 Nuzhat Shehla1,2†Bin Li1,2†
Nuzhat Shehla1,2†Bin Li1,2† Liang Cao1Jianping Zhao3Yuqing Jian1Muhammad Daniyal1Atia-tul Wahab4
Liang Cao1Jianping Zhao3Yuqing Jian1Muhammad Daniyal1Atia-tul Wahab4 Ikhlas A. Khan3
Ikhlas A. Khan3 Duan-fang Liao1
Duan-fang Liao1 Atta-ur Rahman2
Atta-ur Rahman2 M. Iqbal Choudhary1,2,4*
M. Iqbal Choudhary1,2,4* Wei Wang1,2*
Wei Wang1,2*- 1TCM and Ethnomedicine Innovation and Development International Laboratory, Academician Atta-ur-Rahman Belt and Road Traditional Medicine Research Center, School of Pharmacy, Hunan University of Chinese Medicine, Changsha, China
- 2International Center for Chemical and Biological Sciences, H. E. J. Research Institute of Chemistry, University of Karachi, Karachi, Pakistan
- 3National Center for Natural Products Research, Research Institute of Pharmaceutical Sciences, University of Mississippi, Oxford, MS, United States
- 4Dr. Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan
Xuetonglactones A–F (1–6), six unreported highly oxidized lanostane- and cycloartane-type triterpenoids along with 22 known scaffolds (7–28) were isolated from the stems of Kadsura heteroclita (Roxb.) Craib. Compared with previous congeners, xuetonglactone A (1), possesses an unprecedented 20,21-α-epoxide, and xuetonglactone D (4) features an unusual 19-α-hydroperoxyl moiety. The structures and the absolute configurations of the compounds were established by extensive one- and two-dimensional NMR, and electronic circular dichroism (ECD) spectroscopic analysis, with those of 1 and 5 confirmed by single-crystal X-ray diffraction technique. Compounds 1 and 2 exhibited inhibition of iNOS activity in LPS-induced macrophages with IC50 values of 22.0, and 17.0 μg/mL, respectively. While compounds 6, 7, 8, and 24 showed potent cytotoxic activities against human cervical cancer cell lines (HeLa) with the IC50 values of 4.0, 5.8, 5.0, and 6.4 μM, and against human gastric cancer cells (BGC 823) with the IC50 values of 2.0, 5.0, 2.5, and 2.0 μM, respectively. Moreover, plausible biogenetic pathways of (1–6) were also proposed.
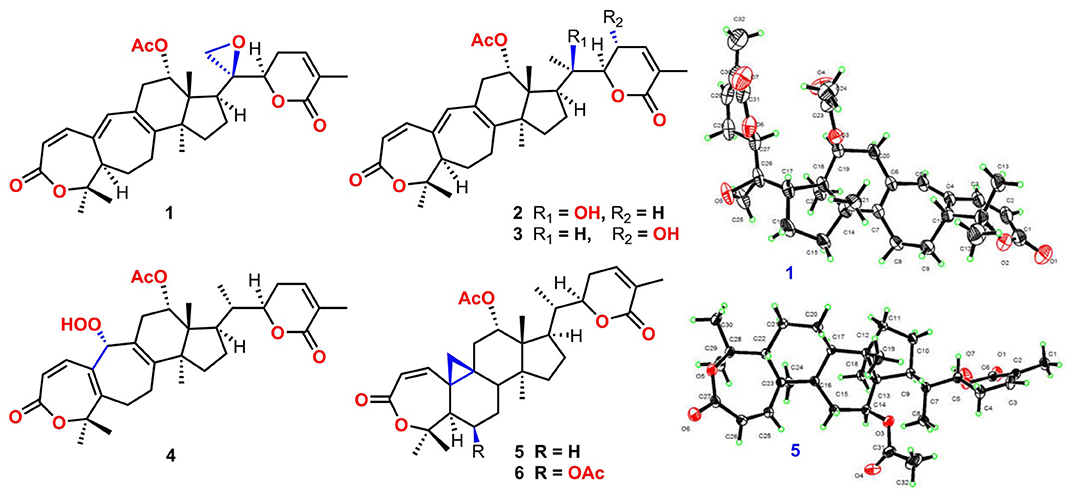
Graphical Abstract. Structures of 1–6.
Introduction
Schisandraceae family has contributed to the novel chemical scaffolds with an array of biological activities in past three decades. The family comprises of around fifty plant species belongs to genus Kadsura and Schisandra that are widely distributed in East, and Southeast Asia. The family has derived significant interest due to its highly oxygenated lanostane- and cycloartane-type triterpenoids, and dibenzocyclooctadiene lignans, along with schinortriterpenoids which are the characteristics isolates (Xiao et al., 2008; Shi et al., 2015). These constituents demonstrated potential pharmacological effects e.g., anti-hepatitis, anti-HIV, anti-inflammatory, anti-cancer, and inhibitory effect in cholesterol biosynthesis (Pu et al., 2008; Liu et al., 2014; Hu et al., 2015; Su et al., 2015).
Kadsura heteroclita (Roxb.) Craib. of the genus Kadsura is a climbing species primarily grows in Southwestern China, has a long history of its folk use in Traditional Chinese Medicine (TCM) (Pu et al., 2008; Liu et al., 2014). The stems of K. heteroclita traditionally known as “Xuetong” has long been consumed for the treatment of rheumatoid arthritis, traumatic injuries, deudenal ulcers, and cancers, particularly by Tujia people living in Wulin mountains area. In “Tujia” dialect “Xue” (blood) herbs are commonly used for the treatment of these diseases by activating the blood circulation, relieving pain and eliminating dampness for centuries (Liu et al., 2016, 2018; Cao et al., 2019). This study aimed to trace back the biologically active chemical constituents responsible for its clinical application contained within the plant species. Recently, we reported the identification of several new sesquiterpenoids, and lignanoids from K. heteroclita and other species of the same genus (Liu et al., 2018; Cao et al., 2019). Our previous pharmacological studies displayed this plant has very good anti-rheumatoid arthritis, anti-inflammatory, and analgesic effects (Yu H. et al., 2019; Yu H. H. et al., 2019).
In course of our continuous efforts to crack the immense diversity in structural frameworks with untapped biological potential, herein four new lanostane-type triterpenoids xuetonglactones A–D (1–4), and two cycloartane-type triterpenoids xuetonglactones E–F (5–6) possessing differently highly oxidized sites, were reported from K. heteroclita. Structurally, xuetonglactones A and D (1, 4) exhibited unique oxidized functionalities, featuring unprecedented 20,21-α-epoxy group in xuetonglactone A, and rare 19-α hydroperoxyl moiety in xuetonglactone D skeletons. The compounds were also evaluated for their cytotoxicity and anti-inflammatory activities. Hence, in this report, the details of isolation, structure elucidation, biological evaluation, and possible biosynthetic pathways of (1–6) were described. The spectroscopic data of 1–6 is presented in the Supplementary Material (Figures S1–S48).
