 Suresh Mandava
Suresh Mandava Jaun Koo
Jaun Koo Hari Krishna Nallapaneni
Hari Krishna Nallapaneni Jongkook Lee
Jongkook Lee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 30 June 2020
Sec. Organic Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00494
This article is part of the Research Topic Synthetic Methodologies for Privileged Natural Product Scaffolds View all 8 articles
The cross-metathesis (CM) of methallyl halides catalyzed using four different ruthenium-based complexes—Grubbs catalyst, Grubbs second-generation catalyst, Hoveyda-Grubbs second-generation catalyst, and Stewart–Grubbs catalyst—was investigated. When methallyl chloride or bromide was reacted with a model substrate containing a benzyl ether group, the Grubbs catalyst, and Grubbs second-generation catalyst did not promote the reaction well. However, the Hoveyda–Grubbs second-generation catalyst and Stewart–Grubbs catalyst afforded the corresponding products in moderate to good yield with moderate E/Z selectivity. Accordingly, several functionalized methallyl halides were prepared by CM. Various functional groups were well-tolerated in this system when the Stewart–Grubbs catalyst was used. To demonstrate the practical utility of our method, methallyl halide CM was successfully employed for the formal total synthesis of a natural product (–)-presphaerene, in which the precursor of the key cyclopentanecarboxylate intermediate was efficiently prepared in three steps.
Currently, methallyl halide moieties are typically introduced by conversion of a preexisting aldehyde via olefination–reduction–halogenation (Phoenix et al., 2008; Tanabe et al., 2014; Kauhl et al., 2016) or Grignard addition–halogenation sequences (Davies et al., 2010; Ghosh and Li, 2011), especially in natural product synthesis. Alternatively, the cross-metathesis (CM) of methallyl halides, which would produce trisubstituted alkenes (Nguen et al., 2017; Xu et al., 2017), has the possibility to serve as a useful synthetic tool for the incorporation of methallyl halide moieties into organic molecules. However, despite its potential, this methodology has been hitherto poorly investigated. Accordingly, very few studies on the CM of methallyl halides using ruthenium-based catalysts I–V (Figure 1) (Schwab et al., 1995; Kingsbury et al., 1999; Scholl et al., 1999; Garber et al., 2000; Stewart et al., 2008) have been published. As rare examples, the Hoveyda–Grubbs second-generation catalyst (III) has been used to promote the CM of methallyl chloride with α-methylene-β-lactam to produce the corresponding tetrasubstituted alkene in moderate yield with moderate selectivity (E/Z, 1:1.6) (Liang et al., 2009). Several functionalized olefins have been shown to smoothly undergo CM with methallyl chloride when ruthenium-based complex V (UmicoreTM M51) is employed as the catalyst (Bilel et al., 2014).
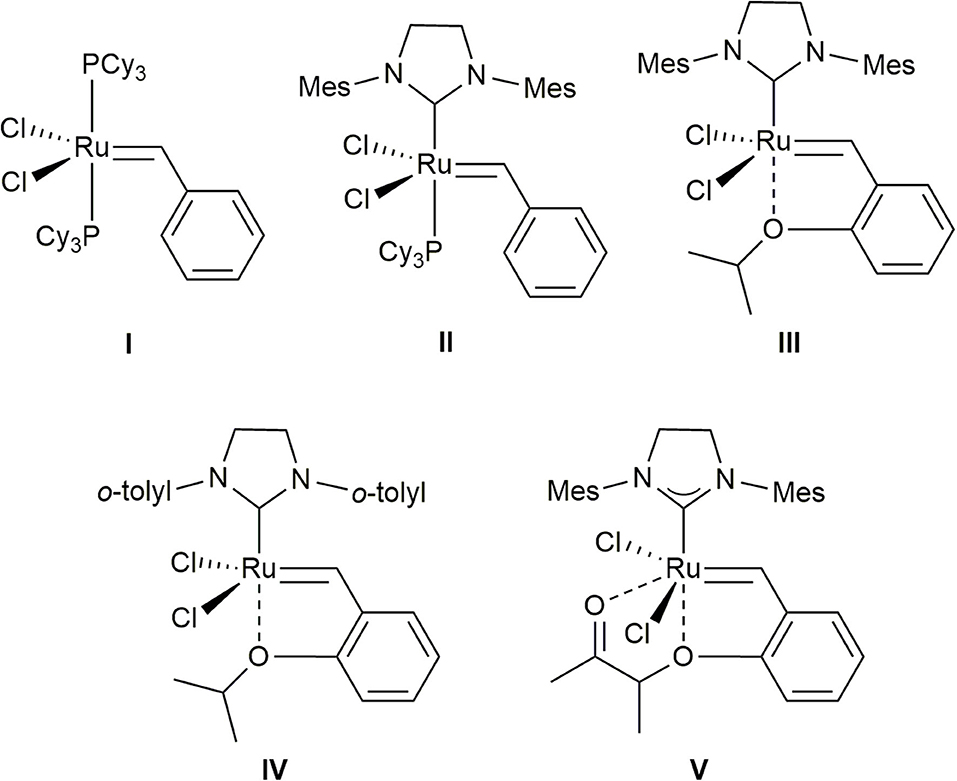
Figure 1. Grubbs catalyst (I), Grubbs second-generation catalyst (II), Hoveyda–Grubbs second-generation catalyst (III), Stewart–Grubbs catalyst (IV), and UmicoreTM M51 (V) considered in this study.
