 Vidhya Chakrapani
Vidhya Chakrapani- 1Howard P. Isermann Department of Chemical and Biological Engineering, Rensselaer Polytechnic Institute, Troy, NY, United States
- 2Department of Physics, Applied Physics, and Astronomy, Rensselaer Polytechnic Institute, Troy, NY, United States
Electrocatalytic reactions such as oxygen evolution (OER) and oxygen reduction reactions (ORR) are one of the most complex heterogeneous charge transfer processes because of the involvement of multiple proton-coupled-electron transfer steps over a narrow potential range and the formation/breaking of oxygen-oxygen bonds. Obtaining a clear mechanistic picture of these reactions on some highly active strongly-correlated oxides such as MnOx, NiOx, and IrOx has been challenging due to the inherent limitations of the common spectroscopic tools used for probing the reactive intermediates and active sites. This perspective article briefly summarizes some of the key challenges encountered in such probes and describes some of unique advantages of confocal near-infrared photoluminescence (NIR-PL) technique for probing surface and bulk metal cation states under in-situ and ex-situ electrochemical polarization studies. Use of this technique opens up a new avenue for studying changes in the electronic structure of metal oxides occurring as a result of perturbation of defect equilibria, which is crucial in a broad range of heterogeneous systems such as catalysis, photocatalysis, mineral redox chemistry, and batteries.
Introduction
Electrocatalytic oxygen evolution and oxygen reduction reactions underpin the efficient operation of many reversible electrochemical energy conversion and storage devices such as solar cells, regenerative fuel cells (Adler, 2004; Gasteiger et al., 2005), and rechargeable metal-air batteries (Cao et al., 2012) that have theoretical energy densities (10–12 kWh/kg) on par with the energy density of gasoline (~13 kWh/kg) (Yang et al., 2011). Consequently, considerable efforts have been dedicated toward improving the energy conversion efficiency of these devices (Badwal et al., 2014; Yoo et al., 2014). However, the main obstacle for achieving this has been the low activity of electrocatalysts employed for the ORR and OER reactions that occur during charge/discharge cycles (Chen et al., 2002). Oxygen reactions are known to be complex charge transfer processes because they involve multiple proton-coupled electron transfer. Electrocatalysts that can efficiently catalyze these reactions are either limited or unstable, which leads to energy losses and decreased lifetime of systems employing them. Some of the best known electrocatalysts for oxygen reactions are Pt, RuO2, and IrO2, but their high cost and low availability is an issue (Horkans and Shafer, 1977; Maiyalagan et al., 2014; Hosseini-Benhangi et al., 2015).
Currently, there is a great interest in the study of Mn-based electrocatalysts for OER/ORR processes. The multi-electron OER process that occurs in nature as a result of photosynthetic water oxidation by green plants, algae, and cyanobacteria has been shown to be catalyzed by a Mn4CaO5 cluster located inside the membrane protein complex called photosystem II (PS-II) (Cox et al., 2014) at a record low overpotential of 160–300 mV at pH = 5.5 with an incident-light-to-fuel conversion efficiency of 16% and turn over frequency (TOF) of 1,000 s−1 (Dau and Zaharieva, 2009; Dau et al., 2010). For comparison, the best benchmarked IrOx and RuO2-based OER catalysts have overpotentials between 300 and 400 mV depending on the pH conditions with a TOF of 0.1–1 s−1 (Hong et al., 2015). One reason for the low TOF of the artificial catalysts compared to the TOF of Mn4CaO5 of PS-II has been postulated to be related to the efficiency of the charge accumulation process, i.e., the ease of formation of higher-valent metal cation states that can stabilize the higher-order oxygen intermediates. Among the first-row transition metals, element Mn has the highest accessible stable valency, which is +7, and also has the largest range of stable oxidation states (Mn2+ to Mn7+). This ability of Mn to support at least five charges is likely why nature chose Mn as the preferred element for the OER process, which requires accumulation of four charges to convert two water molecules to molecular oxygen. IrOx, one of the most active OER catalysts in both acidic and basic electrolytes, is also believed to oxidize H2O through a high-valent Ir5+ intermediate, although it has not yet been conclusively proved. Thus, a knowledge of the nature and charge state of reactive intermediates can help better understand the differences between synthetic and natural electrocatalysts and in turn aid in their better design.
Understanding the mechanism of electrocatalytic reactions, such as water oxidation, requires the knowledge of both the type of reactive intermediates as well as the nature of active sites that enable adsorptive binding. This crucially depends on our ability to characterize the electrocatalyst in their working state. However, no single experimental probe can entirely capture the complexity of such processes, which involves multiple one-electron steps. Instead, the information from multiple probes have to be correlated in order to obtain a global picture.
Experimental techniques to measure metal oxidation states are limited. Common techniques include X-ray absorption (XAS) and photoemission (XPS) spectroscopy (Adarsh et al., 2019). While being extremely valuable for providing structural information, they provide information on the average oxidation state only when used for studying strongly-correlated oxides such as NiOx, and MnOx, while it is known that the working catalyst under polarization is multi-valent (Silvester et al., 1997; Iuzzolino et al., 1998). Similarly, electron paramagnetic resonance (EPR), another powerful technique, only probes paramagnetic metal oxidation states (Iuzzolino et al., 1998; McAlpin et al., 2010). This limitation in probing the dynamic range of the accessible metal valence states especially near the potential of OER/ORR has been the bottleneck in the precise determination of the electronic state of the catalytically active site and its correlation to the electrochemical activity both in PS-II as well as in other synthetic systems.
This perspective article briefly summarizes the benefits of some of the useful spectroscopic tools available for probing valence states, active sites, and reactive intermediates of OER/ORR as well as the limitations of these techniques. In particular, this article elucidates the salient benefits of confocal near-infrared photoluminescence spectroscopy as a relatively novel probe for the use in ex-situ and in-situ electrocatalytic studies and summarizes some of our recent results.
Background
Mechanism of OER/ORR-Nature of Reaction Intermediates
It has now been well-established that OER and ORR on metal oxides proceed via formation of a series of reaction intermediates that involves the binding of the electrolyte ions (typically OH−, H+) and H2O molecules to the metal and oxygen () lattice sites that serve as the catalytic active center for the reaction. For instance, in alkaline environments, OER can be written as
The reverse of this reaction is ORR. The overall reaction, as given by Reaction 1, is generally shown to occur as a series of one-electron intermediates (Bockris and Otagawa, 1983; Goodenough et al., 1990; Dau et al., 2010; Man et al., 2011; McCrory et al., 2013; Mavros et al., 2014; Hong et al., 2015). When the intermediates involve the sequential formation of *OH, *O, *OOH, and *OO (Figure 1A) at the metal cation site (*, active site), which undergoes an increase in the oxidation state, the mechanism is commonly known as the adsorbate evolution mechanism (AEM). This is the preferred mechanism for stable catalysis and generally results in moderate activity (Goodenough et al., 1990; Man et al., 2011; Chen et al., 2017). Experimental studies on active binary oxide catalysts such as IrOx, MnOx, and NiOx have shown that these catalysts show an increase in the average oxidation state (>3) at potentials before OER, which is considered to be the key step for efficient AEM-OER catalysis, while a decrease in the oxidation state (<3) during ORR (Kötz et al., 1983; Augustynski et al., 1984; Kötz and Neff, 1985; Nahor et al., 1991; Mo et al., 2002; Gorlin et al., 2013; Risch et al., 2017). In contrast, if the OER process involves the participation (abstraction) of lattice oxygen atoms that occurs in more covalent oxides [RuO2 (Stoerzinger et al., 2017), many perovskites], which was first proposed by Matsumoto et al. (1977, 1980) and Matsumoto and Sato (1986) then the mechanism is referred to as lattice oxygen-mechanism (LOM) (Matsumoto and Sato, 1986; Mefford et al., 2016). In this reaction scheme (Figure 1B), the oxidation state of the lattice oxygen atoms changes while the metal redox state remains nearly constant. Therefore, identification of the cation oxidation state can provide an insight into the nature of OER/ORR process. Although many different routes can exist (Bockris and Otagawa, 1983; Man et al., 2011; Mavros et al., 2014), as illustrated neatly in recent review articles (Dau et al., 2010; McCrory et al., 2013; Hong et al., 2015) one of the more commonly evoked reaction schemes in basic electrolytes is shown in Figures 1A,B. The key parameter that determines the type of OER process is the hydroxide affinity of the semiconductor/electrolyte interface. Many highly covalent oxides that have a large work function (Hong et al., 2017) exhibit negative hydroxide affinity due to the presence of an e− accumulation at the surface in aqueous electrolyte, which repels the negative OH− from the solid/electrolyte interface. This OH− deficiency at the interface results in insufficient ionic compensation in the oxide during polarization. As a result, the localization of holes at the lattice oxygen site leads to the formation of O− intermediate and a subsequent generation of oxygen vacancy (VO) during OER catalysis (LOM pathway) (Rong et al., 2016; Grimaud et al., 2017). Many in-depth studies on perovskites by Bockris and Otagawa (1983, 1984, 2002), Matsumoto et al. (1980), and Matsumoto and Sato (1986) have pointed out the instability of such perovskites. Consequently, these oxides, while being highly efficient at OER (low Tafel slopes), are unstable in bulk and result in cation leaching and surface amorphization (Lee et al., 2011; May et al., 2012; Chang et al., 2014; Forslund et al., 2018). In contrast, stable AEM materials show positive hydroxide affinity that promotes insertion/adsorption of OH− lattice during OER and therefore supports the subsequent formation of higher-order O intermediates (=O, OOH, OO).
