 Philip M. Punt
Philip M. Punt Lukas M. Stratmann†
Lukas M. Stratmann† Sinem Sevim
Sinem Sevim Guido H. Clever
Guido H. Clever- Faculty of Chemistry and Chemical Biology, TU Dortmund University, Dortmund, Germany
The presence of metal centers with often highly conserved coordination environments is crucial for roughly half of all proteins, having structural, regulatory, or enzymatic function. To understand and mimic the function of metallo-enzymes, bioinorganic chemists pursue the challenge of synthesizing model compounds with well-defined, often heteroleptic metal sites. Recently, we reported the design of tailored homoleptic coordination environments for various transition metal cations based on unimolecular DNA G-quadruplex structures, templating the regioselective positioning of imidazole ligandosides LI. Here, we expand this modular system to more complex, heteroleptic coordination environments by combining LI with a new benzoate ligandoside LB within the same oligonucleotide. The modifications still allow the correct folding of parallel tetramolecular and antiparallel unimolecular G-quadruplexes. Interestingly, the incorporation of LB results in strong destabilization expressed in lower thermal denaturation temperatures Tm. While no transition metal cations could be bound by G-quadruplexes containing only LB, heteroleptic derivatives containing both LI and LB were found to complex CuII, NiII, and ZnII. Especially in case of CuII we found strong stabilizations of up to ΔTm = +34°C. The here shown system represents an important step toward the design of more complex coordination environments inside DNA scaffolds, promising to culminate in the preparation of functional metallo-DNAzymes.
Introduction
Proteins are involved in a vast number of processes ranging from structural and regulatory functions to enzymatic reactions. Roughly half of all proteins depend on metal cations helping to maintain a desired folding or serving as catalytic centers or redox cofactors (Raven et al., 1999; Lu et al., 2009; Rubino and Franz, 2012). Which function the respective metal ion adopts is strongly dependent on its properties, including accessible spin states, oxidation potential, Lewis-acidity, and bioavailability (Holm et al., 1996; Waldron et al., 2009). These properties are further fine-tuned by a well-defined first and second coordination sphere. The former is directly involved in metal coordination and usually consists of mixtures of different donor functionalities. Typically involved in coordination are the amino acids histidine, glutamic/aspartic acid, methionine, cysteine, or the backbone amide groups (Holm et al., 1996; Degtyarenko, 2000; Shook and Borovik, 2010; Valdez et al., 2014). In contrast, the second coordination sphere is not directly involved in metal binding but regulates catalytic processes, proton or electron shuttling, substrate transport, and effects selectivity (Colquhoun et al., 1986; Degtyarenko, 2000; Waldron et al., 2009; Shook and Borovik, 2010; Zhao et al., 2013; Valdez et al., 2014; Cornish et al., 2016).
The design of artificial metallo-enzyme mimics with improved or novel properties is attracting increasing interest, but remains challenging. In the area of preparative bioinorganic chemistry, focus is set on small, multidentate chelate complexes, often requiring tedious multistep syntheses and only covering effects of the first coordination sphere (Samuel et al., 2010; Kanady et al., 2011; Anderson et al., 2013; Dicke et al., 2018). More biologically oriented approaches involve the replacement of natural metal cofactors with metal centers not known in nature. An example is the replacement of hemin in myoglobin with an iridium or rhodium porphyrin complex for enantioselective cyclopropanation reactions (Key et al., 2016; Litman et al., 2018). Another approach is embedding metal cofactors by covalent or non-covalent interactions into empty cavities of usually metal-free proteins. This was successfully applied in a series of examples enabling catalysis of the asymmetric transfer hydrogenation of imines (Wu et al., 2019), ring-closing metathesis (Jeschek et al., 2016), oxime (Drienovská et al., 2018), and hydrazine (Drienovská et al., 2018; Mayer et al., 2019) formation and hydration of alkenes (Drienovská et al., 2017). In contrast to the aforementioned examples, a more bottom up approach is the de novo design of new metallo-proteins by the precise arrangement of certain structural motifs to create a metal binding site (Raven et al., 1999; Lu et al., 2009; Rubino and Franz, 2012). In recent years, a more efficient alternative was developed based on small artificial peptoid structures. Due to their simple accessibility by solid phase synthesis and their capability to form well-ordered secondary structures, many examples were shown for selective metal binding and catalytic applications (Baskin and Maayan, 2016; Knight et al., 2017; Baskin et al., 2018; Ghosh et al., 2018).