Results and Discussion
Compound 1 was purified as a white crystalline solid, and the molecular formula was deduced to be C32H40O7 from HRESI-MS spectrum (positive ion mode) on the basis of [M + Na]+ ion at m/z 559.2671 (559.2672 calculated for C32H40O7 + Na) indicating 13 degrees of unsaturation. A 3,4-secocyclolanostane skeleton was deduced from the 1H- and 13C-NMR chemical shifts data with two α, β-unsaturated lactone rings, one of them being a seven membered ring in this triterpenoidal skeleton. This deduction was also supported by IR absorptions at 1,720, and 1,685 cm−1 for six- and seven-membered unsaturated lactone carbonyls and by UV absorptions (λmax) at 202, and 329 nm, respectively. The 1H-NMR data of 1 (Table 1) showed the presence of six tertiary methyl singlets (3H each, δH 0.71, 1.90, 1.53, 1.41, 1.39, and 2.12), four olefinic methines at δH 6.65 (d, J1, 2 = 12.3 Hz, H-1), 5.82 (d, J2, 1 = 12.0 Hz, H-2), 6.14 (s, H-19), and 6.51 (broad d, J24, 23 = 4.8 Hz, H-24), and two oxygenated methines at δH 5.14 (d, J12β, 11 = 7.4, H-12), and 4.49 (dd, J22, 23β = 12.7 Hz; J22, 23α = 3.7 Hz, H-22). The 13C-NMR data (Table 2) displayed 32 carbon signals attributed to six tertiary methyls (δC 18.6, 16.9, 26.2, 29.3, 27.9, and 21.6), eight methines including four olefinic (δC 143.2, 118.3, 141.7, and 137.6), and two oxygenated methines (δC 71.9, and 78.3), seven methylenes, and eight quaternary carbons, including four olefinic (δC 151.1, 126.4, 140.4, and 128.2), and two oxygenated (δC 80.3, and 58.1) quaternary carbons. Furthermore, it also showed the presence of three carbonyl signals (δC 171.0, δC 166.3, and δC 165.0) corresponding to an acetoxy (C-12), and two lactone (C-3, and C-26) moieties, respectively. The 1H- and 13C-NMR chemical shifts data of 1 showed resemblances with that of known compound heteroclitalactone D (17) (Wang et al., 2006b) with obvious distinctions observed for resonances at C-17, C-20, and C-21. The detailed analysis of the NMR data established the structure of compound 1 bearing unprecedented oxirane in the structure at C-20. The NMR data revealed the presence of an extra methylene (δC 46.88, C-21) with a characteristic proton doublet (δH 2.75; and 2.96, dd, J21a, 21b = 3.3 Hz each, H-21) which supported the presence of additional ring at C-20 as epoxide to fulfill the unsaturation demand in skeleton of 1. Furthermore, the up-field shift of quaternary carbon (δC 58.12) at C-20 suggested the presences of epoxide at this junction, which could be attributed to the steric shielding effect of the strained ring at this position. These assignments were unambiguously confirmed by HMBC experiments, in which the epoxide methylene protons appeared at δH 2.75, and 2.96 (dd, J21a, 21b = 3.3 Hz each, H-21) were correlated with C-20 and C-22, while C-17 methine proton at δH 3.55 (dd, J17, 16a = 10.9, J17, 16b = 7.5 Hz) was correlated with C-12, C-14, C-16, C-18, C-20, and C-21 (Figure 2). This oxidized strained ring along with the presence of three conjugated double bonds, and other oxygenated moieties in the skeleton further supported the structure of 1 is based on a highly oxidized cyclolanostane type triterpenoidal skeleton (Chen et al., 2001; Wang et al., 2006a). Chemical shifts assignments were made on the basis of HSQC, HMBC, and 1H-1H COSY experiments to get the planner structure of 1. The α-configuration of the C-12 (acetoxy group) was concluded based on the ROESY cross peaks between H-12 and CH3-18 (Figure 3). In the ECD spectrum, compound 1 showed a positive Cotton effect at 255 nm (Δε = + 5.64) which was similar to that of schiglausin A (Zou et al., 2012), indicating an R configuration of C-22. Combining the observed ROESY correlations of H-5 with CH3-30, CH3-30 with H-17, H-17 with H-22, the ECD spectrum, and the X-ray diffraction using Cu Kα radiation (Figure 4), the absolute stereochemistry of the seven chiral centers, were determined as 5S, 12S, 13R, 14S, 17R, 20S, and 22R. Thus, the structure of 1 was fully established as shown (Figure 1) and named xuetonglactone A.
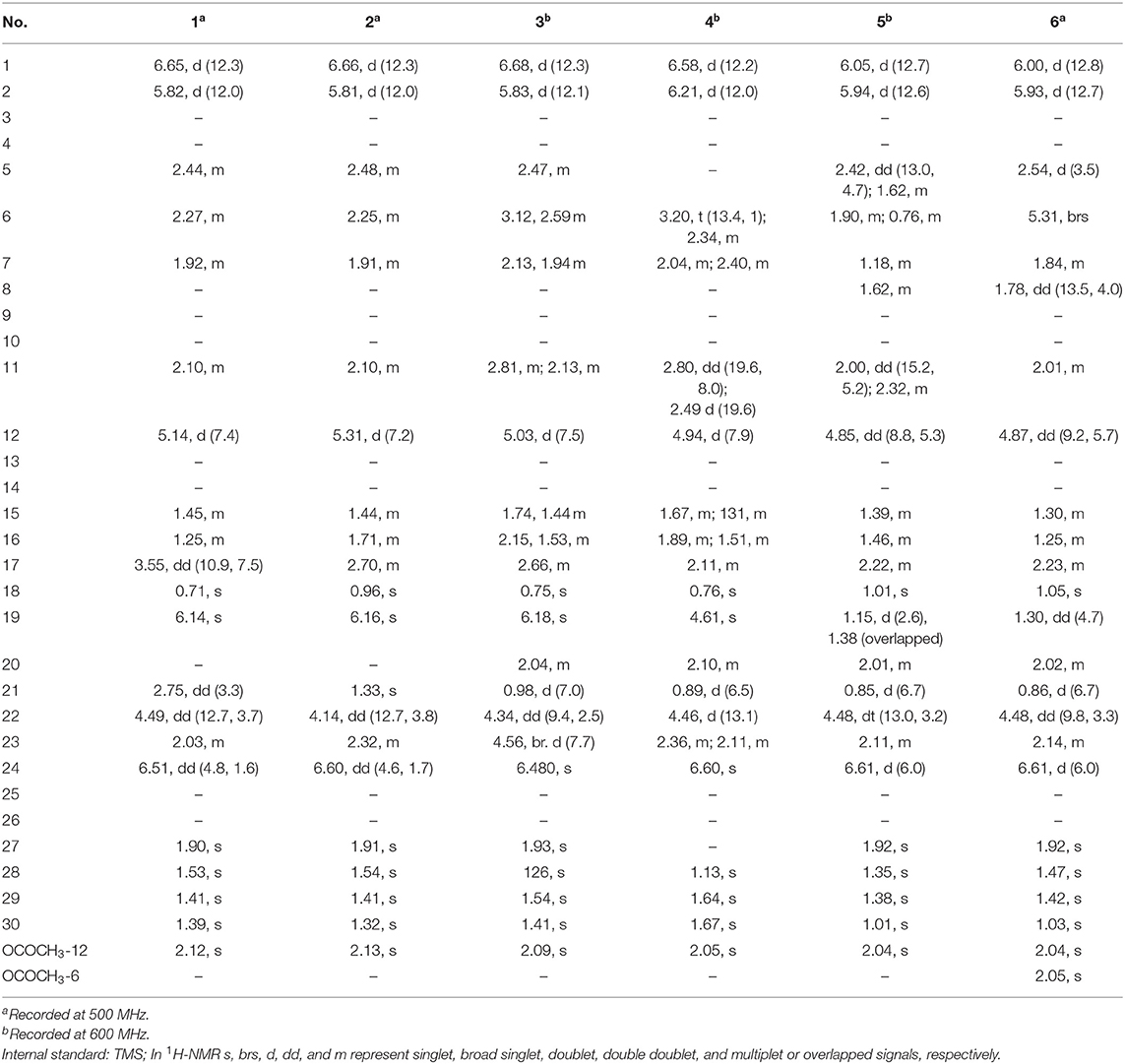
Table 1. 1H NMR data of 1–6 in CDCl3 (δH in ppm, J in Hz within the parenthesis).
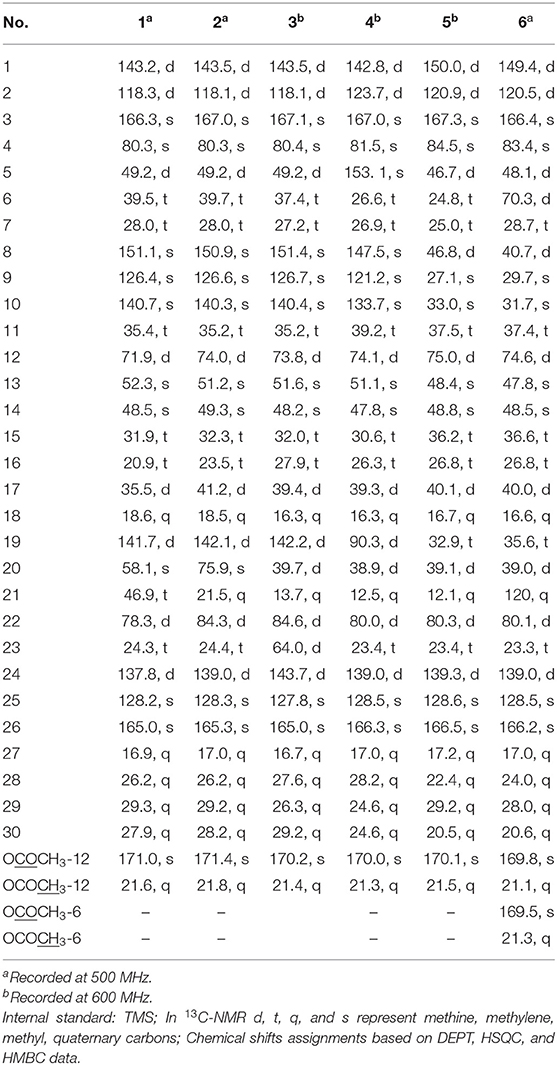
Table 2. 13C-NMR data of 1–6 in CDCl3 (δ in ppm).
