 Anis Ur Rahman1
Anis Ur Rahman1 Guigen Li
Guigen Li Asad Ali
Asad Ali
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 07 July 2020
Sec. Organic Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00523
Herein, we report a protocol for highly efficient hypervalent iodine (III) mediated, group-assisted purification (GAP) method for the regioselectivities and stereoselective aminochlorination of electron-deficient olefins. A series of vicinal chloramines with multifunctionalities were acquired in moderate to excellent yields (45–94%), by merely mixing the GAP auxiliary-anchored substrates with dichloramine T and tosylamide as chlorine/nitrogen sources and iodobenzene diacetate as a catalyst. The vicinal chloramines were obtained without any column chromatographic purification and recrystallization simply by washing the reaction mixture with a minimum amount of common inexpensive solvents and thus avoiding wastage of silica, solvents, time, and labor. The GAP auxiliary is recyclable and reusable. This strategy is easy to handle, cost-effective, greener, sustainable, environmentally benign, and mostly suitable for the syntheses of vicinal haloamines from various electron-deficient alkenes.
Aminohalogenation of olefins is one of the most effective approaches to produce functional α,β-haloamines. These vicinal haloamines are versatile components in organic synthesis as their halogen moieties are labile to cross-coupling and substitution reactions. For example, haloamines are direct precursors to vicinal diamines (Ghorai et al., 2011; Xiong et al., 2014), α,β-dehydroamino acids (Chen et al., 2005), and aziridines (Van and De Kimpe, 2000; Schröder et al., 2017; Thakur et al., 2017). While the aminohalogenation is established for almost five decades now, it still faces shortcomings and limitations related to controlling regioselectivities and stereoselectivities, forming side products, which sometimes makes purification more difficult (Thakur et al., 2003; Bovino and Chemler, 2012).
Several research groups have made great contributions in developing both intermolecular and intramolecular aminohalogenation (Minakata et al., 2006; Li et al., 2007; Michael et al., 2008; Chen et al., 2010; Denmark et al., 2012; Yin et al., 2012; Chemler and Bovino, 2013; Song et al., 2013, 2016; Martínez and Muñiz, 2014; Broggini et al., 2015; Qin et al., 2015; Zhu et al., 2015; Legnani et al., 2018; Cai et al., 2019), in which a series of halogen/nitrogen sources, such as CFBSA (Pu et al., 2016), NCS/MeCN (Tay et al., 2013), NCP (Zhu et al., 2018), NFSI/TMSCl (Arteaga et al., 2018), TsNCl2 (Han et al., 2007; Wu and Wang, 2008; Wei et al., 2009), TsNHCl (Cai et al., 2011), TsNNaCl (Martínez and Muñiz, 2014), 2-NsNNaCl (Li et al., 2001), or the combination of 2-NsNCl2 and 2-NsNHNa (Liu et al., 2006), and so on, were employed for many types of alkene substrates. Most of these aminohalogenation systems take advantage of metals or organic catalysts to give good yields and excellent regioselectivities and diastereoselectivities. The aforementioned synthesis usually involves the use of traditional methods of purification such as column chromatography and recrystallization. Herein, in this report, we would like to present the design of new group-assisted purification (GAP) group-attached olefin substrates and their potentials for aminochlorination enabling ste minimized, greener GAP workup, and purification.
The development of greener methodologies and technologies in chemical and pharmaceutical synthesis is of utmost importance to achieve safer, faster, and economical production outcomes (Trost, 1991; Lee and Robinson, 1995; Schreiber, 2000; Tietze et al., 2008; Anderson, 2012). Recently, our group has established a concept called GAP chemistry for greener synthesis where functionalities of special interest are incorporated into the substrates to facilitate the purification of crude products based on solubility without column chromatography or recrystallization. The pure products can be readily obtained simply by washing the crude mixture with common solvents or cosolvents (Wang et al., 2013; Chennapuram et al., 2014; Dommaraju and Prajapati, 2015; Seifert et al., 2016; Patel et al., 2019). Indeed, GAP chemistry would be the first concept consisting of both chemical aspects (reagent and reaction) and physical aspects (separation and purification). The study on GAP chemistry needs to consider solubility, stability, reactivity, and other properties of GAP reagents and products. More interestingly, we have found that, for some synthesis, GAP functional groups not only can make workup and purification easier but also can increase chemical yields, sometimes resulting in quantitative yields, which is defined as group-assisted synthesis chemistry (Seifert, 2017).
A ubiquitous requirement of GAP methodology is the sufficient solubility of products, which depends on the functional groups of their reactants. The GAP-anchored products have to dissolve in polar solvents such as THF, DCM for further reactions but remain insoluble in less polar solvents such as hexane, ether, and so on. The GAP compounds should exhibit adequate reactivity toward many reactants as well. Chiral GAP groups also control asymmetric addition efficiently. Herein, we report the GAP auxiliaries, which are labile to structural modifications to control the solubility, stability, and chemical reactivity of the products. Besides, these GAP auxiliaries can be easily deprotected via reduction or hydrolysis. Group-assisted purification chemistry has thus shown its potential to revolutionize the pharmaceutical industry as it saves time, manpower, and costs in safer manners.
We initiated the study by investigating GAP auxiliaries and their potential application in GAP chemistry. In our previous work, diphenylphosphine oxide (abbreviated as Dpp) was proven to be a stable GAP candidate. Its synthesis began with the benzylic and phosphine oxidation of commercially available, diphenyl(p-tolyl)phosphine 1 with potassium permanganate to benzoic acid 2. (Scheme 1) Esterification followed by reduction with borohydride gave the first GAP-anchored benzyl alcohol 4, or “dppBnOH,” in high yield (Seifert et al., 2016). Similarly, the condensation of diphenylphosphane 5 with 4-chlorobenzonitrile 6, provided 4-(diphenylphosphanyl)benzonitrile 7, which upon reduction with LiAlH4 resulted in amine 8. Phosphine oxidation of the amine 8 with hydrogen peroxide afforded the second GAP-equipped benzylamine 9 or “dppBnNH2” (Scheme 1) (Hingst et al., 1998; Janssen et al., 2009). The protection of cinnamic acids with dppBnNH2 was both facile and quantitative compared to dppBnOH. The products 11 and 12 could be easily precipitated from an ethyl acetate/petroleum ether solvent mixture, thereby fulfilling the condition of GAP chemistry. The Bndpp group can be easily regained for reuse either by catalytic hydrogenation or by reduction with borohydride in 10% Pd/C.
