product could be obtained in excellent yields (90 and 92%, respectively) (entries 6–7, Table 1). Also, highly polar solvents such as acetone, DMF, and MeCN resulted in the desired product (±)–5a in high yields (70–86%) (entries 9–11, Table 1). Allyltriphenylstannane (Ph3SnCH2CH=CH2) also proved to be an effective nucleophile (entries 12, Table 1). However, low yield (17%) was obtained when allyltrimethylsilane (Me3SiCH2CH=CH2) (entries 13, Table 1) was used as an allyl nucleophile.
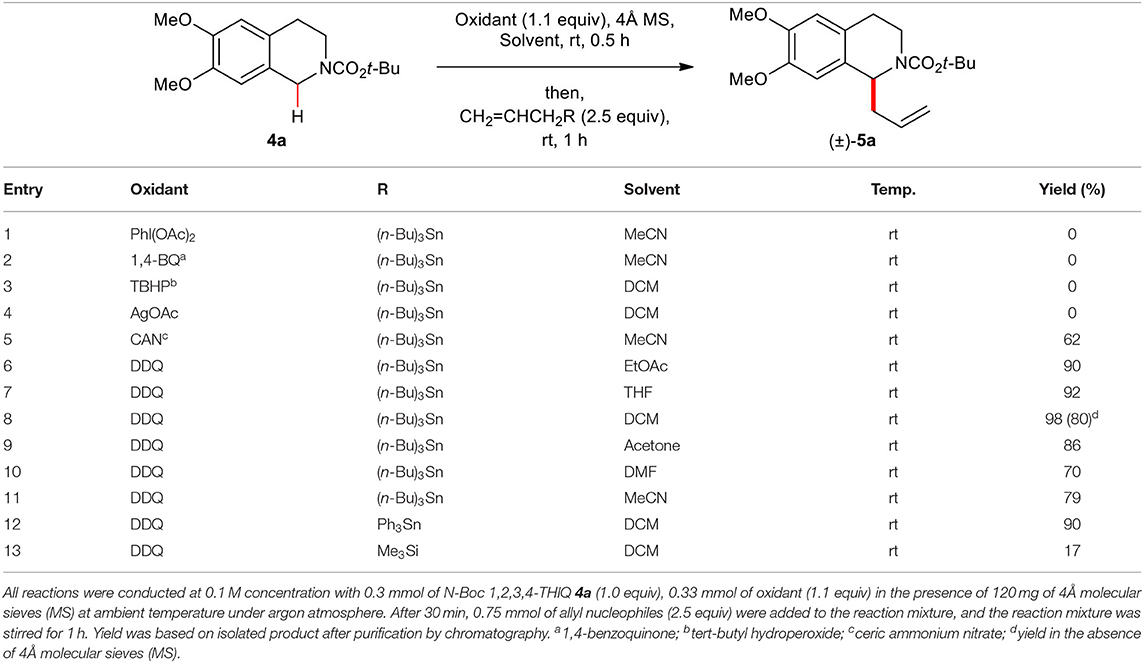
Table 1. Optimization of oxidative C(1)-allylation of N-Boc 1,2,3,4-THIQ 4a with an electron-rich allylating reagent.
With optimized reaction conditions in hand, the scope of the oxidative direct C(sp3)–H functionalization was investigated with a diverse range of electron-rich nucleophiles (Scheme 3). The reactions of methallyltributylstannane [(n-Bu)3SnCH(Me)CH=CH2] and dimethyltributylstannane [(n-Bu)3SnCH2CH=CMe2] provided the corresponding C(1)-allylated products (±)–5b and (±)–5c in 88% and 64% yields, respectively. Also, allenyltributylstannane [(n-Bu)3SnC=C=CH2] provided the desired C(1)-propargylated product (±)–5d in 77% yield. Although a variety of ketones have been widely employed as pro-nucleophiles in cross dehydrogenative coupling (CDC) reactions of N-aryl 1,2,3,4-THIQs, the use of electron-rich silyl enol ethers (Scott et al., 2014) or silyl ketene acetals have rarely been reported. A diverse range of silyl enol ethers and a silyl ketene acetal have been tested in order to expand the scope and utility of this oxidative DDQ-promoted direct C(sp3)–H functionalization of N-acyl 1,2,3,4-THIQs. All of the silyl enol ethers tested so far worked rather well with 4a to provide Mannich products (±)–5e-j in isolated yield ranging from 63 to 91%. The reaction could also be readily expanded to oxidative Friedel–Crafts-type reaction. Under the optimal reaction condition, 4a with 3,5-dimethoxyphenol, 3-dimethylaminophenol and 1-naphthol afforded Friedel–Crafts products (±)-5k (75%), (±)–5l (70%), and (±)–5n (76%) in good yields. In these examples, 4a was coupled to phenols selectively at the ortho-position, while the phenolic-OH is unaffected. Furthermore, we found that N, N-diethylaniline, indole, and 2-methyl furan are good nucleophiles for this oxidative DDQ-promoted direct C(sp3)–H functionalization to afford the corresponding Friedel-Crafts products (±)–5m (78%), (±)–5o (84%), and (±)–5p (55%). To the best of our knowledge, this is the first report using electron-rich nucleophiles in oxidative direct C(sp3)–H functionalization of a N-acyl 1,2,3,4-THIQ.
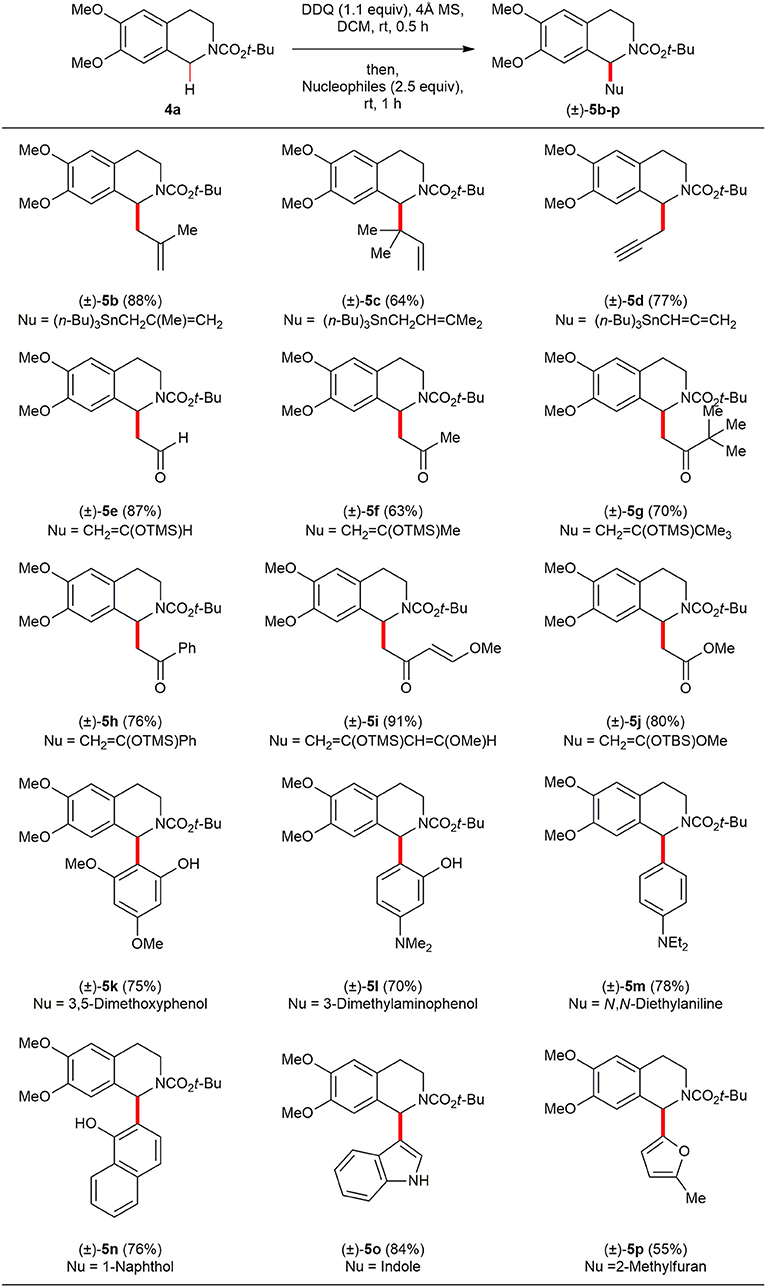
Scheme 3. Reaction scope of N-Boc 1,2,3,4-THIQ 4a and electron-rich nucleophiles. All reactions were conducted at 0.1 M concentration with 0.3 mmol of N-Boc 1,2,3,4-THIQ 4a (1.0 equiv), 0.33 mmol of DDQ (1.1 equiv) in the presence of 120 mg of 4Å MS at ambient temperature under argon atmosphere. After 30 min, 0.75 mmol of nucleophiles were added to the reaction mixture, and the reaction mixture was stirred for 1 h. Yield was based on isolated product after purification by chromatography.
To make this oxidative direct C(sp3)–H functionalization synthetically useful, we explored the use of a broad range of N-acyl/sulfonyl THIQs since installation and liberation of their amine protecting groups are easy and operationally convenient (Scheme 4). The C(1)-allylation reaction of benzyl- (4b), allyl- (4c), methyl- (4d), and ethyl (4e) carbamates under DDQ-promoted oxidative reaction conditions all provided the corresponding C(1)-allylated products (±)–6a–d in good to excellent yields (60–84%). Furthermore, reactions of N-sulfonamides such as N-Ts (4h), N-Ms (4i), N-Ns (4j) generated the corresponding C(1)-allylated products (±)–6g–i in high yields (72–89%). However, amides such as acetamide (4f) and benzamide (4g) proved to be ineffective substrates to afford the corresponding C(1)-allylated products (±)–6e and (±)–6f in low yield (48 and 37%, respectively) under the optimized reaction conditions.
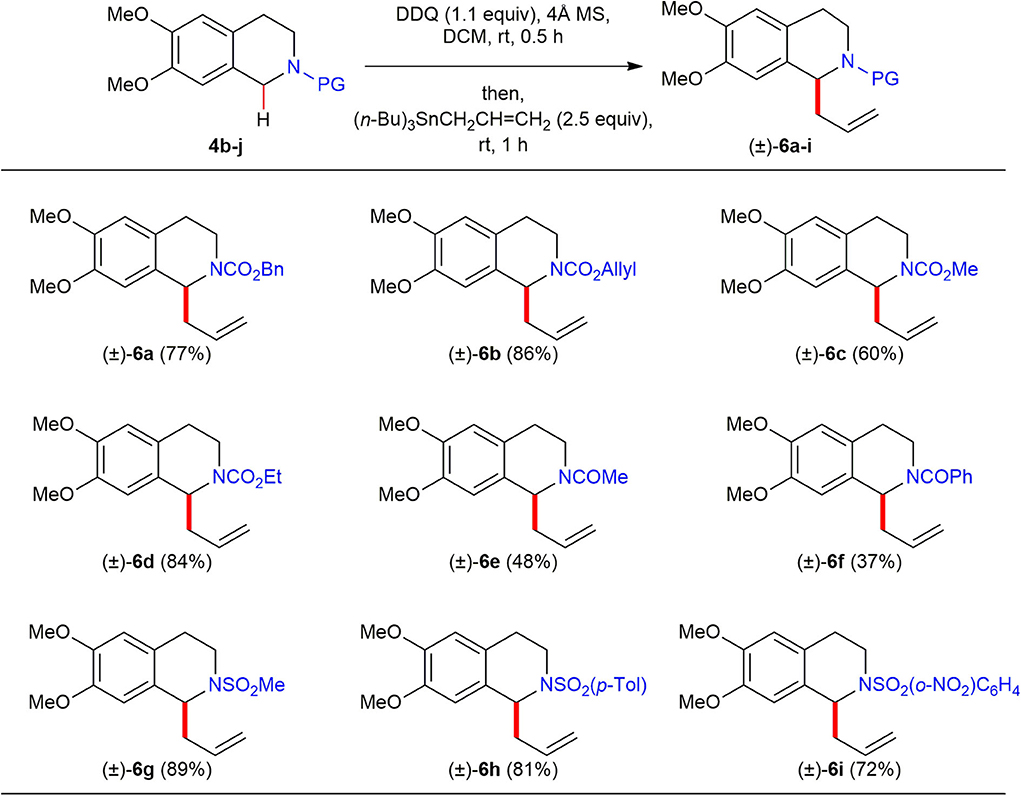
Scheme 4. Reaction scope of N-acyl/sulfonyl 1,2,3,4-THIQs 4b–j and allyltributylstannane. All reactions were conducted at 0.1 M concentration with 0.3 mmol of N-acyl/sulfonyl 1,2,3,4-THIQs 4b–j (1.0 equiv), 0.33 mmol of DDQ (1.1 equiv) in the presence of 120 mg of 4Å MS at ambient temperature under argon atmosphere. After 30 min, 0.75 mmol of allyltributylstannane (2.5 equiv) was added to the reaction mixture, and the reaction mixture was stirred for 1 h. Yield was based on isolated product after purification by chromatography.