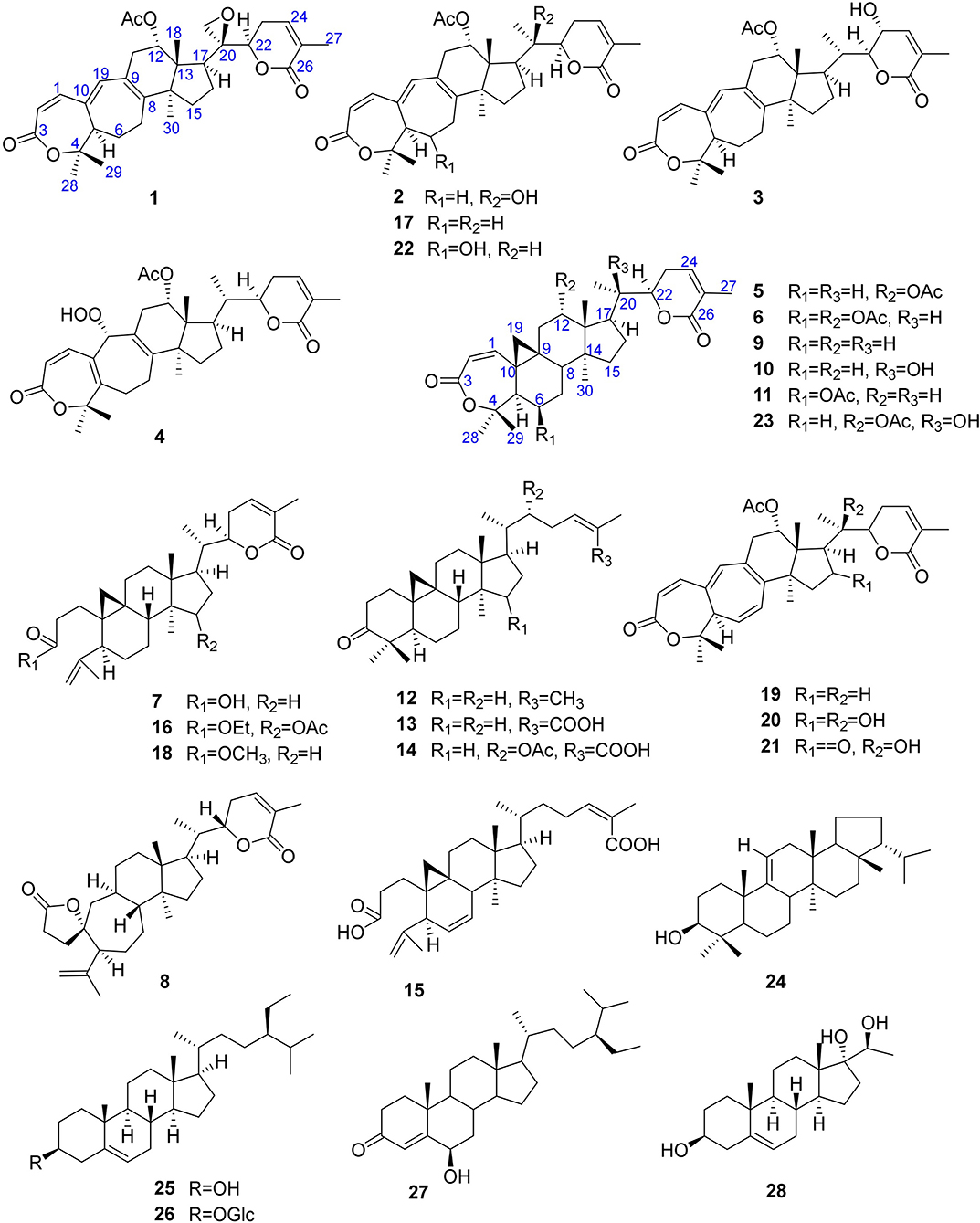
Figure 1. Structures of 1–28.
Compound 2 was obtained as a white amorphous solid. Its molecular formula was determined to be C32H42O7 based on [M + Na]+ ion at m/z 561.2828 (561.2829 calculated for C32H42NaO7) and [M + Cl]− peak at m/z 573.2612 (573.2619 calculated for C32H42O7 + Cl) from its HRESI-MS spectra (positive ion and negative ion modes), corresponding 12 degrees of unsaturation. The 1H-NMR chemical shifts data (Table 1) of 2 showed the presence of seven tertiary methyl singlets (3H each, δH 0.96, 1.32, 1.33, 1.41, 1.54, 1.91, and 2.13). By comparing the NMR chemical shifts data of compound 2 with that of 1 which has a methylene at position C-21, hence the presence of a tertiary methyl (δH 1.33, s, C-21) instead of epoxide, in the skeleton of 2 was inferred. Furthermore, relative configuration on ring C was same as of 1, based on similar ROESY correlations. While the absence of ROESY cross peaks between CH3-21 and CH3-18 thus appearance of cross peaks between CH3-21 and H-17/H-22 (Figure 3) suggested the β-orientation of OH-20. The assignment of the absolute configuration at C-22 was concluded to be R by the similar ECD measurement as that of 1. Therefore, the structure of 2 was established as shown (Figure 1) and named xuetonglactone B accordingly.
Compound 3 was obtained as a white amorphous solid and possess the same molecular formula with 2 as C32H42O7 based on [M + Na]+ ion at m/z 561.2836 (561.2828 calculated for C32H42O7 + Na) from its HRESI-MS spectrum (positive ion mode). The spectroscopic data of 3 (Tables 1, 2) was quite similar with those of 2 except for a secondary methyl (δH 0.98, J21, 20 = 7.0 Hz, H-21) instead of tertiary, and a broad doublet of oxymethine (δH 4.56, J23, 22 = 7.7 Hz, δC 64.0, H-23) instead of methylene protons in the skeleton of 3. These observations were also confirmed by HMBC cross peaks of H-20 with C-23, and that of H-22 with C-17, C-23, and C-21, and C-24. Furthermore, α-configuration of the acetoxy group was determined by the similar cross peaks between H-12 and CH3-18 in NOESY spectrum (Figure 3). A strong negative Cotton effect at 273 nm [Δε (273) = +0.41, MeOH] was observed in the experimental ECD spectrum of 3. The absolute configuration at C-22 was established as S by comparison of its ECD spectrum with analog colossolactone VIII (El Dine et al., 2008). Furthermore, the large coupling constant (9.4 Hz) between H-23 and H-22 indicated an anti-conformation of these two protons (Lakornwong et al., 2014), hence the absolute configuration of these two chiral centers were found to be 22S, and 23R. Consequently, compound 3 was determined and given the trivial name xuetonglactone C.
Compound 4 was isolated as yellow, amorphous solid. Its molecular formula was deduced as C32H42O8 on the basis of [M + Na]+ ion peak at m/z 577.2772 in the HRESI-MS (577.2777 calculated for C32H42O8 + Na), suggesting 12 degrees of unsaturation. Since the NMR resonances of 4 were similar to those of known compound 17 (Wang et al., 2006b) with some obvious discrepancies, therefore detailed comparison of the chemical shifts data revealed that both compounds have similar C/D/E rings system in the skeleton. However, different 1H- and 13C-NMR chemical shifts were observed for C-5, C-6, C-10, and C-19 suggesting the major skeletal difference of 4 corresponds to the rings A/B. The double bond between C-10 and C-19 in 17 shifted in between C-5 (δC 153.1) and C-10 (δC 133.7) in 4. This inference was further supported by the HMBC correlation of H-1 (δH 6.58, d, J1, 2 = 12.2 Hz), H-19 (δH 4.61, s), H3-29 (δH 1.64, s), and H3-30 (δH 1.67, s) with C-5, and of H-2 (δH 6.21, d, J2, 1 = 12.0 Hz) and H-19 with C-10. Furthermore, C-19 was connected with an unusual hydroperoxyl group, which was supported by the carbon resonance observed at δC 90.3 and the methine proton signal at δH 4.61 (1H, s) (Song et al., 2013), which was also confirmed by HMBC cross peaks of H-1 and H-11 to C-19, and H-19 to C-1, C-5, C-8, C-9, C-10, and C-11. α oriented 12-acetoxyl group was ascertained on the basis of NOESY cross peak between H β-12 and CH3-18, the significant NOESY correlations between Hβ-12 /Hβ-11 (δ H 2.82, JHβ−11/Hα−11 = 19.6, JHβ−11/Hβ−12 8.0 Hz), and Hβ-11/Hβ-19 indicated that the hydroperoxyl group should be α-orientated (Figure 3). The absolute configuration 22R could be delineated by similar ECD relationship (Wang et al., 2006b). Hence the structure of 4 was determined and given a trivial name xuetonglactone D accordingly.