Although the CM of methallyl halides could serve as a synthetic shortcut, substituting a single step for the numerous functional-group transformations required for the corresponding conventional sequences, this reaction has not been hitherto successfully applied to natural product synthesis. For instance, an attempt to introduce a methallyl chloride moiety into a complex olefin for the total syntheses of the natural products stephacidins A and B and notoamide B using ruthenium-based catalysts I and II did not afford the corresponding product (Artman III et al., 2007).
Accordingly, we herein investigated the ruthenium-complex-catalyzed CM of methallyl halides with several functionalized olefins and its application to the formal total synthesis of a sphaeroane diterpene, (–)-presphaerene (1) (Figure 2) (Cafieri et al., 1983).
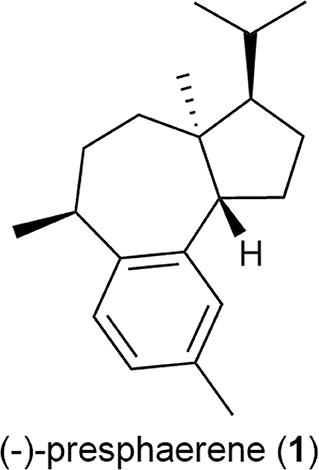
Figure 2. Structure of (–)-presphaerene (1).
Experimental procedures and compound characterization data are provided in the Supplementary Material.
We first attempted the CM of methallyl chloride with commercially available olefin 2 bearing a benzyl ether group in CH2Cl2 employing ruthenium-based catalysts I–IV. While the Grubbs catalyst (I) and Grubbs second-generation catalyst (II) catalyze the reaction poorly, both III and Stewart–Grubbs catalyst (IV) promote the CM of methallyl chloride with 2 in good yield and with moderate E/Z selectivity (Table 1, entry 1). Excess amounts of methallyl chloride were required to reduce homocoupling of olefin 2 and to completely convert 2 to the corresponding product.
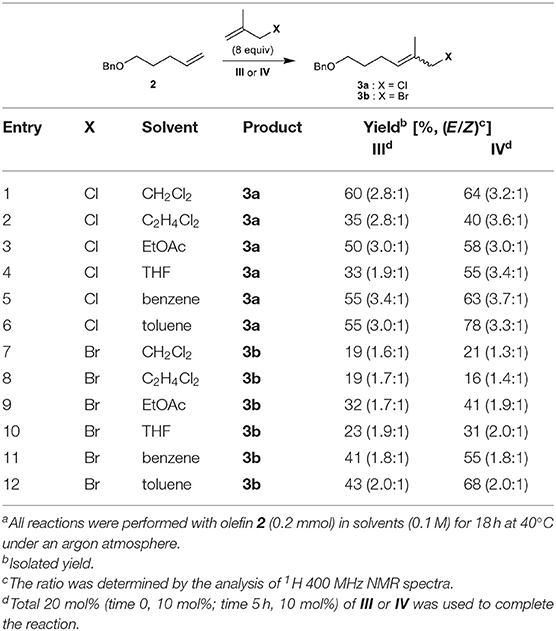
Table 1. Cross-metathesis (CM) of methallyl halides with olefin 2a.