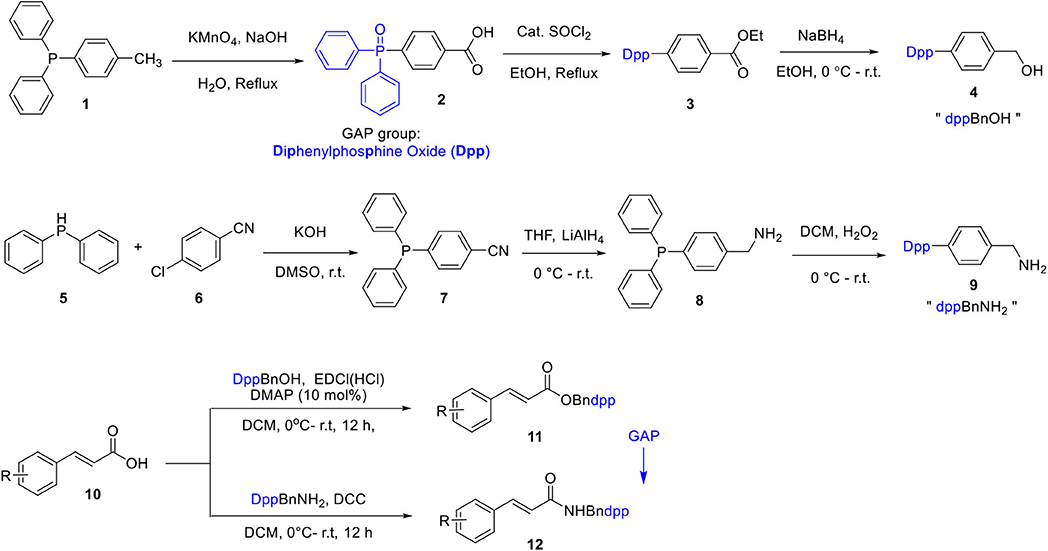
Scheme 1. Synthesis of GAP auxiliaries for cinnamic acid protection.
We further investigated various reaction parameters by subjecting GAP substrates to aminohalogenation reaction. Initially, the GAP-anchored intermediate 11 was subjected to aminochlorination with dichloramine T (4-TsNCl2) (1.5 equiv.) in dichloromethane without any catalyst; however, no product was formed after stirring for 48 h. Then iodobenzene diacetate (PhI(OAc)2 and a series of transition metal catalysts were employed to no avail. Pleasingly, when 1.5 equiv. of tosylamide (4-TsNH2) was added along with 4-TsNCl2 (1.5 equiv.) in the presence of catalyst PhI(OAc)2 (20 mol%), the starting material 11 was consumed in 48 h at room temperature, and a single aminohalogenation product was isolated in a chemical yield of 71% (Table 1, entry 9) with a diastereoselective ratio of 8:1. To improve the yield, we again screened a variety of transition-metal compounds such as ZnCl2, Mn(OAc)2, Pd(OAc)2, FeCl3, and CuI as catalysts for this reaction. The results are depicted in Table 1, which specifies that PhI(OAc)2 (20 mol%) was the potent catalyst for this reaction. It was observed that Mg(OAc)2 gave no product; Pd(OAc)2, FeCl3, and ZnCl2 gave poor yields, whereas CuI and Cu(OTf)2 (Table 1, entry 14 and 15) gave moderately good yields. The next optimization was the addition of another 0.5 equiv. of each 4-TsNCl2 and 4-TsNH2, which increased the yield up to 76% (Table 1, entry 16). Refluxing this reaction mixture further enhanced the yield up to 83% (Table 1, entry 17). However, the yield decreased to 78% when the reaction was refluxed in the absence of 4 Å molecular sieves (Table 1, entry 18).
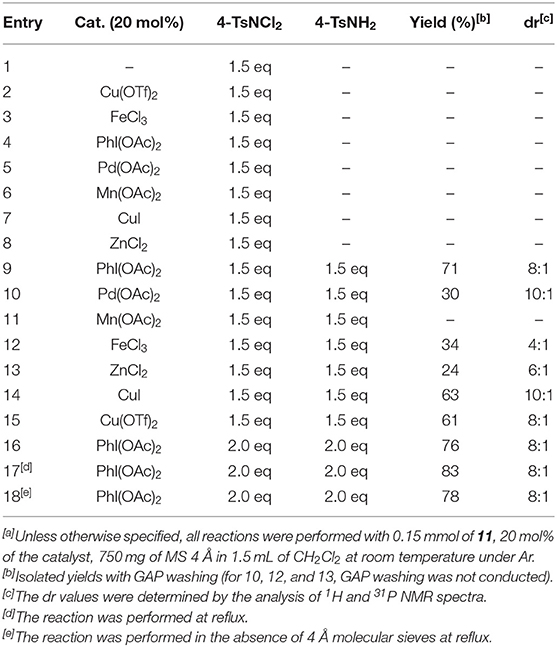
Table 1. Optimization of the reaction conditions[a].