Compound 5 was obtained as white crystalline. The HRESI-MS spectrum of 5 displayed [M + Na]+ ion peak at m/z 547.3047 (547.3036 calculated for C32H44O6 + Na), corresponding to the molecular formula of C32H44O6 indicative of 11 degrees of unsaturation. The IR spectrum showed absorptions at 1,721, and 1,679 cm−1 suggesting two lactone moieties in the skeleton. A 3,4-secocycloartane skeleton was deduced from 1H- and 13C-NMR chemical shifts data (Tables 1, 2). The 1H-NMR spectrum of 5 showed characteristic signals for the cyclopropyl methylene protons at δH 1.15 (d, J19a, 19b = 2.6 Hz), and 1.38 (overlapped), but unlike the known compound 7 (Liu and Huang, 1991), the downfield shift of cyclopropane protons was due to the deshielding effect of conjugated double bond in ring A. The 1H-NMR also displayed six tertiary methyl singlets (δH 1.01, 1.92, 1.35, 1.38, 1.01, and 2.04), and a secondary methyl proton at δH 0.85 (d, J21, 20β = 6.7 Hz, H-21). In 13C-NMR spectrum presence of 32 carbon signals (Table 2) could be assigned to six tertiary (δC 16.7, 17.2, 22.4, 29.2, 20.5, and 21.5), and a secondary methyl (δC 12.1), ten methines, seven methylenes, and nine quaternary carbons including three carbonyl (δC 167.3, 166.5, and 170.1), an oxygenated (δC 84.5), and an olefinic quaternary carbons (δC 128.6), as well as two oxygenated (δC 75.0, and 80.3), and three olefinic methine (δC 150.0, 120.9, and 139.3) signals. In comparison of 3, the major difference in 1H- and 13C-NMR chemical shifts data of 5 was obvious owing to absence of a pair of olefinic carbons and a proton in ring B, thus appearance of a C-19 cyclopropane ring proton doublets (δH 1.15, d, J19a, 19b = 2.6 Hz), and 1.38 (overlapped) corresponding to δC 32.9 instead in the structure. These distinctions were confirmed by the key correlations observed in 1H-1H COSY, and HMBC spectra. In HMBC spectrum CH2-19 protons correlated with C-1, C-10, C-5, C-8, C-9, and C10. Additionally, the 1H-1H COSY interactions between H-11/H12, H-20/H-21, H-20/H-22, H-22/H-23, and H-23/H-24 spin systems were consistent with the unambiguous spectral assignments based on HSQC, and HMBC interactions (Figure 2). Moreover, α-configurations of 12-OCOCH3 was determined by ROESY spectrum (Figure 3). Since a strong positive cotton effect at 272 nm (Δε = +3.31, MeOH) was observed, the absolute configuration at C-22 in 5 was consequently assigned as R-configuration by ECD measurement. The absolute configuration at C-22 in 5, named xuetonglactone E was also further confirmed by single crystal X-ray diffraction technique (Figure 4).
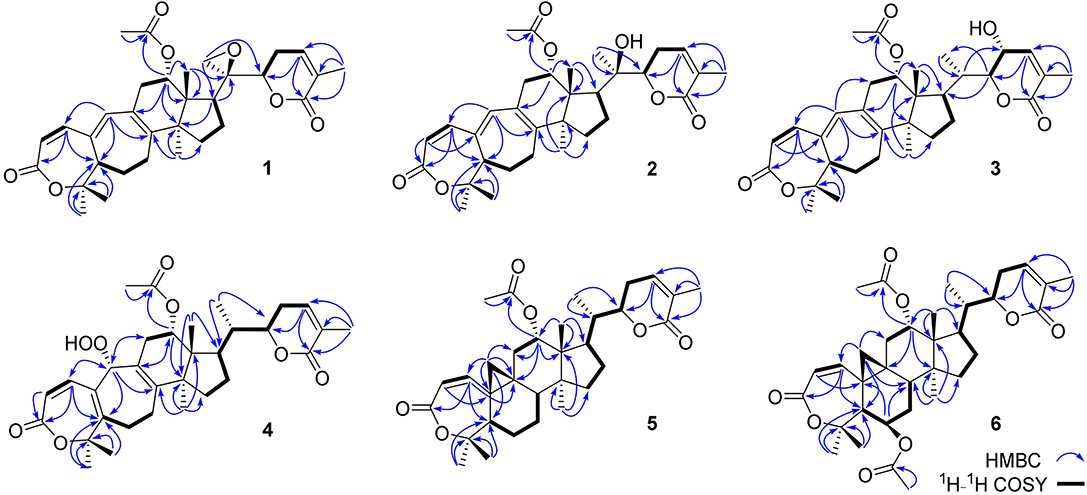
Figure 2. Key HMBC and 1H-1H COSY correlations of 1–6.
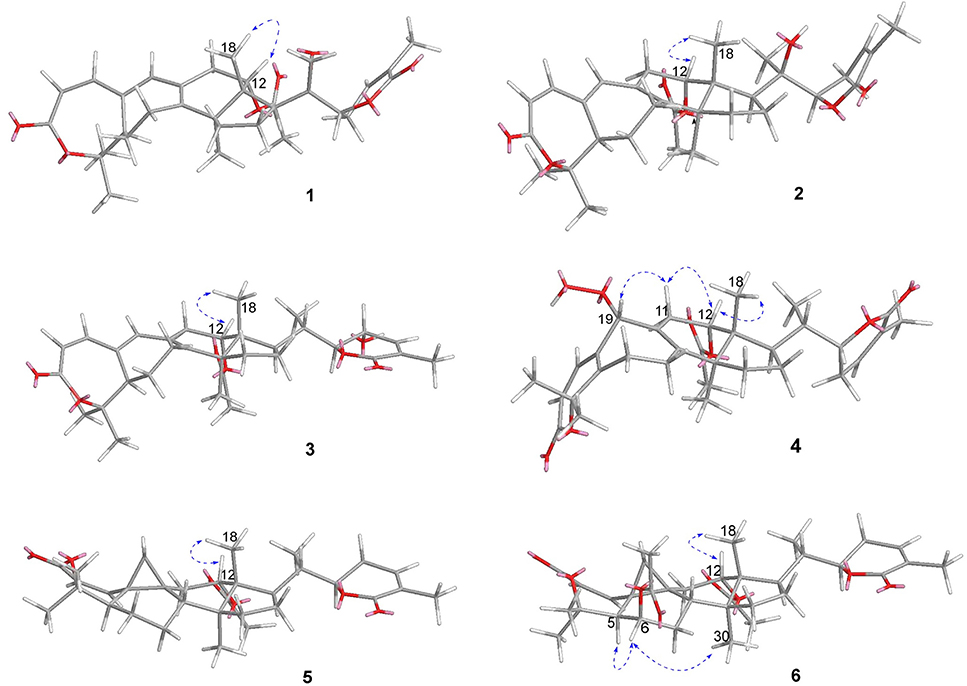
Figure 3. Key ROESY/NOESY correlations of 1–6.
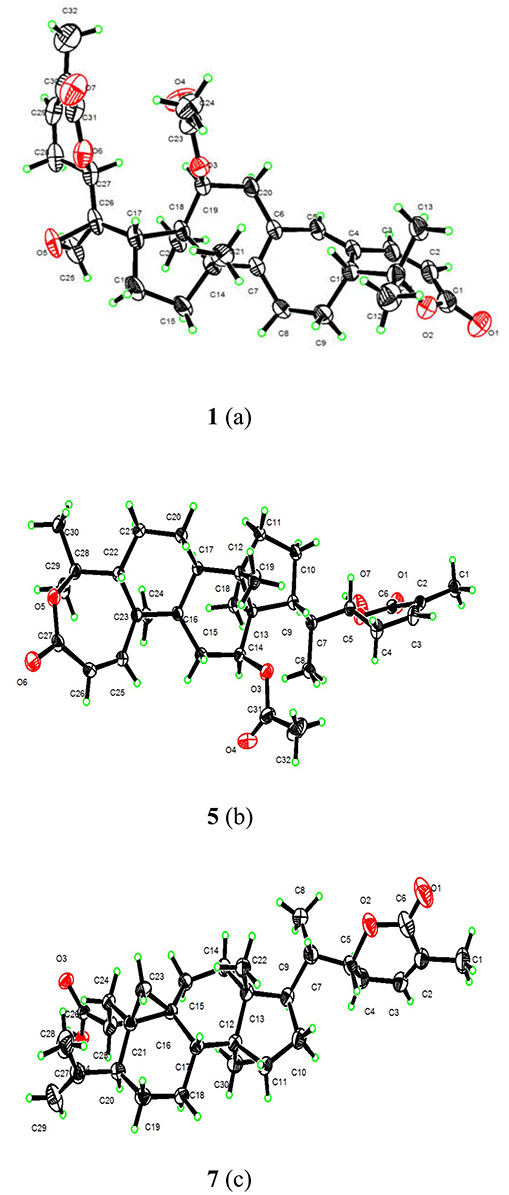
Figure 4. X-ray structures of 1(a), 5(b), and 7(c).