The reaction was then performed in several other solvents, including EtOAc, THF, benzene, and toluene (Table 1, entries 2–6). The methallyl chloride CM of 2 proceeds over IV in benzene and toluene with the highest E/Z selectivity and yield, respectively.
The CM of methallyl bromide with 2 was also conducted in several solvents including CH2Cl2, EtOAc, THF, benzene, and toluene. In contrast to the CM of methallyl chloride, the CM of methallyl bromide proceeds poorly under most conditions assessed (Table 1, entries 7–11). Only catalyst IV in toluene promotes the methallyl bromide CM of 2 smoothly and in good yield with moderate E/Z selectivity (Table 1, entry 12). The relatively poor selectivity compared to the corresponding methallyl chloride CM might be attributed to the instability of the E-form of 3b. Nevertheless, to the best of our knowledge, this constitutes the first example of the incorporation of a methallyl bromide moiety into a functionalized olefin by the CM of methallyl bromide.
The CM of methallyl iodide with 2 was also attempted under a variety of conditions in the presence of III or IV, but the reaction was not observed to any appreciable extent.
Next, we investigated the tolerance of methallyl halide CM to different functional groups, including alkyl, aryl, ester, ketone, hydroxyl, silyl, and epoxy groups, under similar conditions to those employed for the methallyl halide CM of 2.
The methallyl halide CM of commercially or readily available substrates 4a–h (Brimble et al., 2005) is well-catalyzed by IV (Table 2, Table S1). The reactions exhibit moderate selectivity (E/Z, 1.7–4.6:1) in most cases, but the CM of methallyl bromide with both ketone 4d and silyl ether 4f affords the corresponding functionalized methallyl bromides 15d and 15f with poor selectivity (Table S1, entries 4 and 6). The methallyl chloride CM reactions of 4a–h show higher performances than their corresponding methallyl bromide counterparts with respect to both yield and E/Z selectivity. This high performance may be attributed to the better stability of functionalized methallyl chlorides 5a–h than those of the corresponding functionalized methallyl bromides 15a–h considering almost complete conversion of 4a–h.
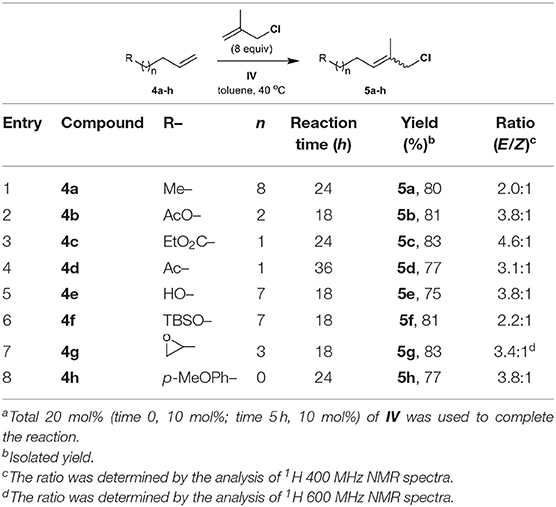
Table 2. CM of methallyl chloride with functionalized olefinsa.
We next directed our attention to the CM of methallyl halides with commercially available olefins that possess nitrogen-containing functional groups, including amide, imide, carbamate, and nitrile groups. In our previous studies, the N,N-dimethylamide group was intolerant to allyl halide CM, but the electron-deficient Weinreb amide group tolerated the reaction catalyzed by III (Yun et al., 2011, 2012).
The methallyl halide CM of olefins bearing amide groups was first examined. Both Weinreb amide and N,N-dimethylamide groups tolerate methallyl halide CM when IV is employed as the catalyst (Table 3, entries 1 and 2), while ruthenium complex III serves as a poor catalyst for the methallyl halide CM of N,N-dimethylamide 6b (data not shown). As expected, the methallyl halide CM of Weinreb amide 6a proceeds more effectively than that of 6b with respect to yield and selectivity (both Table 3 and Table S2, entries 1 and 2). Catalyst IV promotes the CM of methallyl halides with phthalimide 6c in good yield and with moderate E/Z selectivity (both Table 3 and Table S2, entry 3). Both carbamate and nitrile groups tolerate methallyl chloride CM (Table 3, entries 4 and 5) but not methallyl bromide CM (Table S2, entries 4 and 5), most likely due to the instability of functionalized methallyl bromides 16d, e. The methallyl chloride CM reactions of 6a–d are also superior to the corresponding methallyl bromide CM reactions with respect to yield.