Using 20 mol% of PhI(OAc)2 as the catalyst with 2 equiv. of 4-TsNCl2 and 4-TsNH2 at reflux temperature, various solvents such as CHCl3, CH2Cl2, MeCN, THF, Et2O, and toluene were screened, which revealed that CH2Cl2 was found to be the best solvent to give 13 in 83% yield with good stereoselectivity (8:1, Table 2, entries 1 and 12). Moderate yields were obtained in MeCN and DCE solvents. The product 13 was obtained only with great difficulty in toluene; no reaction was observed in THF, Et2O, DMF, and MeOH, whereas only traces were formed in dioxane solvent. A decrease in the reaction time by 24 h led to slightly reduced yield (77%) of the product 13 (Table 2, entry 11); however, the yield did not change with the prolonged reaction time of 72 h (83%, Table 2, entry 12). Finally, we noticed that 20 mol% of PhI(OAc)2 was essential for the reaction as the yield decreased to 27% when 10 mol% of the catalyst was used in the system (Table 2, entry 13).
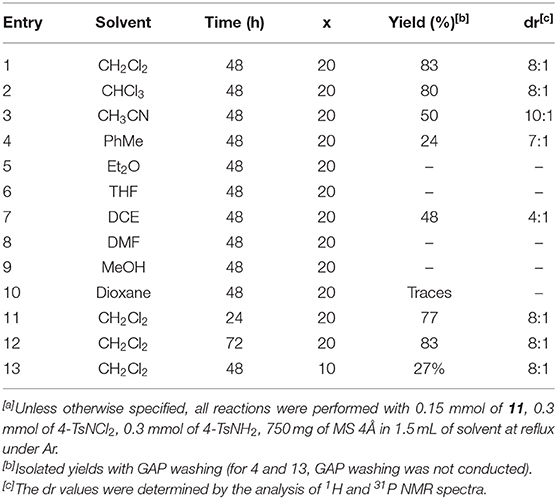
Table 2. Further optimization[a].
With the optimized reaction parameters, the substrate scope of this reaction was thereafter explored with a variety of GAP auxiliary 4 anchored substituted cinnamic acids 11. The results are shown in Scheme 2. Pleasantly, a wide range of functional groups tolerance was observed on the aromatic ring of cinnamic acids including electron-donating groups 13c–13h and electron-withdrawing groups 13i–13o, which provided moderate to high yields (45–94%). As shown in Scheme 2, for those substrates 11 bearing electron-donating groups on the aromatic ring of cinnamic acid, the addition reaction went cleanly to produce the relevant adducts 13 in good yields (Scheme 2, 13c−13h). The highest yield of 94% was obtained for 13e where the aromatic ring has strong electron-donating group MeO at the ortho-position. On the other hand, substrates 11 bearing electron-withdrawing groups such as F, Cl, Br, or NO2 on the aromatic rings gave the resultant products in lower yields under the same conditions (Scheme 2, 13i−13o). The lowest yield of 45% was obtained for 13m, which has a tertiary butyl group at the para-position. Substrates 13n and 13o bearing strong electron-withdrawing groups such as NO2 and Cl did not undergo reaction, whereas substrate 13p with N,N-dimethylamine group at para-position resulted in a complex mixture with traces of desired product.
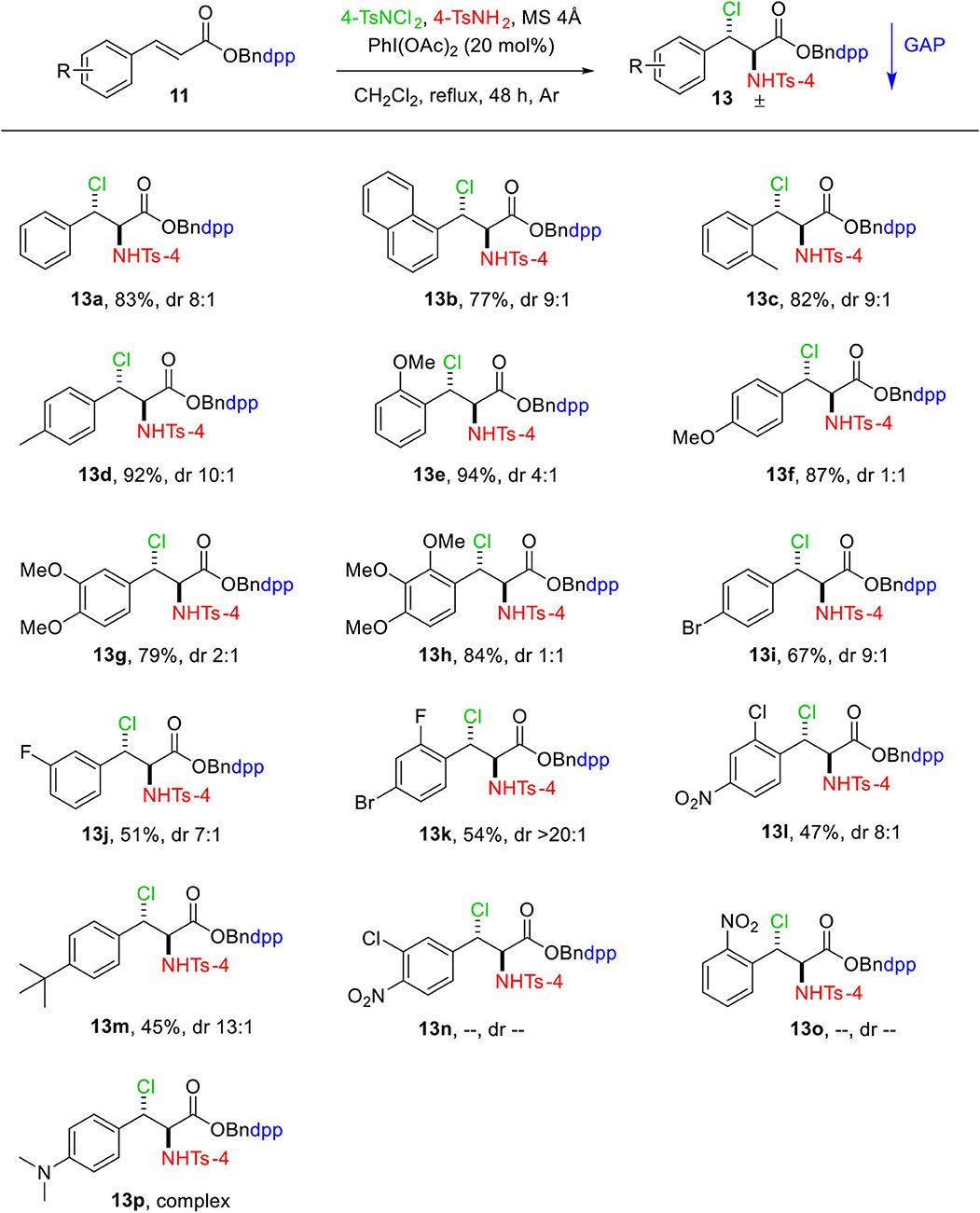
Scheme 2. Substrate scope of aminochlorination of N-(4-(diphenylphosphoryl)benzyl) cinnamates. Unless otherwise specified, all reactions were performed with 0.15 mmol of 11, 0.3 mmol of 4-TsNCl2, 0.3 mmol of 4-TsNH2, 750 mg of MS 4Å in 1.5 mL of DCM at reflux under Ar. The dr values were determined by the analysis of 1H and 31P NMR spectra. Isolated yields with GAP washing.