Compound 6 was purified as white amorphous solid. The molecular formula was assigned as C34H46O8 based on molecular ion peak at 617.2884 m/z [M + Cl]− (617.2881 calculated for C34H46O8 + Cl) in the HRESI-MS spectrum, which was indicative of 12 unsaturation degrees. The 13C-NMR spectrum of 6 exhibited 34 carbon resonances, attributed to seven tertiary, and a secondary methyls, 10 methines, 6 methylenes, and 10 quaternary carbons. Four ester carbonyls (δC 166.4, 166.2, 169.5, 169.8, and) corresponding to C-3, C-26, OCOCH3-6, and OCOCH3-12, respectively were observed in 6. The 1H- and 13C-NMR chemical shifts data of 6 (Tables 1, 2) were extremely similar to those of 5, and the major difference embodied in the chemical shift of C-6 (δH 5.31, δC 70.3) suggesting the appearance of an additional acetyl group at C-6 [δH 2.05 (δC 21.3, 169.5)], thus absence of a methylene, and appearance of an oxygenated methine in 6. Furthermore, configurations of 6-OCOCH3 and 12-OCOCH3 were determined based on the ROESY cross peaks of H-5/H-6/CH3-30/ and H-12/CH3-18, so β- 6-OCOCH3 and α-12-OCOCH3 were inferred (Figure 3). The ECD spectrum of 6 was the same with that of 5, hence C-22 was assigned to the R-configuration, thus the absolute structure was determined as shown (Figure 1), and named xuetonglactone F accordingly.
Xuetongsu (schisanlactone E, 7) (Liu and Huang, 1991) was isolated as colorless crystal. The X-ray diffraction data of 7 was reported for the first time in this report (Figure 4). It was the major compound in “Xuetong” (Wang et al., 2006c). Biosynthetically, it might be the precursor of compounds 1–6, through series of oxidative cleavage via Baeyer-Villiger oxidation, ring expansion, hydroxylation, cyclization, acetoxylation, and epoxidation steps yielded compounds 1–6. A plausible biogenetic route for 1–6 was proposed as shown in Figure 5.
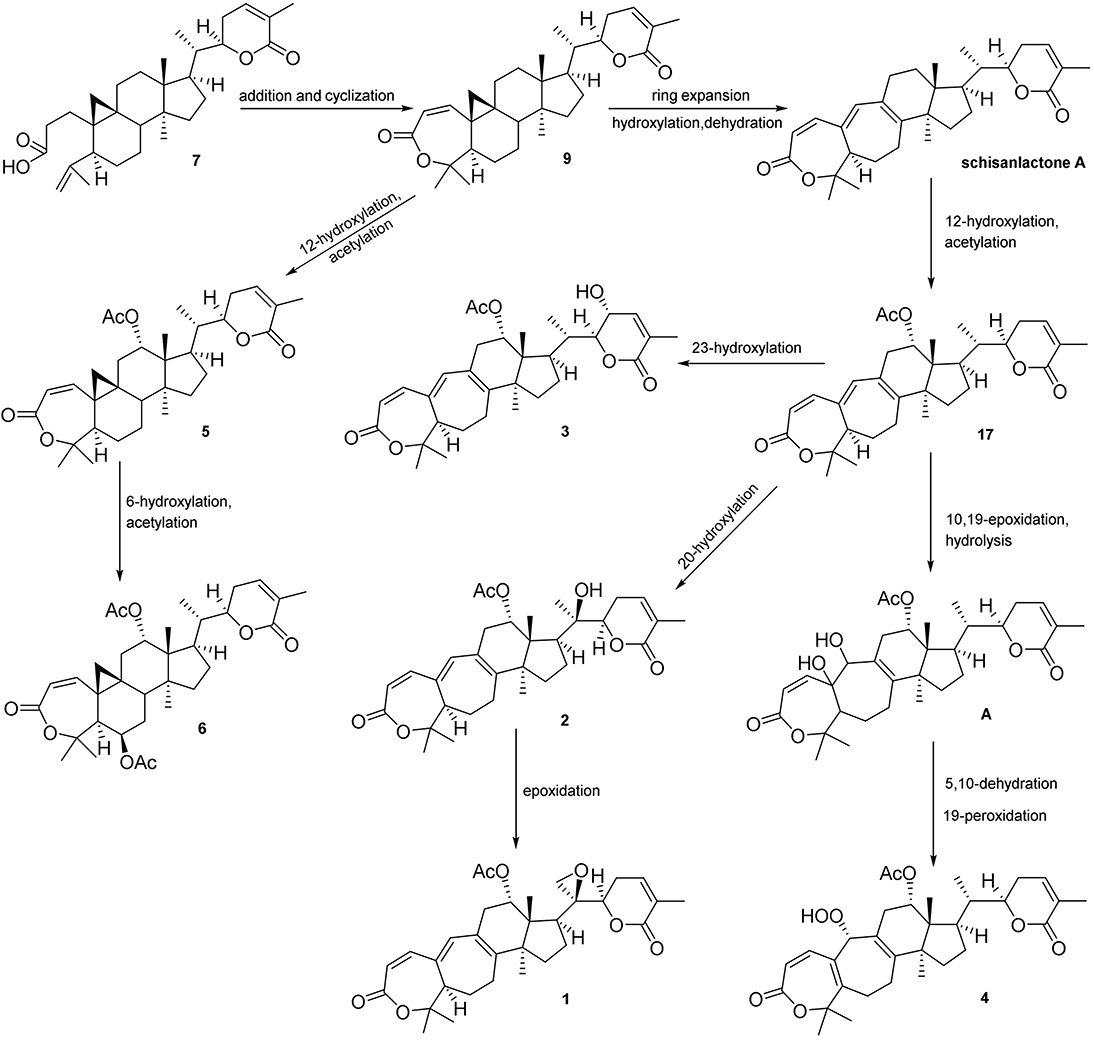
Figure 5. Plausible biosynthetic pathway for 1–6.
Twenty-two known analogous (7–28) were identified by analysis of their spectroscopic data with the reported data for xuetongsu (schisanlactone E, 7) 10.8 g (Liu and Huang, 1991), kadnanolactone A (8) 5.0 mg (Yang et al., 2010), schisanlactone B (9) 25.0 mg (Liu et al., 1983), kadsuphilactone B (10) 6.8 mg (Shen et al., 2005), schisanbilactone A (11) 4.0 mg (Ma et al., 2009), cycloartenone (12) 2.5 g (Pavanasisivam and Sultanbawa, 1973), schisandronic acid (13) 18.2 mg (Li et al., 2003), heteroclic acid (14) 5.6 mg (Wang et al., 2006b), changnanic acid (15) 18.0 mg (Liu and Huang, 1991), heteroclitalactone C (16) 5.5 mg (Wang et al., 2006b), heteroclitalactone D (17) 42.0 mg (Wang et al., 2006b), heteroclitalactone F (18) 7.0 mg (Wang et al., 2006b), heteroclitalactone G (19) 22.0 mg (Wang et al., 2007), heteroclitalactone I (20) 6.5 mg (Wang et al., 2007), heteroclitalactone K (21) 12.5 mg (Wang et al., 2007), heteroclitalactone L (22) 15.3 mg (Wang et al., 2007), heteroclitalactone M (23) 17.3 mg (Wang et al., 2007), sorghumol (24) 12.9 mg (Han et al., 2008), β-sitosterol (25) 100 mg (Chaturvedula and Prakash, 2012), daucosterol (26) 10.0 mg (Rahmana et al., 2009), 6β-hydoxysitostenone (27) 7.6 mg (Liang et al., 2015), and a steroid, trihydoxy pregnene (28) 13.0 mg (Deng et al., 2010). Their structures are presented in Figure 1.
Anti-inflammatory activity of the compounds 1–6 were evaluated for their inhibitory effects against iNOS, and NF-κB activation. Compounds 1 and 2 showed inhibition of iNOS activity in LPS-induced macrophages with the IC50 values of 22.0, and 17.0 μg/mL (Table 3), respectively, while parthenolide was used as control drug (Table 3), unfortunately no inhibitory effects found against NF-κB expression (Zhao et al., 2014). Additionally, cytotoxic activities of all the compounds against HeLa and BGC-823 cancer cell lines were also evaluated (Table 4). Compounds 6, 7, 8, and 24 showed strong cytotoxicities against HeLa cancer cell lines with the IC50 values of 4.0, 5.8, 5.0, and 6.4 μM, and against BGC 823 with the IC50 values of 2.0, 5.0, 2.5, and 2.0 μM, respectively, while compared with paclitaxel as positive control (Table 4; Hayon et al., 2003).

Table 3. Inhibition of iNOS activities of the tested compounds.
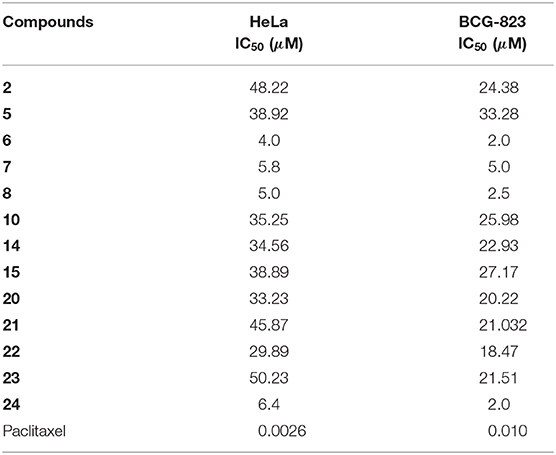
Table 4. Cytotoxicities of the tested compounds on HeLa and BCG-823 cancer cell lines.