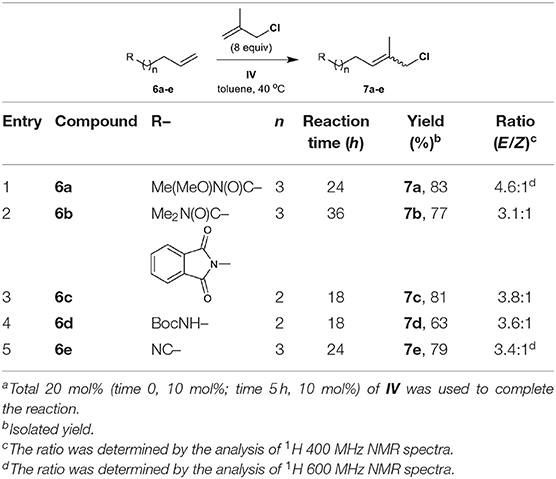
Table 3. CM of methallyl chloride with olefins bearing nitrogen-containing functional groupsa.
Encouraged by these results, we prepared a retrosynthetic plan for the formal total synthesis of 1 in combination with our previously published total synthesis of this natural product (Lee and Hong, 2004) as a means to demonstrate the efficiency of methallyl halide CM. Specifically, the first total synthesis of compound 1, which we published in 2004, proceeds over 19 steps from glyceraldehyde 8 with internal alkylation and intramolecular Friedel–Crafts acylation as key steps, as shown in Figure 3. In this scheme, cyclopentanecarboxylate 11 is cyclized from its precursor 10-E, in which the methallyl bromide moiety is introduced via a Wittig reaction–reduction–bromination sequence from aldehyde 9.
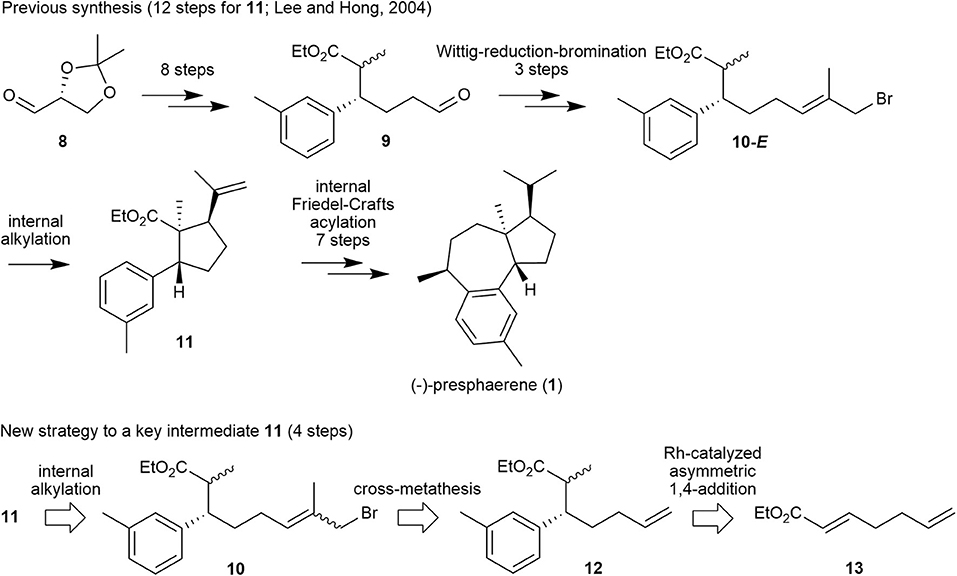
Figure 3. Our previous synthetic approach to (–)-presphaerene (1) and a new retrosynthetic approach to its key intermediate 11.
