 Takahiro Hashishin1
Takahiro Hashishin1 Kazunori Miyamoto
Kazunori Miyamoto
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 04 February 2020
Sec. Organic Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00012
This article is part of the Research Topic New Hypervalent Iodine Reagents for Oxidative Coupling View all 10 articles
Stannylation of calcium carbide followed by Sn–hypervalent iodine(III) exchange reaction cleanly afforded the electrophilic ethynylating agent ethynyl(phenyl)-λ3-iodane in high yield. This two-step method uses very inexpensive materials and is readily operable without any special precautions.
Hypervalent ethynyl(phenyl)-λ3-iodane 1 is an efficient electrophilic ethynylating agent for a variety of nucleophiles (C, N, O, P, As, S, Se, and halides) in the presence or absence of transition metal catalysts (Figure 1A; Ochiai et al., 1990; Stang et al., 1990; Varvoglis, 1992; Ochiai, 2003; Waser, 2016; Yoshimura and Zhdankin, 2016). However, its synthetic utility is restricted by its high cost and heat/moisture-sensitive character: it gradually decomposes at room temperature in air (Ochiai et al., 2003; Yudasaka et al., 2019). Therefore, an inexpensive, rapid, and facile preparation method of 1 has long been highly desired. The current approach to the synthesis of 1 relies on electrophilic Si/Sn–I(III) exchange reaction on acetylenic carbon atoms, and has remained essentially unchanged since the early days. In 1990, Stang and Ochiai independently reported pioneering approaches for the synthesis of 1. Stang et al. prepared ethynyl(phenyl)(triflato)-λ3-iodane (1b) from ethynyl(tributyl)stannane (3) and Tf2O-activated iodosylbenzene (2) (Figure 1B; Stang et al., 1990). On the other hand, a two-step procedure for the synthesis of 1a via [β-(trimethylsilyl)ethynyl](phenyl)-λ3-iodane 5a was developed by Ochiai and co-workers (Figure 1C; Ochiai et al., 1990). Then, in 2011, Kitamura reported another practical stepwise approach using PhI(OAc)2 (6) and bis(tert-butyldimethylsilyl)acetylene (4b) (Figure 1D; Kitamura et al., 2011). Although these methods provide short-step approaches to 1, they have several disadvantages from the viewpoint of cost/safety of reagents. In particular, 3 is still expensive and concentrated aqueous HF is notoriously toxic (Mckee et al., 2014). We report here a safe, low-cost, two-step method for the synthesis of 1a using calcium carbide CaC2 (7) as an ethynyl group source.
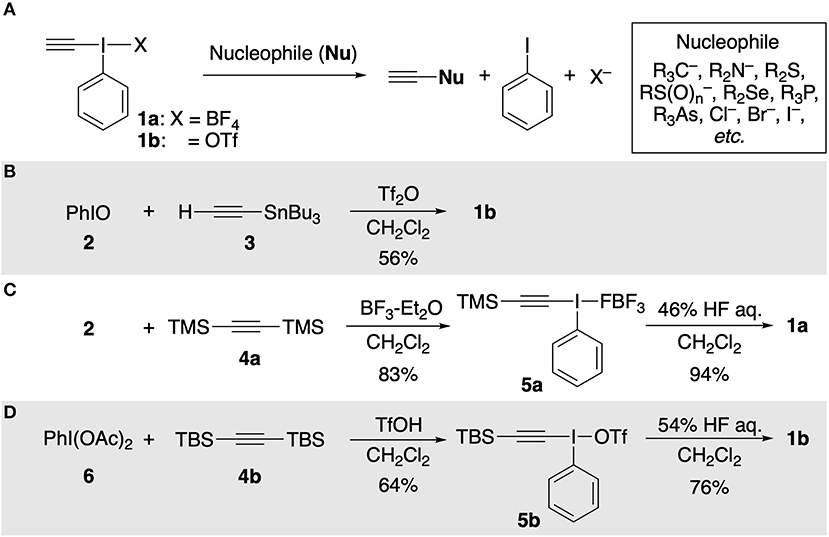
Figure 1. (A) Electrophilic ethynylation of nucleophile with 1. (B) Stang's approach. (C) Ochiai's approach. (D) Kitamura's approach.