Materials and Methods
General Experimental Procedure
Optical rotations were measured on a PerkinElmer 341-MC digital polarimeter, UV spectra were recorded on a TU-1900 spectrophotometer; A Hitachi 260-30 spectrometer was used for scanning IR spectroscopy; Experimental ECD spectra were recorded on a JASCO J-815 Circular Dichroism (CD) Spectropolarimeter; NMR spectra were performed on Bruker ARX-600 spectrometers, and on Agilent DD2-500 NMR spectrometer (500) MHz; HRESIMS were performed on a UPLC/xevo G2 Qtof spectrometer.
Preparative RP-HPLC was conducted on Agilent 1260 Infinity Series equipped with quaternary pump with Eclipse XDB-C18 (5 μm 9.4 × 250 mm) column at flow rate of 2.5 mL/min, at 210 nm UV detection using single wavelength detector. While the separation conditions were optimized on semi-preparative Agilent 1260 HPLC equipped with DAD detector by using Eclipse XDB-C18 (5 μm 4.6 × 250 mm) at flow rate of 1 mL/min. Thin layer Chromatography was performed on TLC aluminum sheets pre-coated with silica gel GF254 (EMD Chemicals, Merck KGaA, Dermstadt, Germany), visualized under UV light of 254 and 365 nm followed by 5% vanilline-H2SO4 reagent, and heat.
Plant Material
The stems of K. heteroclita (Roxb.) Craib. were collected from Hupingshan mountainous region at elevation of 5,971 ft in Shimen County, Hunan, P. R. China, and identified by Prof. Wei Wang from School of Pharmacy, Hunan University of Chinese Medicine. The voucher specimen number (CEL 1280-KH) was deposited to TCM and Ethnomedicine Innovation & Development International Laboratory, School of Pharmacy, Hunan University of Chinese Medicine, Changsha, Hunan, P. R. China.
Extraction and Isolation
Air dried plant material (100 kg) was extracted three times by using 80% ethanol in water under refluxed condition for 3 h each to produce viscous extract. This whole extract was then sequentially partitioned by liquid-liquid extraction (LLE) using non-polar, moderate to high polar organic solvents (pet-ether, chloroform, and n-butanol) against water to obtain wide range of metabolites.
Chloroform extract (353 g) was then subjected to silica gel column chromatography (CC) by gradient elution of solvent system PE-EtOAc (100% PE, 25% EtOAc in PE, 50% EtOAc/PE, 75% EtOAc in PE, 100% EtOAc) followed by EtOAc-MeOH elution. Subsequently the collected fractions were compiled, under the continuous guidance of TLC monitoring system to afford 12 final fractions (Kh-A to Kh-L).
Fraction Kh-C (54.7 g) was subjected to a series of silica gel column chromatography by gradient elution of PE-EtAcO v/v% to afford Kh-C-I to Kh-C-VIII. Fraction Kh-C-I yielded needle like crystals of 12 (35 mg) eluted with 10% EtAcO/PE on silica gel column. Compound 18 (9.0 mg) was isolated as white scaly crystals by silica gel CC using 4% EtAcO/PE mobile phase from Kh-C-III. Fraction Kh-C-V after successive separation afforded white solid of 13 (18.2 mg) eluted with 10% acetone, and 16 (6.0 mg) eluted with 15% acetone in PE, respectively on silica gel column. While Kh-C-VII yielded 25 (150 mg), and 24 (13 mg) by 6% and 10% EtAcO/PE, respectively, thorough silica gel CC.
Fraction Kh-D (42.5 g) after successive chromatography on silica column afforded 10 fractions. Fraction Kh-D-VI was separated on silica gel column and eluted by acetone/PE to give 14 (7.5 mg), and yielded 15 (18.0 mg) as transparent crystals by 6% acetone/CHCl3, furthermore, compound 8 (5.0 mg) was also separated on sephadex LH-20 CC by 1:1 MeOH/CHCl3 from the same sub-fraction. White crystals of 7 (4.0 g) was isolated as major compound by 15% EtAcO/PE on silica gel column.
Fraction Kh-E (25.5 g) was eluted by PE and EtAcO by gradient system. After series of separation 27 (7.6 mg) was isolated on sephadex column by 1:1 MeOH/CHCl3 solvent system from fraction Kh-E-V. While fraction Kh-E-VI yielded compound 19 (22.0 mg) by 50% EtAcO/DCM silica gel CC, and compound 9 (25.0 mg) was purified as feathery substance on sephadex LH-20 by 1:1 MeOH/CHCl3 solvent system from resulting fraction Kh-E-VI-e.
Fraction Kh-F (48.5 g) was subjected to series of silica gel CC using DCM/EtOAc followed by EtOAc/MeOH solvent system of increasing polarity. Compounds 11 (5.0 mg), and 17 (28.00 mg) were isolated on silica gel columns by 30% DCM/PE, and 20% acetone/PE, respectively, from sub-fraction. The resulting fraction Kh-F-X-e was separated on sephadex LH-20 CC using 50% MeOH in CHCl3 to yield sub-fraction Kh-F-X-e-2 (88 mg), which was then further purified by semi-preparative RP-HPLC. The separation conditions were optimized on analytical HPLC equipped with DAD detector. Compound 2 (20.0 mg, retention time = tR 14.03 min), 10 (12.0 mg, retention time = tR 17.52 min), and compound 5 (6.2 mg, retention time = tR 20.53 min) were purified by 75% MeOH in H2O at flow of 2.5 mL/min using 210 nm of UV detection by using Eclipse XDB-C18 (5 μm 9.4 × 250 mm) column. While compound 28 (13.0 mg) was isolated from Kh-F-X-h eluted with 30% EtAcO/DCM on silica gel column.
Fraction Kh-G (42.8 g) was fractionated gradiently, and compound 26 (8.0 mg) was purified as precipitates during fraction collection of 15% EtAcO/DCM.
Fraction Kh-H (55.4 g) was subjected to silica gel CC by gradient elution with PE/EtOAc, and EtOAc/MeOH to yield sub fraction. Further silica gel CC was carried out for sub-fraction Kh-H-IX (6.37 g) using DCM/EtOAc and EtOAc/MeOH solvent system of increased polarity to yield fractions Kh-H-IX-a to Kh-H-IX-f. Compound 1 (10.0 mg), and compound 6 (9.5 mg) were separated on sephadex LH-20 CC eluted with 50% MeOH in CHCl3 from sub-fraction Kh-H-IX-c (155.78 mg), and Kh-H-IX-d-3 (85.70 mg), respectively. While fraction Kh-IX-e (2.4 g) was subjected to successive separations and ultimately compound 21 (12.5 mg, retention time = tR 11.66 min), 22 (15.3 mg, retention time = tR 20.10 min), 23 (10.0 mg, retention time = tR 23.57 min), and compound 20 (6.5 mg, retention time = tR 33.12 min) were purified by preparative HPLC by 55% MeOH//H2O at flow rate of 2.5 mL/min using Zorbax SB-C18 (5 μm 9.4 × 150 mm) column at 210 nm UV detection.
Finally, Fraction I (35.0 g) was chromatographed on silica gel CC eluted with a DCM/MeOH gradient system (99.5:0.5–0:100) to obtain 10 fractions. Kh-I-X (3.0 g) was subjected to silica gel CC eluted with PE/EtOAc (80:20 to 0:100) to give 12 fractions. Fraction Kh-I-X-k was purified on semi-preparative RP-HPLC, with a solvent of MeOH/H2O (3 mL/min, 75:25) at 225 nm, to afford compounds 3 (6.7 mg) and compounds 4 (6.7 mg).
Spectroscopic Data
Xuetonglactone A (1)
Colorless prismatic crystals; + 186.9 (c 2.44, MeOH); ECD 255 nm (Δε = + 5.64); IR νmax 2,967, 1,720, 1,684, 1,599, 1,569, 1,375, 1,291, 1,246, 1,127, 1,101, 1,053, 1,028, 987, 851, 821 cm−1; 1H-NMR (500 MHz, CDCl3) and 13C-NMR (125 MHz, CDCl3) data, see Tables 1, 2, respectively; (+)-HRESIMS m/z 559.2671 [M + Na]+ (calcd for C32H40O7 + Na, 559.2672).
Xuetonglactone B (2)
White amorphous; 242.7 (c 3.94, MeOH); ECD 255 nm (Δε = + 5.80); IR νmax 3,449, 2,952, 1,720, 1,686, 1,669, 1,597, 1,567, 1,373, 1,289, 1,248, 1,127, 1,049, 1,026, 987, 853, 821 cm−1; 1H-NMR (500 MHz, CDCl3) and 13C-NMR (125 MHz, CDCl3) data, see Tables 1, 2, respectively; (+)-HRESIMS m/z 561.2828 [M + Na]+ (calcd for C32H42NaO7 + Na, 561.2829), and (–)-HRESIMS m/z 573.2612 (calcd for C32H42O7 + Cl, 537.2619).