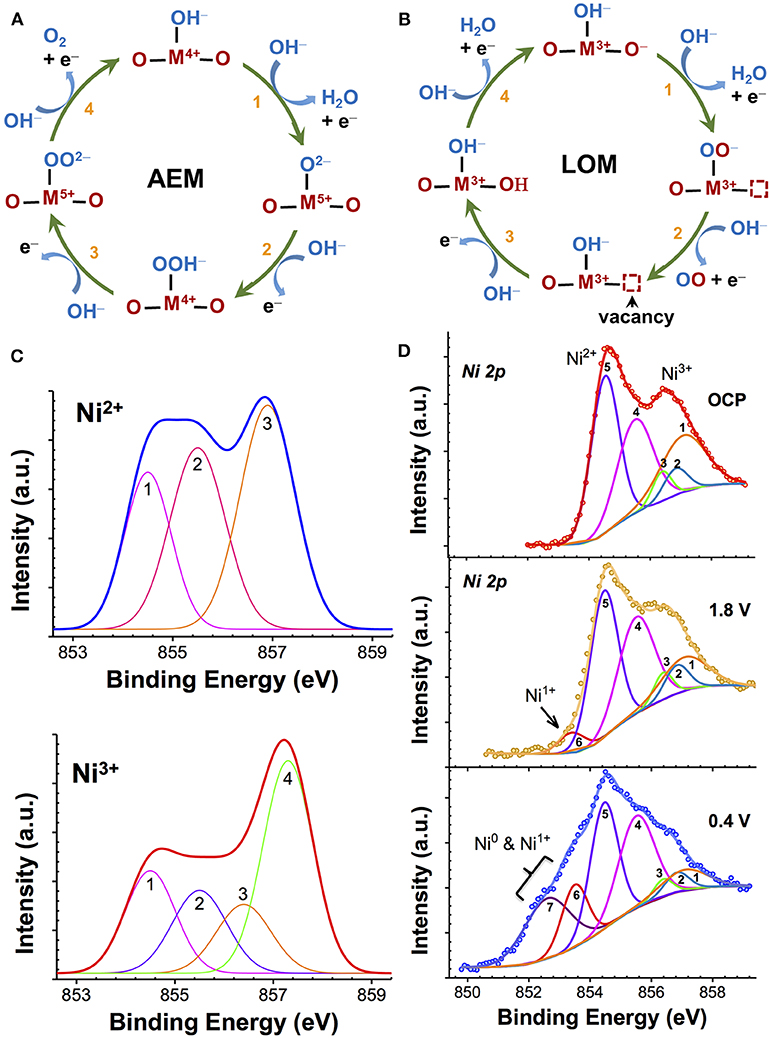
Figure 1. (A, B) Two types of OER mechanisms seen in transition metal oxides. In adsorbate evolution mechanism (AEM), metal cation site (M) is the active site, and undergoes an increase in the oxidation state (A), while in lattice-oxygen mechanism (LOM) a lattice oxygen site is the active site that undergoes redox changes with the resulting formation of a vacancy defect (B); (C) Theoretically-calculated multiplet envelopes of Gupta and Sen (1974, 1975) and McIntyre and coworkers (Grosvenor et al., 2006) for free Ni2+ and Ni3+ ions; and (D) XPS spectra of NiOx showing Ni 2p3/2 core level before (OCP) and after Li+ insertion at potential of 1.8 V and 0.4 V vs. Li/Li+ fitted with Ni2+ and Ni3+ multiplet envelopes, as reported in Wang et al. (2016b).
In addition, pH has a strong effect on the nature of adsorbate intermediates as well as on the charge on the metal cation. The nature of adsorbate intermediates depends strongly on the pH of the electrolyte because the surface charge caused by protonation/deprotonation of surface oxygen-functional species depends on the pH of zero charge (pzc) of the oxide surface relative to the solution pH (Noh and Schwarz, 1989; Pechenyuk, 1999). At pH values higher than the pzc, negatively-charged species accumulate easily on the surface (OH−, OOH−, O2−). Therefore, a lower overpotential is expected for OER in basic solutions than those at acidic/neutral pH (Goodenough et al., 1990; Goodenough and Cushing, 2003; Takashima et al., 2012b) pH can also affect the stability of the metal cation active sites and may lead to their charge disproportionation. pH-dependent studies on δ-MnO2 have shown that the Mn3+ defect site, which is the proposed active site for OER, is unstable at pH <9 and undergoes disproportionation to Mn2+ and Mn4+ defects, whereas Mn3+ is effectively stabilized by the comproportionation of Mn2+ and Mn4+ at higher pH conditions (Takashima et al., 2012a,b).
All-in-all, probing the changes in the anion and cation oxidation states at various potentials preceding OER/ORR steps is necessary for identifying the nature of the active site, type of reactive intermediates, and stability of metal cations, which in turn would help in understanding the overall mechanism of the complex OER/ORR processes.
Common Techniques for Probing Active Sites/Intermediates and Their Limitations
Manganese can exist in oxidation states from −3 to +7, and as a result form many strongly-correlated phases of manganese oxides (MnOx) of more than 30 different polymorphs (Post, 1999; Meng et al., 2014). This makes Mn one of the most complex elements to study. Common techniques for determining the oxidation state of the catalysts are XPS, XAS, EPR, and absorption (UV-Vis) measurements.