Another type of biopolymers forming well-ordered secondary structures are oligonucleotides. In contrast to peptides, RNA and DNA only consist of four nucleotide building blocks, thus reducing the possibilities to create diverse coordination environments for a range of metal cations. To overcome this limitation, different strategies were developed to covalently or non-covalently anchor metal-chelating ligands inside DNA. Roelfes and co-workers pioneered the design of various oligonucleotides capable of Michael-Additions, Carbene transfer, syn-hydrations of alkenes or Diels-Alder reactions (Roelfes and Feringa, 2005; Coquière et al., 2007; Boersma et al., 2010a,b; Rioz-Martínez et al., 2016). Other groups used modified quadruplexes for sequence-specific DNA cleavage, light controlled thrombin catalysis or peroxidase mimicking DNAzymes (Xu et al., 2009; Ali et al., 2019; Wang et al., 2019). A difficulty of this approach lies in the largely unknown exact position and coordination environment of the catalytic centers. This difficulty could be overcome in the field of metal-mediated base pairing, where the hydrogen bonding interaction of canonical base pairs is replaced by metal coordination, leading to highly stabilized DNA structures (Mandal and Müller, 2017). While first examples included only the involvement of canonical bases (Katz, 1963), the field was later expanded by the incorporation of a variety of artificial nucleobases culminating in the development of programmable metal wires inside DNA duplexes (Tanaka et al., 2006; Clever et al., 2007; Mandal et al., 2016; Sandmann et al., 2019). Later, the concept was expanded from duplex to triplex DNA (Tanaka et al., 2002) and i-motifs (Abdelhamid et al., 2018), while we and others started to focus on G-quadruplexes (Miyoshi et al., 2007; Smith et al., 2012; Engelhard et al., 2013). The latter ones form from guanine-rich sequences where four G-residues cyclize to planar G-tetrads via Hoogsteen base pairing. Multiple G-tetrads form a G-quadruplex via π-π stacking interactions. Key to their high stability is the incorporation of a central cation—typically Na+ or K+ (Hänsel-Hertsch et al., 2017; Neidle, 2017). Our group was the first to report CuII-mediated tetramolecular G-quadruplexes based on pyridine and imidazole ligands (Engelhard et al., 2013, 2018b; Punt and Clever, 2019a), aimed at a range of applications. For example, dinuclear systems were employed as CuII-based EPR-rulers for accurate distance measurements (Engelhard et al., 2018a). We later expanded this concept to unimolecular G-quadruplexes, equipped with oligonucleotide loops which form cavities above the G-quadruplex stem in which the metal complexes are embedded (Engelhard et al., 2017). In a recent study, we further showed that these G-quadruplexes can act as robust templates to arrange different numbers of imidazole ligandosides, leading to fine-tuned affinities for a range of transition metal cations with respect to their preferred coordination environments (Punt and Clever, 2019b). While only homoleptic systems were investigated in that study, we herein expand the modular concept to heteroleptic systems with different donor functionalities. We introduce the design of mixed systems with imidazole and benzoate ligands, inspired by metallo-proteins, where the combination of imidazoles and carboxylate is often involved in metal coordination (e.g., in the 2-His-1-carboxylate facial triad) (Greenblatt et al., 1998; Koehntop et al., 2005). We show how this combination affects both, G-quadruplex stability and metal complexation.
Results
In this study we report the incorporation of a new benzoate ligandoside LB in combination with the known imidazole ligandoside LI. Both were incorporated in the (S) configuration as GNA (glycol nucleic acid) building blocks (Zhang et al., 2005, 2006) by solid phase synthesis into tetramolecular and unimolecular G-quadruplexes. The phosphoramidite of LI was synthesized as previously described (Punt and Clever, 2019b). For the new benzoate ligand LB, a literature procedure was adopted (Engelhard et al., 2017). Accordingly, an initial nucleophilic attack of deprotonated solketal to methyl 4-(bromomethyl)benzoate followed by acidic deprotection reaction led to protected benzoate ligandoside (R)-4. Its structure and absolute configuration were confirmed by single-crystal X-ray diffraction (Figure 1). The primary hydroxyl group was DMT-protected (DMT = dimethoxytrityl) followed by a phosphorylation reaction yielding phosphoramidite building block (S)-6. DNA solid phase synthesis was then performed according to standard literature procedures with extended coupling times for the ligandosides LI and LB (see Supplementary Material for details). Coupling efficiencies for LB and LI were typically >99% per step. After solid phase synthesis, oligonucleotides were cleaved from the solid support and deprotected in 0.4 M NaOH in methanol/water (4:1) at 55°C for 16 h. Standard deprotection with concentrated ammonium hydroxide was avoided due to the risk of forming amides instead of carboxylates from the benzoate esters. After reversed-phase HPLC purification, oligonucleotides were desalted and DMT-groups removed using C18 SepPak cartridges and aq. TFA (2%). The oligonucleotides were then lyophilized at stored at −20°C.
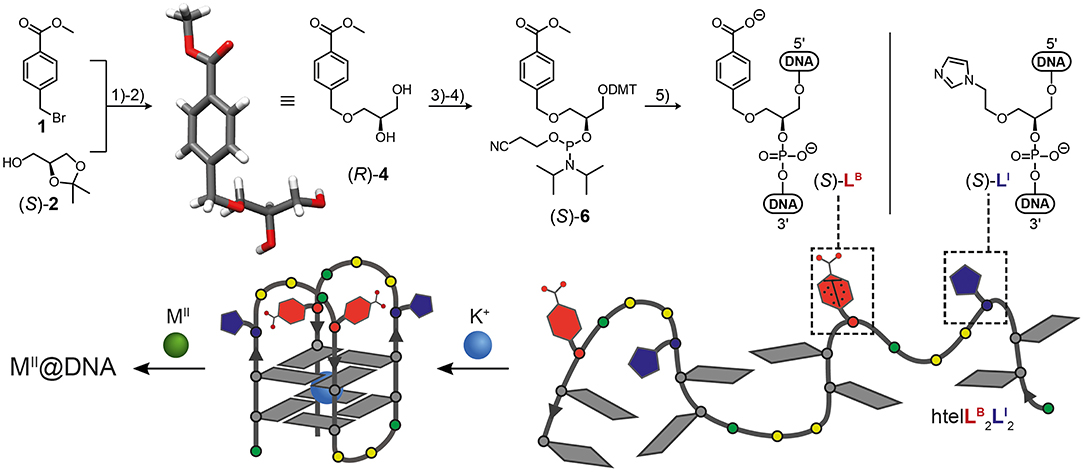
Figure 1. Synthesis of benzoate ligandoside LB and molecular structure of ligandoside LI (top). (1) NaH, CH3CN; (2) CH3COOH, THF/H2O; (3) DMT-Cl, DIPEA, DMAP, THF; (4) CEDIP-Cl, DIPEA, CH2Cl2; (5) automated solid-phase DNA synthesis. The single-crystal X-ray structure of the protected ligandoside (R)-4 is shown. G-quadruplex formation (bottom) of htelLB2LI2 creates a heteroleptic coordination environment for transition metal ions (M = Co, Ni, Cu, Zn). Gray tiles: guanosine; red: ligandoside LB; blue: ligandoside LI; green circles: adenosine; yellow circles: thymidine. DMT = dimethoxytrityl; DIPEA = N,N-diisopropylethylamine; DMAP = N,N-dimethylaminopyridin; CEDIP-Cl = 2-cyanoethyl N,N-diisopropylchlorophosphoramidite.