We further investigated the substrate scope with respect to electronically diverse N-Boc 1,2,3,4-THIQs (Scheme 5). As expected, direct C(1)-allylation of N-Boc 1,2,3,4-THIQs 4k–m bearing electron-donating substituents on the phenyl moiety led to the corresponding C(1)-allylated products (±)–7a–7c in high yields (79–98%). Notably, N-Boc 1,2,3,4-THIQs bearing electron-withdrawing substituents such as fluorine (4n) and bromine (4o) on the phenyl moiety were also tolerated to furnish the corresponding C(1)-allylated products (±)–7d and (±)–7e in high yield (89 and 85%, respectively), which are useful for further diversifications. Also, N-Boc 1,2,3,4-THIQ 4p with no substituents on the phenyl moiety was found to be effective to afford the desired C(1)-allylated product (±)–7r in 92% yield.
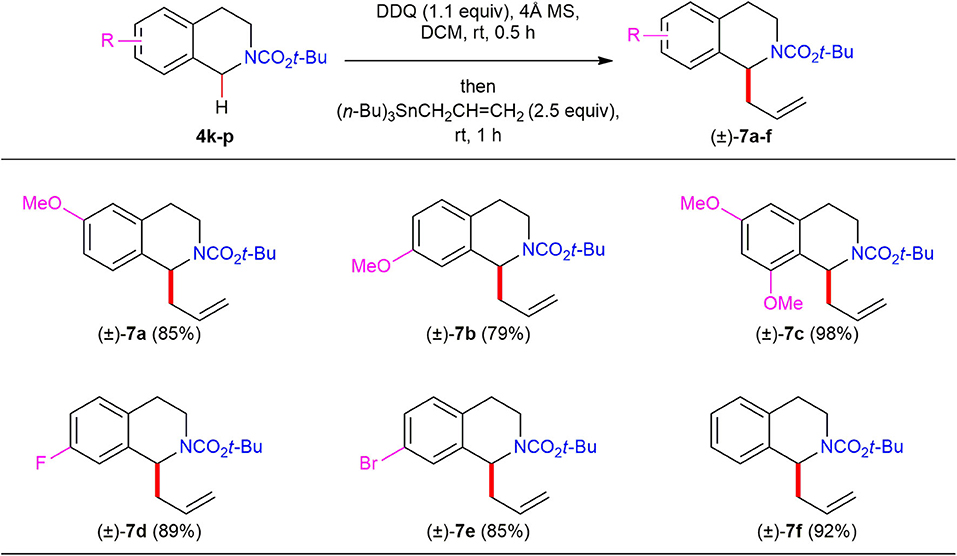
Scheme 5. Reaction scope of electronically diverse N-Boc 1,2,3,4-THIQs 4k–p and allyltributylstannane. All reactions were conducted at 0.1 M concentration with 0.3 mmol of N-Boc 1,2,3,4-THIQs 4k–p (1.0 equiv), 0.33 mmol of DDQ (1.1 equiv) in the presence of 120 mg of 4Å MS at ambient temperature under argon atmosphere. After 30 min, 0.75 mmol of allyltributylstannane (2.5 equiv) was added to the reaction mixture, and the reaction mixture was stirred for 1 h. Yield was based on isolated product after purification by chromatography.
With the desired N-acyl/sulfonyl C(1)-substituted 1,2,3,4-THIQs in hand, a variety of means for liberation of the C(1)-allylated 1,2,3,4-THIQs were investigated (Table 2). The tert-butoxycarbonyl (Boc) group of (±)–5a was cleanly removed under acidic (CF3CO2H) conditions to give free amine (±)–8 in high yield (88%). Alkaline hydrolysis of the methoxycarbonyl group in (±)–6c with KOH by heating at reflux in ethylene glycol furnished free amine (±)–8 in good yield (70%). Also, removal of the 2-nitrobenzenesulfonyl (Ns) group of (±)–6h proceeded smoothly by employing the condition (PhSH and K2CO3) reported by Fukuyama and co-worker (Fukuyama et al., 1995) to afford free amine (±)–8 in high yield (88%).
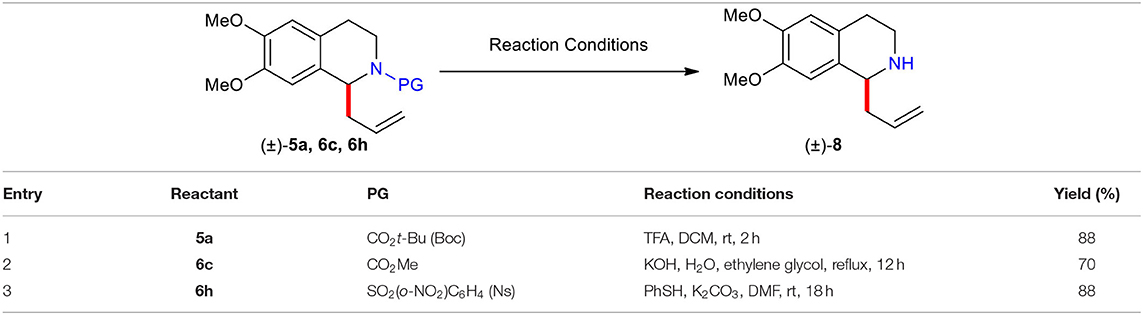
Table 2. Liberation of C(1)-allylated 1,2,3,4-THIQ (±)–8 from (±)–5a, 6c, and 6h.
In order to gain some mechanistic insight into the reaction mechanism, the radical inhibition experiments were conducted. When 2,6-di-tert-butyl-4-methylphenol (BHT) (1.1 equiv) was added to the reaction mixture of N-Boc 1,2,3,4-tetrahydroisoquinoline 4a (1.0 equiv) and DDQ (1.1 equiv), the yield of the desired product (±)–5a was dramatically decreased from 98 to 20% and 79% of the starting material 4a was recovered. This result suggests that a radical cation species might be involved in the reaction. On the basis of the radical inhibition experiments and literature precedents (Muramatsu et al., 2013; Chen et al., 2015), a plausible reaction mechanism for the DDQ-promoted oxidative direct C(sp3)–H functionalization of N-acyl/sulfonyl 1,2,3,4-THIQ 4 was proposed (Scheme 6). N-Acyl/sulfonyl 1,2,3,4-THIQ 4 undergoes a single electron transfer from N-acyl/sulfonyl 1,2,3,4-THIQ 4 to DDQ to generate a radical cation (A). The DDQ radical oxygen then abstracts a H-atom from A, leading to a stable and reactive N-acyl/sulfonyl iminium ion (B). Finally, the trapping the iminium ion (B) with a diverse range of electron-rich nucleophiles afforded the desired N-acyl/sulfonyl C(1)-substituted THIQs (±)–5–7.
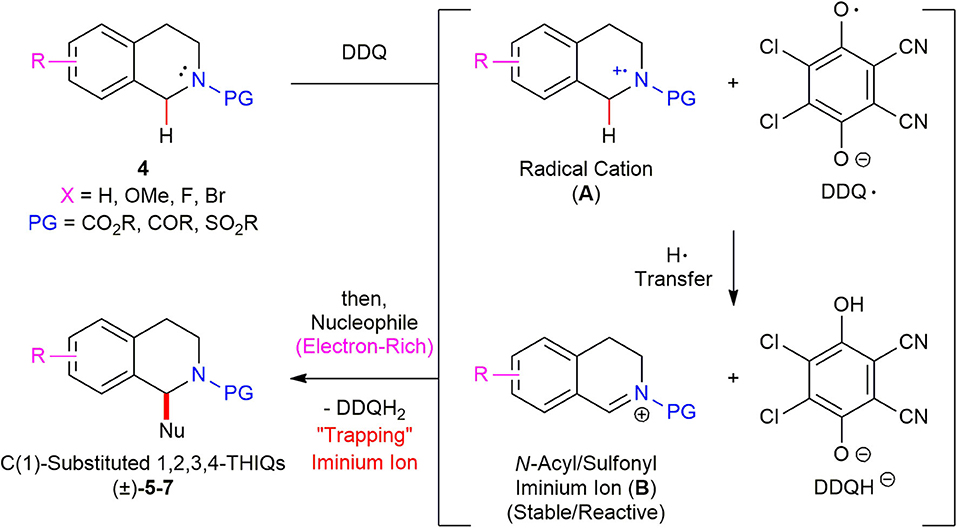
Scheme 6. Proposed reaction mechanism for DDQ-promoted C(1)-allylation of N-acyl/sulfonyl 1,2,3,4-THIQs.
We next turned on our attention to a short and efficient total synthesis of (±)-benzo[a]quinolizidine (10) to prove the synthetic utility of this method (Scheme 7). The oxidative direct C(sp3)–H functionalization of the readily available N-Cbz 1,2,3,4-THIQ 4b (Dunetz et al., 2005; Kim et al., 2018) with CH2=C(OTMS)H afforded aldehyde, which underwent Wittig olefination with a two carbon stabilized ylide Ph3P=CHCO2Me to furnish α,β-unsaturated ester (±)–8 in 79% yield in a one-pot fashion, exhibiting high stereoselectivity (E/Z = 95:5), that is none the less to be rendered in consequential at this stage because the planned hydrogenation/deprotection/ring-closure reaction sequence was envisaged to provide a single product regardless of the olefin geometry. The hydrogenation of the olefin moiety, simultaneous deprotection of the Cbz group on the nitrogen atom of the THIQ framework and ring closure was achieved smoothly by hydrogenation (1 atm) over 10% Pd/C in EtOAc to provide the desired lactam (±)–9 in 85%. Reduction of lactam (±)–9 with LiAlH4 in THF according to Reddy and co-workers (Reddy et al., 2013) afforded (±)-benzo[a]quinolizidine (10) in 77%, whose spectral data were in good agreement with those reported in the literature (Williams et al., 2005; Szawkalo et al., 2007; Reddy et al., 2013; Talk et al., 2016).