X-ray Photoemission Spectroscopy (XPS)
XPS is one of the most common and powerful techniques available today for obtaining both electronic and compositional information of electrocatalysts. Earliest studies employed surface-sensitive XPS on “emersed” electrodes that were polarized ex-situ at different potentials. Pioneering studies of Kolb, Kötz, Neff, and Ross on emersed electrodes have shown that the electrical double layer of the polarized electrode is preserved upon their emersion and the subsequent transfer into an ultrahigh vacuum (UHV) chamber, and therefore can be used for detailed surface-structural studies through X-ray and ultraviolet photoemission studies (Hansen et al., 1980; Rath and Kolb, 1981; Kolb et al., 1983; Kötz et al., 1983, 1986; Wagner and Ross, 1983; Kötz and Neff, 1985). Electrochemical studies on IrOx show that the oxidation state of the metal cation increases with anodic polarization preceding the OER step (Goodenough et al., 1990). On the other hand, results of XPS studies of Kötz et al. on emersed IrOx have not been conclusive. While an increase in the intensity of Ir 4f on higher BE was observed, the authors concluded that a definite correlation of the Ir 4f binding energy (BE) with an Ir oxidation state cannot be made from measured data (Kotz et al., 1984; Kötz and Neff, 1985). From the analysis of the O 1s signal at various anodic potentials, they showed that the contribution of hydroxide species to the overall O 1s signal decreases with anodic polarization.
In the recent decade, advancements in ambient-pressure XPS (APXPS) have enabled in-operando studies of the electrical double layer (Favaro et al., 2016) and have led to the identification of some of the keys intermediates of water oxidation (Yamamoto et al., 2008; Sanchez Casalongue et al., 2014). Further, the use of different X-ray energies has allowed for surface and bulk contributions to be differentiated through variation in the probing depth. In-situ studies of Nilsson and coworkers (Sanchez Casalongue et al., 2014) on IrOx show that the intensity of Ir 4f line increases at higher binding energy (BE) upon polarization at OER potentials along with a decrease in surface hydroxide concentration, which is similar to the results of Kotz et al. (1984) on emersed IrOx electrode. The increase in the high BE signal was attributed to the formation of Ir5+ at the surface and was taken to be evidence for OOH–mediated OER mechanism. In contrast, the comparative XPS studies of amorphous and crystalline IrOx by Pfeifer et al. (2016) indicate that the increased activity of amorphous oxide is due to the presence of both anionic (O−) and cationic (Ir3+) defects in the lattice, which casts doubt on the OOH-mediated OER mechanism in active IrOx.
Limitations when probing correlated-oxides
The determination of the relative contribution of different metal oxidation states (such as Mn2+, Mn3+, and Mn4+) through XPS in strongly-correlated oxides such as MnOx, IrOx, or NiOx is highly complex and challenging because of the overlapping cationic signals (Oswald and Brückner, 2004; Grosvenor et al., 2006; Biesinger et al., 2009). The photoionization process resulting from the X-ray absorption results in the creation of vacancies (holes) in the inner core levels. The strong spin-orbit and electrostatic interactions between the vacancies and the unpaired valence-shell (d) electrons of high spin cation in strongly-correlated systems such as Mn, Fe, and Ni, causes a severe broadening of energy of the photoelectrons (Pal and Gupta, 1982). In a multi-valent sample, the overlapping of broadened peaks of various oxidation state cations makes the assignment of oxidation state to a single identifiable peak binding energy (BE) very difficult. This is demonstrated in Figures 1C,D for NiOx. In such cases, the individual contribution of different metal cations is generally resolved by fitting the main emission peak using theoretically-calculated multiplet envelopes of free ions, such as that predicted by Gupta and Sen (1974, 1975) and successfully applied by Grosvenor et al. (2004, 2006) and elaborated further by Nesbitt and Banerjee (1998) for various MnOx and by us (Wang et al., 2016b) for NiOx. For instance, the multiplet envelope of free Ni2+ and Ni3+ ions, as shown in Figure 1C, are used for XPS fitting (Figure 1D) of a multi-valent NiOx containing Ni3+, Ni2+, Ni+, and Ni0 that was obtained with Li+ ion insertion into stoichiometric NiOx (Wang et al., 2016b). Such a method is highly prone to error when changes in the relative signal intensity of different metal cations with electrochemical polarization under OER must be measured.
In addition to the uncertainty in the assignment of oxidation state to a single defined BE position, XPS studies by Pfeifer et al. (2016) on highly active amorphous IrOx electrocatalyst show that the presence of lower-valent Ir3+ species in predominately Ir4+ cationic framework lead to a reverse BE shift, i.e., it causes an increase in signal intensity on the higher BE side of the Ir 4f line as opposed to the expected lower BE side. These results highlight the need for caution when assigning oxidation states solely based on BE shifts in XPS. Such reverse BE shifts have also been reported in oxides such as AgOx, which has been attributed to the screening of the core-hole by metal s states (Gaarenstroom and Winograd, 1977; Kaspar et al., 2010; Grönbeck et al., 2012).
X-ray Absorption Spectroscopy (XAS) measurements
Oxidation state changes of the catalysts have also been monitored through relatively more bulk-sensitive techniques such as X-ray absorption near-edge (XANES) and extended X-ray absorption fine structure (EXAFS). XAS is an atom-specific probe for both the local geometry of the metal cation as well as its valence state. It can provide important information not only on the valence states but also the interatomic distance, the coordination number and the spread of distances in the first coordination shell. In addition, XAS, unlike XPS measurements, can probe the electrocatalyst under in-operando working conditions in the presence of electrolyte under various polarizations. XAS being a local probe is also advantageous over the X-ray diffraction technique, which cannot resolve crystallites smaller than 5 nm due to the broadening of Bragg peaks. However, XAS is capable of resolving both bulk and nanocrystalline (or amorphous) phases.
In-situ XAS measurements on IrOx electrocatalysts have shown that the average Ir valence increases from 3 at OCP up to 4.8 at the potentials of OER (Hüppauff and Lengeler, 1993; Mo et al., 2002); the non-integer value of valence indicates that the electrocatalyst likely contain mixtures of Ir3+, Ir4+, and Ir5+ states. Similarly, several in-depth characterization studies of various MnOx phases under electrochemical polarizations through XAS measurements have shown that the average oxidation state of Mn increases during OER and decreases to during ORR, as seen from the shifts of the absorption edge to higher or lower energy (Silvester et al., 1997; Gorlin et al., 2013; Risch et al., 2017). Like in XPS, interactions between core holes and valence electrons also results in the broadening of Mn K edge lines in the XAS measurements, which result in the loss of sensitivity and makes resolution of individual chemical states in mixed valent oxide difficult (Manceau et al., 1992). Another disadvantage of XAS is that the technique relies on the calibration of a catalyst spectrum with a spectrum of a reference oxide sample, which in itself may be of mixed valence, as most pure MnOx phases are seldom perfectly stoichiometric. As a result, the nature of active sites for OER on such sites has not been identified with certainty.
Near-Infrared Photoluminescence Spectroscopy
In contrast to XAS and XPS measurements, photoexcitation during PL measurements for different metal cations can be done using a relatively low energy laser and therefore only involves excitation of valence electrons to the higher energy state without core-level excitations. Hence, peak-broadening that limits precise identification of cationic states is not an issue in PL.
PL spectroscopy relies on the detection of the photons emitted from a material when light of sufficient energy is incident upon it. The absorption of the incident photon results in the excitation of electrons from the ground states to higher energy states and their subsequent relaxation back to the ground state by the release of excess energy. When the excess energy is released in the form of photons (radiative transitions), then the process is called photoluminescence. On the other hand, a part of this excess energy from electron transitions to lower energy states also occurs through the release heat or phonons, which is a non-radiative process. Therefore, the intensity of PL spectrum (or quantum efficiency) gives a measure of the relative rates of radiative to non-radiative recombination process. In general, the PL spectrum provides a wealth of information about the electronic structure of the material. The energy of the peak positions denotes the energy difference between the electronic states between which the electron-hole recombination occurs, and therefore can provide information regarding the electronic bands, doping, bulk, and interface trap levels (Gfroerer, 2006). It is a selective and extremely sensitive probe of discrete electronic states. In addition, it is a non-destructive technique that requires very little sample preparation and most importantly, does not require any UHV environment. It can be used to study both electrically conducting and non-conducting phases.