Since LI had already been established in tetramolecular and unimolecular G-quadruplexes, we first investigated the influence of LB in the tetramolecular G-quadruplex (LBG4)4. Clear formation of a parallel G-quadruplex was observed by CD spectroscopy with a positive Cotton effect around ~260 nm (see Figure S25). Thermal denaturation experiments showed a melting temperature Tm of 27°C which was significantly lower compared to previously reported (LIG4)4 (Tm = 36°C; Punt and Clever, 2019b). Since LB and LI are sharing the same backbone modification, we ascribe this destabilization to a repulsive effect between the negatively charged benzoates and phosphates (Figure 2). Next, the interaction of (LBG4)4 with a series of transition metal cations was investigated. In contrast to (LIG4)4 which was shown to complex CuII, NiII, CoII, and ZnII, no signs for metal complexation in (LBG4)4 were observed (see Figures S3, S4). This may be explained by the harder character of the benzoate ligand, competing with hard ligands such as the contained chloride, cacodylate buffer or phosphate backbones. However, even for hard and oxophilic transition metal cations, including GdIII and CeIII, no interactions were found.
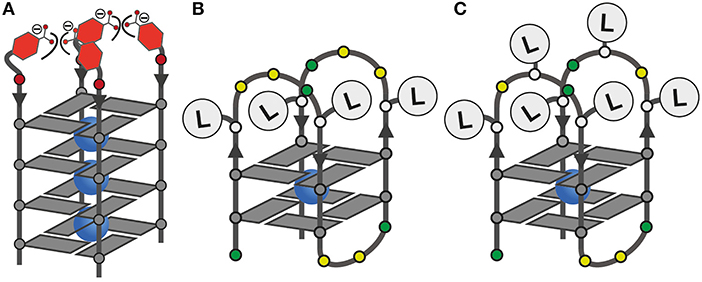
Figure 2. (A) Schematic representation of the tetramolecular G-quadruplex (LBG4)4 with the proposed repulsive effect of negatively charged LB; (B,C) ligand positions in unimolecular G-quadruplexes with four or six incorporated ligands.
Mixing ligands in tetramolecular G-quadruplexes leads to statistical mixtures, which makes it challenging to design distinct heteroleptic coordination environments (see Supplementary Material for details). On the other hand, the folding of unimolecular G-quadruplexes into discrete topologies enables programmable ligand arrangements. Consequently, we moved forward to incorporate LB in unimolecular G-quadruplexes. At first, LB was incorporated four times in htelLB 4. Similar to (LBG4)4, incorporation of LB caused strong destabilization (Tm = 12°C) compared to htelLI4 (Tm = 33°C). Successive replacement of LB with LI was accompanied with a linear increase in stabilization for each replacement (htelLB 3LI Tm = 17°C, htelLB 2LI 2 Tm = 23°C, htelLBLI 3 Tm = 28°C), highlighting the additive destabilizing effect of LB (Figure 3). CD spectroscopy of htelLB 4, htelLB 3LI, htelLB 2LI 2, and htelLBLI 3 showed clear signatures corresponding to an antiparallel G-quadruplex topology with a positive Cotton effect around ~294 nm in all cases (see Figures S26, S27). This is consistent with the previous observations for homoleptic G-quadruplexes containing only LI. Next, the interaction with different transition metal cations was investigated. As for (LBG4)4, for htelLB 4, htelLB 3LI, and htelLB 2LI 2, thermal denaturation experiments showed no signs for interaction with the examined transition metal cations (CuII, NiII, ZnII, CoII, VIVO). Pleasingly, this changed for htelLBLI 3 that showed a weak but distinct stabilization after addition of 1 equiv. of CuII (ΔTm = + 4°C). Additional equivalents resulted in no further stabilization consistent with a specific binding of CuII. CD spectroscopy further confirmed retention of a clear antiparallel topology (see Figures S6, S7, S11–S16).