A major objective of this project was the development of GAP auxiliaries, which could potentially simplify the purification of the haloamine products. Thus, the cinnamic acids 10 were protected with another GAP auxiliary DppBnNH2 9 to afford N-(4-(diphenylphosphoryl)benzyl) cinnamamides 12, (Scheme 1), which were then exposed to aminochlorination reaction. Conditions for this transformation were also optimized (Table 1S, Supporting Information). We found that 20 mol% of PhI(OAc)2, 2.0 equiv. of 4-TsNH2 and 4-TsNCl2, was essential to obtain haloamine product 14a in 73% yield with diastereoselective ratio of 10:1 when refluxed in dichloromethane for 48 h.
We also investigated the scope of this transformation by using a variety of N-(4-(diphenylphosphoryl)benzyl) cinnamamides 12. As illustrated in Scheme 3, the reaction tolerated a wide range of functional groups to provide moderate to high yields (54%−76%). For the substrate having methoxy group at ortho-position on aromatic ring, the highest yield of 76% was obtained (Scheme 3, 14c). The nitro group, on the other hand, lowers the yield considerably under the same conditions (Scheme 3, 14j). The substrate with NO2 group at ortho-position of aromatic ring 14k did not undergo reaction even after 72 h, whereas the reactions for the products 14i and 14l were sluggish and resulted in a complex mixture.
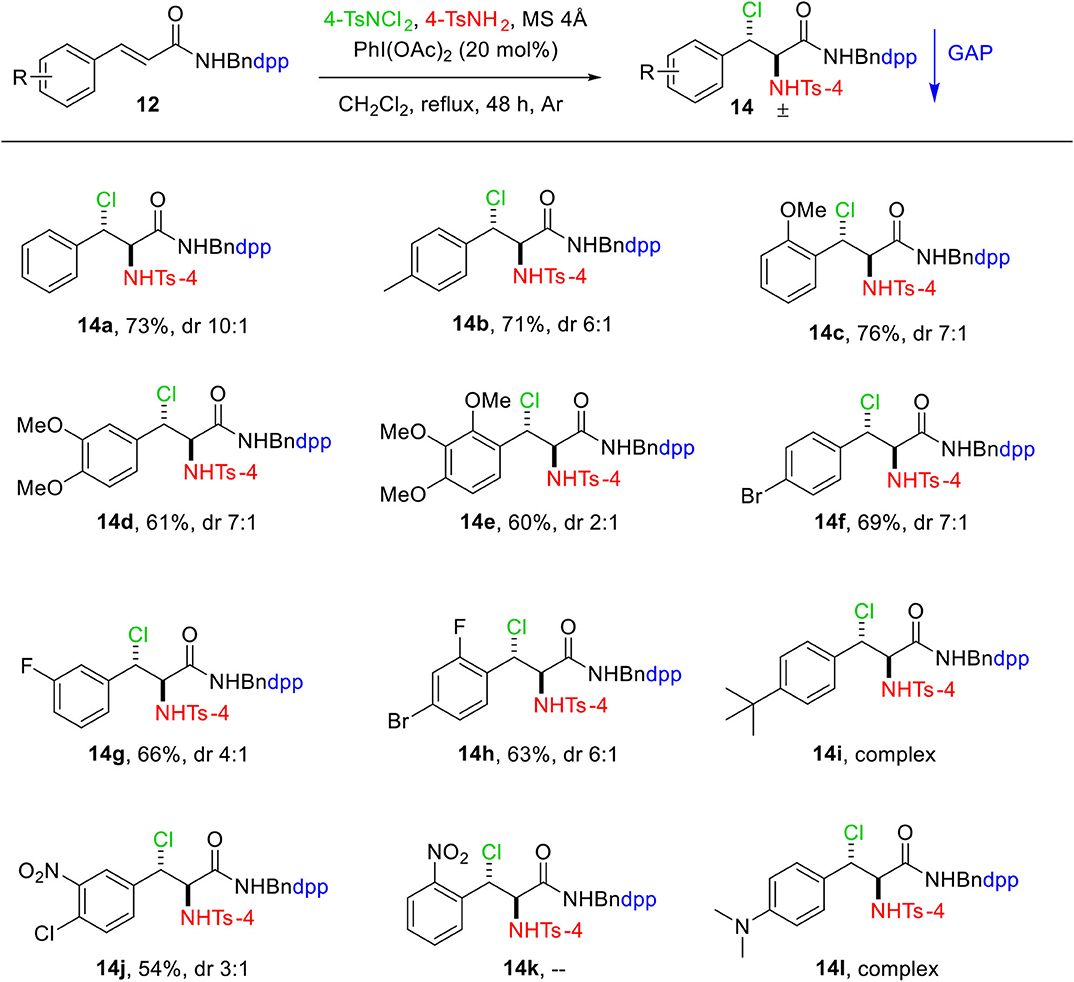
Scheme 3. Substrate scope of aminochlorination of N-(4-(diphenylphosphoryl)benzyl) cinnamamides. Unless otherwise specified, all reactions were performed with 0.15 mmol of 12, 0.3 mmol of 4-TsNCl2, 0.3 mmol of 4-TsNH2, 750 mg of MS 4Å in 1.5 mL of DCM at reflux under Ar. The dr values were determined by the analysis of 1H and 31P NMR spectra. Isolated yields with GAP washing.