The main drawback of the PL technique is that it can only detect radiative emissions. Therefore, information about non-radiative transitions such as that originating from traps or recombination centers must be obtained indirectly. As a result, materials such as amorphous semiconductors, low-crystal quality films, or indirect band gap semiconductors that have low quantum efficiency due to high e−-h+ recombination occurring via non-radiative pathways are difficult to study through commonly used PL instruments. Furthermore, while the intensity of a PL peak is proportional to the density of the radiative states, unlike absorption measurements, the determination of their absolute density (concentration) is not straightforward.
The spectral range of radiative emissions seen in transition metal oxides (TMOs) is shown in Figure 2A (Sherman, 1984). Many TMOs are strongly ionic, and hence have band-to-band or excitonic emissions that lie in the ultraviolet (UV) region. Electronic transitions between ligand (L) to metal charge transfer (MCT) and metal to conduction band (CB) transitions frequently occur in the UV spectral range. The d-d transitions are usually the visible region, and defect-related emissions, both cationic and anionic, in the visible to NIR spectral region. In NiOx, the Ni+1, Ni+2, and Ni+3 have characteristic emission in the 0.7–0.9 eV, while in MnOx, the characteristic emission of various Mn cationic defects (Mn2+-Mn6+) occur in the 0.9–1.9 eV spectral range (Mićić and Draškovi, 1985; Sekiguchi and Adachi, 2015; Silva et al., 2016; Ren and Yang, 2018). Thus, monitoring the changes in emission in the NIR range can provide direct information regarding changes in the oxidation state of the metal cation that occur as a result of electrochemical polarization.
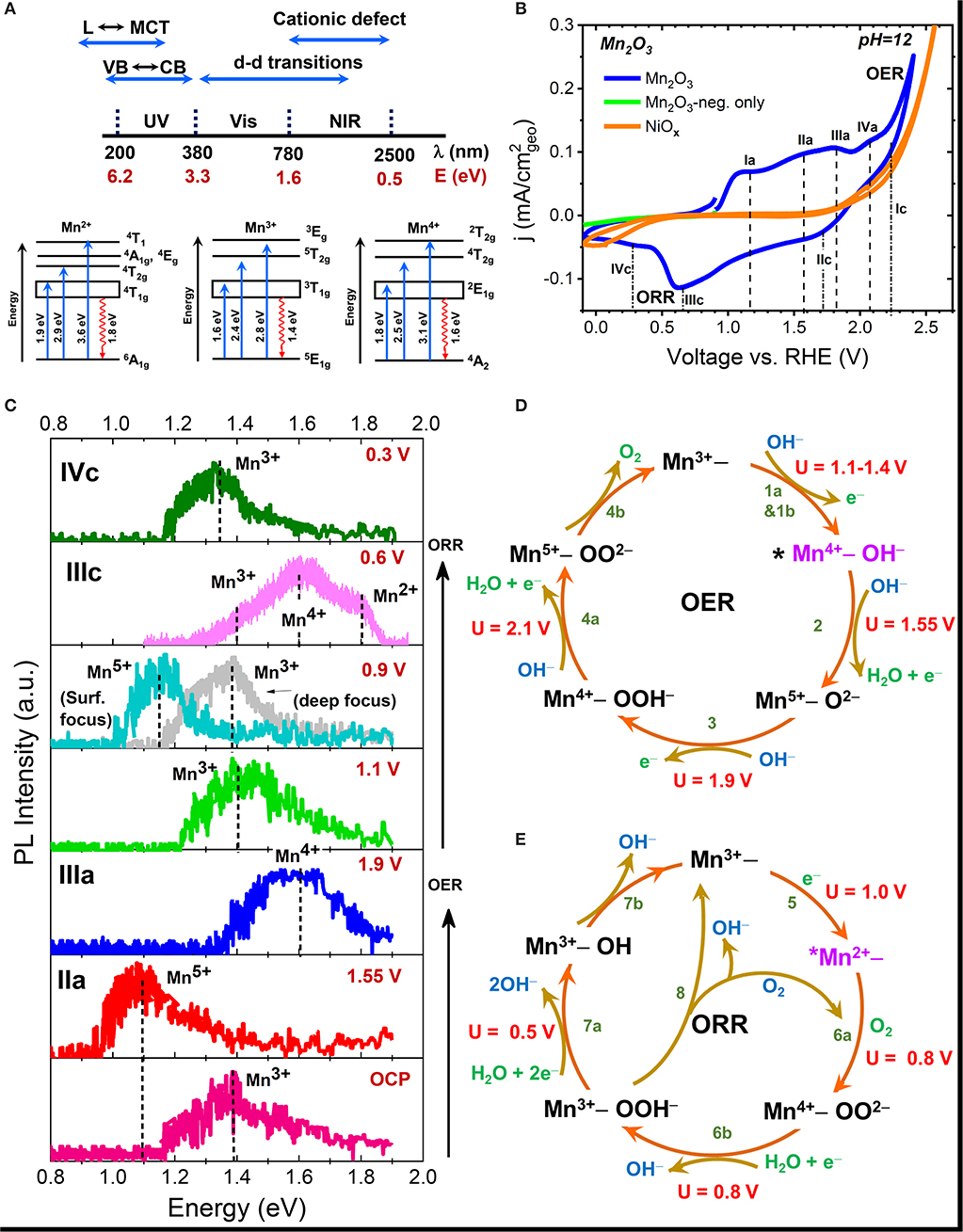
Figure 2. (A) Schematic showing the spectral energy range of various radiative transitions seen in metal oxides along with the various electronic transitions of Mn2+, Mn3+, and Mn4+ cations; (B) Cyclic voltammograms of Mn2O3 and NiOx electrodes obtained in pH = 12 aqueous KOH electrolyte; (C) Ex-situ NIR PL spectra of emersed Mn2O3 electrode after polarization in pH = 12 electrolyte at various peak potential seen in CV. The observed surface shifts in the emission peak reflects the changes in the oxidation state of Mn atom during polarization; and (D,E) Schematic of the mechanistic pathway of the OER (D) and ORR (E) processes on Mn2O3 showing the potential (U)-dependent formation of various intermediates. The * in the figure represents the active site, which in Mn2O3 was deduced to be Mn4+ defect site for OER and Mn2+ site for ORR. Data taken from Roy et al. (2020).
Ultraviolet to visible range PL technique has been successfully used to study high-temperature heterogeneous catalysts. Seminal work in the field was done by Anpo et al., who used visible PL for catalytic studies on metal oxides, and showed the applicability of this technique for obtaining real-time information about various surface chemical phenomena, such as changes in the structure, coordination, and reactivity of metal centers that occur as a result of adsorption of species during catalysis (Anpo et al., 1985, 1988; Anpo and Che, 1999; Matsuoka et al., 2008).
We recently reported through studies (Puntambekar et al., 2016; Wang et al., 2016a,b) on several different oxides and chalcogenides the first use of NIR-PL as a technique for studying various cationic defects under in-operando and ex-situ electrochemical conditions. PL/Raman spectroscopy is a fairly routine technique that is more easily accessible than the synchrotron X-ray facility. Other advantages are that it can be highly surface-sensitive, it is capable of detecting radiative emission from defects at concentrations as low as 1018 cm−3, and with certain precautions can be easily adapted for in-situ catalytic studies that may involve the presence of a metal catalyst (<5 wt%). Without requiring any setup change, the instrument can also obtain Raman spectra. Vibration spectroscopy such as Raman scattering is one of the most sensitive techniques available for structural characterization because of its ability to probe amorphous phases and those with short-range order, such as the highly disordered phases of MnOx. The technique can be directly used to probe the near-neighbor environment of the oxygen coordination around Mn and alkali cation (Julien et al., 2003). Thus, the combined NIR-PL/Raman measurement is uniquely suited to obtain simultaneous electronic and structural information of the catalyst. The spatial resolution of PL/Raman system is usually in micrometers, and therefore cannot provide information on spatial distribution of defects at the nanoscale.