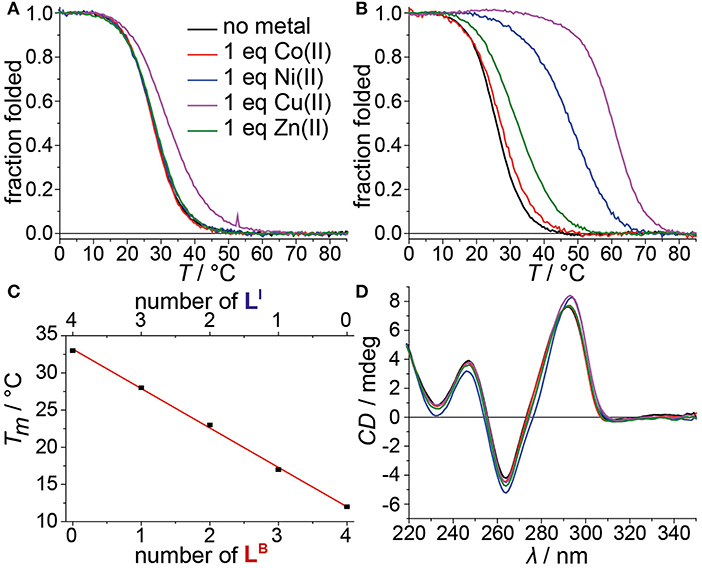
Figure 3. Melting curves of (A) htelLBLI 3 and (B) htelLB 2LI 4 in absence or presence of different transition metal cations. (C) Linear dependence of thermal stabilities of htelLI 4, htelLBLI 3, htelLB 2LI 2, htelLB 3LI, and htelLB 4 depending on the number of incorporated LB. (D) CD spectra of htelLB 2LI 4 in absence or presence of different transition metal cations.
After we could show that at least three imidazole ligands are required to complex CuII, we moved forward to a new series of sequences with six incorporated ligands (htelLB 4LI 2, htelLB 3LI 3, htelLB 2LI 4). Again, the formation of G-quadruplexes with a clear antiparallel topology was observed by CD spectroscopy (see Figures S28, S29). Likewise, comparison of the thermal stabilities showed the destabilizing effect of LB (htelLB 4LI 2 Tm = 17°C, htelLB 3LI 3 Tm = 26°C, htelLB 2LI 4 Tm = 26°C), however, not in the linear fashion as observed for the series htelLB 4−nLI n (n = 0–4). For the examined set of six-ligand-containing sequences, however, direct Tm comparison is not appropriate due to the chosen modification pattern (see Table 1). When investigating the interaction with metal cations, for htelLB 4LI 2, a clear stabilization after addition of CuII (ΔTm = + 6°C) was observed. Considering that for htelLB 2LI 2 almost no stabilization was observed (ΔTm = + 1°C), we conclude that in htelLB 4LI 2 an involvement of one or two ligandosides LB into metal coordination is very likely. When further replacing LB with LI as in htelLB 3LI 3 and htelLB 2LI 4, the CuII-mediated thermal stabilization successively increased from ΔTm = + 9°C (htelLB 3LI 3) to ΔTm = + 34°C (htelLB 2LI 4). This extremely high thermal stabilization is unprecedented for unimolecular G-quadruplexes and much higher compared to the reported G-quadruplexes htelLI 6 (ΔTm = + 18°C) and htelLI 4A (ΔTm = + 23°C) (Punt and Clever, 2019b).
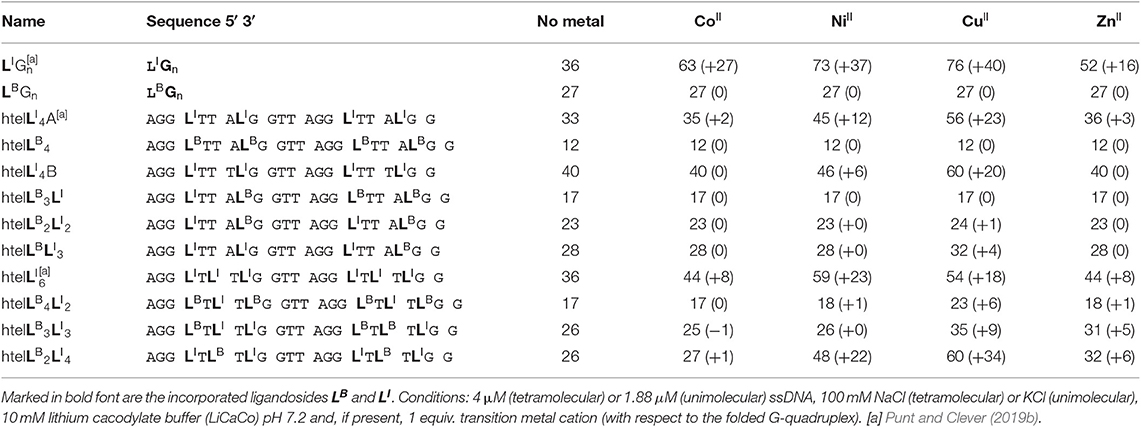
Table 1. Sequences investigated in this study and respective denaturation temperatures Tm (and ΔTm) in absence and presence of 1 equiv. of CuII, NiII, ZnII, CoII (assumed to be oxidized to CoIII under the experimental conditions).
The formation of 1:1 complexes for htelLB 2LI 4 with CuII and NiII was further confirmed by native ESI mass spectrometry. To understand whether a G-quadruplex is folded or unfolded in the gas phase, the intrinsic property of G-quadruplexes is exploited that in their folded state they always bind n−1 potassium ions (where n = number of G-tetrads). For a folded G-quadruplex with two G-tetrads, a main signal corresponding to the adduct with one distinct potassium ion would be expected, followed by a statistical distribution of adducts with further unspecifically bound potassium cations. On the other hand, for an unfolded G-quadruplex, the main signal would correspond to the mass of the DNA strand without potassium ions. The mass spectrum shows a main signal corresponding to [htelLB 2LI 4+Cu+K-7H]4− (Figure 4), thus strongly indicating a folded G-quadruplex coordinating to a CuII or NiII ion in the gas phase (D'Atri et al., 2015; Lecours et al., 2017).