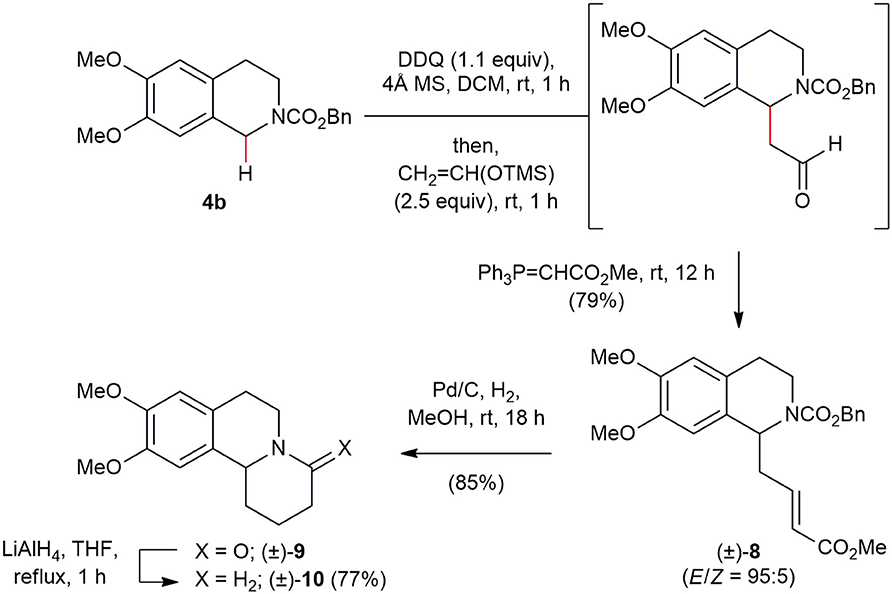
Scheme 7. A concise and efficient 3-step total synthesis of (±)-benzo[a]quinolizidine (11).
In conclusion, N-acyl/sulfonyl 1,2,3,4-THIQ iminium complexes in situ generated by DDQ were found to be very effective and compatible with a wide range of electron-rich nucleophiles. New and useful nucleophiles such as silyl enol ethers and silyl ketene acetals are employed to afford Mannich-type products and use of phenols, heteroaromatics furnished Friedel–Crafts-type products. Further studies are ongoing to expand the synthetic utility of this products to natural product or synthetically useful compounds.
Except as otherwise indicated, reactions were carried out under argon atmosphere in flame- or oven-dried glassware. In aqueous work-up, all organic solutions were dried over sodium sulfate (Na2SO4) or magnesium sulfate (MgSO4), and filtered prior to rotary evaporation at water aspirator pressure. Reactions were monitored by thin layer chromatography (TLC) with 0.25-mm E. Merck pre-coated silica gel plates (Kieselgel 60F254, Merck). Spots were detected by viewing under a UV light, colorizing with charring after dipping in p-anisaldehyde solution with acetic acid and sulfuric acid and ethanol, or ceric ammonium molybdate solution with sulfuric acid and ethanol. Silica gel for flash chromatography (particle size 0.040–0.063 mm) was supplied by E. Merck. Yields refer to chromatographically and spectroscopically pure compounds unless otherwise noted.
All commercial reagents and solvents were purchased from Sigma Aldrich Co. or Tokyo Chemical Industry (TCI) and used as received with the following exceptions. All solvents were freshly purified and dried by standard techniques just before use. Tetrahydrofuran (THF) was distilled from sodium/benzophenone. Dichloromethane (CH2Cl2), acetonitrile (MeCN), N, N-dimethylformamide (Me2NC(=O)H), benzene (C6H6) and toluene (C7H8) were distilled from calcium hydride (CaH2). Methanol (MeOH) was distilled from magnesium sulfate (MgSO4).
1H and 13C spectra were recorded on Varian Mercury-400BB (400 MHz). Chemical shifts are reported as δ value relative to internal chloroform (δ 7.26 for 1H and δ 77.0 for 13C). Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant in Hz, and integration. High resolution mass spectra (HRMS) were recorded on JEOL JMS-700 (FAB or EI) mass spectrometer. High resolution values are calculated to four decimal places from the molecular formula, all found values being within a tolerance of 5 ppm.
To a stirred solution of N-protected 1,2,3,4-tetrahydroisoquinoline (0.30 mmol) in DCM (3.0 mL, 0.1M) was added 4Å molecular sieves (120 mg) at room temperature. After the reaction mixture was stirred for 15 min at room temperature, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (0.45 mmol, 1.1 equiv) was added portionwise and the reaction mixture was stirred at room temperature for 30 min under argon atmosphere. Nucleophile (0.75 mmol, 2.5 equiv) was added dropwise or portionwise at room temperature. The reaction mixture was stirred at room temperature for 1 h under argon atmosphere, then quenched with saturated NaHCO3 solution (10 mL) and the layers were separated. The aqueous layer was extracted with DCM (2 × 25 mL), and the combined organic layer was washed with brine (5 mL), dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash column chromatography on silica gel, using hexanes/EtOAc as eluent, provided the corresponding N-protected 1-substituted-1,2,3,4-tetrahydroisoquinoline.
(±)-tert-Butyl 1-allyl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5a). Yield 98% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 6.60 (s, 2H), 5.80–5.90 (m, 1H), 5.16 (brs, 0.4H), 5.01–5.07 (m, 2.6H), 4.20–4.23 (m, 0.6H), 3.97–4.02 (m, 0.4H), 3.86 (s, 1.2H), 3.84 (1.8H), 3.23–3.28 (m, 0.4H), 3.12–3.18 (m, 0.6H), 2.82–2.90 (m, 1H), 2.64 (t, J = 3.6 Hz, 0.6H), 2.60 (t, J = 3.6 Hz, 0.4H), 2.52 (t, J = 7.2 Hz, 2H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 153.9, (147.0), 146.7, 134.6, (128.7), 128.4, 125.8, (125.5), 116.6, (116.2), 111.0, (110.9), (109.7), 109.4, 79.1, (78.8), 55.5, (55.4), 53.7, (52.8), 41.1, (40.8), (38.0), 36.3, 28.1, (27.9), 27.8; IR (Film) 2975, 1691, 1519, 1422, 1259, 1165 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C19H27NO4: 333.1940; Found 333.1936.
(±)-tert-Butyl 6,7-dimethoxy-1-(2-methylallyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5b). Yield 88% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 6.56–6.60 (m, 2H), 5.27 (dd, J = 8.8, 5.6 Hz, 0.4H), 5.07 (dd, J = 8.8, 5.2 Hz, 0.6H), 4.82 (s, 0.6H), 4.78 (s, 0.4H), 4.68 (s, 1H), 4.24 (dd, J = 13.4, 3.8 Hz, 0.6H), 4.00 (dd, J = 13.6, 3.2 Hz), 3.86 (s, 1.2H), 3.85 (s, 1.8H), 3.13–3.29 (m, 1H), 2.79–2.93 (m, 1H), 2.60–2.64 (m, 1H), 2.49–2.55 (m, 1H), 2.28–2.39 (m, 1H), 1.89 (s, 3H), 1.47 (s, 5.4H), 1.45 (s, 3.6H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 154.5, 147.6, (147.4), 147.0, (142.7), 141.8, (129.9), 129.3, 126.3, (125.9), 114.0, 113.2, 111.5, (111.3), (110.2), 110.1, 79.8, (79.3), (56.1), 55.9, 52.9, (52.1), 45.3, (45.1), (38.0), 36.5, 28.5, (28.3), 28.2, 22.9, (22.6); IR (Film) 2971, 1684, 1516, 1419, 1240, 1161 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C20H30NO4: 348.2175; Found 348.2183.
(±)-tert-Butyl 6,7-dimethoxy-1-(2-methylbut-3-en-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5c). Yield 64% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1:1 mixture of amide rotamers at room temperature) δ 6.74 (s, 0.5H), 6.72 (s, 0.5H), 6.60 (s, 0.5H), 6.58 (s, 0.5H), 5.87 (dd, J = 17.6, 11.2 Hz, 0.5H), 5.83 (dd, J = 16.4, 10.8 Hz, 0.5H), 5.08 (s, 0.5H), 4.97 (s, 0.5H), 4.91–4.95 (m, 2H), 4.18 (ddd, J = 12.8, 7.6, 7.6 Hz, 0.5H), 3.85–3.92 (m, 0.5H), 3.86 (s, 1.5H), 3.85 (s, 1.5H), 3.84 (s, 1.5H), 3.82 (s, 1.5H), 3.52 (ddd, J = 14.8, 7.2, 7.2 Hz, 0.5H), 3.39 (ddd, J = 15.6, 9.6, 6.0 Hz, 0.5H), 2.69–2.88 (m, 2H), 1.49 (s, 4.5H), 1.46 (s, 4.5H), 1.14 (s, 3H), 1.11 (s, 1.5H), 1.08 (s, 1.5H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 155.5, (154.9), (147.7), 147.5, (147.2), 147.0, 146.2, 127.2, (127.0), 126.8, 112.0, 111.5, (111.34), 111.25, (111.1), 79.9, (79.4), (61.7), 61.0, 56.1, (56.0), 55.9, 43.8, 39.6, (38.0), (28.7), 28.6, (27.7), 27.6, 27.5, (27.3), (24.2), 24.0; IR (Film) 2970, 1684, 1517, 1364, 1249, 1160 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C21H32NO4: 362.2331; Found 362.2331.
(±)-tert-Butyl 6,7-dimethoxy-1-(prop-2-yn-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5d). Yield 77% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.4:1 mixture of amide rotamers at room temperature) δ 6.76 (s, 1H), 6.61 (s, 1H), 5.25 (t, J = 6.4 Hz, 0.42H), 5.13 (t, J = 6.4 Hz, 0.58H), 4.14–4.23 (m, 0.58H), 3.90–3.97 (m, 0.42H), 3.87 (s, 2.52H), 3.86 (s, 3.48H), 3.40–3.46 (m, 0.42H), 3.23–3.30 (m, 0.58H), 2.65–2.90 (m, 4H), 2.01 (s, 0.58H), 1.98 (s, 0.42H), 1.51 (s, 5.22H), 1.49 (s, 3.78H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ (154.4), 154.2, 147.7, 147.0, (127.6), 127.4, 126.5, (126.3), 111.2, (111.0), (110.2), 109.9, 81.4, 80.0, (79.7), 70.7, (70.6), 55.9, 55.8, 53.1, (52.4), (39.1), 37.3, 28.5, (28.4), 28.2, 26.6, (26.2); IR (Film) 3287, 2974, 2118, 1690, 1519, 1259 (cm−1); HRMS (FAB-magnetic sector) m/z: {M}+ Calcd for C19H25NO4: 331.1784; Found 331.1779.
(±)-tert-Butyl 6,7-dimethoxy-1-(2-oxoethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5e). Yield 87% as a white foam. 1H NMR (400 MHz, CDCl3, a 1:1 mixture of amide rotamers at room temperature) δ 9.84 (t, J = 3.6 Hz, 1H), 6.62 (s, 3H), 6.60 (s, 3H), 5.61–5.71 (m, 0.5H), 5.46–5.54 (m, 0.5H), 4.15–4.30 (m, 1H), 3.94–4.02 (m, 0.5H), 3.85 (s, 3H), 3.10–3.40 (m, 1H), 2.76–2.96 (m, 3H), 2.68 (t, J = 3.6 Hz, 0.5H), 2.64 (t, J = 3.6 Hz, 0.5H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 199.5, 154.0, (153.2), 147.3, 147.1, 127.3, 125.9, (125.8), 111.0, 109.0, 80.1, (79.6), 55.5, 55.4, 50.8, (49.5), 48.9, 38.3, (36.9), 28.0, (27.7), 27.6; IR (Film): 2975, 1722, 1689, 1519, 1419, 1258, 1163 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C18H25NO5: 335.1733; Found 335.1725.