Xuetonglactone C (3)
White amorphous solid; 108.1 (c 2.44, MeOH); ECD 273 nm (Δε = + 0.41); IR νmax 3,446, 2,929, 1,724, 1,687, 1,656, 1,375, 1,291, 1,249, 1,131, 1,024, 990, 825 cm−1; 1H-NMR (600 MHz, CDCl3) and 13C-NMR (150 MHz, CDCl3) data, see Tables 1, 2, respectively; (+)-HRESIMS m/z 561.2836 [M + Na]+ (calcd for C32H42O7 + Na, 561.2828).
Xuetonglactone D (4)
White amorphous; 41.8 (c 1.67, MeOH); ECD 258 nm (Δε = + 5.53); IR νmax 3,449, 2,988, 2,923, 2,850, 1,724, 1,465, 1,381,1,243, 1,125, 1,032, 988 cm−1; 1H-NMR (600 MHz, CDCl3) and 13C-NMR (150 MHz, CDCl3) data, see Tables 1, 2, respectively; (+)-HRESIMS m/z 577.2772 [M + Na]+ (calcd for C32H42O8 + Na, 577.2777).
Xuetonglactone E (5)
White prismatic crystals; mp 235.1–236.6°C (MeOH); −8.8 (c 0.1, MeOH); ECD 272 nm (Δε = + 3.31); IR νmax 2,946, 1,721, 1,679, 1,558, 1,457, 1,381, 1,243, 1,106, 1,035, 914 cm−1; 1H-NMR (600 MHz, CDCl3) and 13C-NMR (150 MHz, CDCl3) data, see Tables 1, 2, respectively; (+)-HRESIMS m/z 547.3047 [M + Na]+ (calcd for C32H44O6 + Na, 547.3036).
Xuetonglactone F (6)
White amorphous; + 66.7 (c 1.56, MeOH); ECD 266 nm (Δε = + 2.41); IR νmax 2,916, 2,849, 1,736, 1,718, 1,684, 1,459, 1,377, 1,289, 1,239, 1,120, 993, 915, 825 cm−1; 1H-NMR (500 MHz, CDCl3) and 13C-NMR (125 MHz, CDCl3) data, see Tables 1, 2, respectively; (–)-HRESIMS m/z 617.2884 [M + Cl]− (calcd for C34H46O8 + Cl, 617.2881).
X-Ray Crystallographic Analysis
X-ray crystallographic data of 1, 5, and 7 were obtained using a Bruker APEX-II CCD diffractometer with Cu K radiation, = 1.54178 Å The CCDC numbers for 1, 5, and 7 contain the supplementary crystallographic data, which can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Crystal Data for Xuetonglactone A (1)
C32H40O7, M = 536.64, colorless crystals, Orthorhombic, a = 7.1986 (3) Å, b = 13.9377 (6) Å, c = 28.5609 (13) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 2865.6 (2) Å3, s P212121, T = 296 K, Z = 4, μ(Cu Kα) = 0.70 mm−1, 22,617 reflections measured, 5,112 independent reflections (Rint = 0.073). Final R indices [I >2σ(I)]: R1 = 0.052, wR2 = 0.164. Flack parameter: −0.10 (13). CCDC number: 1859825.
Crystal Data for Xuetonglactone E (5)
C32H44O6·H2O, M = 542.69, colorless crystal, Orthorhombic, a = 10.9410 (7) Å, b = 14.5893 (9) Å, c = 18.2354 (11) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 2910.8 (3) Å3, space group P212121, T = 296 K, Z = 4, μ(Cu Kα) = 0.69 mm−1, 31,389 reflections measured, 5,410 independent reflections (Rint = 0.040). Final R indices [I > 2σ(I)]: R1 = 0.036, wR2 = 0.109. Flack parameter: 0.06 (4). CCDC number: 1859823.
Crystal Data for Xuetongsu (7)
4(C30H44O4)·O, M = 1,890.60, colorless crystal, Monoclinic, a = 46.638 (2) Å, b = 7.4805 (4) Å, c = 7.8525 (4) Å, α = 90.00°, β = 91.597(2)°, γ = 90.00°, V = 2738.5 (2) Å3, space group C2, T = 296.15 K, Z = 1, μ(Cu Kα) = 0.59 mm−1, 10,267 reflections measured, 3,797 independent reflections (Rint = 0.098). Final R indices [I > 2s(I)]: R1 = 0.040, wR2 = 0.140. Flack parameter: 0.09 (13). CCDC number: 1859822.
Biological Activity Evaluation
Inhibition of iNOS Activity
The assay was performed in mouse macrophages (RAW264.7) cultured in phenol red-free RPMI medium with 10% bovine calf serum, 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin. The cells were seeded in 96-well plates at the density of 1 × 105 cells/well, and incubated for 24 h for a confluency of 75% or more. The cells were treated with the test compounds, and after 30 min of incubation, lipopolysaccharide (LPS, Sigma-Aldrich, St. Louis, MO, USA) (5 μg/mL) was added and further incubated for 24 h. The activity of iNOS was determined in terms of the concentration of NO by measuring the level of nitrite in the cell culture supernatant using Griess reagent (Sigma-Aldrich, St. Louis, MO, USA). Percent inhibition of nitrite production by the test compound was calculated in comparison to the vehicle control. IC50 values were obtained from dose response curves. Parthenolide was used as the positive control (Zhao et al., 2014).
Cytotoxicity Assay
Cell viability was determined by a MTT assay (Roche Diagnosis, Indianapolis, IN). Briefly, BGC-823 and HeLa cell lines were seeded at 6 × 103 cells/well in 96-well plates. Cells were allowed to adhere for overnight, and then the cells were changed to fresh medium containing various concentrations natural compound dissolved in DMSO. After 48 h incubation, the growth of cells was measured. The effect on cell viability was assessed as the percent cell viability compared with untreated control group, which were arbitrarily assigned 100% viability. The compound concentration required to cause 50% cell growth inhibition (IC50) was determined by interpolation from dose–response curves. All experiments were performed in triplicate, and paclitaxel was used as the positive control (Hayon et al., 2003).
Conclusions
To sum up, four new highly oxygenated lanostane-type triterpenoids xuetonglactones A–D (1–4) and two highly oxygenated cycloartane-type triterpenoids xuetonglactones E–F (5–6), along with 22 known compounds (7–28) were isolated from stems of K. heteroclita. To the best of our knowledge xuetonglactones A (1) endowed with unprecedented 20,21-α-epoxide functionality, and xuetonglactones D (4) possessed rare 19-α hydroperoxyl moiety, their absolute configurations were determined by X-ray diffraction and ECD data analysis. Moreover, bioassays indicated that 1 and 2 showed inhibition of iNOS activity in LPS-induced macrophages, 6, 7, 8, and 24 showed potent cytotoxicities against HeLa and BGC 823 cancer cell lines. Notably, this study has further enriched the chemical diversity of highly oxygenated triterpenoidal skeletons, which might trigger research rigor among synthetic and medicinal chemistry community.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
WW and DL conceived and designed the idea of the study. NS performed the isolation work. BL performed physical data analysis. NS and BL prepared the first draft of the manuscript. LC helped in collection of literature and assisting in crystallization. JZ performed the NMR data acquisition. YJ and AW contributed in analysis of NMR data. MD performed the bioassays of the compounds. MC and AR contributed in revision and final data analyses. IK provided the core facility to acquire NMR and other spectroscopic data. All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (Nos. 81374062, 81703819, and 81673579). Pharmaceutical Open Fund of Domestic Frist-class Disciplines (cultivation) of Hunan Province will pay for open access publication fees of this article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00935/full#supplementary-material
References
Cao, L., Shehla, N., Tasneem, S., Cao, M., Sheng, W., Jian, Y., et al. (2019). New cadinane sesquiterpenes from the stems of Kadsura heteroclita. Molecules 24:1664. doi: 10.3390/molecules24091664
Chaturvedula, V. S. P., and Prakash, I. (2012). Isolation of stigmasterol and β-sitosterol from the dichloromethane extract of Rubus suavissimus. Int. Curr. Pharm. J. 1, 239–242. doi: 10.3329/icpj.v1i9.11613
Chen, Y. G., Qin, G. W., and Xie, Y. Y. (2001). Triterpenoids from medicinal plants of Schisandraceae and their spectroscopic characteristics. Chem. Res. Appl. 13, 363–367.