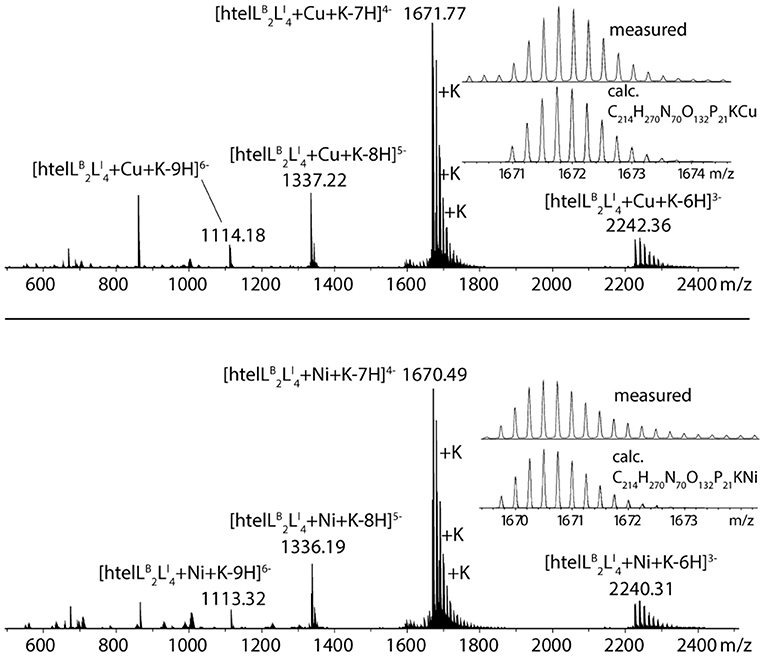
Figure 4. Native ESI-MS spectra of htelLB 2LI 4 in complex with CuII (top) and NiII (bottom).
Jahn-Teller-distorted CuII usually favors the coordination of four strongly associated ligands in a square planar geometry, with two additional ligands more loosely bound in axial positions (Halcrow, 2012). After proving a 1:1 complex for htelLB 2LI 4 and CuII, the question was if all six ligands are participating in metal coordination or if only LI is involved. Therefore, a new sequence htelLI 4B was synthesized where LB was replaced with thymidines. Addition of CuII led to a thermal stabilization of ΔTm = + 20°C, much lower compared to htelLB 2LI 4 (ΔTm = + 34°C). However, when looking at the absolute melting temperature Tm in presence of CuII, one notices that they are the same for both sequences (htelLB 2LI 4 Tm = 60°C, htelLI 4B Tm = 60°C). This could mean that CuII coordination by htelLB 2LI 4 simply compensates the destabilizing effect of LB and no benzoate ligand was involved in CuII coordination. Further studies are required to shed light on this question.
Besides CuII, the addition of ZnII and NiII to htelLB 2LI 4 and htelLB 3LI 3 led to thermal stabilizations. These results were highly intriguing for two reasons. Quadruplex htelLB 2LI 4 was significantly more stabilized with NiII (ΔTm = + 22°C) compared to ZnII (ΔTm = + 6°C). However, in htelLB 3LI 3, the opposite effect was observed, showing a higher stabilization after ZnII addition (ΔTm = + 5°C), while for NiII no complexation was observed. This adds to the established variation of ligand number and position a third layer to our system to fine-tune metal affinities by the introduction of heteroleptic systems. As last question, we were interested whether ZnII in htelLB 3LI 3 is coordinated by one or more benzoates. Interestingly, other sequences shown to complex ZnII (htelLI 4A ΔTm = + 3°C, htelLI 6 ΔTm = + 8°C) always contain at least four counts of LI. Since in htelLB 3LI 3 only three LI were available, we conclude that an involvement of LB in coordination to the ZnII cation is likely.
Conclusion
A new benzoate-based ligandoside LB was established in tetramolecular and unimolecular G-quadruplex structures. Homoleptic G-quadruplex (LBG4)4 was found to form a clear parallel topology. Its thermal stability indicated a strongly destabilizing effect of LB compared to LI which was attributed to an accumulation of negative charges. Also, no interactions between a series of transition metal cations and (LBG4)4 were found. Similarly, for the unimolecular G-quadruplex htelLB 4, a destabilizing effect of LB and no interactions with transition metal cations were observed. The successive replacement of LB with LI in htelLB 3LI, htelLB 2LI 2, htelLBLI 3, and htelLI 4 resulted in a linear increase of the thermal stability. In addition, for htelLBLI 3, a weak thermal stabilization after addition of 1 equiv. CuII indicated specific binding.
When moving to systems with six incorporated ligands, a tremendously high thermal stabilization was observed after addition of CuII to htelLB 2LI 4 (ΔTm = + 34°C). In comparison, for htelLI 4B, addition of CuII resulted in a stabilization of only ΔTm = + 20°C. However, the absolute melting temperatures Tm of htelLB 2LI 4 (Tm = 60°C) and htelLI 4B (Tm = 60°C) are the same, indicating that CuII complexation is rather compensating the destabilizing effect of LB. More interesting were the results for htelLB 2LI 4 and htelLB 3LI 3 after addition of ZnII and NiII, respectively. HtelLB 2LI 4 was significantly more stabilized by NiII (ΔTm = + 22°C) compared to ZnII (ΔTm = + 6°C). However, in htelLB 3LI 3, the opposite effect was observed, showing a higher stabilization after ZnII addition (ΔTm = + 5°C) while for NiII no complexation was found. This expands our toolbox to design tailored binding sites for various transition metal cations. Previously, we had shown to fine-tune coordination environments by varying position and number of ligands. Here, we expand this approach by combining two ligandosides, LB and LI, which we regard as an important step for the design of metal-selective G-quadruplexes with application in diagnostics, selective catalysis, and DNA nanotechnology.