Calcium carbide CaC2 (7) is a widely utilized industrial material that is very inexpensive (0.05 $/g; 3.2 $/mol) (Sigma-Aldrich Co., LLC.). However, its synthetic use has been limited by its poor solubility: it is not soluble in non-reactive common organic solvents (Barber and Sloan, 1961). In recent decades, several approaches using strongly coordinating solvents (DMSO, DMF, etc.) and/or coordinating additives (F−, , HO−, water, etc.) have been reported, for which 7 served as a practical source (Rodygin et al., 2016). It occurred to us that the treatment of 7 with inexpensive chloro(tributyl)stannane (8) (0.3 $/g; 98 $/mol) (Sigma-Aldrich Co., LLC.), followed by Sn–I(III) exchange reaction, might provide more convenient and straightforward access to 1.
Calcium carbide (~80%), ethynyl(tributyl)stannane (95%), bis(tributylstannyl)acetylene (95%) were purchased from Sigma Aldrich and used as received. Chloro(tributyl)stannane (>97%), and boron trifluoride etherate (>98%) were purchased from TCI Japan and used as received. (Diacetoxyiodo)benzene (98+%) was purchased from FUJIFILM Wako Pure Chemical or prepared according to the literature (Watanabe et al., 2018). Potassium carbonate (>99.5%) was purchased from FUJIFILM Wako Pure Chemical and used as received. Iodosylbenzene was prepared from (diacetoxyiodo)benzene according to the literature (Sharefkin and Saltzman, 1963). Anhydrous grade of dimethyl sulfoxide, tetrahydrofuran, and N, N-dimethylformamide was purchased from Kanto Chemical and degassed by purging with argon and/or dried with a solvent purification system containing a one-meter column of activated alumina.
NMR spectra were obtained on a Bruker AVANCE 500 spectrometer. Chemical shifts are expressed in δ (ppm) values. 1H NMR, 13C NMR, and 19F NMR spectra were referenced to tetramethylsilane (0 ppm), CHCl3 (7.26, 77.2 ppm), CHD2CN (1.94 ppm), and CD3CN (118.3 ppm), CFCl3 (0 ppm) as internal standards. IR spectra were obtained on a JASCO FT/IR-4700 spectrometer. Kieselgel 60 (Merck, 230-400 mesh) was used for column chromatography.
These reactions were carried out in a two-necked round bottom flask. In a typical reaction: To a stirred suspension of well-ground calcium carbide (7) (2.48 g, 38.7 mmol) in DMSO (20 mL) were added chloro(tributyl)stannane (8) (3.26 g, 10.0 mmol) and water (0.40 mL, 22.2 mmol) at room temperature under argon. The resulting grayish suspension was warmed to 80°C for 1 h (the disappearance of 8 was monitored by GCMS analysis), then allowed to cool to room temperature. Hexane was added to it, and the organic phase was filtered under reduced pressure through a K2CO3-silica gel (1:9) mixture and transferred to a separating funnel. The combined organic phase was washed with water several times, then filtered, and the filtrate was concentrated under reduced pressure to give an oil, which was further purified by chromatography (ø5 mm) on a column packed with K2CO3-silica gel (1:9). Elution with hexane gave a pale yellow oil (2.51 g). 1H NMR analysis (mesitylene as an internal standard) showed the formation of a mixture of ethynyl(tributyl)stannane (3) (3.4 mmol, 34%) and bis(tributylstannyl)acetylene (9) (2.3 mmol, 45%). Capillary GC analysis (n-dodecane as an internal standard; Bruker BR-5ms column 0.25 mm × 30 m, 100°C) showed different yields of 3 (3.6 mmol, 36%) and 9 (1.60 mmol, 32%), probably reflecting partial decomposition of 9 during the GC analysis. This product mixture was used directly for the synthesis of 1a. Spectroscopic data of 3 and 9 were compared to the authentic samples synthesized according to the literatures (Supplementary Material).