(±)-tert-Butyl 6,7-dimethoxy-1-(2-oxopropyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5f). Yield 63% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.2:1 mixture of amide rotamers at room temperature) δ 6.68 (s, 0.45H), 6.65 (s, 0.55H), 5.61 (s, 0.45H), 5.48 (s, 0.55H), 4.14–4.18 (m, 0.45H), 3.88–3.94 (m, 0.55H), 3.85 (s, 3H), 3.29–3.34 (m, 0.45H), 3.18–3.23 (m, 0.55H), 2.75–2.94 (m, 3H), 2.67 (t, J = 4.0 Hz, 0.55H), 2.63 (t, J = 4.0 Hz, 0.45H), 2.25 (s, 1.35H), 2.19 (s, 1.65H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ (206.3), 206.0, (154.4), 153.9, 147.5, 147.3, 128.5, 126.0, (125.9), 111.2, (109.7), 109.3, 80.3, (79.8), 55.9, 55.8, 51.2, 51.1, (50.5), (38.8), 37.5, 31.2, (30.2), 28.4, (28.1), 27.9; IR (Film): 2976, 1689, 1519, 1418, 1222, 1164 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C19H27NO5: 349.1889; Found 349.1893.
(±)-tert-Butyl 1-(3,3-dimethyl-2-oxobutyl)-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5g). Yield 63% as a white foam. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 6.68 (s, 0.4H), 6.64 (s, 0.6H), 6.59 (s, 1H), 5.62 (t, J = 6.4 Hz, 1H), 4.08–4.12 (m, 0.6H), 3.84 (s, 3H), 3.81 (s, 3H), 3.81–3.84 (m, 0.4H), 3.34–3.43 (m, 0.4H), 3.18–3.28 (m, 0.6H), 2.65–3.03 (m, 4H), 1.47 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 212.3, 154.1, 147.4, 147.2, (129.5), 129.3, 126.0, 111.1, (110.0), 109.5, 80.0, (79.5), 55.8, 50.8, (50.5), 44.9, 44.3, (44.2), (39.6), 38.1, 28.4, 28.1, 26.0; IR (Neat): 2974, 1691, 1517, 1364, 1257, 1220 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C22H33NO5: 391.2359; Found 391.2366.
(±)-tert-Butyl 6,7-dimethoxy-1-(2-oxo-2-phenylethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5h). Yield 76% as a white foam. 1H NMR (400 MHz, CDCl3, a 1:1 mixture of amide rotamers at room temperature) δ 7.95 (d, J = 7.2 Hz, 2H), 7.52–7.60 (m, 1H), 7.47 (t, J = 7.2 Hz, 2H), 6.69 (s, 0.75H), 6.64 (s, 0.25H), 6.61 (s, 1H), 5.69–5.74 (m, 0.25H), 5.66 (dd, J = 6.0, 5.6 Hz, 0.75H), 4.19–4.22 (m, 0.5H), 3.85 (s, 3H), 3.80 (s, 2.25H), 3.75 (s, 0.75H), 3.45–3.50 (m, 1.5H), 3.22–3.34 (m, 2H), 2.69–2.93 (m, 2H), 1.41 (s, 2.25H), 1.30 (s, 6.75H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 197.2, (154.0), 153.6, 147.5, 147.1, (146.9), (136.8), 136.6, 132.8, (132.5), 128.5, 128.3, 128.2, 127.9, 126.0, 111.2, (111.0), (109.9), 109.5, 79.8, (79.14), 55.6, 51.8, (51.2), 46.1, (45.8), (39.3), 37.4, 28.2, 27.9; IR (Film): 2976, 1690, 1518, 1418, 1256, 1164 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C24H29NO5: 411.2046; Found 411.2052.
(±)-(E)-tert-Butyl 6,7-dimethoxy-1-(4-methoxy-2-oxobut-3-en-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5i). Yield 91% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 2:1 mixture of amide rotamers at room temperature) δ 7.65 (d, J = 12.4 Hz, 0.33H), 7.57 (d, J = 12.8 Hz, 0.67H), 6.67 (s, 1H), 6.59 (s, 1H), 5.70 (d, J = 12.4 Hz, 0.33H), 5.59 (d, J = 12.8 Hz, 0.67H), 5.50–5.53 (m, 1H), 4.19–4.22 (m, 0.33H), 3.85–3.90 (m, 0.67H), 3.84 (s, 3H), 3.83 (s, 3H), 3.70 (s, 3H), 3.30–3.40 (m, 0.33H), 3.17–3.22 (m, 0.67H), 2.97 (d, J = 7.2 Hz, 0.33H), 2.93 (d, J = 6.8 Hz, 0.67H), 2.72–2.88 (m, 2H), 2.68 (t, J = 3.6 Hz, 0.67H), 2.64 (t, J = 3.6 Hz, 0.67H), 1.45 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 196.6, 162.9, 154.2, 147.7, 147.4, 128.9, 126.2, 111.4, (110.2) 109.9, 106.3, (105.7), 80.3 (79.8), 57.7, 56.1, (56.0), 51.8, (51.3), 49.0, (39.2), 37.6, 28.5, 28.3; IR (Film): 2975, 1689, 1518, 1419, 1257, 1166 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C21H29NO6: 391.1995; Found 391.1992.
(±)-tert-Butyl 6,7-dimethoxy-1-(2-methoxy-2-oxoethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5j). Yield 80% as a white foam. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 6.66 (s, 0.4H), 6.65 (s, 0.6H), 6.60 (s, 0.6H), 6.59 (s, 0.4H), 5.54 (t, J = 6.0 Hz, 0.4H), 5.46 (t, J = 6.4 Hz, 0.6H), 4.14–4.21 (m, 0.6 H), 3.92–3.99 (m, 0.4H), 3.85 (s, 6H), 3.70 (s, 1.8H), 3.68 (s, 1.2H), 3.30–3.35 (m, 0.4H), 3.16–3.23 (m, 0.6H), 2.63–2.92 (5H), 1.48 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 170.84, (170.77), (154.0), 153.8, 147.6, 147.2, (128.0), 127.8, 126.1, (125.9), 111.2, (111.1), (109.5), 109.2, 79.9, (79.5), (55.78), 55.75, 55.68, (55.64), 51.7, (51.6), 51.5, 51.1, 41.9, (41.4), (38.5), 37.0, 28.2, 28.0, (27.7); IR (Film): 2971, 1739, 1593, 1517, 1418, 1254, 1166 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C19H27NO6: 365.1838; Found 365.1836.
(±)-tert-Butyl 1-(2-hydroxy-4,6-dimethoxyphenyl)-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5k). Yield 75% as a white foam. 1H NMR (400 MHz, CDCl3) δ 10.19 (brs, 1H), 6.60 (s, 1H), 6.29 (s, 1H), 6.24 (d, J = 2.4 Hz, 1H), 6.15 (s, 1H), 5.94 (d, J = 2.4 Hz, 1H), 4.12 (dd, J = 12.8, 5.6 Hz, 1H), 3.86 (s, 3H), 3.78 (s, 3H), 3.64 (s, 3H), 3.53 (dt, J = 12.8, 3.2 Hz, 1H), 3.21 (s, 3H), 2.91 (dt, J = 15.6, 5.6 Hz, 1H), 2.67 (dd, J = 15.6, 2.8 Hz, 1H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 160.7, 159.4, 158.0, 156.3, 147.0, 146.6, 129.0, 125.3, 111.2, 110.6, 108.6, 95.2, 92.8, 81.0, (55.83), 55.76, 55.70, (55.3), 55.2, 55.0, (54.9), 50.1, 39.0, 28.9, 28.3; IR (Film): 3148, 2936, 1644, 1615, 1518, 1428, 1255, 1148 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C24H31NO7: 445.2101; Found 445.2106.
(±)-tert-Butyl 1-(4-(dimethylamino)-2-hydroxyphenyl)-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5l). Yield 70% as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 9.45 (brs, 1H), 6.63 (s, 1H), 6.48 (d, J = 8.8 Hz, 1H), 6.40 (s, 1H), 6.36 (d, J = 2.4 Hz, 1H), 6.29 (s, 1H), 6.08 (dd, J = 8.8, 2.4 Hz, 1H), 3.97 (dd, J = 13.6, 5.6 Hz, 1H), 3.88 (s, 3H), 3.70 (s, 3H), 3.13 (ddd, J = 12.8, 12.8, 3.6 Hz, 1H), 2.88–2.98 (m, 7H), 2.68 (dd, J = 15.6, 2.4 Hz, 1H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 156.5, (156.3), 151.4, 147.7, 147.4, 130.7, 126.9, 126.8, 116.2, 111.0, 110.75, 110.71, 103.6, 100.8, 81.3, (55.94), 55.89, 55.85, 52.0, (40.43), 40.38, 40.31, (40.25), 37.4, 28.5, 28.3; IR (Film): 3198, 2976, 1645, 1518, 1432, 1254 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C24H32N2O5: 428.2311; Found 428.2307.
(±)-tert-Butyl 1-(4-(diethylamino)phenyl)-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5m). Yield 78% as a white foam. 1H NMR (400 MHz, CDCl3, a 1:1 mixture of amide rotamers at room temperature) δ 7.03 (d, J = 8.4 Hz, 2H), 6.94 (s, 1H), 6.56 (d, J = 8.4 Hz, 2H), 6.51 (s, 1H), 6.31 (s, 0.5H), 6.12 (s, 0.5H), 4.11 (s, 0.5H), 3.90 (s, 0.5H), 3.88 (s, 3H), 3.75 (s, 3H), 3.31 (q, J = 7.2 Hz, 4H), 3.05 (s, 0.5H), 2.91 (s, 0.5H), 2.64 (s, 0.5H), 2.61 (s, 0.5H), 1.5 (s, 9H), 1.14 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 154.1, 147.4, 146.9, 146.5, 129.3, 127.5, 126.9, 110.9, 110.7, 79.4, 56.7, 55.7, 55.6, 44.1, (37.5), 36.1, 28.5, 28.1, (12.53), 12.51; IR (Film): 2974, 1687, 1611, 1519, 1220 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C26H36N2O4: 440.2675; Found 440.2679.
(±)-tert-Butyl 1-(1-hydroxynaphthalen-2-yl)-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5n). Yield 76% as a white foam. 1H NMR (400 MHz, CDCl3) δ 10.34 (brs, 1H), 8.44 (dd, J = 6.0, 3.2 Hz, 1H), 7.70 (dd, J = 7.2, 3.2 Hz, 1H), 7.44–7.49 (m, 2H), 7.17 (d, J = 8.8 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 6.68 (s, 1H), 6.56 (s, 1H), 6.33 (s, 1H), 4.05 (dd, J = 13.6, 5.2 Hz, 1H), 3.90 (s, 3H), 3.63 (s, 3H), 3.21 (td, J = 13.2, 3.6 Hz, 1H), 2.99 (td, J = 16.0, 5.6 Hz, 1H), 2.75 (dd, J = 16.0, 2.4 Hz, 1H), 1.49 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.8, 151.9, 148.0, 147.8, 134.1, 127.2, 127.1, 126.8, 126.7, 126.5, 125.8, 125.0, 123.3, 121.1, 118.5, 111.1, 81.8, 55.99, 55.94, 52.5, 38.1, 28.6; IR (Film): 3134, 2976, 1644, 1518, 1432, 1254, 1159 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C26H29NO5: 435.2046; Found 435.2048.
(±)-tert-Butyl 1-(1H-indol-3-yl)-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5o). Yield 84% as a white foam. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 8.10 (s, 1H), 7.88 (s, 0.6H), 7.76 (s, 0.4H), 7.34 (d, J = 8.0 Hz, 1H), 7.19 (dd, J = 8.0, 7.2 Hz, 1H), 7.11 (t, J = 7.2 Hz, 1H), 6.63–6.69 (m, 3.6H), 6.49 (brs, 0.4H), 4.03–4.13 (m, 0.4H), 3.89–3.93 (m, 0.6H), 3.89 (s, 3H), 3.73 (s, 3H), 2.95-3.10 (m, 1H), 2.59–2.63 (m, 1H), 1.60 (s, 3.6H), 1.50 (s, 5.4H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 154.2, 147.5, 146.8, 136.3, 128.1, 126.7, 126.4, 125.1, 121.9, 120.0, 119.4, 118.5, (118.1), 111.2, 111.0, (80.3), 79.5, 55.9, (51.4), 50.3, 37.6, (36.6), 28.6, 28.2, (27.9); IR (Film): 3360, 2975, 1667, 1517, 1422, 1254 (cm−1); HRMS (FAB-magnetic sector) m/z: {M}+ Calcd for C24H28N2O4 408.2049; Found 408.2047.