Data Availability Statement
The datasets generated for this study can be found in the Cambridge Crystallographic Data Center under the CCDC identifier 1961648.
Author Contributions
PP and LS conducted all syntheses and DNA experiments. SS contributed to the tetramolecular systems. LK and CS contributed the X-ray structure of compound 4. PP, LS, and GC designed the study, conceived the experiments, analyzed the data, and authored the manuscript.
Funding
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2033—Projektnummer 390677874. We thank Markus Hüffner for contributing the elemental analyses and also the European Research Council (ERC Consolidator grant 683083, RAMSES) for support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Prof. Herbert Waldmann from the MPI Dortmund is thankfully acknowledged for access to the MALDI-TOF spectrometer.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00026/full#supplementary-material
References
Abdelhamid, M. A., Fábián, L., MacDonald, C. J., Cheesman, M. R., Gates, A. J., and Waller, Z. A. (2018). Redox-dependent control of i-Motif DNA structure using copper cations. Nucleic Acids Res. 46, 5886–5893. doi: 10.1093/nar/gky390
Ali, A., Bullen, G. A., Cross, B., Dafforn, T. R., Little, H. A., Manchester, J., et al. (2019). Light-controlled thrombin catalysis and clot formation using a photoswitchable G-quadruplex DNA aptamer. Chem. Commun. 55, 5627–5630. doi: 10.1039/C9CC01540J
Anderson, J. S., Rittle, J., and Peters, J. C. (2013). Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 501, 84–87. doi: 10.1038/nature12435
Baskin, M., and Maayan, G. (2016). A rationally designed metal-binding helical peptoid for selective recognition processes. Chem. Sci. 7, 2809–2820. doi: 10.1039/C5SC04358A
Baskin, M., Zhu, H., Qu, Z.-W., Chill, J. H., Grimme, S., and Maayan, G. (2018). Folding of unstructured peptoids and formation of hetero-bimetallic peptoid complexes upon side-chain-to-metal coordination. Chem. Sci. 10, 620–632. doi: 10.1039/C8SC03616K
Boersma, A. J., Coquière, D., Geerdink, D., Rosati, F., Feringa, B. L., and Roelfes, G. (2010a). Catalytic enantioselective syn hydration of enones in water using a DNA-based catalyst. Nat. Chem. 2, 991–995. doi: 10.1038/nchem.819
Boersma, A. J., Megens, R. P., Feringa, B. L., and Roelfes, G. (2010b). DNA -based asymmetric catalysis. Chem. Soc. Rev. 39, 2083–2092. doi: 10.1039/b811349c
Clever, G. H., Kaul, C., and Carell, T. (2007). DNA–metal base pairs. Angew. Chem. Int. Ed. 46, 6226–6236. doi: 10.1002/anie.200701185
Colquhoun, H. M., Stoddart, F. J., and Williams, D. J. (1986). Second-sphere coordination – a novel role for molecular receptors. Angew. Chem. Int. Ed. 25, 487–507. doi: 10.1002/anie.198604873
Coquière, D., Feringa, B. L., and Roelfes, G. (2007). DNA-based catalytic enantioselective michael reactions in water. Angew. Chem. Int. Ed. 46, 9308–9311. doi: 10.1002/anie.200703459
Cornish, A. J., Ginovska, B., Thelen, A., da Silva, J. C., Soares, T. A., Raugei, S., et al. (2016). Single-amino acid modifications reveal additional controls on the proton pathway of [FeFe]-hydrogenase. Biochemistry 55, 3165–3173. doi: 10.1021/acs.biochem.5b01044
D'Atri, V., Porrini, M., Rosu, F., and Gabelica, V. (2015). Linking molecular models with ion mobility experiments. Illustration with a rigid nucleic acid structure. J. Mass. Spectrom. 50, 711–726. doi: 10.1002/jms.3590
Degtyarenko, K. (2000). Bioinorganic motifs: towards functional classification of metalloproteins. Bioinformatics 16, 851–864. doi: 10.1093/bioinformatics/16.10.851
Dicke, B., Hoffmann, A., Stanek, J., Rampp, M., Grimm-Lebsanft, B., Biebl, F., et al. (2018). Transferring the entatic-state principle to copper photochemistry. Nat. Chem. 10, 355–362. doi: 10.1038/nchem.2916
Drienovská, I., Alonso-Cotchico, L., Vidossich, P., Lledós, A., Maréchal, J.-D., and Roelfes, G. (2017). Design of an enantioselective artificial metallo-hydratase enzyme containing an unnatural metal-binding amino acid. Chem. Sci. 8, 7228–7235. doi: 10.1039/C7SC03477F
Drienovská, I., Mayer, C., Dulson, C., and Roelfes, G. (2018). A designer enzyme for hydrazone and oxime formation featuring an unnatural catalytic aniline residue. Nat. Chem. 10, 946–952. doi: 10.1038/s41557-018-0082-z
Engelhard, D. M., Meyer, A., Berndhäuser, A., Schiemann, O., and Clever, G. H. (2018a). Di-copper(ii) DNA G-quadruplexes as EPR distance rulers. Chem. Commun. 54, 7455–7458. doi: 10.1039/C8CC04053B