Ethynyl(tributyl)stannane (3) (Stille and Simpson, 1987): a colorless oil; 1H NMR (CDCl3, 500 MHz) δ 2.20 (s, lH), 1.61–1.54 (m, 6H), 1.40–1.29 (m, 6H), 1.02 (t, J = 8.2 Hz, 6H), 0.91 (t, J = 7.3 Hz, 9H). 13C NMR (CDCl3, 125 MHz) δ 96.9, 89.1, 29.0, 27.1, 13.8, 11.2.
Bis(tributylstannyl)acetylene (9) (Brown and Eichler, 2011): a colorless oil; 1H NMR (CDCl3, 500 MHz) δ 1.69–1.48 (m, 12H), 1.40–1.29 (m, 12H), 1.13–0.94 (m, 12H), 0.90 (t, J = 7.3 Hz, 18H). 13C NMR (CDCl3, 125 MHz) δ 116.6, 29.1, 27.1, 13.8, 11.4.
The same procedure was adopted for other conditions shown in Table 1.
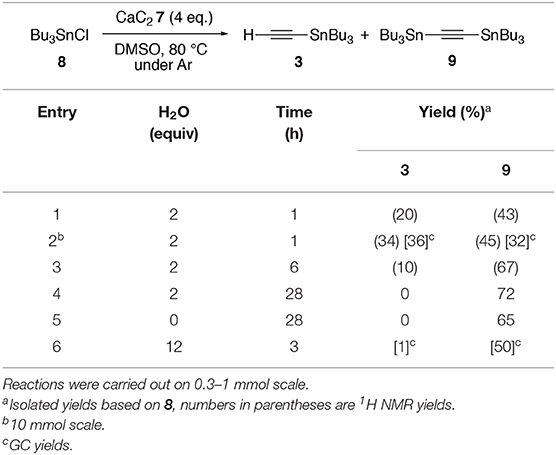
Table 1. Stannylation of CaC2 7 with Bu3SnCl 8.
To a stirred solution of (diacetoxyiodo)benzene (6) (159 mg, 0.49 mmol) in dichloromethane (1 mL) was added BF3-Et2O (130 μL, 1.04 mmol) at −78°C, and then a 60:40 mixture of stannanes 3 and 9 (301 mg, 0.70 mmol) was slowly added. The reaction mixture was stirred at the same temperature for 1 h, then allowed to warm to room temperature, and the solvent was removed under reduced pressure. The resulting pale yellow solid was washed several times with hexane and Et2O at 0°C to give 1a (114 mg, 73%).
Ethynyl(phenyl)(tetrafluoroborato)-λ3-iodane (1a) (Ochiai et al., 1990): a white solid; IR (ATR-FTIR) ν 3,241, 3,080, 2,056, 1,480, 1,442, 1,170–840, 735, 672 cm−1; 1H NMR (CD3CN, 500 MHz) δ 8.18 (d, J = 8.5 Hz, 2H), 7.82 (t, J = 7.6 Hz, 1H), 7.64 (dd, J = 8.5, 7.6 Hz, 2H), 3.89 (s, 1H). 19F NMR (CD3CN, 470 MHz) δ −151.8 (s, 4F). 13C NMR (CDCl3, 125 MHz) δ 136.1, 134.6, 133.8, 116.5, 99.0, 26.7 (see also Supplementary Material).
To a stirred solution of iodosylbenzene (2) (77.6 mg, 0.35 mmol) in dichloromethane (0.7 mL) was added BF3-Et2O (100 μL, 0.77 mmol) at −78°C, and then a 38:62 mixture of stannanes 3 and 9 (218 mg, 0.46 mmol) was slowly added. The reaction mixture was stirred at the same temperature for 1 h, then allowed to warm to room temperature, and the solvent was removed under reduced pressure. The resulting pale brown solid was washed several times with hexane and Et2O at 0°C to give 1a (90.4 mg, 81%); 1H NMR analysis shows this product contained a small amount of impurities. 1H NMR yield: 67% (mesitylene as an internal standard).