We envisioned that methallyl bromide CM could provide a synthetic shortcut to the internal alkylation substrate by substituting several functional-group transformations in the sequence. Specifically, internal alkylation substrate 10 could be prepared from olefin 12, which could be generated from α,β-unsaturated ester 13 by utilizing Rh-catalyzed asymmetric 1,4-addition and subsequent methylation, by methallyl bromide CM.
The synthesis commenced with the preparation of a Michael adduct (Figure 4). Commercially available α,β-unsaturated ester 13 undergoes smooth asymmetric 1,4-addition with lithium m-tolylborate, which is generated in situ from 3-bromotoluene, n-butyllithium and trimethoxyborane, in the presence of Rh(acac)(C2H4)2/(R)-BINAP to produce ester 14 in 85% yield and 88% ee (Takaya et al., 1999; Hayashi, 2001; Hayashi and Yamasaki, 2003). The ee of 14 was determined by chiral HPLC analysis (Phenomenex Lux Cellulose-4 column) of the corresponding alcohol reduced from ester 14. Ester 14 is smoothly methylated to 12 as a diastereomeric mixture (3:2, 1H 400 NMR analysis) in 86% yield.
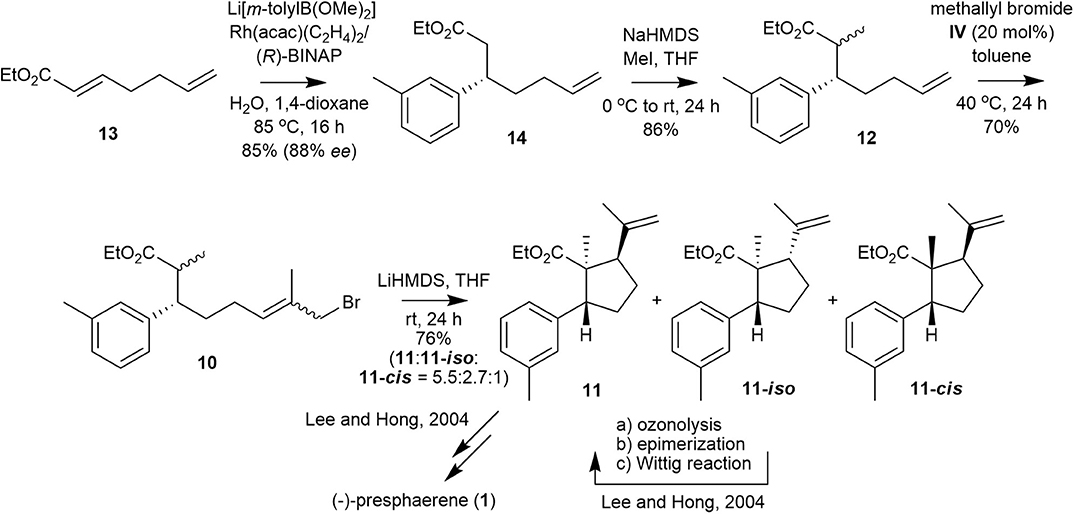
Figure 4. Formal total synthesis of (–)-presphaerene (1).
With olefin 12, we performed methallyl bromide CM to obtain the precursor of cyclopentanecarboxylate 11. Catalyst IV catalyzed the methallyl bromide CM of 12 to afford ω-bromoester 10, which was subjected to the next step without separation of all the isomers, in 70% yield with moderate E/Z selectivity (~3:1, according to 1H 600 MHz NMR analysis). ω-Bromoester 10 was treated with LiHMDS in THF for 24 h at room temperature to yield cyclopentanecarboxylate 11, a key cyclized intermediate in the synthesis of 1, along with 11-iso, which can be converted to 11 as reported in our previous synthesis, and 11-cis in a 76% total yield with 5.5:2.7:1 stereoselectivity (Lee and Hong, 2004).
The lower stereoselectivity and yield reported here compared with those of our previous synthesis (5.5:2.7:1 vs. 9.9:3.3:1, 76 vs. 86%) may be attributed to the poorer internal alkylation of the Z-isomer of 10 (10-Z) compared with that of the E-isomer of 10 (10-E). In our first synthesis of 1, only the E-isomer of 10 (10-E) was used as an internal alkylation substrate.