Engelhard, D. M., Nowack, J., and Clever, G. H. (2017). Copper-induced topology switching and thrombin inhibition with telomeric DNA G-quadruplexes. Angew. Chem. Int. Ed. 56, 11640–11644. doi: 10.1002/anie.201705724
Engelhard, D. M., Pievo, R., and Clever, G. H. (2013). Reversible stabilization of transition-metal-binding DNA G-quadruplexes. Angew. Chem. Int. Ed. 52, 12843–12847. doi: 10.1002/anie.201307594
Engelhard, D. M., Stratmann, L. M., and Clever, G. H. (2018b). Structure–property relationships in CuII-binding tetramolecular G-quadruplex DNA. Chem. Eur. J. 24, 2117–2125. doi: 10.1002/chem.201703409
Ghosh, T., Ghosh, P., and Maayan, G. (2018). A copper-peptoid as a highly stable, efficient, and reusable homogeneous water oxidation electrocatalyst. ACS Catal. 8, 10631–10640. doi: 10.1021/acscatal.8b03661
Greenblatt, H. M., Feinberg, H., Tucker, P. A., and Shoham, G. (1998). Carboxypeptidase a: native, zinc-removed and mercury-replaced forms. Acta Cryst. D54, 289–305. doi: 10.1107/S0907444997010445
Halcrow, M. A. (2012). Jahn–Teller distortions in transition metal compounds, and their importance in functional molecular and inorganic materials. Chem. Soc. Rev. 42, 1784–1795. doi: 10.1039/C2CS35253B
Hänsel-Hertsch, R., Antonio, M., and Balasubramanian, S. (2017). DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell. Bio. 18, 279–284. doi: 10.1038/nrm.2017.3
Holm, R. H., Kennepohl, P., and Solomon, E. I. (1996). Structural and functional aspects of metal sites in biology. Chem. Rev. 96, 2239–2314. doi: 10.1021/cr9500390
Jeschek, M., Reuter, R., Heinisch, T., Trindler, C., Klehr, J., Panke, S., et al. (2016). Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 537, 661–665. doi: 10.1038/nature19114
Kanady, J. S., Tsui, E. Y., Day, M. W., and Agapie, T. (2011). A synthetic model of the Mn3Ca subsite of the oxygen-evolving complex in photosystem II. Science 333, 733–736. doi: 10.1126/science.1206036
Katz, S. (1963). The reversible reaction of Hg (II) and double-stranded polynucleotides a step-function theory and its significance. Biochim. Biophys. Acta 68, 240–253. doi: 10.1016/0926-6550(63)90435-3
Key, H. M., Dydio, P., Clark, D. S., and Hartwig, J. F. (2016). Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 534, 534–537. doi: 10.1038/nature17968
Knight, A. S., Kulkarni, R. U., Zhou, E. Y., Franke, J. M., Miller, E. W., and Francis, M. B. (2017). A modular platform to develop peptoid-based selective fluorescent metal sensors. Chem. Commun. 53, 3477–3480. doi: 10.1039/C7CC00931C
Koehntop, K. D., Emerson, J. P., and Que, L. (2005). The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J. Biol. Inorg. Chem. 10, 87–93. doi: 10.1007/s00775-005-0624-x
Lecours, M. J., Marchand, A., Anwar, A., Guetta, C., Hopkins, S. W., and Gabelica, V. (2017). What stoichiometries determined by mass spectrometry reveal about the ligand binding mode to G-quadruplex nucleic acids. Biochim. Biophys. Acta 1861, 1353–1361. doi: 10.1016/j.bbagen.2017.01.010
Litman, Z. C., Wang, Y., Zhao, H., and Hartwig, J. F. (2018). Cooperative asymmetric reactions combining photocatalysis and enzymatic catalysis. Nature 560, 355–359. doi: 10.1038/s41586-018-0413-7
Lu, Y., Yeung, N., Sieracki, N., and Marshall, N. M. (2009). Design of functional metalloproteins. Nature 460, 855–862. doi: 10.1038/nature08304
Mandal, S., Hebenbrock, M., and Müller, J. (2016). A dinuclear mercury(II)-mediated base pair in DNA. Angew. Chem. Int. Ed. 55, 15520–15523. doi: 10.1002/anie.201608354
Mandal, S., and Müller, J. (2017). Metal-mediated DNA assembly with ligand-based nucleosides. Curr. Opin. Chem. Biol. 37, 71–79. doi: 10.1016/j.cbpa.2017.01.019
Mayer, C., Dulson, C., Reddem, E., Thunnissen, A. W., and Roelfes, G. (2019). Directed evolution of a designer enzyme featuring an unnatural catalytic amino acid. Angew. Chem. Int. Ed. 58, 2083–2087. doi: 10.1002/anie.201813499
Miyoshi, D., Karimata, H., Wang, Z.-M., Koumoto, K., and Sugimoto, N. (2007). Artificial G-wire switch with 2,2′-bipyridine units responsive to divalent metal ions. J. Am. Chem. Soc. 129, 5919–5925. doi: 10.1021/ja068707u
Neidle, S. (2017). Quadruplex nucleic acids as targets for anticancer therapeutics. Nat. Rev. Chem. 1:0041. doi: 10.1038/s41570-017-0041
Punt, P. M., and Clever, G. H. (2019a). Imidazole-modified G-quadruplex DNA as metal-triggered peroxidase. Chem. Sci. 10, 2513–2518. doi: 10.1039/C8SC05020A