We commenced our study by trapping CaC2 7 with 8. Exposure of well-ground 7 (4 equiv) to 8 in DMSO at room temperature did not give any alkynylstannanes. Addition of small amount of water (2 equiv), which has been reported to be effective for the electrophilic trapping of 7, was fruitless (Rodygin et al., 2016). On the other hand, heating at 80°C resulted in smooth consumption of 8 and after 1 h, a 6:4 mixture of 3 and bis(tributylstannyl)acetylene (9) was obtained in 79% yield (Table 1, entry 2). The ratio of 3 and 9 has changed in a range of ca. 3:7–6:4 through several runs, partly due to the reaction scale and the surface area of 7 (entries 1 and 2). Use of longer reaction time increased the ratio of 9 (entries 3 and 4). Under the conditions, the addition of water did not significantly change the yield of 9, but it accelerated the bis-stannylation (entries 4–6). Interestingly, this stannylation did not occur in other aprotic solvents such as THF and DMF, even at elevated temperatures (≤110°C) (Cochran et al., 1990). It should be noted that these alkynylstannanes 3 and 9 could be separated from other organostannane impurities on a short column packed with K2CO3-silica gel (1:9) mixture (Harrowven et al., 2010). Other crystallogen analog, trimethylsilyl chloride did not afford corresponding ethynyl(trimethyl)silanes under optimized conditions, partly because of the more moisture sensitive character of silyl chloride.
Next, we focused on the synthesis of ethynyl-λ3-iodane 1a using a mixture of alkynylstannanes 3 and 9. After screening various reaction conditions, we found an efficient method. Exposure of a 6:4 mixture of 3 and 9 (obtained from the reaction shown in entry 1 in Table 1) to a combination of PhI(OAc)2 6 and BF3-Et2O in dichloromethane at −78°C resulted in smooth Sn–I(III) exchange, and after 1 h, 1a was selectively obtained in 73% yield (Figure 2A). The standard PhIO 2–BF3-Et2O system also afforded 1a in high yield. It should be emphasized that these methods do not require time-consuming work-up. Simple washing of the reaction mixture with hexane and Et2O by decantation gave pure 1a and a mixture of Bu3SnX-type organostannanes thus formed by I(III)–Sn exchange was recovered quantitatively in the supernatant. As we expected, these optimized conditions could also be applied to authentic 3 and 9 individually to provide 1a in moderate to high yields (Figure 2B). In these cases, the combination of I(III)–organostannane pairs (6–3 and 2–9) gave better yields of 1a than opposite pairs (6–9 and 2–3), although the reason remains unclear. From a mechanistic point of view, our results using 9 is somewhat surprising since the Sn–I(III) exchange of 9 with cyano(trifluoromethylsulfonyloxy)iodobenzene (10) selectively affords bis[phenyl(triflato)-λ3-iodanyl]acetylene (11) (Figure 2C; Stang and Zhdankin, 1990, 1991). The moderately electrophilic nature of iodine center of the intermediates such as PhI(OAc)2-BF3 (Izquierdo et al., 2016) or PhIO-BF3 (Ochiai, 2007) might be partly responsible for the selective formation of 1a.
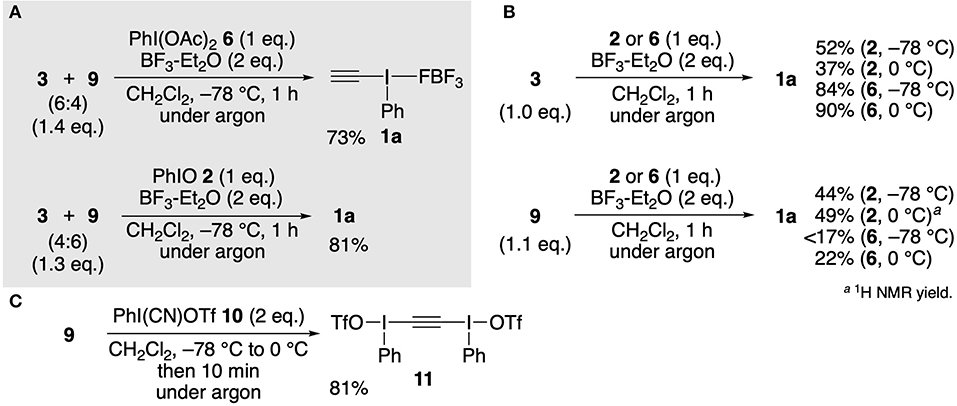
Figure 2. Synthesis of ethynyl(phenyl)-λ3-iodane 1a. (A) Reaction of a mixture of alkynylstannanes 3 and 9 obtained from CaC2 7. (B) Individual reactions of authentic 3 and 9. (C) Stang and Zhdankin's approach.