The practicality of this protocol was determined by performing the reaction on gram scale for the starting material 11a and 12a, which led to the formation of the products 13a and 14a in 80% and 71% yields, respectively (Scheme 4).
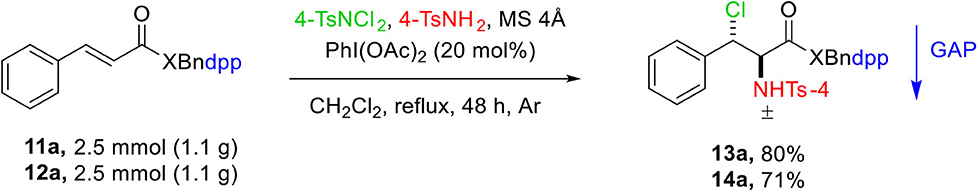
Scheme 4. Gram scale reaction.
The GAP-tailored vicinal chloramine was deprotected in the presence of Pd/C and NaBH4 which afforded Bndpp in 95% yield (Scheme 5). For the purification of the products, the mixture is dissolved in minimal amount of a polar solvent such as ethyl acetate or DCM, and then petroleum ether is added. The GAP auxiliary precipitates in the form of a white solid, which is filtered and washed with petroleum ether. The filtrate is evaporated under vacuum to obtain the desired β-chloroamine as a white product.
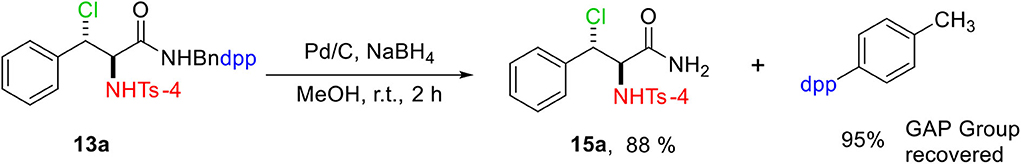
Scheme 5. Group-assisted purification deprotection.
Based on experimental observations and our as well as others' previous works (Li et al., 2001; Wei et al., 2001; Wang and Wu, 2007; Wu and Wang, 2008; Chen et al., 2009), a rational mechanism for the formation of 13 and 14 is illustrated in Scheme 6. 4-TsNCl2 on reaction with 4-TsNH2 may produce N-chloro-p-toluenesulfonamide (4-TsNHCl) 15, which might be oxidized by PhI(OAc)2 to generate intermediate 16 that may either follow cycle A or cycle B. The intermediate 16, in cycle A, could form aziridinium 17 with the double bond of 11 or 12, which in turn is attacked by the dissociated halogen anion from the intermediate 16 at the more electrophilic carbon (beta to carbonyl carbon) to yield compound 18 stereoselectively. Eventually, intermediate 18 together with 4-TsNHCl 15 provides the uttermost haloamine product 13 or 14 and restores intermediate 16. There is a possibility of formation of N-acetoxy-N-halo-p-toluenesulfonamide 19 when phenyl iodide is released as a result of dissociation of the unstable N–I bond of intermediate 16, which could then be the active intermediate for cycle B. Species 19 that forms an equilibrium with nitrenium ion intermediate 20 (Kikugawa et al., 2003; Murata et al., 2008) could react with olefin 11 or 12 to afford aziridinium intermediate 21, which would lead to intermediate 22 following an SN2 nucleophilic attack by the nearby halide anion. Finally, the reaction of the intermediate 22 with 4-TsNHCl gives the final product and regenerate intermediate 19.
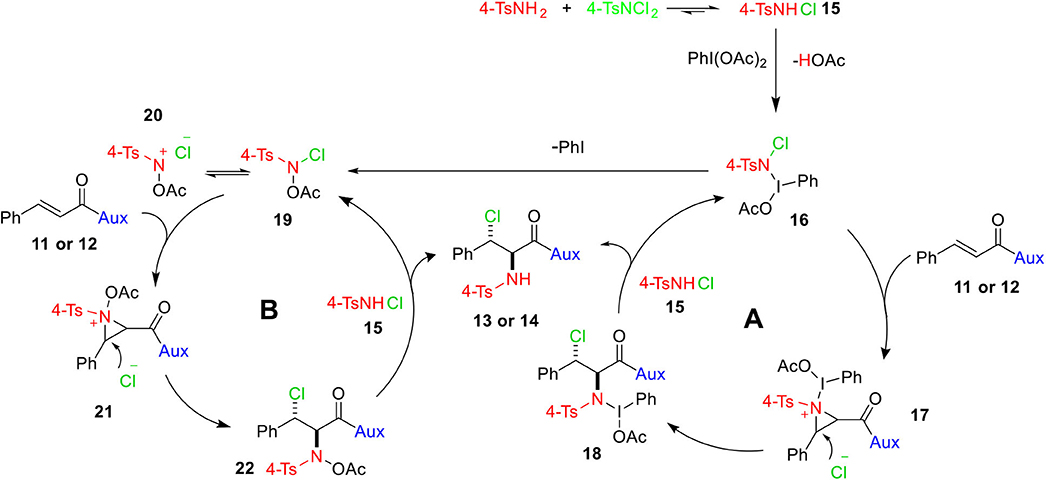
Scheme 6. A plausible mechanism for the syntheses of vicinal chloramines.