Punt, P. M., and Clever, G. H. (2019b). Tailored transition-metal coordination environments in imidazole-modified DNA G-quadruplexes. Chem. Eur. J. 25, 13987–13993. doi: 10.1002/chem.201903445
Raven, J. A., Evans, M. C., and Korb, R. E. (1999). The role of trace metals in photosynthetic electron transport in O2-evolving organisms. Photosynth. Res. 60, 111–150. doi: 10.1023/A:1006282714942
Rioz-Martínez, A., Oelerich, J., Ségaud, N., and Roelfes, G. (2016). DNA-accelerated catalysis of carbene-transfer reactions by a DNA/cationic iron porphyrin hybrid. Angew. Chem. Int. Ed. 55, 14136–14140. doi: 10.1002/anie.201608121
Roelfes, G., and Feringa, B. L. (2005). DNA-based asymmetric catalysis. Angew. Chem. Int. Ed. 44, 3230–3232. doi: 10.1002/anie.200500298
Rubino, J. T., and Franz, K. J. (2012). Coordination chemistry of copper proteins: how nature handles a toxic cargo for essential function. J. Inorg. Biochem. 107, 129–143. doi: 10.1016/j.jinorgbio.2011.11.024
Samuel, A. P., Co, D. T., Stern, C. L., and Wasielewski, M. R. (2010). Ultrafast photodriven intramolecular electron transfer from a zinc porphyrin to a readily reduced diiron hydrogenase model complex. J. Am. Chem. Soc. 132, 8813–8815. doi: 10.1021/ja100016v
Sandmann, N., Bachmann, J., Hepp, A., Doltsinis, N. L., and Müller, J. (2019). Copper(ii)-mediated base pairing involving the artificial nucleobase 3 H -imidazo[4,5- f]quinolin-5-ol. Dalton Trans. 48, 10505–10515. doi: 10.1039/C9DT02043H
Shook, R. L., and Borovik, A. (2010). Role of the secondary coordination sphere in metal-mediated dioxygen activation. Inorg. Chem. 49, 3646–3660. doi: 10.1021/ic901550k
Smith, N. M., Amrane, S., Rosu, F., Gabelica, V., and Mergny, J.-L. (2012). Mercury–thymine interaction with a chair type G-quadruplex architecture. Chem. Commun. 48, 11464–11466. doi: 10.1039/c2cc36481f
Tanaka, K., Clever, G. H., Takezawa, Y., Yamada, Y., Kaul, C., Shionoya, M., et al. (2006). Programmable self-assembly of metal ions inside artificial DNA duplexes. Nat. Nanotechnol. 1, 190–194. doi: 10.1038/nnano.2006.141
Tanaka, K., Yamada, Y., and Shionoya, M. (2002). Formation of silver(I)-mediated DNA duplex and triplex through an alternative base pair of pyridine nucleobases. J. Am. Chem. Soc. 124, 8802–8803. doi: 10.1021/ja020510o
Valdez, C. E., Smith, Q. A., Nechay, M. R., and Alexandrova, A. N. (2014). Mysteries of metals in metalloenzymes. Acc. Chem. Res. 47, 3110–3117. doi: 10.1021/ar500227u
Waldron, K. J., Rutherford, J. C., Ford, D., and Robinson, N. J. (2009). Metalloproteins and metal sensing. Nature 460, 823–830. doi: 10.1038/nature08300
Wang, J., Yue, L., Li, Z., Zhang, J., Tian, H., and Willner, I. (2019). Active generation of nanoholes in DNA origami scaffolds for programmed catalysis in nanocavities. Nat. Commun. 10:4963. doi: 10.1038/s41467-019-12933-9
Wu, S., Zhou, Y., Rebelein, J. G., Kuhn, M., Mallin, H., Zhao, J., et al. (2019). Breaking symmetry: engineering single-chain dimeric streptavidin as host for artificial metalloenzymes. J. Am. Chem. Soc. 141, 15869–15878. doi: 10.1021/jacs.9b06923
Xu, Y., Suzuki, Y., Lönnberg, T., and Komiyama, M. (2009). Human telomeric DNA sequence-specific cleaving by G-quadruplex formation. J. Am. Chem. Soc. 131, 2871–2874. doi: 10.1021/ja807313x
Zhang, L., Peritz, A., and Meggers, E. (2005). A simple glycol nucleic acid. J. Am. Chem. Soc. 127, 4174–4175. doi: 10.1021/ja042564z
Zhang, L., Peritz, A. E., Carroll, P. J., and Meggers, E. (2006). Synthesis of glycol nucleic acids. Synthesis 2006, 645–653. doi: 10.1055/s-2006-926313
Keywords: bioinorganic chemistry, coordination chemistry, DNA, G-quadruplex, DNAzymes
Citation: Punt PM, Stratmann LM, Sevim S, Knauer L, Strohmann C and Clever GH (2020) Heteroleptic Coordination Environments in Metal-Mediated DNA G-Quadruplexes. Front. Chem. 8:26. doi: 10.3389/fchem.2020.00026
Received: 14 November 2019; Accepted: 09 January 2020;
Published: 29 January 2020.
Edited by:
James Tucker, University of Birmingham, United KingdomReviewed by:
Sriram Kanvah, Indian Institute of Technology Gandhinagar, IndiaMiguel Angel Aleman Garcia, Eindhoven University of Technology, Netherlands
Copyright © 2020 Punt, Stratmann, Sevim, Knauer, Strohmann and Clever. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guido H. Clever, Z3VpZG8uY2xldmVyQHR1LWRvcnRtdW5kLmRl
†These authors have contributed equally to this work