In summary, we have developed a safe and inexpensive two-step method for the synthesis of 1a using readily available CaC2 7 as an ethynyl group source. This method not only provides time-/cost-/labor-saving methodology to prepare unstable ethynyl-λ3-iodane 1, but also serves as an effective approach for synthetically useful but costly bis(stannyl)acetylene 9 (Brend'amour et al., 2018).
All datasets generated for this study are included in the article/Supplementary Material.
KM and MU conceived and designed the experiments and wrote the manuscript. TH and TO conducted the experiments. All authors participated in data analyses and discussions.
This work was supported by grants from JSPS KAKENHI (S) (17H06173), KAKENHI (B) (17H03017), NAGASE Science & Technology Development Foundation, and Sumitomo Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00012/full#supplementary-material
Barber, W. A., and Sloan, C. L. (1961). Solubility of calcium carbide in fused salt system. J. Phys. Chem. 65, 2026–2028. doi: 10.1021/j100828a025
Brend'amour, S., Gilmer, J., Bolte, M., Lerner, H. W., and Wagner, M. (2018). C-halogenated 9,10-diboraanthracenes: how the halogen load and distribution influences key optoelectronic properties. Chem. Eur. J. 24, 16910–16915. doi: 10.1002/chem.201804288
Brown, A. E., and Eichler, B. E. (2011). Symmetric diarylacetylenes: one-pot syntheses and solution photoluminescence. Tetrahedron Lett. 52, 1960–1963. doi: 10.1016/j.tetlet.2011.02.062
Cochran, J. C., Lemieux, R. P., Giacobbe, R. C., and Roitstein, A. (1990). Synthesis of bis-stannylacetylenes from calcium carbide; assisted by ultrasound. Synth. React. Inorg. Met. Org. Chem. 20, 251–261. doi: 10.1080/00945719008048132
Harrowven, D. C., Curran, D. P., Kostiuk, S. L., Wallis-Guy, I. L., Whiting, S., Stenning, K. J., et al. (2010). Potassium carbonate–silica: a highly effective stationary phase for the chromatographic removal of organotin impurities. Chem. Commun. 46, 6335–6337. doi: 10.1039/C0CC01328E
Izquierdo, S., Essafi, S., del Rosal, I., Vidossich, P., Pleixats, R., Vallribera, A., et al. (2016). Acid activation in phenyliodine dicarboxylates: direct observation, structures, and implications. J. Am. Chem. Soc. 138, 12747–12750. doi: 10.1021/jacs.6b07999
Kitamura, T., Morshed, M. H., Tsukada, S., Miyazaki, Y., Iguchi, N., and Inoue, D. (2011). Alkynylation of benzotriazole with silylethynyliodonium triflates. Regioselective synthesis of 2-ethynyl-2H-benzotriazole derivatives. J. Org. Chem. 76, 8117–8120. doi: 10.1021/jo2015467
Mckee, D., Thoma, A., Bailey, K., and Fish, J. (2014). A review of hydrofluoric acid burn management. Plast. Surg. 22, 95–98.
Ochiai, M. (2003). “Reactivities, properties and structures,” Topic in Current Chemistry, Vol. 224, ed T. Wirth (Cham: Springer), 5–68.