Typical procedure: Into a dry vial, 11 or 12 (1.5 mmol, 1 eq), 4-TsNCl2 (3 mmol, 2 eq) 4-TsNH2 (3 mmol, 2 eq), PhI(OAc)2 (20 mol%), and freshly activated 4 Å molecular sieves (150 mg) were added. The resulting mixture was capped under argon protection. Anhydrous DCM (3 mL) was injected through a syringe and was refluxed for 48 h. After completion (monitored by TLC), the reaction was quenched with saturated aqueous Na2SO3 solution (2 mL), and DCM (3 × 10 mL) was added to extract the product. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and evaporated to dryness in vacuo. The mixture is dissolved in minimal amount of a polar solvent such as ethyl acetate or DCM, and then petroleum ether is added. The GAP auxiliary precipitates in the form of a white solid, which is filtered and washed with petroleum ether. The filtrate is evaporated under vacuum to obtain the desired β-chloroamine as a white product.
To a stirred solution of 13a or 14a (0.1 g, 0.16 mmol) and 10 wt% Pd/C (10 mg) in MeOH (1 mL), NaBH4 (7.6 mg, 2 equiv.) was added. The 10-mL flask was closed with a rubber septum with an empty (deflated) balloon to avoid the loss of generated hydrogen and overpressure in the flask. After 2 h, the reaction mixture was filtered through Celite, and filtrate was evaporated to dryness and redissolved in EtOAc. Then, the reaction mixture was neutralized with KHSO4. The organic layer was separated, dried over anhydrous Na2SO4, and evaporated to dryness to afford crude GAP auxiliary, which was easily purified using the GAP washing method.
A highly efficient regioselective and stereoselective aminochlorination reaction of electron-deficient olefins anchored with GAP auxiliaries dppBnOH and dppBnNH2 catalyzed by PhI(OAc)2 in dichloromethane with 4-TsNH2 and 4-TsNCl2 as the nitrogen and chlorine sources has been developed. Moderate to good chemical yields and excellent regioselectivity and stereoselectivity have been obtained. The GAP approach has been effectively implemented, which bypasses column chromatography and recrystallization. Pure products have been obtained simply by washing the crude mixtures with inexpensive petroleum solvents or cosolvents to give good to high yields. Besides, the GAP auxiliary is recyclable and reusable.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
GL and AR designed the project. AR, NZ, and WY performed the experiments. AA and PZ analyzed the data. AR, AA, and GL wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to acknowledge the financial support from the National Natural Science Foundation of China (No. 91956110, 21672100) and Robert A. Welch Foundation (D-1361, USA).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00523/full#supplementary-material
Anderson, N. G. (2012). Practical Process Research and Development: A Guide for Organic Chemists. Cambridge, UK: Academic Press.
Arteaga, G. C., Saavedra-Olavarría, J., Almendras, S., Hermosilla-Ibáñez, P., Almodovar, I., and Pérez, E. G. (2018). Copper-catalyzed intermolecular aminochlorination of alkenes. Tetrahedron Lett. 59, 1091–1093. doi: 10.1016/j.tetlet.2018.02.010
Bovino, M. T., and Chemler, S. R. (2012). Catalytic enantioselective alkene aminohalogenation/cyclization involving atom transfer. Angew. Chem. Int. Ed. Engl. 51, 3923–3927. doi: 10.1002/anie.201109044
Broggini, G., Beccalli, E. M., Borelli, T., Brusa, F., Gazzola, S., and Mazza, A. (2015). Intra-intermolecular palladium-catalyzed domino reactions of glycine allylamides for the synthesis of diversely functionalized piperazinones. Eur. J. Org. Chem. 2015, 4261–4268. doi: 10.1002/ejoc.201500386
Cai, Y., Liu, X., Jiang, J., Chen, W., Lin, L., and Feng, X. (2011). Catalytic asymmetric chloroamination reaction of α,β-unsaturated γ-keto esters and chalcones. J. Am. Chem. Soc. 133, 5636–5639. doi: 10.1021/ja110668c
Cai, Y., Liu, X., Zhou, P., and Feng, X. (2019). Asymmetric catalytic halofunctionalization of α,β-unsaturated carbonyl compounds. J. Org. Chem. 84, 1–13. doi: 10.1021/acs.joc.8b01951
Chemler, S. R., and Bovino, M. T. (2013). Catalytic aminohalogenation of alkenes and alkynes. ACS Catal 3, 1076–1091. doi: 10.1021/cs400138b
Chen, D., Guo, L., Liu, J., Kirtane, S., Cannon, J. F., and Li, G. (2005). Functionalization of alpha,beta-unsaturated esters and ketones: a facile and highly stereoselective one-pot approach to N-protected alpha,beta-dehydroamino acid derivatives. Org. Lett. 7, 921–924. doi: 10.1021/ol050002u
Chen, Z.-G., Wang, Y., Wei, J.-F., Zhao, P.-F., and Shi, X.-Y. (2010). K3PO4-catalyzed regiospecific aminobromination of β-nitrostyrene derivatives with N-bromoacetamide as aminobrominating agent. J. Org. Chem. 75, 2085–2088. doi: 10.1021/jo9026879
Chen, Z.-G., Wei, J.-F., Wang, M.-Z., Zhou, L.-Y., Zhang, C.-J., and Shi, X.-Y. (2009). Aluminium powder-catalyzed regio- and stereoselective aminobromination of α,β-unsaturated carbonyl compounds and simple olefins with the p-toluenesulfonamide/ N-bromosuccinimide (TsNH2-NBS) system. Adv. Synth. Catal. 351, 2358–2368. doi: 10.1002/adsc.200900343
Chennapuram, M., Emmadi, N. R., Bingi, C., Nanubolu, J. B., and Atmakur, K. (2014). Group-assisted purification (GAP) chemistry for dihydrofurans: water as a medium for catalyst free synthesis in a one pot four component reaction. Green Chem. 16, 3237–3246. doi: 10.1039/c4gc00388h
Denmark, S. E., Kuester, W. E., and Burk, M. T. (2012). Catalytic, asymmetric halofunctionalization of alkenes—a critical perspective. Angew. Chem. Int. Ed. Engl. 51, 10938–10953. doi: 10.1002/anie.201204347
Dommaraju, Y., and Prajapati, D. (2015). A highly efficient group-assisted purification method for the synthesis of poly-functionalized pyrimidin-5-yl-pyrroles via one-pot four-component domino reaction. Mol. Divers. 19, 173–187. doi: 10.1007/s11030-014-9547-1
Ghorai, M. K., Sahoo, A. K., and Kumar, S. (2011). Synthetic route to chiral tetrahydroquinoxalines via ring-opening of activated aziridines. Org. Lett. 13, 5972–5975. doi: 10.1021/ol2023906
Han, J.-L., Zhi, S.-J., Wang, L.-Y., Pan, Y., and Li, G. (2007). CuCl-catalyzed regio- and stereoselective aminohalogenation of α,β-unsaturated nitriles. Eur. J. Org. Chem. 2007, 1332–1337. doi: 10.1002/ejoc.200600902
Hingst, M., Tepper, M., and Stelzer, O. (1998). Nucleophilic phosphanylation of fluoroaromatic compounds with carboxyl, carboxymethyl, and aminomethyl functionalities – an efficient synthetic route to amphiphilic arylphosphanes. Eur. J. Org. Chem. 1998, 73–82.