(±)-tert-Butyl 6,7-dimethoxy-1-(5-methylfuran-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (5p). Yield 55% as a white foam. 1H NMR (400 MHz, CDCl3, a 2:1 mixture of amide rotamers at room temperature) δ 6.63 (s, 1H), 6.62 (s, 1H), 6.24 (s, 0.33H), 6.07 (s, 0.67H), 5.82 (s, 1H), 5.81 (s, 1H), 4.20 (s, 0.67H), 4.05 (s, 0.33H), 3.87 (s, 3H), 3.80 (s, 3H), 3.03–3.31 (m, 1H), 2.84–2.96 (m, 1H), 2.67 (s, 0.67H), 2.63 (s, 0.33H), 2.24 (s, 3H), 1.51 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 154.2, 153.2, 151.4, 147.8, 147.0, 126.9, (125.0), 111.2, 111.1, (110.7), 108.9, 105.9, 105.5, 79.7, 55.85, (55.81), 55.7, 52.1, (51.4), (38.7), 37.1, 28.4, 28.1, 13.6; IR (Film): 2976, 1694, 1519, 1415, 1254 (cm−1); HRMS (FAB-magnetic sector) m/z: {M}+ Calcd for C21H27NO5 373.1889; Found 373.1894.
(±)-Benzyl 1-allyl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (6a). Yield 77% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.2:1 mixture of amide rotamers at room temperature) δ 7.30–7.37 (m, 5H), 6.61 (s, 0.45H), 6.59 (s, 0.55H), 6.57 (s, 0.55H), 6.56 (s, 0.45H), 5.82–5.92 (m, 0.45H), 5.71–5.82 (m, 0.55H), 5.16–5.22 (m, 2H), 4.97–5.12 (m, 3H), 4.27 (dd, J = 13.2, 3.2 Hz, 0.55H), 4.08–4.11 (m, 0.45H), 3.84 (s, 6H), 3.31–3.38 (m, 0.45H), 3.21–3.28 (m, 0.55H), 2.79–2.94 (m, 1H), 2.65 (dd, J = 11.6, 2.4 Hz, 1H), 2.51–2.59 (m, 2H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 155.1, 147.5, (147.4), 147.1, (136.7), 136.5, 134.7, (134.6), (128.7), 128.4, 128.2, (127.9), 127.8, 127.7, (127.5), 126.0, (125.7), 117.3, (117.1), 111.4, (111.2), 67.1, (66.9), (55.9), 55.8, 54.0, 41.4, (41.2), (38.3), 37.6, (28.3), 28.0; IR (Film) 2934, 1691, 1516, 1426, 1214 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C22H26NO4 368.1862; Found 386.1867.
(±)-Allyl 1-allyl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (6b). Yield 86% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.2:1 mixture of amide rotamers at room temperature) δ 6.58–6.61 (m, 2H), 5.95 (ddd, J = 16.0, 10.8, 5.6 Hz, 1H), 5.78–5.87 (m, 1H), 5.31 (dd, J = 17.2, 6.4 Hz, 0.55H), 5.20–5.22 (m, 1.1H), 5.11 (t, J = 6.8 Hz, 0.45H), 5.03–5.07 (m, 2.9H), 4.55–4.67 (m, 2H), 4.25 (dd, J = 12.8, 3.6 Hz, 0.55H), 4.01 (dd, J = 8.0, 3.6 Hz, 0.45H), 3.85 (s, 6H), 3.34 (dt, J = 10.0, 4.0 Hz, 0.45H), 3.23 (dt, J = 9.6, 4.0 Hz, 0.55H), 2.81–2.93 (m, 1H), 2.63–2.67 (m, 1H), 2.56–2.57 (m, 2H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 155.3, 147.9, (147.8), 147.4, (135.1), 134.9, (133.3), 133.2, (129.1), 128.7, 126.3, (126.0), 117.7, 117.6, (117.4), 117.2, 111.7, (111.5), (110.2), 110.0, 66.3, (66.2), 56.3, 56.2, 54.3, 41.8, (41.5), (38.5), 37.9, (28.6), 28.3; IR (Film) 2934, 1691, 1516, 1431, 1256, 1214 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C18H23NO4 317.1627; Found 317.1623.
(±)-Methyl 1-allyl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (6c). Yield 60% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.2:1 mixture of amide rotamers at room temperature) δ 6.58–6.61 (m, 2H), 5.78–5.90 (m, 1H), 5.19 (t, J = 9.6 Hz, 0.45H), 5.02–5.06 (m, 2.55H), 4.23–4.25 (m, 0.45H), 4.01–4.02 (m, 0.55H), 3.85 (s, 6H), 3.71 (s, 3H), 3.20–3.33 (m, 1H), 2.82–2.93 (m, 1H), 2.66 (t, J = 3.6 Hz, 0.55H), 2.62 (t, J = 3.6 Hz, 0.45H), 2.53–2.55 (m, 2H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 155.7, 147.4, (147.3), 147.0, 134.7, (134.5), 128.7, (128.4), 125.9, (125.6), 117.1, (117.0), 111.3, (111.1), 109.8, (109.6), 55.9, 55.8, 53.8, 52.4, 41.3, (41.0), (38.1), 37.4, 28.1, (27.8); IR (Film) 2953, 1699, 1520, 1449, 1258, 1220 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C17H21NO4 291.1471; Found 291.1466.
(±)-Ethyl 1-allyl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (6d). Yield 84% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 6.58 (brs, 2H), 5.82–5.86 (m, 1H), 5.19 (t, J = 7.2 Hz, 0.4H), 5.07–5.10 (m, 0.6H), 5.02–5.07 (m, 2H), 4.03–4.26 (m, 3H), 3.85 (s, 6H), 3.30 (dt, J = 11.2, 2.8 Hz, 0.4H), 2.86 (dt, J = 12.4, 4.0 Hz, 0.6H), 2.66 (t, J = 2.8 Hz, 0.6H), 2.62 (t, J = 3.2 Hz, 0.4H), 2.54 (brs, 2H), 1.28 (t, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 155.1, 147.2, (147.1), 146.8, (134.7), 134.4, (128.6), 128.3, 125.8, (125.5), 117.0, (116.7), 111.1, (110.9), (109.6), 109.3, 60.99, (60.97), (55.7), 55.6, 53.5, 41.2, 40.9, (37.8), 37.0, (28.0), 27.8, 14.6; IR (Film) 2934, 1689, 1516, 1427, 1215, 1098 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C17H23NO4 305.1627; Found 305.1625.
(±)-1-(1-Allyl-6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethanone (6e). Yield 48% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 6.62 (s, 0.6H), 6.61(s, 0.4H), 6.592 (s, 0.4H), 6.585 (s, 0.6H), 5.80–5.90 (m, 0.6H), 5.61–5.64 (m, 0.4H), 5.13–5.17 (m, 0.8H), 5.00–5.04 (m, 1.2H), 4.76 (dd, J = 9.2, 5.2 Hz, 0.6H), 4.71 (dd, J = 8.4, 4.8 Hz, 0.4H), 3.87 (s, 1.2H), 3.86 (s, 1.2H), 3.85 (s, 1.8H), 3.84 (s, 1.8H), 3.79 (ddd, J = 8.8, 5.6, 3.6 Hz, 0.4H), 3.53 (ddd, J = 14.8, 13.2, 4.4 Hz, 0.6H), 3.04 (dt, J = 12.0, 4.4 Hz, 0.4H), 2.87 (dt, J = 10.8, 5.6 Hz, 1H), 2.77 (t, J = 4.0 Hz, 0.6H), 2.73 (t, J = 4.0 Hz, 0.4H), 2.49–2.67 (m, 3H), 2.16 (s, 3H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ (168.7), 168.5, (147.4), 147.1, 146.9, (146.8), 134.6, (133.5), 128.5, (127.8), (125.9), 124.9, (118.2), 116.6, (111.1), 110.7, 109.7, (109.2), (56.6), (55.7), 55.6, 55.5, 51.0, (41.0), 40.7, 40.3, (34.6), 28.4, (27.4), (21.8), 21.6; IR (Film) 2927, 1632, 1514, 1428, 1255, 1220, 1120 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C16H21NO3 275.1521; Found 275.1524.
(±)-(1-Allyl-6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)(phenyl)methanone (6f). Yield 37% as a white foam. 1H NMR (400 MHz, CDCl3, a 4:1 mixture of amide rotamers at room temperature) δ 7.33–7.42 (m, 5H), 6.69 (s, 0.8H), 6.64 (s, 0.2H), 6.57 (s, 0.8H), 6.38 (s, 0.2H), 5.96–6.06 (m, 0.8H), 5.81 (dd, J = 8.8, 4.8 Hz, 0.8H), 5.57–5.65 (m, 0.2H), 5.03–5.13 (m, 2.2H), 4.84 (dd, J = 13.6, 6.0 Hz, 0.2H), 4.73–4.76 (m, 0.2H), 3.87 (s, 2.4H), 3.85 (s, 2.4H), 3.79 (s, 1.2H), 3.73–3.77 (m, 1H), 3.45 (dt, J = 12.0, 4.0 Hz, 1H), 3.24 (dt, J = 12.0, 4.0 Hz, 0.2H), 3.04–3.14 (m, 0.2H), 2.42–2.87 (m, 6.2H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ (170.7), 170.4, (147.9), 147.6, 136.6, (136.4), 135.0, (133.9), 129.2, (128.5), 128.4, (128.3), (126.9), 126.4, (125.9), 124.9, (118.4), 117.2, (111.6), 111.2, 110.0, (109.3), (57.4), 56.03, 55.93, 50.9, (41.8), 41.5, 41.2, (35.4), 29.1, (27.8); IR (Film) 2933, 1626, 1515, 1428, 1255, 1223, 1118 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C21H23NO3 337.1678; Found 337.1679.
(±)-1-Allyl-6,7-dimethoxy-2-(methylsulfonyl)-1,2,3,4-tetrahydroisoquinoline (6g). Yield 89% as a white foam. 1H NMR (400 MHz, CDCl3) δ 6.59 (s, 1H), 6.57 (s, 1H), 5.85–5.95 (m, 1H), 5.13 (s, 1H), 5.10 (d, J = 5.6 Hz, 1H), 4.81 (dd, J = 8.0, 5.6 Hz, 1H), 3.94 (dd, J = 14.4, 6.4 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.46 (ddd, J = 16.8, 12.0, 4.8 Hz, 1H), 2.98 (ddd, J = 17.2, 12.0, 6.8, 1H), 2.77 (s, 3H), 2.65–2.70 (m, 1H), 2.52–2.62 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 147.9, 147.4, 134.6, 127.6, 124.5, 117.7, 111.6, 109.7, 56.0, 55.9, 55.6, 41.8, 40.1, 38.8, 26.7; IR (Film) 2935, 1611, 1516, 1316, 1247, 1163, 1120 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C15H21NO4S 311.1189; Found 311.1191.