Ochiai, M. (2007). Stoichiometric and catalytic oxidations with hypervalent organo-λ3-iodanes. Chem. Rec. 7, 12–23. doi: 10.1002/tcr.20104
Ochiai, M., Ito, T., Takaoka, Y., Masaki, Y., Kunishima, M., Tani, S., et al. (1990). Synthesis of ethynyl(phenyl)iodonium tetrafluoroborate. A new reagent for ethynylation of 1,3-dicarbonyl compounds. J. Chem. Soc. Chem. Commun. 1990, 118–119. doi: 10.1039/C39900000118
Ochiai, M., Miyamoto, K., Suefuji, T., Sakamoto, S., Yamaguchi, K., and Shiro, M. (2003). Synthesis, characterization, and reaction of ethynyl(phenyl)-λ3-iodane complex with [18]crown-6. Angew. Chem. Int. Ed. 42, 2191–2194. doi: 10.1002/anie.200250866
Rodygin, K. S., Werner, G., Kucherov, F. A., and Ananikov, V. P. (2016). Calcium carbide: a unique reagent for organic synthesis and nanotechnology. Chem. Asian J. 11, 965–976. doi: 10.1002/asia.201501323
Sharefkin, J. G., and Saltzman, H. (1963). Iodosobenzene. Org. Synth. 43:60. doi: 10.15227/orgsyn.043.0060
Stang, P. J., Arif, A. M., and Crittell, C. M. (1990). Ethynyl(phenyl)iodonium triflate, [HCCIPh][OSO2CF3]: preparation, spectral properties, mechanism of formation and X-ray molecular structure. Angew. Chem. Int. Ed. Engl. 29, 287–288. doi: 10.1002/anie.199002871
Stang, P. J., and Zhdankin, V. V. (1990). Bis[phenyl[(perfluoroalkanesulfonyl)oxy]iodo]acetylene, PhI+CCI+Ph•2RF, and 1,4-bis[phenyl[(perfluoroalkanesulfonyl)oxy]iodo]-1,3-butadiyne, PhI+CC-CCI+Ph•2 RF. J. Am. Chem. Soc. 112, 6437–6438. doi: 10.1021/ja00173a064
Stang, P. J., and Zhdankin, V. V. (1991). Preparation and chemistry of PhI+CCI+Ph•2−OTf, bis[phenyl[[(trifluoromethyl)sulfonyl]oxy]iodo]acetylene, a novel difunctional acetylene, bis(iodonium) species and a stable C2-transfer agent. J. Am. Chem. Soc. 113, 4571–4576. doi: 10.1021/ja00012a028
Stille, J. K., and Simpson, J. H. (1987). Stereospecific palladium-catalyzed coupling reactions of vinyl iodides with acetylenic tin reagents. J. Am. Chem. Soc. 109, 2138–2152. doi: 10.1021/ja00241a035
Waser, J. (2016). “Alkynylation with hypervalent iodine reagents,” Topic in Current Chemistry, Vol. 373, ed T. Wirth (Cham: Springer), 187–222.
Watanabe, A., Miyamoto, K., Okada, T., Asawa, T., and Uchiyama, M. (2018). Safer synthesis of (diacetoxyiodo)arenes using sodium hypochlorite pentahydrate. J. Org. Chem. 83, 14262–14268. doi: 10.1021/acs.joc.8b02541
Yoshimura, A., and Zhdankin, V. V. (2016). Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 116, 3328–3435.
Keywords: hypervalent, iodine, stannane, calcium carbide, ethynyl
Citation: Hashishin T, Osawa T, Miyamoto K and Uchiyama M (2020) Practical Synthesis of Ethynyl(phenyl)-λ3-Iodane Using Calcium Carbide as an Ethynyl Group Source. Front. Chem. 8:12. doi: 10.3389/fchem.2020.00012
Received: 27 November 2019; Accepted: 07 January 2020;
Published: 04 February 2020.
Edited by:
Toshifumi Dohi, Ritsumeikan University, JapanReviewed by:
Akira Yoshimura, University of Minnesota Duluth, United StatesCopyright © 2020 Hashishin, Osawa, Miyamoto and Uchiyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazunori Miyamoto, a21peWFAbW9sLmYudS10b2t5by5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.