We obtained the highly functionalized cyclopentanecarboxylate 11, a key intermediate of 1 that was previously synthesized from glyceraldehyde 8 via 12 steps and in 16% overall yield, from heptadienoate 13 via four steps in 23% overall yield. Thus, 1 may be obtained via 11 steps using a combination of our previous and new syntheses, and the overall yield for 11 via this new synthesis is improved by 44% with respect to that by our previous synthesis.
In summary, we have prepared a number of functionalized methallyl halides via CM of methallyl halides as promoted by the Hoveyda–Grubbs second-generation catalyst (III) and the Stewart–Grubbs catalyst (IV) in moderate to good yield with moderate E/Z selectivity. In most instances of methallyl halides CM, IV was superior to III. Several nitrogen-containing functional groups tolerated the CM of methallyl halides when IV was employed. Unlike the catalyst III, IV also efficiently catalyzed the methallyl halide CM of an olefin bearing an N,N-dimethyl amide group.
The practicality of this method was demonstrated by accomplishing a formal total synthesis of (–)-presphaerene (1). Accordingly, we believe that the CM of methallyl halide represents an excellent and practical alternative to general olefination–reduction–halogenation and Grignard addition–halogenation of aldehydes for the incorporation of methallyl halide groups in natural product synthesis.
All datasets presented in this study are included in the article/Supplementary Material.
JL designed the experiments and supervised the research. SM, JK, and HN carried out the synthesis. SM and JH performed the preparative-LC purification and spectroscopic analyses. HP and JL offered guidance on the project. The manuscript was written by SM, HP, and JL and the final version was edited and approved by all the contributing authors.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT; NRF-2018R1A2B6001314).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Central Laboratory of Kangwon National University for providing us with technical assistance on the spectroscopic experiments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00494/full#supplementary-material
Artman, III G. D., Grubbs, A. W., and Williams, R. M. (2007). Concise, asymmetric, stereocontrolled total synthesis of stephacidins A, B and notoamide B. J. Am. Chem. Soc. 129, 6336–6342. doi: 10.1021/ja070259i
Bilel, H., Hamdi, N., Zagrouba, F., Fischermeister, C., and Bruneau, C. (2014). Terminal conjugated dienes via a ruthenium catalyzed cross-metathesis/elimination sequence: application to renewable resources. Catal. Sci. Technol. 4, 2064–2071. doi: 10.1039/C4CY00315B
Brimble, M. A., Flowers, C. L., Hutchinson, J. K., Robinson, J. E., and Sidford, M. (2005). Synthesis of the phthalide-containing anti-Helicobacter pylori agents CJ-13,015, CJ-13,102, CJ-13,103, CJ-13,104 and CJ-13,108. Tetrahedron 61, 10036–10047. doi: 10.1016/j.tet.2005.08.027
Cafieri, F., Ciminiello, P., Santacroce, C., and Fattorusso, E. (1983). Three diterpenes from the red alga Sphaerococcus coronopifolius. Phytochemistry 22, 1824–1825. doi: 10.1016/S0031-9422(00)80282-4
Davies, J. J., Krulle, T. M., and Burton, J. W. (2010). Total synthesis of 7,11-cyclobotryococca-5,12,26-triene using an oxidative radical cyclization as a key step. Org. Lett. 12, 2738–2741. doi: 10.1021/ol100794k
Garber, S. B., Kingsbury, J. S., Gray, B. L., and Hoveyda, A. H. (2000). Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J. Am. Chem. Soc. 122, 8168–8179. doi: 10.1021/ja0151448
Ghosh, A. K., and Li, J. (2011). A stereoselective synthesis of (+)-herboxidiene/GEX1A. Org. Lett. 13, 66–69. doi: 10.1021/ol102549a