Distinguishing Surface vs. Bulk States Through Confocal Microscopy
The penetration depth of the excitation light, which is a measure of thickness of material that is probed, in any material depends strongly on the energy of the incident light and is given by the inverse of the wavelength-dependent absorption coefficient. The surface sensitivity of the PL technique depends both on the wavelength of light used as well as on the electronic structure of the material. When direct band gap semiconductors are probed using UV or an above-band gap energy excitation source, the penetration depth is small, spanning tens to hundreds of nanometers. While the diffusion of photoexcited charge carrier would tend to increase the probe depth, the PL spectrum in such a case is quite sensitive to surface effects and recombination. In contrast, penetration depth can span several tens of microns when absorption is weaker as in the case of indirect band gap semiconductors or when excitation is done using a sub-band gap energy light source. Here, PL signal is dominated by bulk recombination processes. Therefore, spectra obtained using multiple excitation wavelengths can help distinguish surface vs. bulk states. Another strategy is to utilize a confocal PL microscope with a three-dimensional (3D) optical resolution capability. Confocal microscopes work on the principle of point excitation in the specimen (diffraction limited spot) and point detection of the resulting fluorescence signal. In most confocal setups, 3D resolution is achieved through the use of a pinhole placed in front of the detector such that signal originating from an in-focus plane passes freely through the pinhole and is imaged by the detector, whereas light coming from out-of-focus planes is physically blocked from passing through the pinhole and reaching the detector. Raster scanning the specimen one point at a time permits thin optical sections to be collected by simply changing the z-focus. The resulting images can be stacked to produce a 3D fluorescence image of the catalysts. In this way, one can obtain depth-dependent changes in the PL spectrum that can be used to distinguish bulk vs. surface intermediates without sample destruction or movement.
Ex-situ Electrocatalytic Studies of OER/ORR on Manganese Oxides
In our recent work (Roy et al., 2020), we demonstrated the successful application of ex-situ NIR-PL technique to track the OER and ORR intermediates and identify the nature of catalytic sites on the most active MnOx phase, namely Mn2O3. Mn2O3 is one of the very few bifunctional electrocatalyst capable of catalyzing both OER and ORR processes. Such catalysts are of tremendous importance in renewable electrochemical devices such as metal-air batteries, regenerative fuel cells, and electrolyzers.
Successful application of NIR-PL for the detection of reaction intermediates on a wide variety of electrocatalysts requires development of a database of characteristic emission energies of different metal cations. For the case of Mn cation, reference spectra obtained with mineral samples (or binary oxides) of known composition gives the characteristic peak emissions of various oxidation states: Mn2+ at 1.8 eV, Mn3+ at 1.35 eV, Mn4+ at 1.6 eV, and Mn5+ at 1.1 eV, and are consistent with the values reported in literature (Sherman, 1984; Hozoi, 2003; Xiao et al., 2017; Cao et al., 2018). Readers can refer to the supporting information of (Roy et al., 2020) for further details. Figure 2A shows the schematic of electronic transitions giving rise to the characteristic absorption and emission signals (Hozoi, 2003) of various Mn cations. Figure 2B shows the cyclic voltammogram of a Mn2O3 electrode in Ar-purged electrolyte along with the labels for all the various potentials of peak currents. Oxygen evolution was observed at potentials more positive than 1.9 V vs. reversible hydrogen electrode (RHE). The reduction of oxygen evolved during the anodic scan can be seen at potentials more negative than 0.7 V. Very little current is observed when the electrode is polarized in the cathodic direction alone. Figure 2C shows the results of ex-situ NIR-PL of the same electrode after polarization at various peak potentials during a linear potential scan from OCP to OER potential and then from OER potential to ORR potential in pH = 12 KOH electrolyte. All spectra were recorded using a 633 nm laser light focused on the sample surface through a confocal microscope at very low laser power to avoid spot damage due to localized laser heating. PL results confirm that the peaks seen in the voltammograms during the anodic scan correspond to the formation of Mn4+, Mn5+ oxidation states while a decrease in the oxidation state to Mn3+ and Mn2+ is observed at cathodic ORR potentials. More detailed studies in combination with gravimetric and in-situ absorption measurements revealed Mn2+ as the dominant active site for the ORR process. Measurements of spectra under different z-focus (Figure 2C) revealed that the observed formation of different Mn valence states is restricted to the surface in Mn2O3.
While our results are generally in agreement with the trend seen in XAS measurements (Risch et al., 2017), we also distinctly observed co-existing valence states together with the observation of Mn5+ as an intermediate of OER during electrocatalysis, as predicted by theoretical calculations (Fernando et al., 2016). The observation of potential-dependent formation of various metal charged states enabled us to map the mechanistic pathways of both OER and ORR processes (Figures 2D,E), including the identification of the active sites (Roy et al., 2020). This is shown schematically in Figure 2D for the OER process on the Mn4+ active site, and in Figure 2E for the ORR process on the Mn2+ active site. The knowledge was used to develop a band structure framework that correlates electrochemical activity with the formation energy of various metal cation intermediates. Nakamura et al. attributed one reason for the lower electrocatalytic OER efficiency of MnO2 and Fe2O3 catalysts compared to IrOx to the charge disproportionation of high-valent states into lower-valent cations in the former oxides during OER, in contrast to the stabilization of higher-valent (Ir5+) state in the latter oxide (Ooka et al., 2017). Our results show for the first time that Mn2O3, which is the most active OER/ORR catalyst within the MnOx system, also catalyzes OER through a high-valent Mn5+ intermediate. However, comparison of electrocatalytic activity rates indicate that Mn2O3 is still less active than IrOx. In this regard, time-dependent PL studies on the relative formation of higher valent states can shed light on the kinetics of high-valent intermediate formation and is currently underway in our group.
In summary, one bottleneck for identifying the key intermediate metal cation states using X-ray based absorption and photoemission techniques is the broadening and overlapping of the individual peaks of various metal cations due to the interactions of core hole and d electrons that makes it challenging to resolve the individual valence states in the multivalent electrocatalyst of strongly-correlated oxides. It is shown here that confocal NIR-PL spectroscopy is uniquely suited to study multi-valent states of correlated oxides under in-situ and ex-situ electrochemical conditions. In addition, the technique can also resolve bulk vs. surface active sites with three-dimensional optical resolution through a confocal microscope setup. When coupled with Raman technique, it can be a powerful tool for obtaining simultaneous structural and electronic changes occurring in the electrocatalyst in the working state.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary files.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by National Science Foundation.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Rensselaer Polytechnic Institute (RPI) and National Science Foundation, DMR award (No: 1709649) for providing the financial support for this work.
References
Adarsh, K. S., Chandrasekaran, N., and Chakrapani, V. (2019). In-situ spectroscopic techniques as critical evaluation tools for electrochemical carbon dioxide reduction: a mini review. Front. Chem. 8:137. doi: 10.3389/fchem.2020.00137
Adler, S. B. (2004). Factors governing oxygen reduction in solid oxide fuel cell cathodes. Chem. Rev. 104, 4791–4844. doi: 10.1021/cr020724o
Anpo, M., Aikawa, N., Kubokawa, Y., Che, M., Louis, C., and Giamello, E. (1985). Photoluminescence and photocatalytic activity of highly dispersed titanium oxide anchored onto porous vycor glass. J. Phys. Chem. 89, 5017–5021. doi: 10.1021/j100269a025
Anpo, M., and Che, M. (1999). Applications of photoluminescence techniques to the characterization of solid surfaces in relation to adsorption, catalysis, and photocatalysis. Adv. Catal. 44, 119–257. doi: 10.1016/S0360-0564(08)60513-1
Anpo, M., Kondo, M., Che, M., and Louis, C. (1988). Application of dynamic photoluminescence spectroscopy to catalysis -differences between anchored and impregnated Mo/SiO2 catalysts. J. Lumin. 40, 829–830. doi: 10.1016/0022-2313(88)90457-7
Augustynski, J., Koudelka, M., Sanchez, J., and Conway, B. (1984). ESCA study of the state of iridium and oxygen in electrochemically and thermally formed iridium oxide films. J. Electroanal. Chem. Inter. Electrochem. 160, 233–248.