Janssen, M., Müller, C., and Vogt, D. (2009). ‘Click' dendritic phosphines: design, synthesis, application in suzuki coupling, and recycling by nanofiltration. Adv. Synth. Catal. 351, 313–318. doi: 10.1002/adsc.200900058
Kikugawa, Y., Nagashima, A., Sakamoto, T., Miyazawa, E., and Shiiya, M. (2003). Intramolecular cyclization with nitrenium ions generated by treatment of N-acylaminophthalimides with hypervalent iodine compounds: formation of lactams and spiro-fused lactams. J. Org. Chem. 68, 6739–6744. doi: 10.1021/jo0347009
Lee, S. A., and Robinson, G. (1995). Process Development: Fine Chemicals From Grams to Kilograms. Oxford, UK: Oxford University Press Oxford.
Legnani, L., Prina-Cerai, G., Delcaillau, T., Willems, S., and Morandi, B. (2018). Efficient access to unprotected primary amines by iron-catalyzed aminochlorination of alkenes. Science 362, 434–439. doi: 10.1126/science.aat3863
Li, G., Kotti, S. R. S. S., and Timmons, C. (2007). Recent development of regio- and stereoselective aminohalogenation reaction of alkenes. Eur. J. Org. Chem. 2007, 2745–2758. doi: 10.1002/ejoc.200600990
Li, G., Wei, H.-X., and Kim, S. H. (2001). Unexpected copper-catalyzed aminohalogenation reaction of olefins using N-halo-N-metallo-sulfonamide as the nitrogen and halogen sources. Tetrahedron 57, 8407–8411. doi: 10.1016/S0040-4020(01)00847-X
Liu, J., Wang, Y., and Li, G. (2006). Regio- and stereoselective synthesis of anti-1,3-diaryl-3-chloro- 2-(o-nitrophenylsulfonylamino)-3-propan-1-ones through catalytic aminohalogenation reaction of α,β-unsaturated ketones. Eur. J. Org. Chem. 2006, 3112–3115. doi: 10.1002/ejoc.200600020
Martínez, C., and Muñiz, K. (2014). A versatile metal-free intermolecular aminochlorination of alkenes. Adv. Synth. Catal. 356, 205–211. doi: 10.1002/adsc.201300880
Michael, F. E., Sibbald, P. A., and Cochran, B. M. (2008). Palladium-catalyzed intramolecular chloroamination of alkenes. Org. Lett. 10, 793–796. doi: 10.1021/ol702922c
Minakata, S., Yoneda, Y., Oderaotoshi, Y., and Komatsu, M. (2006). Unprecedented CO2-promoted aminochlorination of olefins with chloramine-T. Org. Lett. 8, 967–969. doi: 10.1021/ol0600178
Murata, K., Tsukamoto, M., Sakamoto, T., Saito, S., and Kikugawa, Y. (2008). Hydrazidohydroxylation of styrenes with N-acetylaminophthalimide using phenyliodine(III) bis(trifluoroacetate) (PIFA). Synthesis 2008, 32–38. doi: 10.1055/s-2007-1000819
Patel, D. M., Vala, R. M., Sharma, M. G., Rajani, D. P., and Patel, H. M. (2019). A practical green visit to the functionalized [1,2,4]Triazolo[5,1-b]quinazolin-8(4H)one scaffolds using the group-assisted purification (GAP) chemistry and their pharmacological testing. Chemistry Select 4, 1031–1041. doi: 10.1002/slct.201803605
Pu, X.-Q., Zhao, H.-Y., Lu, Z.-H., He, X.-P., and Yang, X.-J. (2016). Aminochlorination of alkenes with CFBSA. Eur. J. Org. Chem. 2016, 4526–4533. doi: 10.1002/ejoc.201600709
Qin, Q., Ren, D., and Yu, S. (2015). Visible-light-promoted chloramination of olefins with N-chlorosulfonamide as both nitrogen and chlorine sources. Org. Biomol. Chem. 13, 10295–10298. doi: 10.1039/C5OB01725D
Schreiber, S. L. (2000). Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287, 1964–1969. doi: 10.1126/science.287.5460.1964
Schröder, S. P., van de Sande, J. W., Kallemeijn, W. W., Kuo, C.-L., Artola, M., van Rooden, E. J., et al. (2017). Towards broad spectrum activity-based glycosidase probes: synthesis and evaluation of deoxygenated cyclophellitol aziridines. Chem. Commun. 53, 12528–12531. doi: 10.1039/C7CC07730K
Seifert, C. W. (2017). New synthetic methodology for chiral amines and peptides via GAP technology (Ph.D. thesis). Texas Tech University.