Hayashi, T. (2001). Rhodium-catalyzed asymmetric 1,4-addition of organoboronic acids and their derivatives to electron deficient olefins. Synlett 2001, 879–887. doi: 10.1055/s-2001-14657
Hayashi, T., and Yamasaki, K. (2003). Rhodium-catalyzed asymmetric 1,4-addition and its related asymmetric reactions. Chem. Rev. 103, 2829–2844. doi: 10.1021/cr020022z
Kauhl, U., Andernach, L., Weck, S., Sandjo, L. P., Jacob, S., Thines, E., et al. (2016). Total synthesis of (–)-hymenosetin. J. Org. Chem. 81, 215–228. doi: 10.1021/acs.joc.5b02526
Kingsbury, J. S., Harrity, J. P. A., Bonitatebus, P. J., and Hoveyda, A. H. (1999). A recyclable Ru-based metathesis catalyst. J. Am. Chem. Soc. 121, 791–799. doi: 10.1021/ja983222u
Lee, J., and Hong, J. (2004). First synthesis and structural elucidation of (–)-presphaerene. J. Org. Chem. 69, 6433–6440. doi: 10.1021/jo049351c
Liang, Y., Raju, R., Le, T., Taylor, C. D., and Howell, A. R. (2009). Cross-metathesis of α-methylene-β-lactams: the first tetrasubstituted alkenes by CM. Tetrahedron Lett. 50, 1020–1022. doi: 10.1016/j.tetlet.2008.12.060
Nguen, T. T., Koh, M. J., Mann, T. J., Schrock, R. R., and Hoveyda, A. H. (2017). Synthesis of E- and Z-trisubstituted alkenes by catalytic cross-metathesis. Nature 552, 347–357. doi: 10.1038/nature25002
Phoenix, S., Reddy, M. S., and Deslongchamps, P. (2008). Total synthesis of (+)-cassaine via transannular Diels-Alder reaction. J. Am. Chem. Soc. 130, 13989–13995. doi: 10.1021/ja805097s
Scholl, M., Ding, S., Lee, C. W., and Grubbs, R. H. (1999). Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1, 953–956. doi: 10.1021/ol990909q
Schwab, P., France, M. B., Ziller, J. W., and Grubbs, R. H. (1995). A Series of well-defined metathesis catalysts–synthesis of [RuCl2(=CHR′)(PR3)2] and its reactions. Angew. Chem. Int. Ed. Engl. 34, 2039–2041. doi: 10.1002/anie.199520391
Stewart, I. C., Douglas, C. J., and Grubbs, R. H. (2008). Increased efficiency in cross-metathesis reactions of sterically hindered olefins. Org. Lett. 10, 441–444. doi: 10.1021/ol702624n
Takaya, Y., Senda, T., Kurushima, H., Ogasawara, M., and Hayashi, T. (1999). Rhodium-catalyzed asymmetric 1,4-addition of arylboron reagents to α,β-unsaturated esters. Tetrahedron Asymmetry 10, 4047–4056. doi: 10.1016/S0957-4166(99)00417-6
Tanabe, Y., Sato, E., Nakajima, N., Ohkubo, A., Ohno, O., and Suenaga, K. (2014). Total synthesis of biselyngbyolide A. Org. Lett. 16, 2858–2861. doi: 10.1021/ol500996n
Xu, C., Liu, Z., Torker, S., Shen, X., Xu, D., and Hoveyda, A. H. (2017). Synthesis of Z- or E-trisubstituted allylic alcohols and ethers by kinetically controlled cross-metathesis with a Ru catechothiolate complex. J. Am. Chem. Soc. 139, 15640–15643. doi: 10.1021/jacs.7b10010
Yun, J. I., Kim, D., and Lee, J. (2011). Cross-metathesis of allyl halides with olefins bearing an α-alkoxy amide group. Tetrahedron Lett. 52, 1928–1930. doi: 10.1016/j.tetlet.2011.02.043
Keywords: cross-metathesis, methallyl halide, Stewart–Grubbs catalyst, total synthesis, presphaerene
Citation: Mandava S, Koo J, Hwang J, Nallapaneni HK, Park H and Lee J (2020) Cross-Metathesis of Methallyl Halides: Concise Enantioselective Formal Total Synthesis of (–)-Presphaerene. Front. Chem. 8:494. doi: 10.3389/fchem.2020.00494
Received: 06 April 2020; Accepted: 14 May 2020;
Published: 30 June 2020.
Edited by:
Jiyong Hong, Duke University, United StatesReviewed by:
Xiao-Wei Liang, Duke University, United StatesCopyright © 2020 Mandava, Koo, Hwang, Nallapaneni, Park and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jongkook Lee, amtsQGthbmd3b24uYWMua3I=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.