Badwal, S. P., Giddey, S. S., Munnings, C., Bhatt, A. I., and Hollenkamp, A. F. (2014). Emerging electrochemical energy conversion and storage technologies. Front. Chem. 2:79. doi: 10.3389/fchem.2014.00079
Biesinger, M. C., Payne, B. P., Lau, L. W. M., Gerson, A., and Smart, R. S. C. (2009). X-ray Photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 41, 324–332. doi: 10.1002/sia.3026
Bockris, J. O., and Otagawa, T. (1983). Mechanism of oxygen evolution on perovskites. J. Phys. Chem. 87, 2960–2971. doi: 10.1021/j100238a048
Bockris, J. O. M., and Otagawa, T. (1984). The electrocatalysis of oxygen evolution on perovskites. J. Electrochem. Soc. 131, 290–302.
Bockris, J. O. M., and Otagawa, T. (2002). Mechanism of oxygen evolution on perovskites. J. Phys. Chem. 87, 2960–2971
Cao, R., Cheng, X., Zhang, F., Su, L., Chen, T., Ao, H., et al. (2018). Enhanced luminescence properties of MgTO3:Mn4+ red-emitting phosphor by adding Ge4+ ion and H3BO3. J. Mater. Sci. 29, 13005–13010. doi: 10.1007/s10854-018-9421-5
Cao, R., Lee, J.-S., Liu, M., and Cho, J. (2012). Recent progress in non-precious catalysts for metal-air batteries. Adv. Energy Mater. 2, 816–829. doi: 10.1002/aenm.201200013
Chang, S. H., Danilovic, N., Chang, K.-C., Subbaraman, R., Paulikas, A. P., Fong, D. D., et al. (2014). Functional links between stability and reactivity of strontium ruthenate single crystals during oxygen evolution. Nat. Commun. 5:4191. doi: 10.1038/ncomms5191
Chen, G., Bare, S. R., and Mallouk, T. E. (2002). Development of supported bifunctional electrocatalysts for unitized regenerative fuel cells. J. Electrochem. Soc. 149, A1092–A1099. doi: 10.1149/1.1491237
Chen, X., Aschaffenburg, D., and Cuk, T. (2017). One-electron intermediates of water oxidation & the role of solvation in their stability. J. Mater. Chem. A 5, 11410–11417. doi: 10.1039/C7TA02240A
Cox, N., Retegan, M., Neese, F., Pantazis, D. A., Boussac, A., and Lubitz, W. (2014). Electronic structure of the oxygen-evolving complex in photosystem II prior to OO bond formation. Science 345, 804–808. doi: 10.1126/science.1254910
Dau, H., Limberg, C., Reier, T., Risch, M., Roggan, S., and Strasser, P. (2010). The mechanism of water oxidation: from electrolysis via homogeneous to biological catalysis. Chem. Cat. Chem. 2, 724–761. doi: 10.1002/cctc.201000126
Dau, H., and Zaharieva, I. (2009). Principles, efficiency, and blueprint character of solar-energy conversion in photosynthetic water oxidation. Acc. Chem. Res. 42, 1861–1870. doi: 10.1021/ar900225y
Favaro, M., Jeong, B., Ross, P. N., Yano, J., Hussain, Z., Liu, Z., et al. (2016). Unravelling the electrochemical double layer by direct probing of the solid/liquid interface. Nat. Commun. 7:12695. doi: 10.1038/ncomms12695
Fernando, A., Haddock, T., and Aikens, C. M. (2016). Theoretical investigation of water oxidation on fully saturated Mn2O3 and Mn2O4 complexes. J. Phys. Chem. A 120, 2480–2492. doi: 10.1021/acs.jpca.6b02280
Forslund, R. P., Hardin, W. G., Rong, X., Abakumov, A. M., Filimonov, D., Alexander, C. T., et al. (2018). Exceptional electrocatalytic oxygen evolution via tunable charge transfer interactions in La0.5Sr1.5Ni1−−xFexO4±δ Ruddlesden-Popper oxides. Nat. Commun. 9:3150. doi: 10.1038/s41467-018-05600-y
Gaarenstroom, S. W., and Winograd, N. (1977). Initial and final state effects in the ESCA spectra of cadmium and silver oxides. J. Chem. Phys. 67, 3500–3506. doi: 10.1063/1.435347
Gasteiger, H. A., Kocha, S. S., Sompalli, B., and Wagner, F. T. (2005). Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Cat. B Environ. 56, 9–35. doi: 10.1016/j.apcatb.2004.06.021
Gfroerer, T. H. (2006). Photoluminescence in Analysis of Surfaces and Interfaces. Encyclopedia of Analytical Chemistry: Applications, Theory And Instrumentation. New York, NY: John Wiley & Sons.
Goodenough, J. B., and Cushing, B. L. (2003). Handbook of Fuel Cells – Fundamentals, Technology and Applications. Chichester: Wiley.
Goodenough, J. B., Manoharan, R., and Paranthaman, M. (1990). Surface protonation and electrochemical activity of oxides in aqueous solution. J. Am. Chem. Soc. 112, 2076–2082.
Gorlin, Y., Lassalle-Kaiser, B., Benck, J. D., Gul, S., Webb, S. M., Yachandra, V. K., et al. (2013). In situ X-ray absorption spectroscopy investigation of a bifunctional manganese oxide catalyst with high activity for electrochemical water oxidation and oxygen reduction. J. Am. Chem. Soc. 135, 8525–8534. doi: 10.1021/ja3104632
Grimaud, A., Diaz-Morales, O., Han, B., Hong, W. T., Lee, Y.-L., Giordano, L., et al. (2017). Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9:457–465. doi: 10.1038/nchem.2695
Grönbeck, H., Klacar, S., Martin, N. M., Hellman, A., Lundgren, E., and Andersen, J. N. (2012). Mechanism for reversed photoemission core-level shifts of oxidized Ag. Phys. Rev. B 85:115445. doi: 10.1103/PhysRevB.85.115445
Grosvenor, A. P., Biesinger, M. C., Smart, R. S. C., and McIntyre, N. S. (2006). New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 600, 1771–1779. doi: 10.1016/j.susc.2006.01.041
Grosvenor, A. P., Kobe, B. A., Biesinger, M. C., and McIntyre, N. S. (2004). Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Inter. Anal. 36, 1564–1574. doi: 10.1002/sia.1984
Gupta, R. P., and Sen, S. K. (1974). Calculation of multiplet structure of core p-vacancy levels. Phys. Rev. B 10, 71–77.
Gupta, R. P., and Sen, S. K. (1975). Calculation of multiplet structure of core p-vacancy levels. II. Phys. Rev. B 12, 15–19.
Hansen, W. N., Kolb, D. M., Rath, D. L., and Wille, R. (1980). An ESCA study on emersed electrodes. J. Electroanal. Chem. 110, 369–373. doi: 10.1016/s0022-0728(80)80391-3
Hong, W. T., Risch, M., Stoerzinger, K. A., Grimaud, A., Suntivich, J., and Shao-Horn, Y. (2015). Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404–1427. doi: 10.1039/C4EE03869J
Hong, W. T., Stoerzinger, K. A., Lee, Y.-L., Giordano, L., Grimaud, A., Johnson, A. M., et al. (2017). Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energy Environ. Sci. 10, 2190–2200. doi: 10.1039/C7EE02052J
Horkans, J., and Shafer, M. (1977). An investigation of the electrochemistry of a series of metal dioxides with rutile-type structure: MoO2, WO2, ReO2, RuO2, OsO2, and IrO2. J. Electrochem. Soc. 124, 1202–1207.