Deng, Y. R., Wei, Y. P., Yin, F., Yang, H., and Wang, Y. (2010). A new cardenolide and two new pregnane glycosides from the root barks of Periploca sepium. Hel. Chim. Acta. 93, 1602–1609. doi: 10.1002/hlca.200900320
El Dine, R. S., El Halawany, A. M., Ma, C. M., and Hattori, M. (2008). Anti-HIV-1 protease activity of lanostane triterpenes from the Vietnamese mushroom Ganoderma colossum. J. Nat. Prod. 71, 1022–1026. doi: 10.1021/np8001139
Han, M. H., Yang, X. W., and Jin, Y. P. (2008). Novel triterpenoid acyl esters and alkaloids from Anoectochilus roxburghii. Phytochem. Anal. 19, 438–443. doi: 10.1002/pca.1070
Hayon, T., Dvilansky, A., Shpilberg, O., and Nathan, I. (2003). Appraisal of the MTT-based assay as a useful tool for predicting drug chemosensitivity in leukemia. Leuk Lymphoma 44, 1957–1962. doi: 10.1080/1042819031000116607
Hu, Z. X., Shi, Y. M., Wang, W. G., Li, X. N., Du, X., Liu, M., et al. (2015). Kadcoccinones A–F, new biogenetically related lanostane-type triterpenoids with diverse skeletons from Kadsura coccinea. Org. Lett. 17, 4616–4619. doi: 10.1021/acs.orglett.5b02360
Lakornwong, W., Kanokmedhakul, K., Kanokmedhakul, S., Kongsaeree, P., Prabpai, S., Sibounnavong, P., et al. (2014). Triterpene lactones from cultures of Ganoderma sp. KM01. J. Nat. Prod. 77, 1545–1553. doi: 10.1021/np400846k
Li, R. T., Han, Q. B., Zhao, A. H., and Sun, H. D. (2003). Micranoic acids A and B: two new octanortriterpenoids from Schisandra micrantha. Chem. Pharm. Bull. 51, 1174–1176. doi: 10.1248/cpb.51.1174
Liang, X. U., Wang, X. H., Luo, R. Y., Lu, S. Q., Gou, Z. J., Wang, M. A., et al. (2015). Secondary metabolites of rice sheath blight pathogen Rhizoctonia solani Kühn and their biological activities. J Integr. Agric. 14, 80–87. doi: 10.1016/S2095-3119(14)60905-9
Liu, J., Qi, Y., Lai, H., Zhang, J., Jia, X., Liu, H., et al. (2014). Genus Kadsura, a good source with considerable characteristic chemical constituents and potential bioactivities. Phytomedicine 21, 1092–1097. doi: 10.1016/j.phymed.2014.01.015
Liu, J. S., and Huang, M. F. (1991). Isolation and structures of schisanlactone E and changnanic acid. Acta. Chim. Sin. 49, 502–506.
Liu, J. S., Huang, M. F., Ayer, W. A., and Bigam, G. (1983). Schisanlactone B, a new triterpenoid from a Schisandra sp. Tetrahedron Lett. 24, 2355–2358. doi: 10.1016/S0040-4039(00)81923-1
Liu, Y., Yang, Y., Tasneem, S., Hussain, N., Daniyal, M., Yuan, H., et al. (2018). Lignans from Tujia ethnomedicine Heilaohu: chemical characterization and evaluation of their cytotoxicity and antioxidant activities. Molecules 23:2147. doi: 10.3390/molecules23092147
Liu, Y., Zhao, J., Chen, Y., Li, W., Li, B., Jian, Y., et al. (2016). Polyacetylenic oleanane-type triterpene saponins from the roots of Panax japonicus. J. Nat. Prod. 79, 3079–3085. doi: 10.1021/acs.jnatprod.6b00748
Ma, W., He, J., Li, L., and Qin, L. (2009). Two new triterpenoids from the stems of Schisandra bicolor. Helv. Chim. Acta. 92, 2086–2091. doi: 10.1002/hlca.200900245
Pavanasisivam, G., and Sultanbawa, M. U. S. (1973). Cycloartenyl acetate, cycloartenol and cycloartenone in the bark of Artocarpus species. Phytochemistry 12, 2725–2726. doi: 10.1016/0031-9422(73)85088-5
Pu, J. X., Yang, L. M., Xiao, W. L., Li, R. T., Lei, C., Gao, X. M., et al. (2008). Compounds from Kadsura heteroclita and related anti-HIV activity. Phytochemistry 69, 1266–1272. doi: 10.1016/j.phytochem.2007.11.019
Rahmana, S. M. M., Muktaa, Z. A., and Hossainb, M. A. (2009). Isolation and characterization of β-sitosterol-D-glycoside from petroleum extract of the leaves of Ocimum sanctum L. Asn. J. Food, Agro-Industry. 2, 39–43.
Shen, Y. C., Lin, Y. C., Chiang, M. Y., Yeh, S. F., Cheng, Y. B., and Liao, C. C. (2005). Kadsuphilactones A and B, two new triterpene dilactones from Kadsura philippinensis. Org. Lett. 7, 3307–3310. doi: 10.1021/ol051155k
Shi, Y. M., Xiao, W. L., Pu, J. X., and Sun, H. D. (2015). Triterpenoids from the Schisandraceae family: an update. Nat. Prod. Rep. 32, 367–410. doi: 10.1039/C4NP00117F
Song, Q. Y., Jiang, K., Zhao, Q. Q., Gao, K., Jin, X. J., and Yao, X. J. (2013). Eleven new highly oxygenated triterpenoids from the leaves and stems of Schisandra chinensis. Org. Biomol. Chem. 11, 1251–1258. doi: 10.1039/c2ob27115j
Su, W., Zhao, J., Yang, M., Yan, H. W., Pang, T., Chen, S. H., et al. (2015). A coumarin lignanoid from the stems of Kadsura heteroclita. Bioorg. Med. Chem. Lett. 25, 1506–1508. doi: 10.1016/j.bmcl.2015.02.022
Wang, W., Liu, J., Han, J., Xu, Z., Liu, R., Liu, P., et al. (2006b). New triterpenoids from Kadsura heteroclita and their cytotoxic activity. Planta Med. 72, 450–457. doi: 10.1055/s-2005-916263
Wang, W., Liu, J., Yang, M., Sun, J., Wang, X., Liu, R., et al. (2006c). Simultaneous determination of six major constituents in the stems of Kadsura heteroclita by LC-DAD. Chromatographia 64, 297–302. doi: 10.1365/s10337-006-0031-7
Wang, W., Liu, J. Z., Ma, X. C., Yang, M., Wang, W. X., Xu, Z. R., et al. (2006a). Three new cyclolanostane triterpenoids from the ethanol extract of the stems of Kadsura heteroclita. Helv. Chim. Acta. 89, 1888–1893. doi: 10.1002/hlca.200690180
Wang, W., Xu, Z., Yang, M., Liu, R., Wang, W., Liu, P., et al. (2007). Structural determination of seven new triterpenoids from Kadsura heteroclita by NMR techniques. Magn. Reson. Chem. 45, 522–526. doi: 10.1002/mrc.2000
Xiao, W. L., Li, R. T., Huang, S. X., Pu, J. X., and Sun, H. D. (2008). Triterpenoids from the Schisandraceae family. Nat. Prod. Rep. 25, 871–891. doi: 10.1039/b719905h
Yang, J. H., Wen, J., Du, X., Li, X. N., Wang, Y. Y., Li, Y., et al. (2010). Triterpenoids from the stems of Kadsura ananosma. Tetrahedron. 66, 8880–8887. doi: 10.1016/j.tet.2010.09.059
Yu, H., Zeng, R., Lin, Y., Li, X., Tasneem, S., Yang, Z., et al. (2019). Kadsura heteroclita stem suppresses the onset and progression of adjuvant-induced arthritis in rats. Phytomedicine 58:152876. doi: 10.1016/j.phymed.2019.152876
Yu, H. H., Lin, Y., Zeng, R., Li, X., Zhang, T., Tasneem, S., et al. (2019). Analgesic and anti-inflammatory effects and molecular mechanisms of Kadsura heteroclita stems, an anti-arthritic Chinese Tujia ethnomedicinal herb. J. Ethnopharmacol. 238:111902. doi: 10.1016/j.jep.2019.111902
Zhao, J., Khan, S. I., Wang, M., Vasquez, Y., Yang, M. H., Avula, B., et al. (2014). Octulosonic acid derivatives from Roman chamomile (Chamaemelum nobile) with activities against inflammation and metabolic disorder. J. Nat. Prod. 77, 509–515. doi: 10.1021/np400780n
Keywords: xuetonglactones, highly oxidized, lanostane triterpenoids, Kadsura heteroclita, cytotoxicity
Citation: Shehla N, Li B, Cao L, Zhao J, Jian Y, Daniyal M, Wahab A, Khan IA, Liao D, Rahman A, Choudhary MI and Wang W (2020) Xuetonglactones A–F: Highly Oxidized Lanostane and Cycloartane Triterpenoids From Kadsura heteroclita Roxb. Craib. Front. Chem. 7:935. doi: 10.3389/fchem.2019.00935
Received: 25 November 2019; Accepted: 23 December 2019;
Published: 21 January 2020.
Edited by:
Toshio Morikawa, Kindai University, JapanReviewed by:
Sebastiano Di Pietro, University of Pisa, ItalyZhengxi Hu, Huazhong University of Science and Technology, China
Copyright © 2020 Shehla, Li, Cao, Zhao, Jian, Daniyal, Wahab, Khan, Liao, Rahman, Choudhary and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Iqbal Choudhary, aXFiYWwuY2hvdWRoYXJ5QGljY3MuZWR1; Wei Wang, d2FuZ3dlaTQwMkBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work