(±)-1-Allyl-6,7-dimethoxy-2-tosyl-1,2,3,4-tetrahydroisoquinoline (6h). Yield 81% as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.53 (s, 1H), 6.38 (s, 1H), 5.83 (dddd, J = 17.6, 10.4, 7.2, 7.2 Hz, 1H), 5.05 (d, J = 10.4 Hz, 1H), 5.04 (d, J = 17.6 Hz, 1H), 4.97 (t, J = 6.8 Hz, 1H), 3.85 (s, 3H), 3.81–3.83 (m, 1H), 3.78 (s, 3H), 3.43 (ddd, J = 16.4, 10.8, 5.6 Hz, 1H), 2.41–2.60 (m, 4H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 147.7, 147.2, 143.0, 137.9, 134.6, 129.3, 127.7, 127.0, 124.9, 117.6, 111.3, 109.7, 56.1, 56.0, 55.9, 42.2, 39.2, 26.4, 21.6; IR (Film) 2935, 1517, 1325, 1228, 1157 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C21H25NO4S 387.1504; Found 387.1504.
(±)-1-Allyl-6,7-dimethoxy-2-((2-nitrophenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline (6i). Yield 72% as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.95(dd, J = 7.6, 1.6 Hz, 1 H), 7.53–7.64 (m, 3H), 6.60 (s, 1H), 6.48 (s, 1H), 5.73 (ddd, J = 17.2, 10.0, 7.2 Hz, 1H), 5.02 (d, J = 16.8 Hz, 1H), 5.00 (d, J = 10.0 Hz, 1H), 4.94 (d, J = 9.6 Hz, 1H), 4.04 (dd, J = 12.8, 5.6 Hz, 1H), 3.86 (s, 3H), 3.81 (s, 3H), 3.53 (ddd, J = 14.8, 12.0, 4.8 Hz, 1H), 2.76 (ddd, J = 16.8, 12.0, 6.4 Hz, 1H), 2.62–2.63 (m, 1H), 2.57 (dd, J = 15.2, 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 147.8, 147.7, 147.2, 134.1, 134.0, 133.3, 131.5, 130.3, 127.6, 124.5, 123.9, 117.8, 111.3, 109.6, 56.6, 56.0, 55.8, 41.9, 39.4, 27.2; IR (Film) 2937, 1542, 1518, 1350, 1247, 1163, 1120 (cm−1); HRMS (FAB-magnetic sector) m/z: {M}+ Calcd for C20H22N2O6S 418.1199; Found 418.1196.
(±)-tert-Butyl 1-allyl-6-methoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (7a). Yield 85% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 7.03 (d, J = 8.0 Hz, 1H), 6.65 (brs, 1H), 5.78–5.88 (m, 1H), 5.17–5.20 (m, 0.4H), 4.99–5.05 (m, 2.6H), 4.16–4.20 (m, 0.6H), 3.92–3.96 (m, 0.4H), 3.78 (s, 3H), 3.26–3.31 (m, 0.4H), 3.14–3.21 (m, 0.6H), 2.80–2.95 (m, 1H), 2.72 (t, J = 4.0 Hz, 0.6H), 2.68 (t, J = 4.0 Hz, 0.4H), 2.45–2.56 (m, 2H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 157.9, 154.6, 135.7, (135.5), 135.1, (129.5), 129.3, (128.2), 127.9, 117.2, (116.9), 113.3, 112.4, (112.0), 79.8, (79.5), 55.3, 54.2, (53.3), 41.9, (41.6), (38.6), 36.8, (29.2), 29.1, 28.6; IR (Film) 2974, 1685, 1418, 1232, 1159 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C18H26NO3 304.1913; Found 304.1913.
(±)-tert-Butyl 1-allyl-7-methoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (7b). Yield 79% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 7.00–7.05 (m, 1H), 6.73–6.75 (m, 1H), 6.65 (d, J = 2.4 Hz, 1H), 5.79–5.89 (m, 1H), 5.20–5.22 (m, 0.4H), 5.01–5.07 (m, 2.6H), 4.18–4.20 (m, 0.6H), 3.93–3.97 (m, 0.4H), 3.79 (s, 3H), 3.24–3.30 (m, 0.4H), 3.13–3.20 (m, 0.6H), 2.76–2.90 (m, 1H), 2.68 (t, J = 4.0 Hz, 0.6H), 2.64 (t, J = 4.0 Hz, 0.4H), 2.54 (t, J = 7.6 Hz, 2H),1.48 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 157.6, (154.7), 154.5, (138.3), 138.1, (135.1), 135.0, 129.8, (129.5), 126.4, (126.2), 117.3, (116.9), 112.6, (112.1), 112.0, 79.8, (79.4), 55.4, 54.8, (53.9), 41.6, (41.3), (38.9), 37.1, 28.6, (28.0), 27.9; IR (Film) 2975, 1686, 1420, 1249, 1159 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C18H26NO3 304.1913; Found 304.1907.
(±)-tert-Butyl 1-allyl-6,8-dimethoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (7c). Yield 98% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 6.31 (d, J = 2.4 Hz, 0.6H), 6.28 (d, J = 2.4 Hz, 0.4H), 6.25 (d, J = 2.0 Hz, 0.6H), 6.22 (d, J = 2.0 Hz, 0.4H), 5.81–5.94 (m, 1H), 5.40 (dd, J = 9.6, 4.0 Hz, 0.4H), 5.20 (dd, J = 9.6, 3.2 Hz, 0.6H), 4.94–5.07 (m, 2H), 4.20 (ddd, J = 13.2, 6.0, 1.6 Hz, 0.6H), 3.95 (ddd, J = 12.8, 6.4, 3.2 Hz, 0.4H), 3.82 (s, 1.8H), 3.78 (s, 4.2H), 3.29 (ddd, J = 14.8, 10.4, 4.4 Hz, 0.4H), 3.18 (ddd, J = 13.2, 11.6, 4.4 Hz, 0.6H), 2.78–2.93 (m, 1H), 2.59–2.68, m, 2H), 2.28–2.36 (m, 1H), 1.47 (s, 5.4H), 1.45 (s, 3.6H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 158.9, (158.8), (156.7), 156.5, 154.7, (136.2), 136.0, 135.9, (119.3), 118.9, 116.3, (115.8), 104.2, (104.1), 96.5, (96.3), 79.6, (79.2), 55.4, 55.3, 50.0, (49.0), 38.8, (38.7), (37.8), 36.1, (29.0), 28.9, 28.6; IR (Film) 2975, 1686, 1420, 1249, 1159 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C19H28NO4 334.2018; Found 334.2016.
(±)-tert-Butyl 1-allyl-7-fluoro-3,4-dihydroisoquinoline-2(1H)-carboxylate (7d). Yield 89% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 7.06–7.07 (m, 1H), 6.82–6.86 (m, 2H), 5.81–5.83 (m, 1H), 5.22 (brs, 0.4H), 5.03–5.06 (m, 2H), 4.21 (d, J = 12.0 Hz, 0.6H), 3.97 (d, J = 10.4 Hz, 0.4H), 3.25–3.27 (m, 0.4H), 3.14–3.19 (m, 0.6H), 2.83–2.86 (m, 1H), 2.72 (t, J = 3.6 Hz, 0.6H), 2.67 (t, J = 3.6 Hz, 0.4H), 2.51–2.59 (m, 2H), 1.48 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 162.1, 159.7, 138.9, 134.7, 130.4, 130.1, 117.7, (117.4), 113.9, 113.6, (113.4), 54.7, (53.8), 41.6, (41.3), (38.7), 37.1, 28.7, (28.3), 28.2; IR (Film) 2976, 1688, 1413, 1246, 1161, 1114 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C17H23FNO2 292.1713; Found 292.1713.
(±)-tert-Butyl 1-allyl-7-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (7e). Yield 85% as a white foam. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 7.27 (brs, 2H), 6.98–7.00 (m, 1H), 5.76–5.85 (m, 1H), 5.21 (m, 0.4H), 5.04–5.08 (m, 2.6H), 4.19–4.22 (m, 0.6H), 3.96–3.99 (m, 0.4H), 3.23–3.28 (m, 0.4H), 3.11–3.18 (m, 0.6H), 2.81–2.89 (m, 1H), 2.70 (t, J = 3.2 Hz, 0.6H), 2.66 (t, J = 3.2Hz, 0.4H), 2.52 (d, J = 8.0 Hz, 2H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ (154.6), 154.5, (139.5), 139.3, 134.6, 133.5, (133.2), 130.7, (130.4), (130.0), 129.8, 129.6, 119.5, 117.7, (117.4), 41.6, (41.3), (38.4), 36.7, 28.7, (28.5), 28.3; IR (Film) 2975, 1687, 1412, 1230, 1159 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C17H23BrNO2 352.0912; Found 352.0915.
(±)-tert-Butyl 1-allyl-3,4-dihydroisoquinoline-2(1H)-carboxylate (7f). Yield 92% as a colorless oil. 1H NMR (400 MHz, CDCl3, a 1.5:1 mixture of amide rotamers at room temperature) δ 7.10–7.15 (m, 4H), 5.81–5.87 (m, 1H), 5.24 (brs, 0.4H), 5.01–5.06 (m, 2.6H), 4.18–4.21 (m, 0.6H), 3.94 (s, 0.4H), 3.17–3.31 (m, 1H), 2.89–2.91 (m, 1H), 2.75 (t, J = 4.0 Hz, 0.6H), 2.71 (t, J = 4.0 Hz, 0.4H), 2.54 (t, J = 7.2 Hz, 2H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ 154.4, 136.9, 134.93, 134.85, 134.2, (134.0), 128.8, (128.5), 126.8, (126.3), 125.7, 117.1, (116.7), 79.5, (79.2), 54.5, (53.7), 41.6, (41.3), (38.5), 36.9, 28.5; IR (Film) 2978, 1694, 1422, 1166, 1124 (cm−1); HRMS (FAB-magnetic sector) m/z: {M+H}+ Calcd for C17H24NO2 274.1807; Found 274.1807.
To a stirred solution of (±)–5a (100.0 mg, 0.30 mmol) in DCM (3.0 mL) was added TFA (0.69 mL, 3.0 mmol) at room temperature. The reaction mixture was stirred for 2 h at room temperature under argon atmosphere and then quenched with saturated NaHCO3 (5 mL) and the layers were separated and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, EtOAc/MeOH/Et3N = 15:1:0.1) to afford (±)–8 (61.6 mg, 0.26 mmol) as a colorless oil.
To a stirred solution of (±)–6c (87.4 mg, 0.30 mmol) in ethylene glycol/H2O [3.0 mL, 1:1 (v/v)] was added KOH (168.3 mg, 3.0 mmol) at room temperature. The reaction mixture was heated at reflux for 12 h under argon atmosphere and cooled to room temperature and then quenched with saturated NH4Cl (5 mL) and the layers were separated and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, EtOAc/MeOH/Et3N = 15:1:0.1) to afford (±)-8 (49.0 mg, 0.21 mmol) as a colorless oil.
To a stirred solution of (±)–6h (125.5 mg, 0.30 mmol) in DMF (3.0 mL) was added PhSH (0.09 mL, 0.90 mmol) and K2CO3 (124.4 mg, 0.90 mmol) at room temperature. The reaction mixture was stirred for 12 h at room temperature under argon atmosphere and then quenched with saturated NaHCO3 (5 mL) and the layers were separated and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, EtOAc/MeOH/Et3N = 15:1:0.1) to afford (±)–8 (61.6 mg, 0.26 mmol) as a colorless oil. 1H NMR (400MHz, CDCl3) δ 6.64 (s, 1H), 6.56 (s, 1H), 5.83 (dddd, J = 16.8, 10.4, 7.6, 6.8 Hz, 1H), 5.12–5.20 (m, 2H), 3.99 (dd, J = 8.8, 3.6 Hz, 1H), 3.84 (s, 6H), 3.22 (ddd, J = 12.4, 4.8, 4.8 Hz, 1H), 2.95 (ddd, J = 12.4, 7.6, 4.8 Hz, 1H), 2.72–2.79 (m, 1H), 2.60–2.70 (m, 2H), 2.45–2.53 (m, 1H); 13C NMR (100MHz, CDCl3) δ 147.1, 146.9, 135.4, 130.2, 127.2, 117.8, 111.6, 108.9, 56.0, 55.8, 54.7, 41.1, 40.8, 29.5; IR (Film) 2932, 1510, 1464, 1355, 1258, 1112 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C14H19NO2 233.1416; Found 233.1416.