Hosseini-Benhangi, P., Garcia-Contreras, M. A., Alfantazi, A., and Gyenge, E. L. (2015). Method for enhancing the bifunctional activity and durability of oxygen electrodes with mixed oxide electrocatalysts: potential driven intercalation of potassium. J. Electrochem. Soc. 162, F1356–F1366. doi: 10.1149/2.0561512jes
Hüppauff, M., and Lengeler, B. (1993). Valency and structure of iridium in anodic iridium oxide films. J. Electrochem. Soc. 140, 598–602.
Iuzzolino, L., Dittmer, J., Dörner, W., Meyer-Klaucke, W., and Dau, H. (1998). X-ray absorption spectroscopy on layered photosystem II membrane particles suggests manganese-centered oxidation of the oxygen-evolving complex for the S0-S1, S1-S2, and S2-S3 transitions of the water oxidation cycle. Biochem. 37, 17112–17119.
Julien, C., Massot, M., Baddour-Hadjean, R., Franger, S., Bach, S., and Pereira-Ramos, J. (2003). Raman spectra of birnessite manganese dioxides. Solid State Ion. 159, 345–356. doi: 10.1016/S0167-2738(03)00035-3
Kaspar, T. C., Droubay, T., Chambers, S. A., and Bagus, P. S. (2010). Spectroscopic Evidence for Ag(iii) in highly oxidized silver films by X-ray photoelectron spectroscopy. J. Phys. Chem. C 114, 21562–21571. doi: 10.1021/jp107914e
Kolb, P. M., Rath, D. L., Wille, R., and Hansen, W. N. (1983). An ESCA study on the electrochemical double layer of emersed electrodes. Ber. Bunsenges. Phys. Chem. 87, 1108–1113. doi: 10.1002/bbpc.19830871206
Kötz, E., Neff, H., and Müller, K. (1986). A UPS, XPS and work function study of emersed silver, platinum and gold electrodes. J. Electroanal. Chem. Inter. Electrochem. 215, 331–344.
Kötz, E. R., and Neff, H. (1985). Anodic iridium oxide films: an UPS study of emersed electrodes. Surf. Sci. 160, 517–530. doi: 10.1016/0039-6028(85)90791-5
Kötz, R., Lewerenz, H. J., and Stucki, S. (1983). XPS studies of oxygen evolution on Ru and RuO2 anodes. J. Electrochem. Soc. 130, 825–829. doi: 10.1149/1.2119829
Kotz, R., Neff, H., and Stucki, S. (1984). Anodic iridium oxide films. J. Electrochem. Soc. 131, 72.
Lee, Y.-L., Kleis, J., Rossmeisl, J., Shao-Horn, Y., and Morgan, D. (2011). Prediction of solid oxide fuel cell cathode activity with first-principles descriptors. Energy Environ. Sci. 4, 3966–3970. doi: 10.1039/C1EE02032C
Maiyalagan, T., Jarvis, K. A., Therese, S., Ferreira, P. J., and Manthiram, A. (2014). Spinel-type lithium cobalt oxide as a bifunctional electrocatalyst for the oxygen evolution and oxygen reduction reactions. Nat. Commun. 5:3949. doi: 10.1038/ncomms4949
Man, I. C., Su, H.-Y., Calle-Vallejo, F., Hansen, H. A., Martínez, J. I., Inoglu, N. G., et al. (2011). Universality in oxygen evolution electrocatalysis on oxide surfaces. Chem. Cat. Chem. 3, 1159–1165. doi: 10.1002/cctc.201000397
Manceau, A., Gorshkov, A. I., and Drits, V. A. (1992). Structural chemistry of Mn, Fe, Co, and Ni in manganese hydrous oxides; part i, information from XANES spectroscopy. Am. Miner. 77, 1133–1143.
Matsumoto, Y., and Sato, E. (1986). Electrocatalytic properties of transition metal oxides for oxygen evolution reaction. Mater. Chem. Phys. 14, 397–426.
Matsumoto, Y., Yamada, S., Nishida, T., and Sato, E. (1980). Oxygen evolution on La1−x Sr x Fe1−y CoyO3 series oxides. J. Electrochem. Soc. 127, 2360–2364.
Matsumoto, Y., Yoneyama, H., and Tamura, H. (1977). Influence of the nature of the conduction band of transition metal oxides on catalytic activity for oxygen reduction. J. Electroanal. Chem. 83, 237–243.
Matsuoka, M., Kamegawa, T., and Anpo, M. (2008). “Photoluminescence spectroscopy and its application to the characterization of active sites and reaction dynamics in catalysis,” Handbook of Heterogeneous Catalysis, eds G. Ertl, H. Knözinger, F. Schüth, and J. Weitkamp (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA).
Mavros, M. G., Tsuchimochi, T., Kowalczyk, T., McIsaac, A., Wang, L.-P., and Voorhis, T. V. (2014). What can density functional theory tell us about artificial catalytic water splitting? Inorg. Chem. 53, 6386–6397. doi: 10.1021/ic5002557
May, K. J., Carlton, C. E., Stoerzinger, K. A., Risch, M., Suntivich, J., Lee, Y.-L., et al. (2012). Influence of oxygen evolution during water oxidation on the surface of perovskite oxide catalysts. J. Phys. Chem. Lett. 3, 3264–3270. doi: 10.1021/jz301414z
McAlpin, J. G., Surendranath, Y., Dinca, M., Stich, T. A., Stoian, S. A., Casey, W. H., et al. (2010). EPR evidence for Co (IV) species produced during water oxidation at neutral pH. J. Am. Chem. Soc. 132, 6882–6883. doi: 10.1021/ja1013344
McCrory, C. C. L., Jung, S., Peters, J. C., and Jaramillo, T. F. (2013). Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987. doi: 10.1021/ja407115p
Mefford, J. T., Rong, X., Abakumov, A. M., Hardin, W. G., Dai, S., Kolpak, A. M., et al. (2016). Water electrolysis on La1−−xSrxCoO3−−δ perovskite electrocatalysts. Nat.Commun. 7:11053. doi: 10.1038/ncomms11053
Meng, Y., Song, W., Huang, H., Ren, Z., Chen, S.-Y., and Suib, S. L. (2014). Structure–property relationship of bifunctional MnO2 nanostructures: highly efficient, ultra-stable electrochemical water oxidation and oxygen reduction reaction catalysts identified in alkaline media. J. Am. Chem. Soc. 136, 11452–11464. doi: 10.1021/ja505186m
Mićić, R., and Draškovi,ć, B. (1985). Some photoluminescence properties of Mn2+ in magnesium bromide. Phys. Stat. Sol. 128, 489–494. doi: 10.1002/pssb.2221280214
Mo, Y., Stefan, I. C., Cai, W.-B., Dong, J., Carey, P., and Scherson, D. A. (2002). In situ iridium LIII-edge X-ray absorption and surface enhanced raman spectroscopy of electrodeposited iridium oxide films in aqueous electrolytes. J. Phys. Chem. B 106, 3681–3686. doi: 10.1021/jp014452p
Nahor, G., Hapiot, P., Neta, P., and Harriman, A. (1991). Changes in the redox state of iridium oxide clusters and their relation to catalytic water oxidation: radiolytic and electrochemical studies. J. Phys. Chem. 95, 616–621.