Seifert, C. W., Paniagua, A., White, G. A., Cai, L., and Li, G. (2016). GAP Peptide Synthesis via Design of New GAP Protecting Group: An Fmoc/tBu Synthesis of Thymopentin Free from Polymers, Chromatography and Recrystallization. Eur. J. Org. Chem. 2016, 1714–1719. doi: 10.1002/ejoc.201600026
Song, L., Luo, S., and Cheng, J.-P. (2013). Catalytic intermolecular haloamidation of simple alkenes with N-halophthalimide as both nitrogen and halogen source. Org. Lett. 15, 5702–5705. doi: 10.1021/ol402726d
Song, L., Luo, S., and Cheng, J.-P. (2016). Visible-light promoted intermolecular halofunctionalization of alkenes with N-halogen saccharins. Org. Chem. Front. 3, 447–452. doi: 10.1039/C5QO00432B
Tay, D. W., Tsoi, I. T., Er, J. C., Leung, G. Y. C., and Yeung, Y.-Y. (2013). Lewis basic selenium catalyzed chloroamidation of olefins using nitriles as the nucleophiles. Org. Lett. 15, 1310–1313. doi: 10.1021/ol400249x
Thakur, R., Rawal, G. K., and Vankar, Y. D. (2017). Synthesis of chiral aziridines from glycals: application in the synthesis of a piperidine–azepine fused derivative. Eur. J. Org. Chem. 2017, 4235–4241. doi: 10.1002/ejoc.201700624
Thakur, V. V., Talluri, S. K., and Sudalai, A. (2003). Transition metal-catalyzed regio- and stereoselective aminobromination of olefins with TsNH2 and NBS as nitrogen and bromine sources. Org. Lett. 5, 861–864. doi: 10.1021/ol027530f
Tietze, L., Brasche, G., and Gericke, K. (2008). Domino Reactions in Organic Synthesis 2006. Weinheim: Wiley-VCH. doi: 10.1002/9783527609925
Trost, B. M. (1991). The atom economy–a search for synthetic efficiency. Science 254, 1471–1477. doi: 10.1126/science.1962206
Van, T. N., and De Kimpe, N. (2000). Synthesis of chiral cis-1,2,3-trisubstituted aziridines. Tetrahedron 56, 7299–7304. doi: 10.1016/S0040-4020(00)00627-X
Wang, G.-W., and Wu, X.-L. (2007). Mechanochemical aminochlorination of electron-deficient olefins with chloramine-T promoted by (Diacetoxyiodo)benzene. Adv. Synth. Catal. 349, 1977–1982. doi: 10.1002/adsc.200700020
Wang, H., Liu, X., Feng, X., Huang, Z., and Shi, D. (2013). GAP chemistry for pyrrolyl coumarin derivatives: a highly efficient one-pot synthesis under catalyst-free conditions. Green Chem. 15, 3307–3311. doi: 10.1039/c3gc41799a
Wei, H.-X., Kim, S. H., and Li, G. (2001). The first transition metal–ligand complex-catalyzed regioselective and stereoselective aminohalogenation of cinnamic esters. Tetrahedron 57, 3869–3873. doi: 10.1016/S0040-4020(01)00228-9
Wei, J.-F., Zhang, L.-H., Chen, Z.-G., Shi, X.-Y., and Cao, J.-J. (2009). KI-catalyzed aminobromination of olefins with TsNH2-NBS combination. Org. Biomol. Chem. 7, 3280–3284. doi: 10.1039/b904789a
Wu, X.-L., and Wang, G.-W. (2008). Aminohalogenation of electron-deficient olefins promoted by hypervalent iodine compounds. Eur. J. Org. Chem. 2008, 6239–6246. doi: 10.1002/ejoc.200800842
Xiong, Y., Qian, P., Cao, C., Mei, H., Han, J., Li, G., et al. (2014). One-pot stereoselective synthesis of α,β-differentiated diamino esters via the sequence of aminochlorination, aziridination and intermolecular SN2 reaction. Beilstein J. Org. Chem. 10, 1802–1807. doi: 10.3762/bjoc.10.189
Yin, G., Wu, T., and Liu, G. (2012). Highly selective palladium-catalyzed intramolecular chloroamination of unactivated alkenes by using hydrogen peroxide as an oxidant. Chem. A Euro. J. 18, 451–455. doi: 10.1002/chem.201102776
Zhu, C.-L., Tian, J.-S., Gu, Z.-Y., Xing, G.-W., and Xu, H. (2015). Iron(II)-catalyzed asymmetric intramolecular olefin aminochlorination using chloride ion. Chem. Sci. 6, 3044–3050. doi: 10.1039/C5SC00221D
Keywords: aziridinium, diastereoselectivity, iodobenzene diacetate, nitrogen/halogen source, protecting groups
Citation: Rahman AU, Zarshad N, Zhou P, Yang W, Li G and Ali A (2020) Hypervalent Iodine (III) Catalyzed Regio- and Diastereoselective Aminochlorination of Tailored Electron Deficient Olefins via GAP Chemistry. Front. Chem. 8:523. doi: 10.3389/fchem.2020.00523
Received: 01 May 2020; Accepted: 21 May 2020;
Published: 07 July 2020.
Edited by:
Zhendong Jin, University of Iowa, United StatesReviewed by:
Dengfu Lu, Huazhong University of Science and Technology, ChinaCopyright © 2020 Rahman, Zarshad, Zhou, Yang, Li and Ali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guigen Li, Z3VpZ2VuLmxpQHR0dS5lZHU=; Asad Ali, YXNhZGFsaUBhd2t1bS5lZHUucGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.