(±)-(E)-Benzyl 6,7-dimethoxy-1-(4-methoxy-4-oxobut-2-en-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (9). To a stirred solution of 4b (120.5 mg, 0.37 mmol) in DCM (3.70 mL) was added 4Å molecular sieves (160 mg) at room temperature. After the reaction mixture was stirred for 15 min at room temperature, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (91.9 mg, 0.40 mmol) was added portionwise and the reaction mixture was stirred at room temperature for 30 min under argon atmosphere. CH2=CH(OTMS) (0.14 mL, 0.92 mmol) was added dropwise at room temperature and the reaction mixture was stirred at room temperature for 1 h under argon atmosphere. Ph3P=CO2Me (213.4 mg, 0.63 mmol) was added portionwise and the reaction mixture was stirred at room temperature for 12 h and then diluted with hexanes (5.0 mL) and concentrated in vacuo. Purification of the crude residue by flash chromatography on silica gel, using hexanes/EtOAc (4:1 to 3:1) as elutant, provided (±)–8 (124.4 mg, 0.29 mmol, E/Z = 95:5) as a white foam. 1H NMR (400 MHz, CDCl3, a 1:1 mixture of amide rotamers at room temperature) δ 7.29–7.38 (m, 5H), 6.93–7.00 (m, 1H), 6.59 (s, 0.5H), 6.57 (s, 0.5H), 6.56 (s, 0.5H), 6.51 (s, 0.5H), 5.82 (d, J = 16.0 Hz, 0.5H), 5.80 (d, J = 16.0 Hz, 0.5H), 5.29 (t, J = 6.8 Hz, 0.5H), 5.17 (t, J = 6.8 Hz, 0.5H), 5.16 (d, J = 4.0 Hz, 1H), 5.12 (d, J = 4.0 Hz, 1H), 4.24–4.28 (m, 0.5H), 4.02–4.07 (m, 0.5H), 3.85 (s, 3H), 3.83 (s, 1.5H), 3.82 (s, 1.5H), 3.71 (s, 1.5H), 3.70 (s, 1.5H), 3.29–3.36 (m, 0.5H), 3.17–3.25 (m, 0.5H), 2.78–2.94 (m, 1H), 2.64–2.74 (m, 3H);); 13C NMR (100 MHz, CDCl3, a rotameric mixture, resonances for minor rotamer are enclosed in parenthesis) δ (166.3), 166.2, (155.2), 155.0, 147.8, (147.7), 147.3, (144.9), 144.8, (136.6), 136.2, 128.4, 128.1, (128.0), (127.8), 127.6, 127.4, 126.1 (125.9), 123.24, (123.18), 111.4, (111.3), (109.7), 109.4, 67.6, (67.2), 56.0, (55.9), (53.63), 53.59, 51.5, 39.8, (39.4), (38.8), 37.8, (28.2), 27.9; IR (Film) 2937, 1542, 1517, 1348, 1246, 1162 (cm−1); HRMS (FAB-magnetic sector) m/z: {M}+ Calcd for C24H27NO6 425.1838; Found 425.1837.
(±)-9,10-Dimethoxy-2,3,6,7-tetrahydro-1H-pyrido[2,1-a]isoquinolin-4(11bH)-one (9). To a stirred solution of (±)-8 (30.5 mg, 0.072 mmol) in EtOAc (2.4 mL) was added 10% Pd/C (3.1 mg) at room temperature. The reaction mixture was stirred under H2 atmosphere for 18 h, then filtered through a pad of Celite 545 and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel, using hexanes/EtOAc (1:5.5) as elutant, provided (±)–9 (16.0 mg, 0.061 mmol) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 6.67 (s, 1H), 6.61 (s, 1H), 4.88 (ddd, J = 12.4, 4.4, 2.4 Hz, 1H), 4.61 (dd, J = 10.8, 4.4 Hz, 1H), 3.86 (s, 6H), 2.91 (dt, J = 12.0, 3.6 Hz, 1H), 2.80 (dt, J = 12.0, 2.8 Hz, 1H), 2.50–2.66 (m, 3H), 2.37 (ddd, J = 18.0, 12.0, 6.8 Hz, 1H), 1.79–1.92 (m, 1H), 1.79–1.90 (m, 1H) 1.62–1.72 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 168.7, 147.3, 147.2, 128.7, 126.8, 111.2, 107.9, 56.4, 55.8, 55.6, 39.4, 32.0, 30.7, 28.3, 19.4; IR (Film) 3454, 2936, 1635, 1515, 1257, 1225 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C15H19NO3 261.1365; Found 261.1363.
(±)-9,10-Dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinoline (10). To a stirred solution of (±)–9 (70.0 mg, 0.268 mmol) in THF (2.4 mL) was LiAlH4 (152.5 mg, 4.018 mmol) at 0 °C. The reaction mixture was heated to reflux for 1 h under argon atmosphere, then cooled to room temperature and then quenched with saturated Rochelle's salt (5 mL) and the layers were separated and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane/EtOAc/Et3N = 1:9:0.1) to afford (±)–10 (51.0 mg, 0.206 mmol) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.68 (s, 1H), 6.56 (s, 1H), 3.84 (s, 6H), 3.04–3.15 (m, 2H), 2.91–2.99 (m, 2H), 2.60 (dd, J = 16.0, 3.6 Hz, 1H), 2.50 (dt, J = 11.2, 4.0 Hz, 1H), 2.32 (dd, J = 11.2, 4.0 Hz, 1H), 2.23–2.28 (m, 1H), 1.90–1.95 (m, 1H), 1.66–1.77 (m, 2H), 1.37–1.54 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 147.3, 147.1, 130.4, 126.8, 111.6, 108.3, 63.4, 57.0, 56.2, 56.0, 53.0, 31.7, 29.3, 25.7, 25.3; IR (Film) 3423, 2933, 1602, 1510, 1259, 1225 (cm−1); HRMS (EI-magnetic sector) m/z: {M}+ Calcd for C15H21NO2 247.1572; Found 247.1574.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
DL contributed conception and design of the study. HY and DL have been involved in the synthesis of all compounds with the help of HK. S-HB and DL analyzed the results and wrote the paper. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge financial support of this work by the Basic Science Research Program through the National Research Fund of Korea (NRF) funded by the Ministry of Science and ICT and the Ministry of Education (NRF-2016R1A2B1012930 and NRF-2018R1D1A1A02086359).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00629/full#supplementary-material
Akinboye, E. S., and Bakare, O. (2011). Biological activities of emetine. Open Nat. Prod. J. 4, 8–15. doi: 10.2174/1874848101104010008
Alagiri, K., Devadig, P., and Prabhu, K. R. (2012a). Molybdenum trioxide catalyzed oxidative cross-dehydrogenative coupling of benzylic sp3C–H bonds: synthesis of α-aminophosphonates under aerobic conditions. Tetrahedron Lett. 53, 1456–1459. doi: 10.1016/j.tetlet.2012.01.031
Alagiri, K., Devadig, P., and Prabhu, K. R. (2012b). CDC reactions of N-aryl tetrahydroisoquinolines using catalytic amounts of DDQ: C-H activation under aerobic condirions Chem. Eur. J. 18, 5160–5164. doi: 10.1002/chem.201200100
Alagiri, K., Kumara, G. S. R., and Prabhu, K. R. (2011). An oxidative cross-dehyderogenative-coupling reaction in water using molecular oxygen as the oxidant: vanadium catalyzed indolation of tetrahydroisoquinoliens. Chem. Commun. 47, 11781–11789. doi: 10.1039/c1cc15050b
Baslé, O., and Li, C.-J. (2008). Copper-catalyzed oxidative sp3 C–H bond arylation with aryl boronic acids. Org. Lett. 10, 3661–3663. doi: 10.1021/ol8012588
Bentley, K. W. (2001). β-Phenylethylamines and the isoquinoline alkaloids. Nat. Prod. Rep. 18, 148–170. doi: 10.1039/a909672h
Chen, Q., Zhou, J., Wang, Y., Wang, C., Liu, X., Xu, Z., et al. (2015). Transition-metal-free dehydrosilative difluoroamidation of tetrahydroisoquinolines under mild conditions. Org. Lett. 17, 4212–4215. doi: 10.1021/acs.orglett.5b01997
Chen, W., Zheng, H., Pan, X., Xie, Z., Zan, X., Sun, B., et al. (2014). A metal-free cross-dehydrogenative coupling of N-carbamoyl tetrahydroisoquinoline by sodium persulfate. Tetrahedron Lett. 55, 2879–2882. doi: 10.1016/j.tetlet.2014.03.094
Chrzanowska, M., and Rozwadowska, M. D. (2004). Asymmetric synthesis of isoquinoline alkaloids. Chem. Rev. 104, 3341–3370. doi: 10.1021/cr030692k
Denmark, S. E., and Fu, J. (2003). Catalytic Enantioselective addition of allylic organometallic reagents to aldehydes and ketones. Chem. Rev. 103, 2763–2794. doi: 10.1021/cr020050h
Dhineshkumar, J., Lamani, M., Alagiri, K., and Prabhu, K. R. (2013). A versatile C–H functionalization of tetrahydroisoquinolines catalyzed by iodine at aerobic conditions. Org. Lett. 15, 1092–1095. doi: 10.1021/ol4001153
Dubs, C., Hamashima, Y., Sasamoto, N., Seidel, T. M., Suzuki, S., Hashizume, D., et al. (2008). Mechanistic studies on the catalytic asymmetric Mannich-type reaction with dihydroisoquinolines and development of oxidative Mannich-type reactions starting from tetrahydroisoquinolines. J. Org. Chem. 73, 5859–5871. doi: 10.1021/jo800800y
Dunetz, J. R., Ciccolini, R. P., Fröling, M., Paap, S. M., Allen, A. J., Holmes, A. B., et al. (2005). Pictet–Spengler reactions in multiphasic supercritical carbon dioxide/CO2-exopanded liquid media. In situ generation of carbamates as a strategy for reactions of amines in supercritical carbon dioxide. Chem. Commun. 4465–4467. doi: 10.1039/b508151c
Freeman, D. B., Furst, L., Condie, A. G., and Stephenson, C. R. J. (2012). Functionally diverse ncleophilic trapping of iminium intermediates generated utilizing visible light. Org. Lett. 14, 94–97. doi: 10.1021/ol202883v
Fu, P. F., and Harvey, R. G. (1978). Dehydrogenation of polycyclic hydroaromatic compounds, Chem. Rev. 78, 317–361. doi: 10.1021/cr60314a001
Fukuyama, T., Jow, C.-K., and Cheung, M. (1995). 2- and 4-Nitrobenzenesulfonamides: Exceptionally versatile means for preparation of secondary amines and protection of amines. Tetrahedron Lett. 36, 6373–6374. doi: 10.1016/0040-4039(95)01316-A
Ghobrial, M., Schünrch, M., and Mihovilovic, M. D. (2011). Direct functionalization of (un)protected tetrahydroisoquinoline and isochroman under iron and copper catalysis: Two metal, two mechanisms. J. Org. Chem. 76, 8781–8793. doi: 10.1021/jo201511d
Girard, N., and Hurvois, J.-P. (2007). Anodic cyanation of C-4 hydroxylated piperidines: total synthesis of (±)-alkaloid 241D, Tetrahedron Lett. 48, 4097–4099. doi: 10.1016/j.tetlet.2007.04.010
Girard, N., Hurvois, J.-P., Toupet, L., and Moinet, C. (2005). Anodic cyanation of (–)-N-phenyl-2-methylpiperidine: a short synthesis of (+)-solenopsin A and (+)-isosolenopsin A. Synth. Commun. 35, 711–723. doi: 10.1081/SCC-200050370