Nesbitt, H., and Banerjee, D. (1998). Interpretation of XPS Mn (2p) spectra of Mn oxyhydroxides and constraints on the mechanism of MnO2 precipitation. Am. Mineral. 83, 305–315.
Noh, J. S., and Schwarz, J. A. (1989). Estimation of the point of zero charge of simple oxides by mass titration. J. Colloid Interface Sci. 130, 157–164. doi: 10.1016/0021-9797(89)90086-6
Ooka, H., Takashima, T., Yamaguchi, A., Hayashi, T., and Nakamura, R. (2017). Element strategy of oxygen evolution electrocatalysis based on in situ spectroelectrochemistry. Chem. Comm. 53, 7149–7161. doi: 10.1039/C7CC02204B
Oswald, S., and Brückner, W. (2004). XPS depth profile analysis of non-stoichiometric NiO films. Surf. Interface Anal. 36, 17–22. doi: 10.1002/sia.1640
Pal, S., and Gupta, R. (1982). Calculation of the multiple structure of core p-vacancy levels in the crystal field. Phys. Rev. B 26:35.
Pechenyuk, S. I. (1999). The use of the pH at the point of zero charge for characterizing the properties of oxide hydroxides. Russ. Chem. Bull. 48, 1017–1023. doi: 10.1007/bf02495994
Pfeifer, V., Jones, T. E., Velasco Vélez, J. J., Massu,é, C., Arrigo, R., Teschner, D., et al. (2016). The electronic structure of iridium and its oxides. Surf. Interface Anal. 48, 261–273. doi: 10.1002/sia.5895
Post, J. E. (1999). Manganese oxide minerals: crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. U.S.A. 96, 3447–3454.
Puntambekar, A., Wang, Q., and Chakrapani, V. (2016). Electrochemical charging of CdSe quantum dots: effects of adsorption versus intercalation. ACS Nano 10,10988–10999. doi: 10.1021/acsnano.6b05779
Rath, D. L., and Kolb, D. M. (1981). Continuous work function monitoring for electrode emersion. Surf. Sci. 109, 641–647. doi: 10.1016/0039-6028(81)90432-5
Ren, H., and Yang, F. (2018). Influence of Mn2+ on the up-conversion emission performance of Mn2+, Yb3+, Er3+: ZnWO4 green phosphors. J. Mater. Sci. doi: 10.1007/s10854-018-9053-9
Risch, M., Stoerzinger, K. A., Han, B., Regier, T. Z., Peak, D., Sayed, S. Y., et al. (2017). Redox processes of manganese oxide in catalyzing oxygen evolution and reduction: an in situ soft X-ray absorption spectroscopy study. J. Phys. Chem. C 121, 17682–17692. doi: 10.1021/acs.jpcc.7b05592
Rong, X., Parolin, J., and Kolpak, A. M. (2016). A fundamental relationship between reaction mechanism and stability in metal oxide catalysts for oxygen evolution. ACS Cat. 6, 1153–1158. doi: 10.1021/acscatal.5b02432
Roy, I., Wang, Q., and Chakrapani, V. (2020). Nature of reaction intermediates and origin of bifunctionality in manganese oxide. J. Phys. Chem. C 124, 5286–5299. doi: 10.1021/acs.jpcc.0c00714
Sanchez Casalongue, H. G., Ng, M. L., Kaya, S., Friebel, D., Ogasawara, H., and Nilsson, A. (2014). In situ observation of surface species on iridium oxide nanoparticles during the oxygen evolution reaction. Angew. Chem. 126, 7297–7300. doi: 10.1002/ange.201402311
Sekiguchi, D., and Adachi, S. (2015). Synthesis and photoluminescence spectroscopy of BaGeF6:Mn4+ red phosphor. Opt. Mater. 42, 417–422. doi: 10.1016/j.optmat.2015.01.039
Sherman, D. M. (1984). The electronic structures of manganese oxide minerals. Am. Mineral. 69, 788–799.
Silva, A. S., Lourenco, S. A., and Dantas, N. O. (2016). Mn concentration-dependent tuning of Mn2+ d emission of Zn1−xMnxTe nanocrystals grown in a glass system. Phys. Chem. Chem. Phys. 18, 6069–6076. doi: 10.1039/C5CP06802A
Silvester, E., Manceau, A., and Drits, V. A. (1997). Structure of synthetic monoclinic Na-rich birnessite and hexagonal birnessite: II. results from chemical studies and EXAFS spectroscopy. Am. Miner. 82, 962–978.
Stoerzinger, K. A., Diaz-Morales, O., Kolb, M., Rao, R. R., Frydendal, R., Qiao, L., et al. (2017). Orientation-dependent oxygen evolution on RuO2 without lattice exchange. ACS Energy Lett. 2, 876–881. doi: 10.1021/acsenergylett.7b00135
Takashima, T., Hashimoto, K., and Nakamura, R. (2012a). Inhibition of charge disproportionation of MnO2 electrocatalysts for efficient water oxidation under neutral conditions. J. Am. Chem. Soc. 134, 18153–18156. doi: 10.1021/ja306499n
Takashima, T., Hashimoto, K., and Nakamura, R. (2012b). Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts. J. Am. Chem. Soc. 134, 1519–1527. doi: 10.1021/ja206511w
Wagner, F. T., and Ross, P. N. (1983). Thickness of electrolyte layers on emersed Pt electrodes. J. Electrochem. Soc. 130, 1789–1791.
Wang, Q., Brier, M., Joshi, S., Puntambekar, A., and Chakrapani, V. (2016a). Defect induced burstein-moss shift in reduced V2O5 nanostructures. Phys. Rev. B 94:245305. doi: 10.1103/PhysRevB.94.245305
Wang, Q., Puntambekar, A., and Chakrapani, V. (2016b). Vacancy-induced semiconductor-insulator-metal transitions in non-stoichiometric nickel and tungsten oxides. Nano Lett. 16, 7067–7077. doi: 10.1021/acs.nanolett.6b03311
Xiao, W., Lei, F., Yin, L., Shi, Y., Xie, J., and Zhang, L. (2017). Solid state synthesis, luminescent properties and energy transfer from Eu2+ to Mn2+ in red phosphor BaMg2Si2O7:Eu2+,Mn2+. Solid State Sci. 72, 116–123. doi: 10.1016/j.solidstatesciences.2017.08.008
Yamamoto, S., Bluhm, H., Andersson, K., Ketteler, G., Ogasawara, H., Salmeron, M., et al. (2008). In situ x-ray photoelectron spectroscopy studies of water on metals and oxides at ambient conditions. J. Phys. Condens. Matter 20:184025. doi: 10.1088/0953-8984/20/18/184025
Yang, Z., Zhang, J., Kintner-Meyer, M. C., Lu, X., Choi, D., Lemmon, J. P., et al. (2011). Electrochemical energy storage for green grid. Chem. Rev. 111, 3577–3613. doi: 10.1021/cr100290v
Keywords: electrocatalysis, OER, oxidation state, photoluminescence spectroscopy (PL), intermediates and mechanism, X-ray photoemission spectroscopy (XPS), X-ray absorption spectroscopy (XAS)
Citation: Chakrapani V (2020) Probing Active Sites and Reaction Intermediates of Electrocatalysis Through Confocal Near-Infrared Photoluminescence Spectroscopy: A Perspective. Front. Chem. 8:327. doi: 10.3389/fchem.2020.00327
Received: 21 January 2020; Accepted: 31 March 2020;
Published: 28 April 2020.
Edited by:
Simelys Hernández, Politecnico di Torino, ItalyReviewed by:
Michael Nolan, University College Cork, IrelandDong Guohui, Shaanxi University of Science and Technology, China
Copyright © 2020 Chakrapani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vidhya Chakrapani, Y2hha3J2QHJwaS5lZHU=