Girard, N., Gautier, C., Malassene, R., Hurvois, J.-P., Moinet, C., and Toupet, L. (2004). Dearomatization of N-phenyl-2,6-dialkylpiperidines: Practical synthesis of (±)-solenopsin A and (±)-dihydropinidine. Synlett 2005–2009. doi: 10.1055/s-2004-830884
Hari, D. P., and König, B. (2011). Eosin Y catalyzed visible light oxidative C–C and C–P bond formation. Org. Lett. 13, 3852–3855. doi: 10.1021/ol201376v
Hickin, J. A., Ahmed, A., Fucke, K., Ashcroft, M., and Jones, K. (2014). The synthesis and structure revision of NSC-134754. Chem. Commum. 50, 1238–1240. doi: 10.1039/C3CC48189A
Kim, H. P., Yu, H., Kim, H., Kim, S.-H., and Lee, D. (2018). DDQ-promoted mild and efficient metal-free oxidative α-cyanation of N-acyl/sulfonyl 1,2,3,4-tetrahydroisoquinolines. Molecules 23, 3223–3235. doi: 10.3390/molecules23123223
Klussmann, M., and Sureshkumar, D. (2011). Catalytic oxidative coupling reactions for the formation of carbon-carbon bonds without carbon-metal intermediates. Synthesis 353–369. doi: 10.1055/s-0030-1258303
Li, C.-J. (2009). Cross-dehydrogenative coupling (CDC): exploring C–C bond formations beyond functional group transformations. Acc. Chem. Res. 42, 335–344. doi: 10.1021/ar800164n
Liu, X., Sun, S., Meng, Z., Lou, H., and Liu, L. (2015). Organocatalytic asymmetric C–H vinylation and arylation of N-acyl tetrahydroisoquinolines. Org. Lett. 17, 2396–2399. doi: 10.1021/acs.orglett.5b00909
Luo, W., Yang, J.-D., and Cheng, J.-P. (2020). Toward rational understandings of α-C-H functionalization: Energetic studies of representative tertiary amines. iScience 23:100851. doi: 10.1016/j.isci.2020.100851
Ma, Y., Zhang, G., Zhang, J., Yang, D., and Wang, R. (2014). Organocatalyzed asymmetric oxidative coupling of α-Csp3-H of tertiary amines to α,β-unsaturated γ-butyrolactam: Synthesis of MBH-type products. Org. Lett. 16, 5358–5361. doi: 10.1021/ol5025597
Murahashi, S., Komiya, N., and Terai, H. (2005). Ruthenium-catalyzed oxidative cyanation of tertiary amines with hydrogen peroxide and sodium cyanide. Angew. Chem. Int. Ed. 44, 6931–6933. doi: 10.1002/anie.200501496
Murahashi, S., Komiya, N., Terai, H., and Nakae, T. (2003). Aerobic ruthenium-catalyzed oxidative cyanation of tertiary amines with sodium cyanide. J. Am. Chem. Soc. 125, 15312–15313. doi: 10.1021/ja0390303
Muramatsu, W., Nakano, K., and Li, C.-J. (2013). Simple and direct sp3 C–H bond arylation of tetrahydroisoquinolines and isochromans via 2,3-dichloro-5,6-dicyano-1,4-benzoquinone oxidation under mild conditions. Org. Lett. 15, 3650–3653. doi: 10.1021/ol401534g
Nobuta, T., Tada, N., Fujiya, A., Kariya, A., Miura, T., and Itoh, A. (2013). Molecular iodine catalyzed cross-dehydrogenative coupling reaction between two sp3C–H bonds using hydrogen peroxide. Org. Lett. 15, 574–577. doi: 10.1021/ol303389t
Reddy, N. S. S., Reddy, B. J. M., and Reddy, B. V. S. (2013). A convergent and stereoselective total synthesis of (–)-crispine A, (–)-benzo[a]quinolizidine and (–)-salsolidine. Tetrahedron Lett. 54, 4228–4231. doi: 10.1016/j.tetlet.2013.05.132
Richter, H., Frohlich, R., Daniliuc, C.-G., and Mancheño, O. G. (2012). Mild metal-free tandem α-alkylation.cyclization of N-benzyl carbamates with simple olefins. Angew. Chem., Int. Ed. 51, 8656–8660. doi: 10.1002/anie.201202379
Rinehart, K. L. (2000). Antitumor compounds from tunicates. Med. Res. Rev. 20, 1–27. doi: 10.1002/(SICI)1098-1128(200001)20:1<1::AID-MED1>3.0CO;2-A
Rohlmann, R., and Mancheño, O. G. (2013). Metal-free oxidative C(sp3)–H bond couplings as valuable synthetic tools for C–C bond formations. Synlett 24, 6–10. doi: 10.1002/chin.201312261
Scheuermann, C. J. (2010). Beyond traditional cross couplings: the scope of the cross dehydrogenative coupling reaction. Chem. Asian. J. 5, 436–451. doi: 10.1002/asia.200900487
Scott, J. D., and Williams, R. M. (2002). Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics, Chem. Rev. 102, 1669–1730. doi: 10.1021/cr010212u
Scott, M., Sud, A., Boess, E., and Klussmann, M. (2014). Reaction progress kinetic analysis of a copper-catalyzed aerobic oxidative coupling reaction with N-phenyl tetrahydroisoquinoline. J. Org. Chem. 79, 12033–12040. doi: 10.1021/jo5018876
Segal, M. S., Goldstein, M. M., and Attinger, E. O. (1957). The use of noscapine (narcotine) as an antitussive agent. Chest 32, 305–309. doi: 10.1378/chest.32.3.305
Shen, Y., Li, M., Wang, S., Zhan, T., Tan, Z., and Guo, C.-C. (2009). An efficient copper-catalyzed oxidative Mannich reaction between tertiary amines and methyl ketones. Chem. Commun. 953–955. doi: 10.1039/b819657e
Su, W., Yu, J., Li, Z., and Jiang, Z. (2011) Solvent-free cross-dehydrogenative coupling reactions under high speed ball-milling conditions applied to the synthesis of functionalized tetrahydroisoquinolines. J. Org. Chem. 76, 9144–9150. doi: 10.1021/jo2015533
Sud, A, Sureshkumar, D., and Klussmann, M. (2009). Oxidative coupling of amines and ketones by combined vanadium- and organocatalysis. Chem. Commun. 3169–3171. doi: 10.1039/b901282f
Sun, S., Li, C., Floreancig, P. E., Lou, H., and Liu, L. (2015). Highly enantioselective catalytic cross-dehydrogenative coupling of N-carbamoyl tetrahydroisoquinolines and terminal alkynes. Org. Lett. 17, 1684–1687. doi: 10.1021/acs.orglett.5b00447
Szawkalo, J., Czarnocki, S. J., Zawadzka, A., Wojtasiewicz, K., Leniewski, A., Maurin, J. K., et al. (2007). Enantioselective synthesis of some tetrahydroisoquinoline and tetrahydro-β-carboline alkaloids. Tetrahedron 18, 406–413. doi: 10.1016/j.tetasy.2007.01.014
Talk, R. A., Duprerray, A., Li, X., and Coldham, I. (2016). Synthesis of substituted tetrahydroisoquinolines by lithiation then electrophilic quench. Org. Biomol. Chem. 14, 4908–4917. doi: 10.1039/C6OB00577B
Tsang, A. S.-K., Ingram, K., Keiser, J., Hibbert, D. B., and Todd, M. H. (2013). Enhancing the usefulness of cross dehydrogenative coupling reactions with a removable protecting group. Org. Biomol. Chem. 11, 4921–4924. doi: 10.1039/c3ob40503f
Tsang, A. S.-K., and Todd, M. (2009). Facile synthesis of vicinal diamines via oxidation of N-phemyltetrahydroisoqunolines with DDQ. Tetrahedron Lett. 50, 1199–1202. doi: 10.1016/j.tetlet.2008.12.101
Walker, D., and Hiebert, J. D. (1967). 2,3-Dichloro-5,6-dicyanobenzoquinone and its reactions, Chem. Rev. 67, 153–195. doi: 10.1021/cr60246a002
Wang, H., Li, X., Wu, F., and Wan, B. (2012). Direct oxidative phosphonylation of amines under metal-free conditions. Tetrahedron Lett. 53, 681–683. doi: 10.1016/j.tetlet.2011.11.120
Wendlandt, A. E., and Stahl, S. S. (2015). Quinone-catalyzed selective oxidation of organic molecules. Angew. Chem. Int. Ed. 54, 14638–14658. doi: 10.1002/anie.201505017
Williams, G. D., Wade, C. E., and Wills, M. (2005). One-pot formation of nitrogen-containing heterocyclic ring systems using a deprotection–cyclisation–asymmetric reduction sequence. Chem. Commun. 4735–4737. doi: 10.1039/b509231k
Xie, Z., Liu, L., Chen, W., Zheng, H., Xu, Q., Yuan, H., et al. (2014). Practical metal-free C(sp3)–H functionalization: Construction of structurally diverse a-substituted N-benzyl and N-allyl carbamates. Angew. Chem. Int. Ed. 53, 3904–3908. doi: 10.1002/anie.201310193
Xie, Z., Zan, X., Sun, S., Pan, X., and Liu, L. (2016). Organocatalytic enantioselective cross-dehydrogenative coupling of N-carbamoyl cyclic amines with aldehydes. Org. Lett. 18, 3944–3947. doi: 10.1021/acs.orglett.6b01625
Yan, C., Liu, Y., and Wang, Q. (2014). Mild and highly efficient metal-free oxidative acyanation of N-acyl/sulfonyl tetrahydroisoquinolines. RSC Adv. 4, 60075–60078. doi: 10.1039/C4RA12922A
Yan, C., Liu, Y., and Wang, Q. (2015). Direct C–H allylation of N-acyl/sulfonyl tetrahydroisoquinolines and analogues. Org. Lett. 17, 5714–5717. doi: 10.1021/acs.orglett.5b03042
Yeung, C. S., and Dong, V. M. (2011). Catalytic dehydrogenative cross-coupling: forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 111, 1215–1292. doi: 10.1021/cr100280d
Yoo, W.-J., and Li, C.-J. (2010). Cross-dehydrogenative coupling reactions of sp3-hybridized C–H bonds. Top. Curr. Chem. 292, 281–302. doi: 10.1007/128_2009_17
Yu, J., Li, Z., Jia, K., Jiang, Z., Liu, M., and Su, W. (2013). Fast, solvent-free asymmetric alkynylation of prochiral sp3 C–H bonds in a ball mill for the preparation of optically active tetrahydroisoquinoline derivatives. Tetrahedron Lett. 54, 2006–2009. doi: 10.1016/j.tetlet.2013.02.007
Keywords: tetrahydroisoquinoline, oxidation, DDQ, electron-rich, natural products
Citation: Yu H, Kim H, Baek S-H and Lee D (2020) Direct and Efficient C(sp3)–H Functionalization of N-Acyl/Sulfonyl Tetrahydroisoquinolines (THIQs) With Electron-Rich Nucleophiles via 2,3-Dichloro-5,6-Dicyano-1,4-Benzoquinone (DDQ) Oxidation. Front. Chem. 8:629. doi: 10.3389/fchem.2020.00629
Received: 04 April 2020; Accepted: 17 June 2020;
Published: 29 July 2020.
Edited by:
Nam-Jung Kim, Kyung Hee University, South KoreaReviewed by:
Andrea Gualandi, University of Bologna, ItalyCopyright © 2020 Yu, Kim, Baek and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seung-Hoon Baek, c2hiYWVrQGFqb3UuYWMua3I=; Dongjoo Lee, ZG9uZ2pvb0Bham91LmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.